1. Introduction
Dengue is an arthropod-borne disease that is transmitted to humans through the bites of female
Aedes aegypti or
Aedes albopictus mosquitos which have been infected with a dengue virus (DENV). The DENV can be classified into four genetically related but antigenically distinct serotypes, designated as DENV 1–4. Infection by all dengue serotypes causes a wide spectrum of diseases, ranged from asymptomatic/mild febrile illness to severe clinical manifestations that can lead to life-threatening dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS) [
1]. Dengue has been declared to be endemic in over 100 countries (primarily in the tropical and subtropical regions) and continue to be a global threat to public health. It has been estimated that 390 million dengue infections occur annually across the world and contribute to approximately 25,000 casualties [
2]. Dengue cases remain prevalent and high in the tropical and subtropical regions, predominantly in Southeast Asia [
3]. Currently, there is no antiviral drug for dengue treatment. The current preventive measures against dengue are directed towards vector control, which offer a temporary solution.
Vaccination remains the most effective way for long-term prevention and control of dengue infections. The first licensed dengue vaccine, Dengvaxia
® (CYD-TDV) was first introduced into the clinical setting in December 2015. The vaccine efficacy and safety profile, however, varied according to the vaccination recipient’s serostatus and age. The use of CYD-TDV, a live attenuated virus (LAVs)-based vaccine had an increased risk of clinically severe dengue in seronegative (those who were without prior dengue infection) vaccinated individuals. Notably, a higher risk of hospitalization was observed among children ages between 2–5 years [
4]. This unprecedented situation that hampers the use of CYD-TDV has been associated with a phenomenon called antibody-dependent enhancement (ADE) [
5]. ADE occurs when the raised antibodies were insufficient to completely neutralize the virus, and these sub-neutralizing antibodies instead facilitate the virus entry into host cells, which subsequently enhanced viral loads and disease severity. The use of LAV as vaccine antigen was not favorable as a natural immune response against DENV viruses was found to be predominantly directed against membrane protein (prM/M), which cross-reacted among DENV serotypes at suboptimal neutralizing effect, thus suggesting its involvement in ADE induction [
6].
As such, a newer generation of tetravalent dengue vaccine focusing on inducing a specific type of immune response is an emerging strategy to minimize the risk of ADE development. Peptide-based vaccines appear as suitable candidates for dengue vaccine development as they can elicit epitope-specific antibodies and immune responses, thus eliminating the risk of vaccine-associated adverse effects, such as autoimmune and/or allergenic reactions. In addition to antibody responses, cell-mediated immunity has a profound role in conferring protection against DENV. T cell epitopes are responsible for the generation of long-lasting cellular immunity capable of eradicating circulating viruses and virus-infected cells [
7]. It has been evidenced that DENV infection in a group of mice with cytotoxic T cells (CTLs) depletion reduced the mice’s ability to clear DENV and resulted in considerably greater viral loads in the serum, spleen and brain compared to undepleted mice [
8]. On the other hand, the activation of T helper (Th) cells is essential for effective vaccination as it plays a central role in maintaining and priming both effector CTLs and B cells function in promoting memory immune responses [
9]. In pursuit of these immunological rationales, we have designed a multi-epitope peptide dengue vaccine that can induce the activation of CTLs, Th and B cells. The respective antigenic B cell (E
383−397aa) and T cell (E
345−359aa) peptide epitopes were derived from the domain III region of the DENV-2 E protein (EDIII) (NCG strain) [
10]. The known Th (E
352−368aa) peptide epitope was derived from DENV-2 EDIII of Jamaica strain and has been previously reported to enhance the neutralizing antibody response that cross-reacted against DENV serotypes [
11]. EDIII is known as the prime target of highly potent DENV neutralizing antibodies as it possesses a receptor-binding region [
12]. Thus, cellular and humoral immunity directed towards the EDIII region can confer protection against DENV with minimal risk of ADE development. The multi-epitope polypeptide was further conjugated to two copies of palmitic acid (C16) by using lysine residue as the branching core at the N-terminal, named LipoDV (
Figure 1a). Peptide lipidation is a prominent strategy to improve peptide’s immunogenicity due to the recognition of lipid tail by toll-like receptor 2 (TLR2) [
13,
14].
