
Candidates for Repurposing as Anti-Virulence Agents Based on the Structural Profile Analysis of Microbial Collagenase Inhibitors

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets Preparation
2.2. Molecular Descriptors
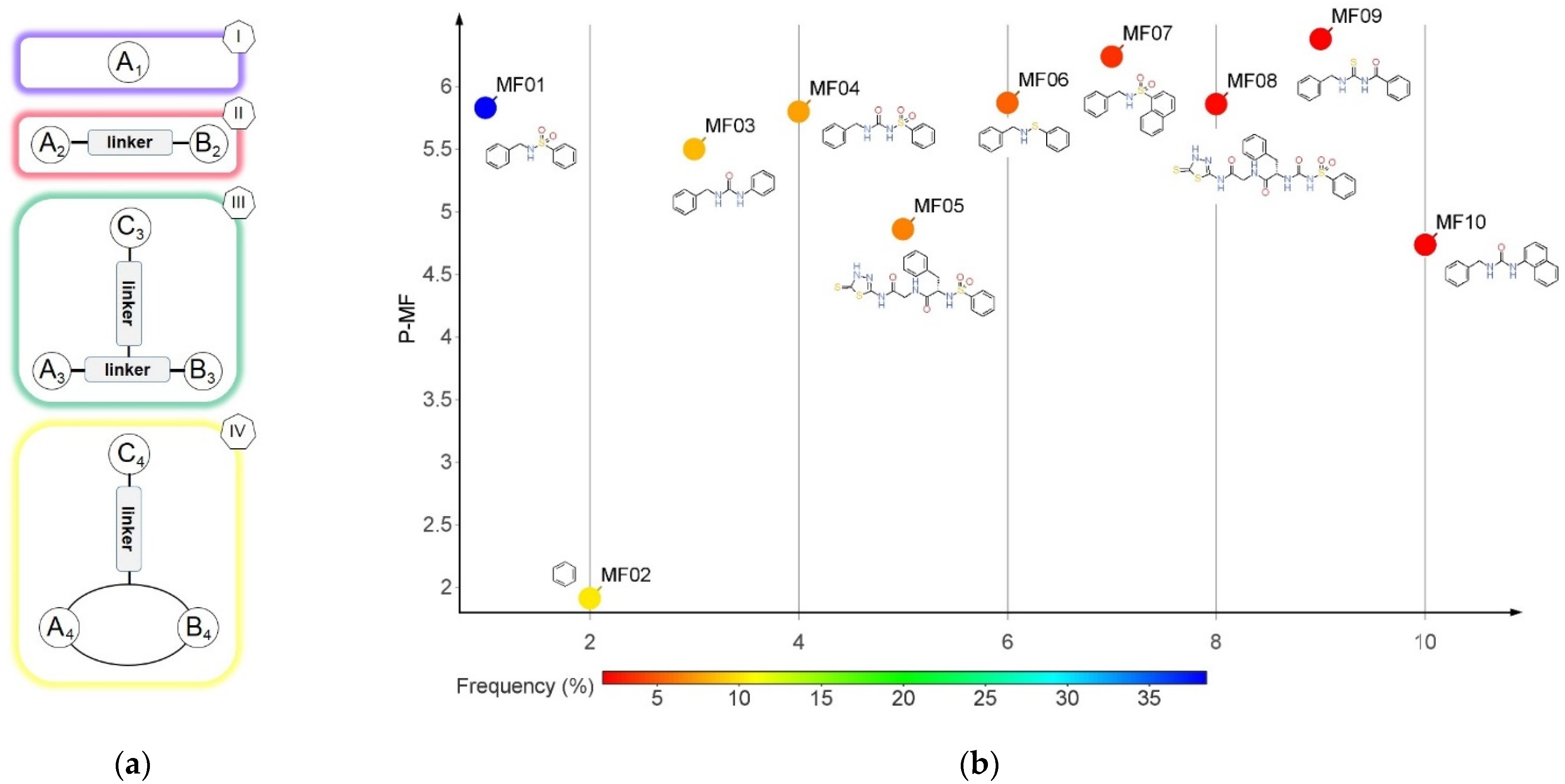
2.3. Bemis-Murcko Skeletons and Murcko Frameworks Analysis
2.4. Plain Ring Analysis
2.5. Repurposing Study
2.6. Homology Modeling
2.7. Molecular Docking
3. Results
3.1. Datasets
3.2. Murcko Frameworks Profile
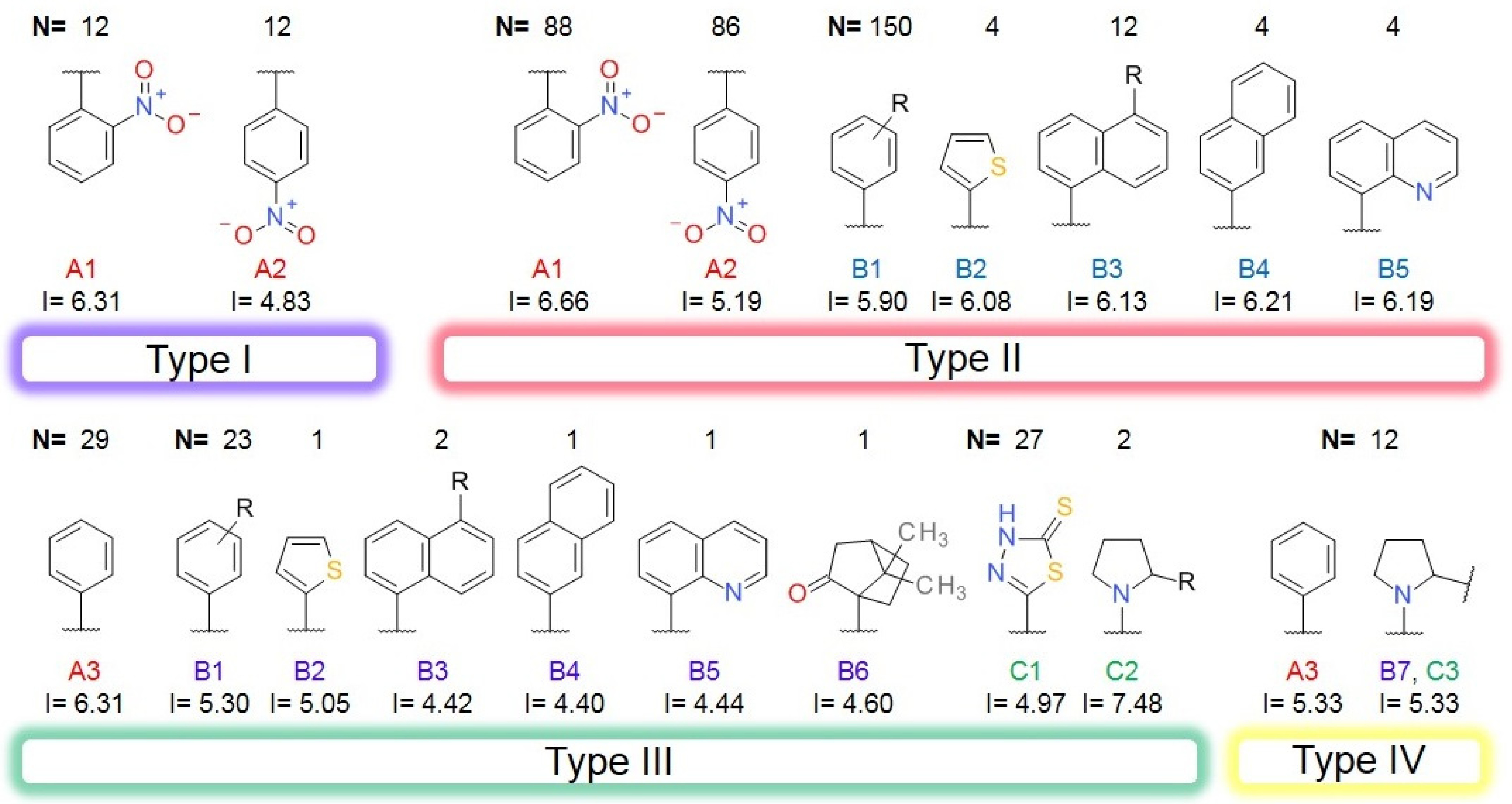
3.3. Bemis-Murcko Skeletons Profile
3.4. Plain Ring Analysis
3.5. Classification Model
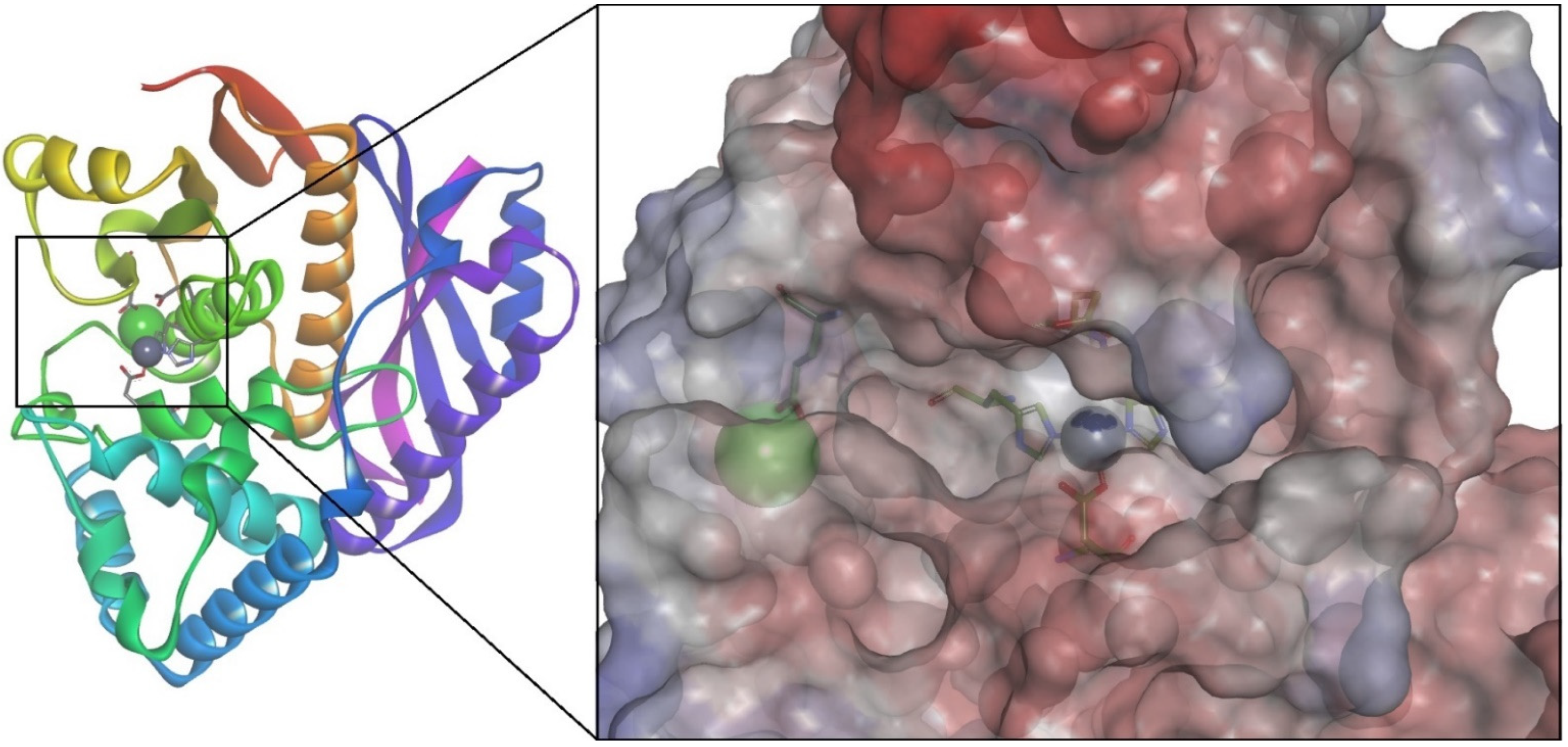
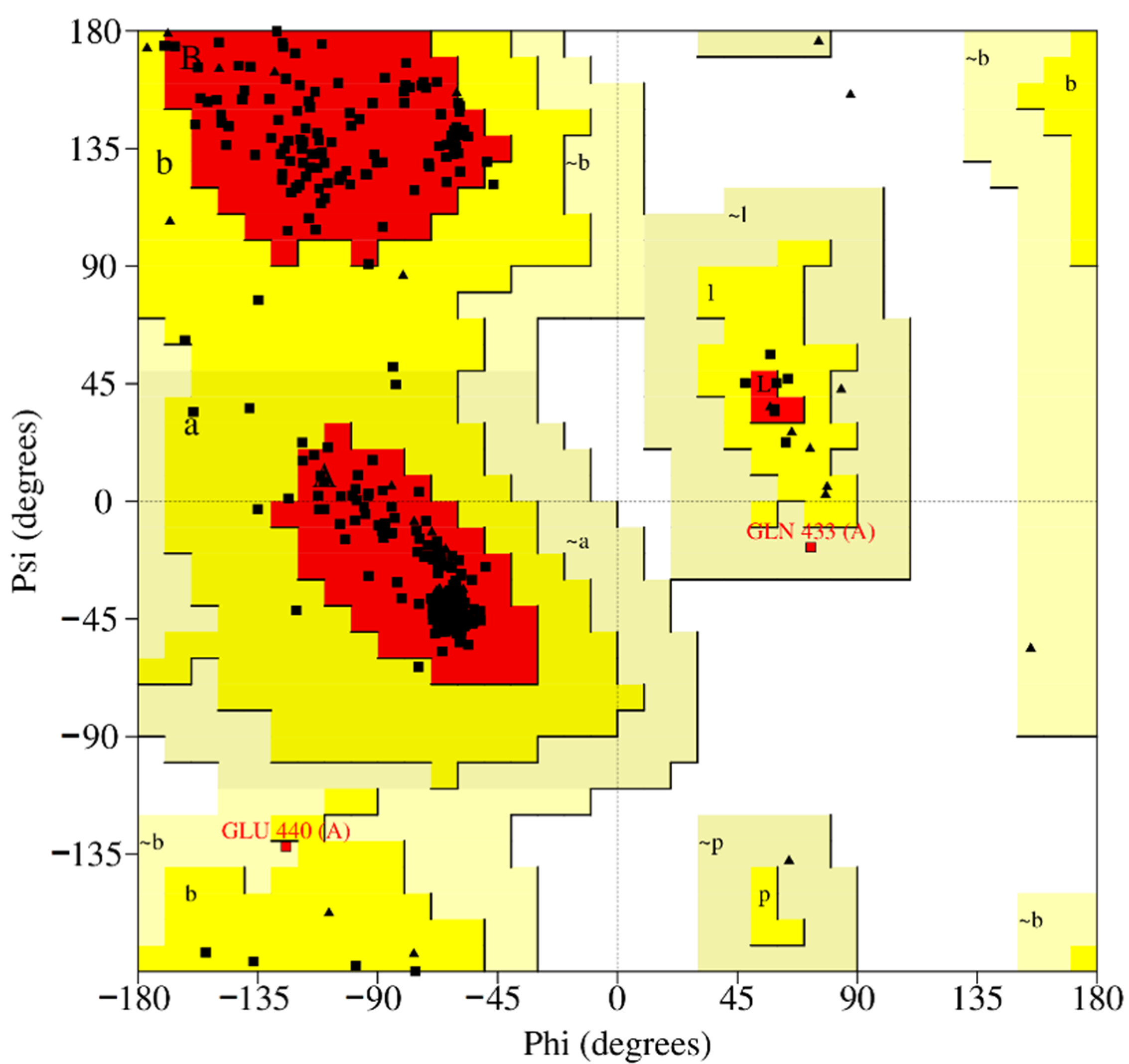
3.6. Homology Modeling and Molecular Docking of ColA Inhibitors
3.7. Repurposing Study
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Medina, E.; Pieper, D.H. Tackling threats and future problems of multidrug-resistant bacteria. In Current Topics in Microbiology and Immunology; Springer: Cham, Switzerland, 2016; Volume 398, pp. 3–33. [Google Scholar] [CrossRef]
- Obolski, U.; Stein, G.Y.; Hadany, L. Antibiotic Restriction Might Facilitate the Emergence of Multi-drug Resistance. PLoS Comput. Biol. 2015, 11, e1004340. [Google Scholar] [CrossRef]
- Escaich, S. Antivirulence as a new antibacterial approach for chemotherapy. Curr. Opin. Chem. Biol. 2008, 12, 400–408. [Google Scholar] [CrossRef]
- Cegelski, L.; Marshall, G.R.; Eldridge, G.R.; Hultgren, S.J. The biology and future prospects of antivirulence therapies. Nat. Rev. Microbiol. 2008, 6, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Ogawara, H. Possible drugs for the treatment of bacterial infections in the future: Anti-virulence drugs. J. Antibiot. 2021, 74, 24–41. [Google Scholar] [CrossRef]
- Krueger, E.; Brown, A.C. Inhibition of bacterial toxin recognition of membrane components as an anti-virulence strategy. J. Biol. Eng. 2019, 13, 4. [Google Scholar] [CrossRef]
- Nitulescu, G.; Margina, D.; Zanfirescu, A.; Olaru, O.T.; Nitulescu, G.M. Targeting bacterial sortases in search of anti-virulence therapies with low risk of resistance development. Pharmaceuticals 2021, 14, 415. [Google Scholar] [CrossRef]
- Escajadillo, T.; Nizet, V. Pharmacological targeting of pore-forming toxins as adjunctive therapy for invasive bacterial infection. Toxins 2018, 10, 542. [Google Scholar] [CrossRef] [Green Version]
- Åberg, V.; Sellstedt, M.; Hedenström, M.; Pinkner, J.S.; Hultgren, S.J.; Almqvist, F. Design, synthesis and evaluation of peptidomimetics based on substituted bicyclic 2-pyridones-Targeting virulence of uropathogenic E. coli. Bioorganic Med. Chem. 2006, 14, 7563–7581. [Google Scholar] [CrossRef]
