
Oxyresveratrol Inhibits TNF-α-Stimulated Cell Proliferation in Human Immortalized Keratinocytes (HaCaT) by Suppressing AKT Activation

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Cell Culture and TNF-α Treatment
2.3. MTT Assay
2.4. Cell Counting
2.5. Western Blot Analysis
2.6. Immunofluorescence Study
2.7. Colony Forming Assay
2.8. Cell Cycle Analysis by Flow Cytometry
2.9. Statistical Analysis
3. Results
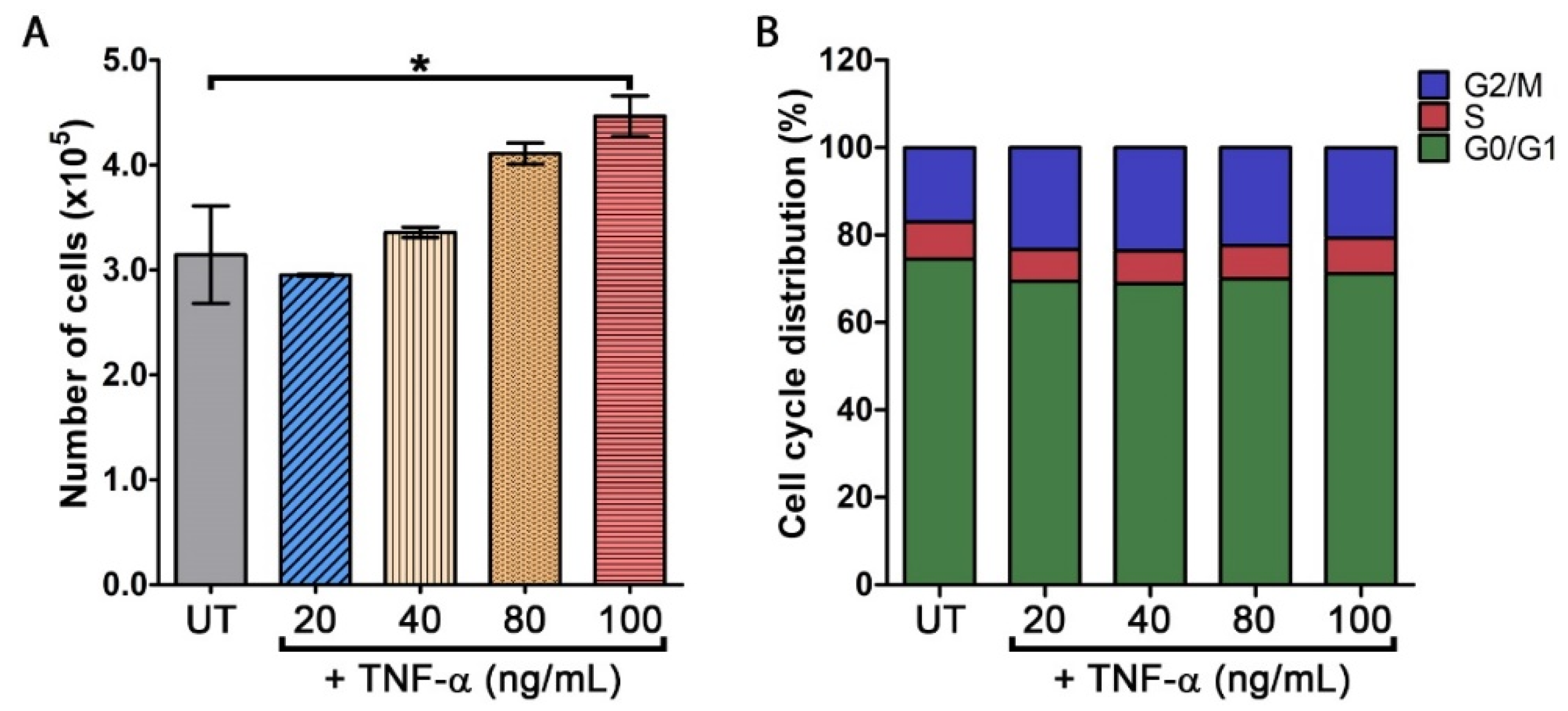
3.1. TNF-α Stimulates an Increase in Keratinocyte Cell Number
3.2. Oxyresveratrol (OXY) Inhibits TNF-α-Induced Keratinocyte Proliferation
3.3. OXY Blocks TNF-α-Induced Phosporylation of AKT and GSK3-β
3.4. OXY Suppresses TNF-α-Induced HaCaT Colony Expansion and Causes Cell Cycle Arrest
3.5. OXY Inhibits TNF-α-Induced Expression of Proliferation and Survival Markers in HaCaT Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Christophers, E. Psoriasis-epidemiology and clinical spectrum. Clin. Exp. Dermatol. 2001, 26, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Lowes, M.A.; Bowcock, A.M.; Krueger, J.G. Pathogenesis and therapy of psoriasis. Nature 2007, 445, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Kaplan, D.H.; Barker, J. Mechanisms of Disease: Psoriasis. N. Engl. J. Med. 2009, 361, 496–509. [Google Scholar] [CrossRef]
- Yarilina, A.; Park-Min, K.H.; Antoniv, T.; Hu, X.; Ivashkiv, L.B. TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat. Immunol. 2008, 9, 378–387. [Google Scholar] [CrossRef]
- Gibellini, L.; De Biasi, S.; Bianchini, E.; Bartolomeo, R.; Fabiano, A.; Manfredini, M.; Ferrari, F.; Albertini, G.; Trenti, T.; Nasi, M.; et al. Anti-TNF-alpha Drugs Differently Affect the TNFalpha-sTNFR System and Monocyte Subsets in Patients with Psoriasis. PLoS ONE 2016, 11, e0167757. [Google Scholar] [CrossRef]
- Nograles, K.E.; Davidovici, B.; Krueger, J.G. New Insights in the Immunologic Basis of Psoriasis. Semin. Cutan. Med. Surg. 2010, 29, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, W.-M. Tumor necrosis factor. Cancer Lett. 2013, 328, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Ritsu, M.; Kawakami, K.; Kanno, E.; Tanno, H.; Ishii, K.; Imai, Y.; Maruyama, R.; Tachi, M. Critical role of tumor necrosis factor-α in the early process of wound healing in skin. J. Dermatol. Dermatol. Surg. 2017, 21, 14–19. [Google Scholar] [CrossRef] [Green Version]
- Sugarman, B.J.; Aggarwal, B.B.; Hass, P.E.; Figari, I.S.; Palladino, M.A., Jr.; Shepard, H.M. Recombinant human tumor necrosis factor-alpha: Effects on proliferation of normal and transformed cells in vitro. Science 1985, 230, 943–945. [Google Scholar] [CrossRef]
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M. Tumour necrosis factor signalling in health and disease. F1000Research 2019, 8, F1000. [Google Scholar] [CrossRef]
- Piura, B.; Medina, L.; Rabinovich, A.; Dyomin, V.; Levy, R.S.; Huleihel, M. Distinct expression and localization of TNF system in ovarian carcinoma tissues: Possible involvement of TNF-α in morphological changes of ovarian cancerous cells. Anticancer Res. 2014, 34, 745–752. [Google Scholar] [PubMed]