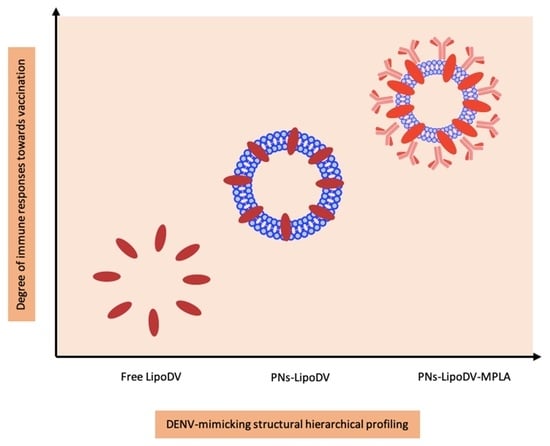
Peptide vaccines represent an alternative strategy for a safer dengue vaccine development due to their minimalistic antigenic composition. However, the removal of ‘danger signal’ components that are typically possessed by a pathogen reduce the prospective recognition by immune cells which relatively lead to poor efficacy of peptide vaccine. Thus, the use of a vaccine delivery system incorporated with potent immunostimulants is necessary to trigger the desired immune response against the peptide antigen. Herein, we reported the development of a novel multifunctional nanoparticle platform that emulated the external structural and functional traits of a DENV for a safe and efficient presentation of the peptide antigen to the immune system (
Figure 1b). DENV virion structure is characterized as spherical nanoparticles. The core of DENV consists of viral genome-nucleocapsid complexes that are surrounded by a well-organized lipid bilayer membrane, the viral envelope. The surface of the viral envelop is embedded by the prM/M and E proteins that are critical for the maturation and attachment of DENV particles to host cells, respectively. In this DENV-mimicking platform, the virus envelope was mimicked by the self-assembled polymersome made up of amphiphilic di-block copolymer poly(butadiene)-poly(ethylene oxide) (PBD-PEO). Due to the increased bilayer thickness, polymersomes are generally more chemically stable and less permeable than conventional liposomes [
15]. The prM/M functionality was emulated by the incorporation of monophosphoryl-lipid A (MPLA) while the E protein was resembled by the constructed lipidated antigen, LipoDV. MPLA is a well-known TLR4 agonist which significantly induces the maturation of antigen presenting cells for efficient processing of vaccine antigen [
16]. The two lipid tails anchored by LipoDV recapitulated the functionalities of protein E in inducing host membrane cells permeation and cell receptor binding. The key physicochemical properties such as size, morphology and surface charge of the fabricated nanoparticles were characterized. The capacity of the nanoparticle-based vaccine carrier to be taken up by APCs was evaluated in vitro. Finally, to address our question of whether the formulated DENV-mimicry nanocarrier platform would serve as a potent vaccine delivery system, several dengue vaccine candidates; LipoDV alone and its encapsulation within lipophilic bilayer of polymersome with (PNs-LipoDV-MPLA)/without (PNs-LipoDV) the presence of MPLA were assessed for their safety and immunogenicity effects in mice, particularly in inducing humoral and cell-mediated immune responses. The structure-activity relationship analysis revealed that dengue vaccine candidate, PNs-LipoDV-MPLA which was closely resembled DENV structural morphology induced strong antibody responses that were regulated by a balanced Th1/Th2 polarization and exhibited a satisfactory safety profile.
2. Materials and Methods
2.1. Materials
All chemicals and reagents were at the highest analytical grade and were used directly without any further purification unless otherwise stated. PBD32-PEO21 di-block copolymers were obtained from Polymer Source (Montreal, QC, Canada). Dimethyl sulfoxide (DMSO), thiazolyl blue tetrazolium bromide (MTT), phosphate buffer saline (PBS), TritonTM X-100, MES buffer, lysis buffer and anti-Mouse IgG conjugated to horseradish peroxidase were purchased from Sigma-Aldrich (Saint Louis, MO, USA). Eagle’s Minimum Essential Medium (EMEM) and IMDM Glutamax medium Fetal Bovine Serum (FBS) were purchased from Addexbio (San Diego, CA, USA). Fetal Bovine Serum (FBS) and penicillin/streptomycin were purchased from Gibco (Jenks, OK, USA). 2-mercaptoethanol (50 µM) and SnakeSkinTM dialysis tubing (7K MWCO) were supplied by Thermo Fisher Scientific (Waltham, MA, USA). CD11c was purchased from eBioscience (San Diego, CA, USA), while F4/80 was obtained from BioLegend, Pacific, (San Diego, CA, USA).