- Wu, H.; Song, Z.; Hentzer, M.; Andersen, J.B.; Molin, S.; Givskov, M.; Høiby, N. Synthetic furanones inhibit quorum-sensing and enhance bacterial clearance in Pseudomonas aeruginosa lung infection in mice. J. Antimicrob. Chemother. 2004, 53, 1054–1061. [Google Scholar] [CrossRef] [Green Version]
- Holst, O. Structure of the Lipopolysaccharide Core Region. In Bacterial Lipopolysaccharides; Springer: Vienna, Austria, 2011; pp. 21–39. [Google Scholar]
- Dickey, S.W.; Cheung, G.Y.C.; Otto, M. Different drugs for bad bugs: Antivirulence strategies in the age of antibiotic resistance. Nat. Rev. Drug. Discov. 2017, 16, 457–471. [Google Scholar] [CrossRef]
- Bhagwat, P.K.; Dandge, P.B. Collagen and collagenolytic proteases: A review. Biocatal. Agric. Biotechnol. 2018, 15, 43–55. [Google Scholar] [CrossRef]
- Duarte, A.S.; Correia, A.; Esteves, A.C. Bacterial collagenases—A review. Crit. Rev. Microbiol. 2016, 42, 106–126. [Google Scholar] [CrossRef]
- Schönauer, E.; Brandstetter, H. Inhibition and activity regulation of bacterial collagenases. Curr. Top. Med. Chem. 2016, 22, 69–94. [Google Scholar]
- Zhang, Y.Z.; Ran, L.Y.; Li, C.Y.; Chen, X.L. Diversity, structures, and collagen-degrading mechanisms of bacterial collagenolytic proteases. Appl. Environ. Microbiol. 2015, 81, 6098–6107. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, N.D.; Barrett, A.J.; Bateman, A. MEROPS: The peptidase database. Nucleic Acids Res. 2009, 38, D227–D233. [Google Scholar] [CrossRef]
- Eckhard, U.; Schönauer, E.; Brandstetter, H. Structural basis for activity regulation and substrate preference of clostridial collagenases G, H, and T. J. Biol. Chem. 2013, 288, 20184–20194. [Google Scholar] [CrossRef] [Green Version]
- Harrington, D.J. Bacterial collagenases and collagen-degrading enzymes and their potential role in human disease. Infect. Immun. 1996, 64, 1885–1891. [Google Scholar] [CrossRef] [Green Version]
- French, M.F.; Bhown, A.; Van Wart, H.E. Identification of Clostridium histolyticum collagenase hyperreactive sites in type I, II, and III collagens: Lack of correlation with local triple helical stability. J. Protein Chem. 1992, 11, 83–97. [Google Scholar] [CrossRef]
- Yoshihara, K.; Matsushita, O.; Minami, J.; Okabe, A. Cloning and nucleotide sequence analysis of the colH gene from Clostridium histolyticum encoding a collagenase and a gelatinase. J. Bacteriol. 1994, 176, 6489–6496. [Google Scholar] [CrossRef] [Green Version]