- Furue, K.; Ito, T.; Tsuji, G.; Kadono, T.; Furue, M. Psoriasis and the TNF/IL23/IL17 axis. G. Ital. Dermatol. Venereol. 2019, 154, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Pirowska, M.; Obtułowicz, A.; Lipko-Godlewska, S.; Goździalska, A.; Podolec, K.; Wojas-Pelc, A. The level of proinflammatory cytokines: Interleukins 12, 23, 17 and tumor necrosis factor α in patients with metabolic syndrome accompanying severe psoriasis and psoriatic arthritis. Postepy Dermatol. Alergol. 2018, 35, 360–366. [Google Scholar] [CrossRef]
- Gudjonsson, J.E.; Johnston, A.; Sigmundsdottir, H.; Valdimarsson, H. Immunopathogenic mechanisms in psoriasis. Clin. Exp. Immunol. 2004, 135, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Zheng, W.; Dong, Y.; Wang, J.; Garstka, M.A.; Li, R.L.; An, J.G.; Ma, H.Q. Serum levels of adipokines and cytokines in psoriasis patients: A systematic review and meta-analysis. Oncotarget 2018, 9, 1266–1278. [Google Scholar] [CrossRef] [Green Version]
- Arnett, H.A.; Mason, J.; Marino, M.; Suzuki, K.; Matsushima, G.K.; Ting, J.P.Y. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat. Neurosci. 2001, 4, 1116–1122. [Google Scholar] [CrossRef]
- Wang, X.H.; Hong, X.; Zhu, L.; Wang, Y.T.; Bao, J.P.; Liu, L.; Wang, F.; Wu, X.T. Tumor necrosis factor alpha promotes the proliferation of human nucleus pulposus cells via nuclear factor-kappaB, c-Jun N-terminal kinase, and p38 mitogen-activated protein kinase. Exp. Biol. Med. 2015, 240, 411–417. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.; Charles, K.A.; Mantovani, A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 2005, 7, 211–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egberts, J.H.; Cloosters, V.; Noack, A.; Schniewind, B.; Thon, L.; Klose, S.; Kettler, B.; von Forstner, C.; Kneitz, C.; Tepel, J.; et al. Anti-tumor necrosis factor therapy inhibits pancreatic tumor growth and metastasis. Cancer Res. 2008, 68, 1443–1450. [Google Scholar] [CrossRef] [Green Version]
- Scott, K.A.; Moore, R.J.; Arnott, C.H.; East, N.; Thompson, R.G.; Scallon, B.J.; Shealy, D.J.; Balkwill, F.R. An anti-tumor necrosis factor-alpha antibody inhibits the development of experimental skin tumors. Mol. Cancer Ther. 2003, 2, 445–451. [Google Scholar]
- Wu, S.; Boyer, C.M.; Whitaker, R.S.; Berchuck, A.; Wiener, J.R.; Weinberg, J.B.; Bast, R.C., Jr. Tumor necrosis factor alpha as an autocrine and paracrine growth factor for ovarian cancer: Monokine induction of tumor cell proliferation and tumor necrosis factor alpha expression. Cancer Res. 1993, 53, 1939–1944. [Google Scholar]