2.2. Synthetic Peptide
LipoDV(C16K(C16)LITVNPIVTEKSPVNIERHVLGRLITVNPIVTEPGQLKLNWFKKGSS-NH2) and B cell peptide epitope (E383−397aa: EPGQLKLNWFKKGSS) were synthesized and purified by Bankpeptide Co., Ltd. (Hefei, China). The chemical identity of the peptides was confirmed by the observed molecular weight (LipoDV: MW 5956.25 g/mol and E383−397aa: MW 1718.90 g/mol) as determined by ESI-MS. Both peptides were obtained at a purity greater than 95% as measured by analytical RP-HPLC based on the area under the curve analysis.
2.3. Polymersome-Based Nanoparticle Vaccine Formulation
The polymersome nanoparticles (PNs) vaccine were prepared via film rehydration method with slight modifications [
17]. Briefly, 3.0 mg of PBD
32-PEO
21 diblock copolymer was dissolved in 0.6 mL of chloroform (CHCl3) in a round bottom flask. LipoDV (1 mg in 1 mL acetonitrile) was added to the above polymer solution to prepare LipoDV-loaded PNs (PNs-LipoDV). The solvent mixture was evaporated under reduced pressure to remove the organic phase at 37 °C until a thin polymer film formed on the wall of the flask and dried overnight in a desiccator. Then, the polymer film was rehydrated with 600 µL prewarmed sterile Milli-Q water and stirred at 25 °C. The resulted cloudy dispersion was extruded using a mini extruder (Avanti
® Mini-Extruder, Avanti Polar Lipid, Alabaster, AL, USA) through a 400 nm polycarbonate membrane for 20 times in both directions followed by another series of 20 times extrusion using a 100 nm polycarbonate membrane. The formulation of PNs loaded with both LipoDV and MPLA (PNs-LipoDV-MPLA) was prepared in a similar protocol to PNs-LipoDV, with the only difference was being that 3 µg of MPLA was added dropwise to the PBD
32-PEO
21 suspension before LipoDV was added. The concentration of MPLA was finalized when the particle surface charge reached the maximum negative value of zeta potential (fully saturated).
2.4. Physiochemical Characterization
The formulated nanoparticles vaccines were characterized for hydrodynamic particle size, polydispersity index (PDI) and zeta potential via dynamic light scattering technique at a backscattering angle of 173° at 25 °C in folded capillary cuvettes by a using Malvern Zetasizer Nano ZS (Malvern, UK). The obtained data were processed using Malvern’s Zetasizer software 6.2 and expressed as an average of at least three measurements for each batch. Zeta potential and particle size values were also used as the indication of complete coverage of the nanoparticle’s functionalization with MPLA.
2.5. Morphological Analysis
The morphology of the nanoparticles vaccines was characterized by using a Talos L120C Transmission Electron Microscope (TEM, Thermo Fisher, Waltham, MA, USA) at an accelerating voltage of 100 kV. In brief, a drop of diluted nanoparticles solution was placed in a 200-mesh carbon-coated grid for 5 min. Upon particles settlement in the grid, negative staining was performed using 2.5% uranyl acetate before capturing the microscopic image.
2.6. Encapsulation Efficiencies
The encapsulation efficiency (EE) of vaccine antigen, LipoDV inside the nanoparticles was measured by using the dialysis method. Briefly, nanoparticles solution was added to a dialysis tubing (SnakeSkin
TM) of a molecular weight cut-off = 7 kDa. The sample (containing free LipoDV) was collected after 2 h of dialysis. The standard calibration curve of LipoDV (20–120 µg/mL) was plotted and the quantification of the loaded LipoDV was carried out by UV-vis spectroscopy at 280 nm. The percentage of EE was calculated using the following formula:
2.7. Ex Vivo Hemolysis Assay
Hemolysis assay was carried out using fresh human red blood cells as described elsewhere with slight modification [
18]. In brief, a fresh human blood sample was obtained and placed in a heparinized tube to prevent coagulation. The blood sample was centrifuged at 750×
g for 15 min to extract the serum and washed three times with sterile PBS. The same volume of sterile PBS was added after the serum was removed to produce a 1% suspension of red blood cells. Then, 100 µL of tested compounds were added into the microcentrifuge tube at various concentrations of 1 mg/mL, 0.5 mg/mL and 0.124 mg/mL, along with 100 µL of red blood cells suspension. Triton X-200 with a final concentration of 0.5% (vol/vol) in sterile PBS was used as a positive control while sterile PBS was used as a negative control. The tubes were incubated for 2 h at 37 °C in an S1500 shaker (Stuart Scientific, Nottingham, UK). Then, the tubes were centrifuged at 750×
g for 15 min after incubation, and 50 µL of supernatant was extracted in triplicates and transferred onto a 96-well plate. The absorbance was measured at 540 nm by using a spectrophotometric microplate reader (NanoQuant Infinite M200 Pro, Tecan, Switzerland). The percentage of hemolysis activity was calculated using the following equation:
AbsCompound represent the value of nanoparticle formulations, AbsPositive represents value of positive control, and AbsNegative represents value of negative control.