- Eckhard, U.; Schönauer, E.; Ducka, P.; Briza, P.; Nüss, D.; Brandstetter, H. Biochemical characterization of the catalytic domains of three different clostridial collagenases. Biol. Chem. 2009, 390, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Van Damme, L.; Cox, N.; Callens, C.; Dargatz, M.; Flügel, M.; Hark, S.; Thiemann, F.; Pelzer, S.; Haesebrouck, F.; Ducatelle, R.; et al. Protein Truncating Variants of colA in Clostridium perfringens Type G Strains. Front. Cell. Infect. Microbiol. 2021, 11, 348. [Google Scholar] [CrossRef]
- Miyoshi, S.I.; Nitanda, Y.; Fujii, K.; Kawahara, K.; Li, T.; Maehara, Y.; Ramamurthy, T.; Takeda, Y.; Shinoda, S. Differential gene expression and extracellular secretion of the collagenolytic enzymes by the pathogen Vibrio parahaemolyticus. FEMS Microbiol. Lett. 2008, 283, 176–181. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, J.; Okuda, K. Vibrio collagenase. In Handbook of Proteolytic Enzymes, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 1, pp. 414–415. ISBN 9780080984155. [Google Scholar]
- Beecher, D.J.; Olsen, T.W.; Somers, E.B.; Wong, A.C.L. Evidence for contribution of tripartite hemolysin BL, phosphatidylcholine-preferring phospholipase C, and collagenase to virulence of Bacillus cereus endophthalmitis. Infect. Immun. 2000, 68, 5269–5276. [Google Scholar] [CrossRef] [Green Version]
- Hoppe, I.J.; Brandstetter, H.; Schönauer, E. Biochemical characterisation of a collagenase from Bacillus cereus strain Q1. Sci. Rep. 2021, 11, 4187. [Google Scholar] [CrossRef]
- Kato, T.; Takahashi, N.; Kuramitsu, H.K. Sequence analysis and characterization of the Porphyromonas gingivalis prtC gene, which expresses a novel collagenase activity. J. Bacteriol. 1992, 174, 3889–3895. [Google Scholar] [CrossRef] [Green Version]
- Kavermann, H.; Burns, B.P.; Angermüller, K.; Odenbreit, S.; Fischer, W.; Melchers, K.; Haas, R. Identification and characterization of Helicobacter pylori genes essential for gastric colonization. J. Exp. Med. 2003, 197, 813–822. [Google Scholar] [CrossRef]
- Han, H.J.; Taki, T.; Kondo, H.; Hirono, I.; Aoki, T. Pathogenic potential of a collagenase gene from Aeromonas veronii. Can. J. Microbiol. 2008, 54, 1–10. [Google Scholar] [CrossRef]
- Carlson, S.A.; McCuddin, Z.P.; Wu, M.T. SlyA regulates the collagenase-mediated cytopathic phenotype in multiresistant Salmonella. Microb. Pathog. 2005, 38, 181–187. [Google Scholar] [CrossRef]
- Rani, S.; Pooja, K. Elucidation of structural and functional characteristics of collagenase from Pseudomonas aeruginosa. Process Biochem. 2018, 64, 116–123. [Google Scholar] [CrossRef]