- Aksamitiene, E.; Kiyatkin, A.; Kholodenko, B.N. Cross-talk between mitogenic Ras/MAPK and survival PI3K/Akt pathways: A fine balance. Biochem. Soc. Trans. 2012, 40, 139–146. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markman, B.; Dienstmann, R.; Tabernero, J. Targeting the PI3K/Akt/mTOR Pathway-Beyond Rapalogs. Oncotarget 2010, 1, 530–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, T.L.; Wulf, G.; Burga, L.; Cantley, L.C. Cell-to-Cell Variability in PI3K Protein Level Regulates PI3K-AKT Pathway Activity in Cell Populations. Curr. Biol. 2011, 21, 173–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamcheu, J.C.; Chaves-Rodriquez, M.I.; Adhami, V.M.; Siddiqui, I.A.; Wood, G.S.; Longley, B.J.; Mukhtar, H. Upregulation of PI3K/AKT/mTOR, FABP5 and PPAR beta/delta in Human Psoriasis and Imiquimod-induced Murine Psoriasiform Dermatitis Model. Acta Derm.-Venereol. 2016, 96, 854–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamcheu, J.C.; Pal, H.C.; Siddiqui, I.A.; Adhami, V.M.; Ayehunie, S.; Boylan, B.T.; Noubissi, F.K.; Khan, N.; Syed, D.N.; Elmets, C.A.; et al. Prodifferentiation, Anti-Inflammatory and Antiproliferative Effects of Delphinidin, a Dietary Anthocyanidin, in a Full-Thickness Three-Dimensional Reconstituted Human Skin Model of Psoriasis. Skin Pharmacol. Phys. 2015, 28, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Un, X.R.; Meng, X.M.; Lin, M. Phosphoinositide-3 Kinase/Protein Kinase-B/Mammalian Target of Rapamycin Pathway in Psoriasis Pathogenesis. A Potential Therapeutic Target? Acta Derm.-Venereol. 2014, 94, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Saoncella, S.; Tassone, B.; Deklic, E.; Avolio, F.; Jon, C.; Tornillo, G.; De Luca, E.; Di Iorio, E.; Piva, R.; Cabodi, S.; et al. Nuclear Akt2 opposes limbal keratinocyte stem cell self-renewal by repressing a FOXO-mTORC1 signaling pathway. Stem Cells 2014, 32, 754–769. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, H.; Ma, S. Up-regulation of phosphatidylinositol 3-kinase in psoriatic lesions. Chin. Med. J. 1999, 112, 1097–1100. [Google Scholar] [PubMed]
- Wang, H.; Ran, L.W.; Hui, K.; Wang, X.Y.; Zheng, Y. Expressions of survivin, PI3K and AKT in keratinocytes in skin lesions and their pathogenic role in psoriasis vulgaris. Nan Fang Yi Ke Da Xue Xue Bao 2017, 37, 1512–1516. [Google Scholar] [PubMed]
- Madonna, S.; Scarponi, C.; Pallotta, S.; Cavani, A.; Albanesi, C. Anti-apoptotic effects of suppressor of cytokine signaling 3 and 1 in psoriasis. Cell Death Dis. 2012, 3, e334. [Google Scholar] [CrossRef] [Green Version]
- Malemud, C.J. The PI3K/Akt/PTEN/mTOR pathway: A fruitful target for inducing cell death in rheumatoid arthritis? Future Med. Chem. 2015, 7, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Burger, C.; Shirsath, N.; Lang, V.; Diehl, S.; Kaufmann, R.; Weigert, A.; Han, Y.Y.; Ringel, C.; Wolf, P. Blocking mTOR Signalling with Rapamycin Ameliorates Imiquimod-induced Psoriasis in Mice. Acta Derm. Venereol. 2017, 97, 1087–1094. [Google Scholar] [CrossRef] [Green Version]