2.8. MTT Cytotoxic Assay
The in vitro cytotoxicity activity of the constructed PNs vaccines was evaluated against human embryonic kidney cell (HEK293) based on established protocol [
15]. Briefly, HEK293 was cultured in EMEM supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were plated in a clear 96-well plate (5.0 × 10
4 cells/well) and maintained for 24 h at 37 °C with 5% CO
2 in a humidified chamber for cell attachment. PNs were prepared and serially diluted in DMEM to give concentrations ranging from 0.25 to 1 mg/mL. Then, 200 µL of each diluted solution was added to each wells containing attached cells in triplicate after the used media were removed and incubated with 5% CO
2 at 37 °C for 48 h. After the incubation, the media were removed and 20 μL of 5 mg/mL MTT solutions in PBS were added to each well and further incubated for 4 h with 5% CO
2 at 37 °C. The media was then replaced by 100 µL of dimethyl sulfoxide (DMSO) and assiduously mixed to solubilize the formed formazan crystal. DMSO alone was used as a blank and the untreated cells were used as a negative control. The absorbance was measured using NanoQuant infinite M200 Pro spectrophotometric microplate reader (Tecan, Männedorf, Switzerland) at 570 nm and the viability of the cells was calculated using the following formula:
2.9. Antigen-Presenting Cell Uptake Assay
This assay was performed based on a previously published protocol with some modifications [
19]. Under aseptic conditions, spleens were dissected from female Balb/c mice. The isolated spleens were harvested and homogenized by passing through a cell strainer to dissociate into a single cell suspension. The red blood cells were lysed using erylysis buffer (pH 7.2–7.4, sterilized). The resultant splenocytes were seeded into a 12-well plate (cell density: 2 × 10
5 cells/well) in phenol-free IMDM Glutamax medium, supplemented with 10% FBS, 50 µM 2-mercaptoethanol, 100 IU/mL penicillin, and 100 µg/mL streptomycin. Then, the tested compounds; PNs-based vaccine candidates were labelled with Dil (1,1 dioctadecyl-3,3,3′,3-tetramethylinocarbocynine perchlorate). The Dil-labelled vaccine compounds at a concentration of 50 μM concentration were then added to the wells and incubated for 24 h. Cells adhering to the plates were scraped and incubated with Fc-block for 20 min at 4 °C. After incubation, the plates were centrifuged and the supernatants were discarded and resuspended in FACS buffer (PBS, 0.02% sodium azide, 0.5% bovine serum albumin (BSA)) containing CD11c and F4/80 antibodies at 4 °C for 30 min. The cells were centrifuged again and then analyzed using the LSR II flow cytometer (BD FacsCanto II, Franklin Lakes, NJ, USA) in a 0.5 mL FACS buffer (PBS, 0.02% sodium azide, 0.5% BSA) (
Supplementary Figure S1). The fluorescence intensity of dendritic cells and macrophages that were treated with normal saline was used as a control. The actual uptake was calculated as the percentage of cells for Dil and CD11c or Dil and F4/80.
2.10. Mice Immunization Study
All of the mice handling protocols and guidelines were carried out according to University Kebangsaan Malaysia Animal Ethics Committee (UKMAEC, Ref No: FF/2020/FAZREN/25-MAR./1097-APR.-2020-APR.-2021). Female Balb/c mice (6-week-old) were purchased from UKM Laboratory Animal Research Unit (LARU). All mice were maintained under pathogen-free conditions with free access to food and water. Mice were divided into five groups with six mice in each group (n = 6). Each mouse was immunized via subcutaneous injection into the loose skin over the neck region on day 0, followed by two booster doses on day 10 and 20 with different vaccine candidates (free LipoDV, PNs-LipoDV and PNs-LipoDV-MPLA) at a dose of 30 µg of LipoDV immunogen at a volume of 50 µL. Each mouse in the positive control group was immunized with 30 µg of LipoDV emulsified in Complete Freund’s Adjuvant (CFA) to a total volume of 50 µL and boosted with 30 µg of LipoDV in 50 µL of normal saline in similar immunization interval. Negative control mice were immunized with 50 μL of normal saline.