- Zhao, H.; Li, X.; Johnson, D.E.; Mobley, H.L.T. Identification of protease and rpoN-associated genes of uropathogenic Proteus mirabilis by negative selection in a mouse model of ascending urinary tract infection. Microbiology 1999, 145, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Popov, S.G.; Popova, T.G.; Hopkins, S.; Weinstein, R.S.; MacAfee, R.; Fryxell, K.J.; Chandhoke, V.; Bailey, C.; Alibek, K. Effective antiprotease-antibiotic treatment of experimental anthrax. BMC Infect. Dis. 2005, 5, 25. [Google Scholar] [CrossRef] [Green Version]
- Kassegne, K.; Hu, W.; Ojcius, D.M.; Sun, D.; Ge, Y.; Zhao, J.; Yang, X.F.; Li, L.; Yan, J. Identification of collagenase as a critical virulence factor for invasiveness and transmission of pathogenic leptospira species. J. Infect. Dis. 2014, 209, 1105–1115. [Google Scholar] [CrossRef] [Green Version]
- Schönauer, E.; Kany, A.M.; Haupenthal, J.; Hüsecken, K.; Hoppe, I.J.; Voos, K.; Yahiaoui, S.; Elsässer, B.; Ducho, C.; Brandstetter, H.; et al. Discovery of a Potent Inhibitor Class with High Selectivity toward Clostridial Collagenases. J. Am. Chem. Soc. 2017, 139, 12696–12703. [Google Scholar] [CrossRef] [Green Version]
- Konstantinović, J.; Yahiaoui, S.; Alhayek, A.; Haupenthal, J.; Schönauer, E.; Andreas, A.; Kany, A.M.; Müller, R.; Koehnke, J.; Berger, F.K.; et al. N-Aryl-3-mercaptosuccinimides as Antivirulence Agents Targeting Pseudomonas aeruginosa Elastase and Clostridium Collagenases. J. Med. Chem. 2020, 63, 8359–8368. [Google Scholar] [CrossRef]
- Voos, K.; Schönauer, E.; Alhayek, A.; Haupenthal, J.; Andreas, A.; Müller, R.; Hartmann, R.W.; Brandstetter, H.; Hirsch, A.K.H.; Ducho, C. Phosphonate as a Stable Zinc-Binding Group for “Pathoblocker” Inhibitors of Clostridial Collagenase H (ColH). ChemMedChem 2021, 16, 1257–1267. [Google Scholar] [CrossRef]
- Leão, C.; Borges, A.; Simões, M. Nsaids as a drug repurposing strategy for biofilm control. Antibiotics 2020, 9, 591. [Google Scholar] [CrossRef]
- Pérez-Moraga, R.; Forés-Martos, J.; Suay-García, B.; Duval, J.L.; Falcó, A.; Climent, J. A COVID-19 drug repurposing strategy through quantitative homological similarities using a topological data analysis-based framework. Pharmaceutics 2021, 13, 488. [Google Scholar] [CrossRef]
- Mihai, D.P.; Nitulescu, G.M.; Ion, G.N.D.; Ciotu, C.I.; Chirita, C.; Negres, S. Computational drug repurposing algorithm targeting trpa1 calcium channel as a potential therapeutic solution for multiple sclerosis. Pharmaceutics 2019, 11, 446. [Google Scholar] [CrossRef] [Green Version]