- Chamcheu, J.C.; Adhami, V.M.; Esnault, S.; Sechi, M.; Siddiqui, I.A.; Satyshur, K.A.; Syed, D.N.; Dodwad, S.M.; Chaves-Rodriquez, M.I.; Longley, B.J.; et al. Dual Inhibition of PI3K/Akt and mTOR by the Dietary Antioxidant, Delphinidin, Ameliorates Psoriatic Features In Vitro and in an Imiquimod-Induced Psoriasis-Like Disease in Mice. Antioxid. Redox Signal. 2017, 26, 49–69. [Google Scholar] [CrossRef] [Green Version]
- Frigerio, E.; Colombo, M.D.; Franchi, C.; Altomare, A.; Garutti, C.; Altomare, G.F. Severe psoriasis treated with a new macrolide: Everolimus. Br. J. Dermatol. 2007, 156, 372–374. [Google Scholar] [CrossRef] [PubMed]
- Likhitwitayawuid, K. Oxyresveratrol: Sources, Productions, Biological Activities, Pharmacokinetics, and Delivery Systems. Molecules 2021, 26, 4212. [Google Scholar] [CrossRef] [PubMed]
- Hankittichai, P.; Buacheen, P.; Pitchakarn, P.; Na Takuathung, M.; Wikan, N.; Smith, D.R.; Potikanond, S.; Nimlamool, W. Artocarpus lakoocha Extract Inhibits LPS-Induced Inflammatory Response in RAW 264.7 Macrophage Cells. Int. J. Mol. Sci. 2020, 21, 1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hankittichai, P.; Lou, H.J.; Wikan, N.; Smith, D.R.; Potikanond, S.; Nimlamool, W. Oxyresveratrol Inhibits IL-1β-Induced Inflammation via Suppressing AKT and ERK1/2 Activation in Human Microglia, HMC3. Int. J. Mol. Sci. 2020, 21, 6054. [Google Scholar] [CrossRef]
- Choi, D.H.; Hwang, H.S. Anti-inflammation activity of brazilin in TNF-α induced human psoriasis dermatitis skin model. Appl. Biol. Chem. 2019, 62, 46. [Google Scholar] [CrossRef] [Green Version]
- Takuathung, M.N.; Potikanond, S.; Sookkhee, S.; Mungkornasawakul, P.; Jearanaikulvanich, T.; Chinda, K.; Wikan, N.; Nimlamool, W. Anti-psoriatic and anti-inflammatory effects of Kaempferia parviflora in keratinocytes and macrophage cells. Biomed. Pharmacother. 2021, 143, 112229. [Google Scholar] [CrossRef] [PubMed]
- Thaklaewphan, P.; Ruttanapattanakul, J.; Monkaew, S.; Buatoom, M.; Sookkhee, S.; Nimlamool, W.; Potikanond, S. Kaempferia parviflora extract inhibits TNF-α-induced release of MCP-1 in ovarian cancer cells through the suppression of NF-κB signaling. Biomed. Pharmacother. 2021, 141, 111911. [Google Scholar] [CrossRef]
- Wang, X.; Lin, Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leng, H.; Pu, L.; Xu, L.J.; Shi, X.; Ji, J.; Chen, K. Effects of aloe polysaccharide, a polysaccharide extracted from Aloe vera, on TNF-alpha-induced HaCaT cell proliferation and the underlying mechanism in psoriasis. Mol. Med. Rep. 2018, 18, 3537–3543. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.W.; Deng, X.E.; Wang, J.D.; Li, Q. Baicalin Inhibits Cell Proliferation and Inflammatory Cytokines Induced by Tumor Necrosis Factor alpha (TNF-alpha) in Human Immortalized Keratinocytes (HaCaT) Human Keratinocytes by Inhibiting the STAT3/Nuclear Factor kappa B (NF-kappa B) Signaling Pathway. Med. Sci. Monit. 2020, 26, e919392-1. [Google Scholar] [CrossRef]
- van der Vos, K.E.; Eliasson, P.; Proikas-Cezanne, T.; Vervoort, S.J.; van Boxtel, R.; Putker, M.; van Zutphen, I.J.; Mauthe, M.; Zellmer, S.; Pals, C.; et al. Modulation of glutamine metabolism by the PI(3)K-PKB-FOXO network regulates autophagy. Nat. Cell Biol. 2012, 14, 829–837. [Google Scholar] [CrossRef]