2.11. Serum Collection and Dissection of Selected Organs
Blood samples were collected via tail bleed on days 9 and 19, while blood samples on day 27 were collected by cardiac puncture. Blood was centrifuged at 10,000× g for 10 min to separate the serum. The collected serum samples were stored at −80 °C until further analysis. On day 27, mice were euthanized, and the specimens including spleen tissues, liver and kidney were collected under aseptic conditions and fixed in 4% v/v buffered formalin for preservation until further and were kept at −80 °C for subsequent analysis.
2.12. Evaluation of Antigen-Specific Systemic IgG Antibodies
The collected serum samples were used for the analysis of B cell epitope (E
383-397aa)-specific serum IgG antibodies using an enzyme linked immunosorbent assay (ELISA). The assay was performed using DuoSet ELISA Ancillary Reagent Kit (DY008; R&D Systems, Minneapolis, MN, USA) as previously reported [
20]. Briefly, the polycarbonate plates were coated with diluted E
383-397aa peptide antigen to 0.5 mg/mL in ELISA coating buffer. Then, the plates were washed using washing buffer before being blocked with 150 μL of blocking buffer (3% blocking buffer in PBS with 0.05% Tween-20). A 1:100 concentration of serum to reagent diluent (0.1% BSA in PBS, with 0.05% Tween-20) was serially diluted 1:2 down the plate and incubated for 2 h at 37 °C. Then secondary antibody (antimouse-IgG conjugated to horseradish peroxidase mixture with hydrogen peroxide and tetramethylbenzidine) was diluted to 1:3000 before being added to the plates. 100 μL of color reagent was added to each well and the plate was incubated for 20 min at room temperature in the dark according to the manufacturer’s instructions. After the incubation, 50 μL of stop solution was added and the plate was read using a spectrophotometer (NanoQuant infinite M200 Pro, Tecan, Männedorf, Switzerland) at 450 nm. The antibody titer was expressed as the lowest dilution that gave an absorbance of >3 standard deviations (SD) above the mean absorbance of negative control wells (contains negative control mouse serum). Statistical analysis (
p < 0.05 = statistical significance) was determined using one-way ANOVA followed by Turkey post hoc test using GraphPad Prism Software 8.0, (San Diego, CA, USA).
2.13. Spleen Cell Culture and Cytokines Quantification
The aseptically dissected spleen tissues on day 27th (7 days after final immunization) were immediately homogenized using a sonicator (Model MSX-Q125220, Qsonica, Newtown, CT, USA) at 25 HZ for 5 min for cytokine secretion analysis without restimulation with antigen. Cells were collected and washed using cold PBS. 50 μL of RIPA buffer was added per 1 mg of tissue to lyse the red blood cells. The supernatants were isolated by centrifugation at 16,000 rpm for 10 min at 4 °C. The supernatants were diluted to 50 mg/protein with RIPA buffer and 150 µL of each sample was inserted onto a round-bottom 96-well plate. The cytokines (IFN-γ, IL-2 and IL-4) expression was identified using commercially available MILLIPLEX® Multiplex Assays Using Luminex® kit (Merck Millipore, Watford, UK) according to the manufacturer’s protocol. Each sample was assayed in triplicate, and the cytokine standards and quality controls were supplied by the manufacturer and standardized on each plate as a referral point.
2.14. In Vivo General Toxicity Analysis
The immunized mice were monitored closely throughout the experiment period for any toxicity signs and symptoms. Bodyweight (recorded at every predetermined time interval) and mortality were observed throughout the immunization point.
2.15. Histology Analysis
The histological examinations of livers and kidneys of each experimental group were performed based on an established protocol [
21]. In brief, the isolated organs were fixed in 10% buffered formalin. Then, the tissue blocks were immersed in paraffin wax and routine sections were stained with hematoxylin and eosin. The hematoxylin-eosin (HE) stained sections were examined by light microscopy on an Olympus BX40-B microscope at a magnification of 40×.