- Ion, G.N.D.; Mihai, D.P.; Lupascu, G.; Nitulescu, G.M. Application of molecular framework-based data-mining method in the search for beta-secretase 1 inhibitors through drug repurposing. J. Biomol. Struct. Dyn. 2019, 37, 3674–3685. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34 (Suppl. S1), D668–D672. [Google Scholar] [CrossRef]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Bemis, G.W.; Murcko, M.A. The Properties of Known Drugs. 1. Molecular Frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef]
- Ion, G.N.D.; Olaru, O.T.; Nitulescu, G.; Olaru, I.I.; Tsatsakis, A.; Burykina, T.I.; Spandidos, D.A.; Nitulescu, G.M. Improving the odds of success in antitumoral drug development using scoring approaches towards heterocyclic scaffolds. Oncol. Rep. 2020, 44, 589–598. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Land, H.; Humble, M.S. YASARA: A Tool to Obtain Structural Guidance in Biocatalytic Investigations. In Protein Engineering; Humana Press: New York, NY, USA, 2018; pp. 43–67. [Google Scholar]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [Green Version]
- Pontius, J.; Richelle, J.; Wodak, S.J. Deviations from Standard Atomic Volumes as a Quality Measure for Protein Crystal Structures. J. Mol. Biol. 1996, 264, 121–136. [Google Scholar] [CrossRef] [Green Version]
- Bowie, J.U.; Lüthy, R.; Eisenberg, D. A Method to Identify Protein Sequences That Fold into a Known Three-Dimensional Structure. Science 1991, 253, 164–170. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open chemical toolbox. J. Cheminform. 2011, 3, 32. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Cao, C.; Li, Z. Approach to estimation and prediction for Normal Boiling Point (NBP) of alkanes based on a novel Molecular Distance-Edge (MDE) vector, γ. J. Chem. Inf. Comput. Sci. 1998, 38, 387–394. [Google Scholar] [CrossRef]
- Gramatica, P.; Corradi, M.; Consonni, V. Modelling and prediction of soil sorption coefficients of non-ionic organic pesticides by molecular descriptors. Chemosphere 2000, 41, 763–777. [Google Scholar] [CrossRef]
- Eron, S.J.; MacPherson, D.J.; Dagbay, K.B.; Hardy, J.A. Multiple Mechanisms of Zinc-Mediated Inhibition for the Apoptotic Caspases-3, -6, -7, and -8. ACS Chem. Biol. 2018, 13, 1279–1290. [Google Scholar] [CrossRef] [Green Version]
- Lo, S.Y.; Goulet, D.L.; Fraaz, U.; Siemann, S. Effect of pH and denaturants on the fold and metal status of anthrax lethal factor. Arch. Biochem. Biophys. 2020, 692, 108547. [Google Scholar] [CrossRef]
- Boschi, A.; Uccelli, L.; Martini, P. A Picture of Modern Tc-99m Radiopharmaceuticals: Production, Chemistry, and Applications in Molecular Imaging. Appl. Sci. 2019, 9, 2526. [Google Scholar] [CrossRef] [Green Version]