- Xu, N.; Lao, Y.; Zhang, Y.; Gillespie, D.A. Akt: A double-edged sword in cell proliferation and genome stability. J. Oncol. 2012, 2012, 951724. [Google Scholar] [CrossRef]
- Kim, J.E.; Son, J.E.; Jeong, H.; Joon Kim, D.; Seo, S.K.; Lee, E.; Lim, T.G.; Kim, J.R.; Chen, H.; Bode, A.M.; et al. A Novel Cinnamon-Related Natural Product with Pim-1 Inhibitory Activity Inhibits Leukemia and Skin Cancer. Cancer Res. 2015, 75, 2716–2728. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Nimlamool, W.; Miller, C.J.; Lou, H.J.; Turk, B.E. Rational Redesign of a Functional Protein Kinase-Substrate Interaction. ACS Chem. Biol. 2017, 12, 1194–1198. [Google Scholar] [CrossRef]
- Burmester, G.R.; Panaccione, R.; Gordon, K.B.; McIlraith, M.J.; Lacerda, A.P. Adalimumab: Long-term safety in 23 458 patients from global clinical trials in rheumatoid arthritis, juvenile idiopathic arthritis, ankylosing spondylitis, psoriatic arthritis, psoriasis and Crohn’s disease. Ann. Rheum. Dis. 2013, 72, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Menter, A.; Thaci, D.; Wu, J.J.; Abramovits, W.; Kerdel, F.; Arikan, D.; Guo, D.; Ganguli, A.; Bereswill, M.; Camez, A.; et al. Long-Term Safety and Effectiveness of Adalimumab for Moderate to Severe Psoriasis: Results from 7-Year Interim Analysis of the ESPRIT Registry. Dermatol. Ther. 2017, 7, 365–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florek, A.G.; Wang, C.J.; Armstrong, A.W. Treatment preferences and treatment satisfaction among psoriasis patients: A systematic review. Arch. Dermatol. Res. 2018, 310, 271–319. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wikan, N.; Hankittichai, P.; Thaklaewphan, P.; Potikanond, S.; Nimlamool, W. Oxyresveratrol Inhibits TNF-α-Stimulated Cell Proliferation in Human Immortalized Keratinocytes (HaCaT) by Suppressing AKT Activation. Pharmaceutics 2022, 14, 63. https://doi.org/10.3390/pharmaceutics14010063
Wikan N, Hankittichai P, Thaklaewphan P, Potikanond S, Nimlamool W. Oxyresveratrol Inhibits TNF-α-Stimulated Cell Proliferation in Human Immortalized Keratinocytes (HaCaT) by Suppressing AKT Activation. Pharmaceutics. 2022; 14(1):63. https://doi.org/10.3390/pharmaceutics14010063
Chicago/Turabian StyleWikan, Nitwara, Phateep Hankittichai, Phatarawat Thaklaewphan, Saranyapin Potikanond, and Wutigri Nimlamool. 2022. "Oxyresveratrol Inhibits TNF-α-Stimulated Cell Proliferation in Human Immortalized Keratinocytes (HaCaT) by Suppressing AKT Activation" Pharmaceutics 14, no. 1: 63. https://doi.org/10.3390/pharmaceutics14010063
APA StyleWikan, N., Hankittichai, P., Thaklaewphan, P., Potikanond, S., & Nimlamool, W. (2022). Oxyresveratrol Inhibits TNF-α-Stimulated Cell Proliferation in Human Immortalized Keratinocytes (HaCaT) by Suppressing AKT Activation. Pharmaceutics, 14(1), 63. https://doi.org/10.3390/pharmaceutics14010063