2.16. Stability of Polymersome Nanoparticle
The stability of the blank PNs (either with/without the presence of MPLA) was analyzed for seven months under different conditions: 25 °C ± 2 °C (at room temperature), 4 °C (in the refrigerator) and 40 °C (at elevated temperature). The samples were taken in specified intervals for analysis of charge and size.
4. Discussion
Dengue is considered a serious and continuing threat to the global community, particularly in tropical and sub-tropical climates regions. There are no specific antiviral agents is currently available to treat dengue. The only licensed dengue vaccine, Dengvaxia is not clinically satisfactory due to lack of safety profile. Epidemiology evidence associates the use of Dengvaxia with ADE development in dengue-seronegative recipients [
22]. Thus, the development of an effective dengue vaccine against all four different DENV serotypes and minimizing the risk of ADE development at the same time remain a challenge. Collective clinical and experimental data demonstrated that both neutralizing antibodies and T-cell oriented immune responses are necessary for vaccine-mediated protection against dengue [
5].
In pursuit of addressing the problems, we have designed and constructed a multi-epitope peptide vaccine by fusing the B cell, Th and T cell epitopes in a single polypeptide chain bearing two copies of palmitic acid (LipoDV). The utilization of these minimal antigenic epitopes in vaccine design would enable the productions of highly targeted humoral and cellular immune responses, thus eliminating the risk of adverse reactions, including disease worsening condition via ADE mechanism. However, to be optimally effective, such peptide vaccines need to be administered with a potent vaccine delivery system. One of the attractive approaches in designing an efficient vaccine delivery system is by mirroring the morphology and pathogenic features of the targeted pathogen. Herein, we have formulated our novel nanoscale dengue vaccine delivery platform technology to mimic the structural features of DENV (
Figure 1). LipoDV resembled the functionality of DENV E protein as the principal target for neutralizing antibodies induction and focal determinant for facilitating virus entry into the host cell membrane. LipoDV was immobilized in the hydrophobic region of the PBD-PEO polymersome nanoparticles that represent the DENV envelope. The PBD-PEO polymersome was functionalized with MPLA, an established immune-activating ligand that mimics the role of DENV M protein in maturing the virion particle during cellular infection. The DENV-mimicking nanoparticle vaccine formulation, PNs-LipoDV-MPLA was further tested for immunogenicity effect in mice, alongside PNs-LipoDV and free LipoDV for systematic structural variation comparison.
During the dehydration-rehydration process of PNs fabrication, LipoDV was solubilized alongside the PBD-PEO polymer to achieve its intercalation within the hydrophobic bilayer of the polymersome structure. It has been demonstrated that the localization of vaccine antigen on the surface of liposome bilayer induced better recognition by B cells compared to its entrapment inside the aqueous core [
23]. The high EE of LipoDV may be contributed by the non-covalent hydrophobic interaction of the anchoring palmitic acids with the hydrophobic bilayers of polymersomes. Both PNs-LipoDV-MPLA and PNs-LipoDV were successfully fabricated with a uniform-size nanoscale dimension particle of approximately 130 nm as determined via DLS and TEM. The polymersome structure was visible and confirmed by TEM imaging (
Figure 2). The additional incorporation of MPLA enhanced the stability of the polymersome as indicated by the increase in a negative value of zeta potential. It seems that MPLA exerts a similar impact to cholesterol in improving the stability of conventional liposome bilayer [
24]. In the long-term stability study, both blank PNs (either with/without MPLA) retained acceptable structural integrity changes during the 7 months at all tested conditions, with PNs containing MPLA exhibiting a greater stability profile in terms of homogeneity distributions. Notably, at 40 °C, the particle size of NPs functionalized with MPLA was observed to be smaller while had an increased in PDI and negative value of zeta potential. This situation may be explained by the greater complexation between MPLA and the PBD
32-PEO
21 polymer which occurs over the time. The complexation may lead to a reduced particles size but affecting the uniformity of size distribution pattern.