- Marie, S.; Hernández-Lozano, I.; Breuil, L.; Saba, W.; Novell, A.; Gennisson, J.L.; Langer, O.; Truillet, C.; Tournier, N. Validation of pharmacological protocols for targeted inhibition of canalicular MRP2 activity in hepatocytes using [99mtc]mebrofenin imaging in rats. Pharmaceutics 2020, 12, 486. [Google Scholar] [CrossRef]
- Zwama, M.; Nishino, K. Ever-Adapting RND Efflux Pumps in Gram-Negative Multidrug-Resistant Pathogens: A Race against Time. Antibiotics 2021, 10, 774. [Google Scholar] [CrossRef]
- Jamloki, A.; Karthikeyan, C.; Hari Narayana Moorthy, N.S.; Trivedi, P. QSAR analysis of some 5-amino-2-mercapto-1,3,4-thiadiazole based inhibitors of matrix metalloproteinases and bacterial collagenase. Bioorganic Med. Chem. Lett. 2006, 16, 3847–3854. [Google Scholar] [CrossRef]
- Baranauskiene, L.; Škiudaitė, L.; Michailovienė, V.; Petrauskas, V.; Matulis, D. Thiazide and other Cl-benzenesulfonamide-bearing clinical drug affinities for human carbonic anhydrases. PLoS ONE 2021, 16, e0253608. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathogen | Disease | Collagenase | MEROPS ID | Class | Substrate | Reference |
|---|---|---|---|---|---|---|
| Clostridium histolyticum | Clostridial myonecrosis | Collagenase H | M09.003 | Class II | Type I, II, and III collagens | [20,21] |
| Collagenase G/A | M09.002 | Class I | Type I, II, and III collagens | |||
| Clostridium tetani | Tetanus | Collagenase col T | M09.005 | [22] | ||
| Clostridium perfringens | Clostridial myonecrosis | Collagenase G/A (or collagenase A g.p.) | M09.002 | Class I | Type I collagen, Pz peptide, azocoll | [23] |
| Vibrio alginolyticus, Vibrio parahaemolyticus, Vibrio vulnificus | Cellulitis, septicemia | Collagenase V | M09.001 | Class III collagenases | Gelatin, casein, collagen, synthetic substrate | [24,25] |
| Vibrio mimicus, Vibrio parahaemolyticus, Vibrio cholerae | Gastroenteritis | VMC peptidase | M09.004 | Class II collagenases | Type I, II, III collagens, gelatin, Cbz-GPLGP, Cbz-GPGGPA | [14,24,25] |
| B. cereus | Periodontal disease, endophthalmitis | Collagenases Q1 | M09.002/M09.003 | Class I collagenases/ class II | Type I, II, III collagens | [26,27] |
| Porphyromonas gingivalis | Periodontal disease | Collagenase (Porphyromonas type) | U32.001 | Not applicable | Soluble and reconstituted fibrillar or heat-denatured type I collagen, | [28] |
| Helicobacter pylori | Gastro-duodenal ulcer | Collagenase (Helicobacter type) | U32.002 | Not characterized | Not characterized | [29] |
| Aeromonas veronii | arthritis, gastroenteritis, meningitis, septicemia | Collagenase (Salmonella type) | U32.003 | Not characterized | Not characterized | [30,31] |
| Salmonella sp. | Salmonellosis (associated with abomasitis, peritonitis and polyserositis) | |||||
| Escherichia Coli | Urinary infections, digestive infections, etc. | YhbV | U32.A.01 | Not characterized | Not characterized | [17] |
| Code | PR01 | PR02 | PR03 | PR04 | PR05 | PR06 |
|---|---|---|---|---|---|---|
| Structure | 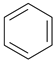 |  |  |  |  | 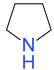 |
| P-PR | 0.368 | 4.971 | 1.063 | 0.281 | 0.291 | 0.065 |
| Frequency (%) BCI | 100.00 | 11.30 | 7.95 | 2.09 | 2.09 | 0.84 |
| Frequency (%) DCY | 78.47 | 0.56 | 2.20 | 2.18 | 2.11 | 4.79 |
| OD (BCI vs. DCY) | 1.27 | 20.00 | 3.62 | 0.96 | 0.99 | 0.17 |
| Code | Type | Cutoff | Descriptor’s Mathematical Representation |
|---|---|---|---|
| MDEO-11 | MDE | >1.194 | Molecular distance edge between all primary oxygens |
| AATS5s | Autocorrelation | >3.315 | Average Broto–Moreau autocorrelation—lag 5, weighted by I-state |
| AATSC0s | Autocorrelation | >2.008 | Average centered Broto–Moreau autocorrelation—lag 0, weighted by I-state |
| AATS0s | Autocorrelation | >5.239 | Average Broto–Moreau autocorrelation—lag 0/weighted by I-state |
| ASP-0 | ChiPath | >0.714 | Average simple path, order 0 |
| maxHBint5 | Electrotopological State Atom Type | - | Maximum E-State descriptors of strength for potential Hydrogen Bonds of path length 5 |
| meanI | Electrotopological State Atom Type | >2.370 | Mean intrinsic state values I |
| MAXDN | Electrotopological State Atom Type | >2.490 | Maximum negative intrinsic state difference in the molecule |
| Method | Model | Template | ERRAT (Overall Quality Factor) | VERIFY3D (3D-1D Score >0.2% Residues) | PROVE (Buried Outlier Protein Atoms Total, %) | Residues in Most Favored Regions (%) | Residues in Disallowed Regions (%) |
|---|---|---|---|---|---|---|---|
| SWISS-MODEL | S1 | 2Y3U | 96.7016 | 97.93 | 4.7 | 93.2 | 0.2 |
| S2 | 4ARE | 97.1299 | 98.51 | 4.2 | 92.9 | 0.5 | |
| S3 | 4AR9 | 96.5517 | 96.12 | 4.1 | 91.9 | 0.0 | |
| S4 | 5O7E | 95.0954 | 92.80 | 4.6 | 92.5 | 0.0 | |
| YASARA | Y1 | 4ARE | 98.3824 | 98.41 | 3.1 | 93.4 | 0.3 |
| Y2 | 2Y3U | 99.1098 | 97.41 | 3.3 | 94.2 | 0.3 | |
| Y3 | 4AR9 | 98.7310 | 99.02 | 4.5 | 92.6 | 0.0 | |
| Y4 | 5IKU | 76.9874 | 90.44 | 3.5 | 86.9 | 0.0 | |
| Y5 | hybrid | 97.3529 | 99.42 | 2.5 | 93.6 | 0.5 |
| Code | Name | TS | Target |
|---|---|---|---|
| DB08498 | 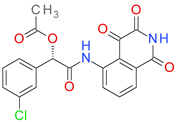 | 6.11 | • Caspase-3 |
| DB08497 | 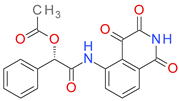 | 6.11 | • Caspase-3 |
| DB01689 | 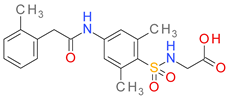 | 5.15 | • Aldose reductase |
| DB07030 |  | 5.15 | • Aldose reductase |
| DB07556 | 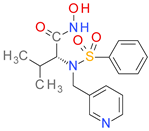 | 5.15 | • Macrophage metalloelastase • Interstitial collagenase |