The principal concept of incorporating vaccine antigens into nanoparticles is to promote their uptake by the APCs for efficient immune cells processing. The most targeted APCs in vaccine design are DCs and macrophages due to their central role in inducing the activation of the adaptive arm of the immune system. Our uptake analysis data showed that both nanovaccine formulations; PNs-LipoDV-MPLA and PNs-LipoDV were taken up efficiently by the studied APCs, although the uptake level was higher in DCs compared to macrophages (
Figure 3). This may be attributed to the particle size of the PNs vaccines which was in the range of 120–130 nm. It was well established that DCs preferentially uptake virus-sized particles (20–200 nm) while macrophages take up more of the larger particles which is comparable to bacterial size (0.5–5 μm) [
25]. In comparison to PNs-LipoDV, the uptake level of PNs-LipoDV-MPLA by DCs was statistically significantly reduced. The greater density of negative surface charge of PNs-LipoDV-MPLA may trigger a stronger repulsive force with the negatively charged cell membranes of DCs. A similar outcome on how the particle surface charge influences their uptake by APCs was also observed in other studies [
26,
27]. However, higher uptake by DCs does not warrant the initiation of immune responses [
26]. A variety of other immune cascades, such as maturation of APCs, down-regulation of antigen internalization and surface expression of costimulatory molecules that govern the potentiation of immune response productions.
Therefore, the immunogenicity and adjuvanting potency of the vaccine candidates were evaluated in vivo. BALB/c mice were used in this study as they are highly responsive in inducing humoral immune response without compromising the activation of T-cell mediated immunity. The potent immunogenicity of our lead vaccine candidate, PNs-LipoDV-MPLA that was designed to mimic the structural morphology of DENV virion was reflected by the significantly higher antigen-specific antibody titers than PNs-LipoDV and free LipoDV (
Figure 4b). However, the antibody productions of the mice group immunized with PNs-LipoDV-MPLA was comparatively lower than the positive group; LipoDV adjuvanted with CFA. CFA is regarded as the gold standard for adjuvants used in pre-clinical vaccine development; however, it is too toxic for clinical utility [
13,
28]. The toxicity of CFA was documented in the histopathology examinations of liver and kidney tissues obtained from mice immunized with CFA-adjuvanted antigen (
Figure 7). The presence of activated Kupffer cells in the liver tissue indicated the formation of granulomas, which was highly associated with the administration of CFA [
29]. Similarly, the mice group receiving CFA as vaccine adjuvant showed signs of nephrotic syndrome which was associated with CFA. In contrast to PNs-LipoDV-MPLA, there was no significant abnormal histopathological changes were observed in the examined organs as compared to the negative control.
Other than humoral immune response, the ability to induce cell-mediated immune response has been appraised as a critical attribute to DENV infection. Hence, we assessed whether our DENV-mimicking PNs-LipoDV-MPLA could demonstrate similar functional traits to DENV. The cytokine responses induced among tested vaccine candidates were evaluated. The polarity of the immune responses was monitored by quantifying the marker cytokines secreted by Th1 ((IFN-γ and IL-2) and Th2 (IL-4) cells. The results demonstrated that the secretion of IFN-γ was significantly elevated in splenocytes from mice vaccinated with PNs-LipoDV-MPLA in comparison to PNs-LipoDV and free LipoDV, while the secretion of IL-2 and IL-4 were comparable among the tested groups (
Figure 5). The obtained data indicated that our designated vaccine antigen, LipoDV possess self-adjuvating properties. The strategy in anchoring two copies of palmitic acid (C16) to the multi-epitope peptide antigen significantly promoted humoral immune response induction as indicated by high-level IL-4 cytokines secretion, as well as the antigen-specific antibodies productions. The self-adjuvanting property of lipopeptides as vaccine antigen was well observed in various studies [
13,
30,
31,
32]. It was documented that the induction of IFN-γ is essential for host reaction in conferring substantial protection against DENV virus infection [
33]. As the level of IFN-γ secretion was the highest in mice immunized with PNs-LipoDV-MPLA, our strategy in utilizing MPLA as immunostimulatory molecules enhanced the polarization of the Th1 cell. The adjuvanting property of MPLA was primarily biased towards the induction of cell-mediated immune response as reported in previous studies [
34,
35,
36]. Thus, our collective data demonstrated that immunization with PNs-LipoDV-MPLA induced a potent humoral and cell-mediated immune response with enhanced IFN-γ secretions which emulated DENV infections.
Importantly, all the tested vaccine candidates, especially the lead formulation (PNs-LipoDV-MPLA) were found to be non-toxic to the normal cells and did not induce hemolysis. Additionally, the in vivo toxicity evaluations showed that no symptoms of illness (including body weight changes), local reaction at the injection site and death were observed in mice immunized with our formulated dengue vaccines, thus confirming their safety.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}