| DB06989 |  | 5.15 | • Receptor-type tyrosine-protein phosphatase beta |
| DB07290 | 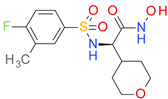 | 5.15 | • Anthrax lethal factor endopeptidase |
| DB03124 | 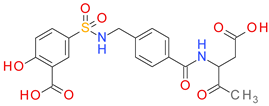 | 5.15 | • Caspase-3 |
| DB04659 |  | 5.15 | • Lactase-phlorizin hydrolase |
| DB08229 |  | 5.15 | • Caspase-3 |
| Code | LE (kcal/mol Per Heavy Atom) | ΔG (kcal/mol) | pKd (M) | No. of Contacts |
|---|---|---|---|---|
| DB08498 | 0.3483 | −9.751 | 7.148 | 18 |
| DB08497 | 0.3476 | −9.386 | 6.880 | 18 |
| DB01689 | 0.3545 | −9.572 | 7.016 | 18 |
| DB07030 | 0.3472 | −8.68 | 6.363 | 17 |
| DB07556 | 0.2664 | −7.192 | 5.272 | 16 |
| DB06989 | 0.2938 | −8.226 | 6.030 | 15 |
| DB07290 | 0.3643 | −8.379 | 6.142 | 16 |
| DB03124 | 0.3114 | −9.965 | 7.304 | 21 |
| DB04659 | 0.3082 | −8.013 | 5.874 | 16 |
| DB08229 | 0.3352 | −9.386 | 6.880 | 19 |
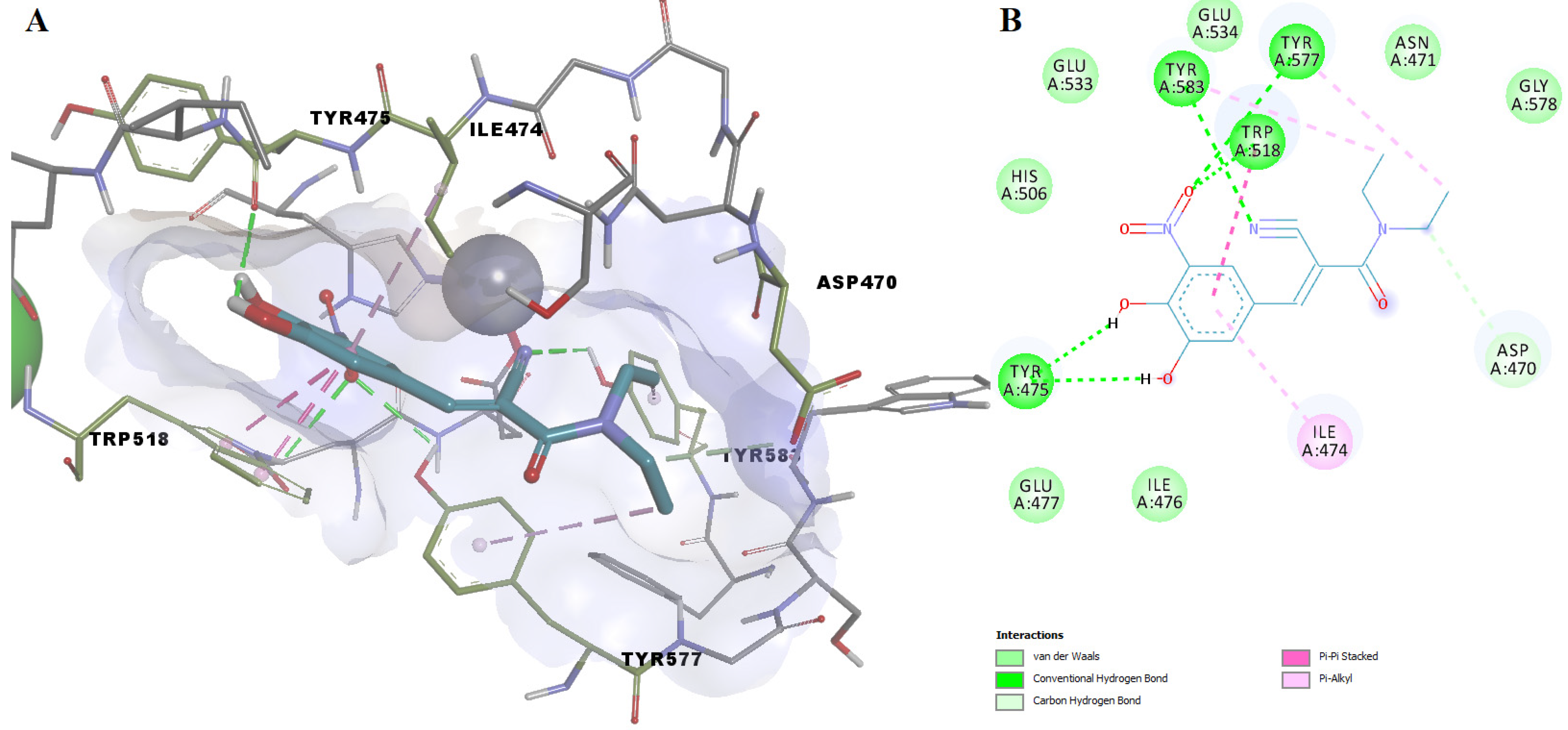
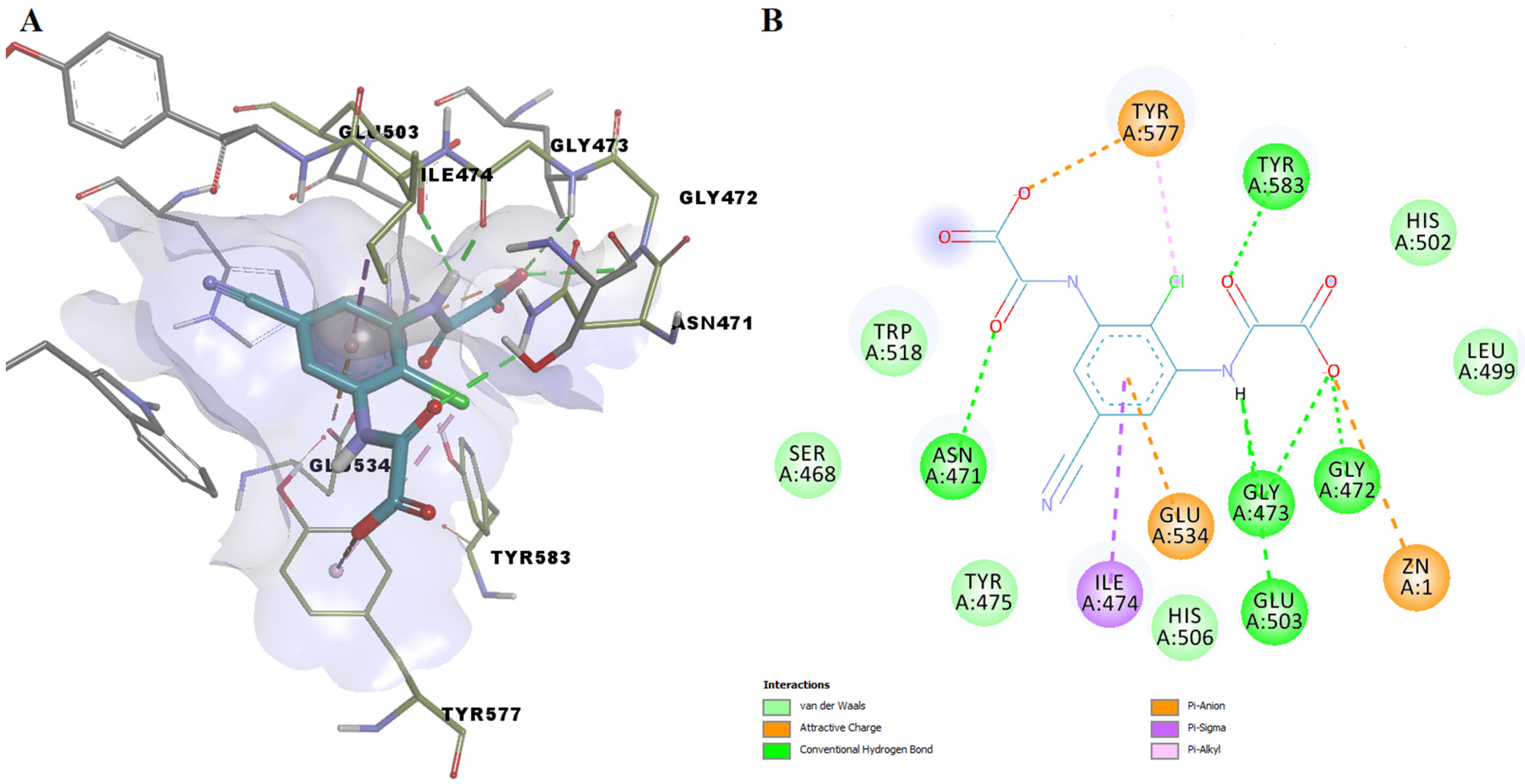
| Name | Code | TS | LE (kcal/mol Per Heavy Atom) | ΔG (kcal/mol) | pKd (M) | H-Bonding Residues | H-Bond Length (Å) |
|---|---|---|---|---|---|---|---|
| Benzthiazide | DB00562 | 2.85 | 0.3669 | −9.539 | 6.992 | Gly578 | 2.155 |
| Tyr583 | 2.246 | ||||||
| Asp470 | 1.839 | ||||||
| Asp470 | 2.328 | ||||||
| Glu503 | 2.095 | ||||||
| Entacapone | DB00494 | 1.96 | 0.3521 | −7.746 | 5.678 | Trp518 | 2.987 |
| Tyr577 | 2.891 | ||||||
| Tyr583 | 2.690 | ||||||
| Tyr475 | 1.908 | ||||||
| Tyr475 | 2.592 | ||||||
| Lodoxamide | DB06794 | 1.96 | 0.3744 | −7.862 | 5.763 | Asn471 | 2.647 |
| Gly472 | 2.720 | ||||||
| Gly473 | 2.844 | ||||||
| Gly473 | 1.732 | ||||||
| Tyr583 | 1.753 | ||||||
| Glu503 | 2.870 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nitulescu, G.; Nitulescu, G.M.; Zanfirescu, A.; Mihai, D.P.; Gradinaru, D. Candidates for Repurposing as Anti-Virulence Agents Based on the Structural Profile Analysis of Microbial Collagenase Inhibitors. Pharmaceutics 2022, 14, 62. https://doi.org/10.3390/pharmaceutics14010062
Nitulescu G, Nitulescu GM, Zanfirescu A, Mihai DP, Gradinaru D. Candidates for Repurposing as Anti-Virulence Agents Based on the Structural Profile Analysis of Microbial Collagenase Inhibitors. Pharmaceutics. 2022; 14(1):62. https://doi.org/10.3390/pharmaceutics14010062
Chicago/Turabian StyleNitulescu, Georgiana, George Mihai Nitulescu, Anca Zanfirescu, Dragos Paul Mihai, and Daniela Gradinaru. 2022. "Candidates for Repurposing as Anti-Virulence Agents Based on the Structural Profile Analysis of Microbial Collagenase Inhibitors" Pharmaceutics 14, no. 1: 62. https://doi.org/10.3390/pharmaceutics14010062