
Photo-Oxidation of Therapeutic Protein Formulations: From Radical Formation to Analytical Techniques

 , ,
, ,
Abstract
:1. Introduction
2. Measurement of Light Intensity
3. Photo-Oxidation and Oxygen
3.1. Singlet Oxygen (1O2)
3.2. Superoxide Anions (·O2−)
3.3. Hydrogen Peroxide (H2O2)
3.4. Hydroxyl Radicals (·OH)
3.5. Carbon-Centered Radicals (R·), Alkoxyl Radicals (RO·), Peroxyl Radicals (ROO·), and Organic Hydroperoxides (ROOH)
4. Photo-Oxidation of Proteins via ROS and Detection of Specific Modifications
4.1. Structural Perturbations Due to Photo-Oxidation
4.2. Detection of Specific Modifications
4.2.1. Tryptophan Derivatives
4.2.2. Tyrosine
4.2.3. Phenylalanine
4.2.4. Methionine
4.2.5. Cysteine
4.2.6. Histidine
4.2.7. Lysine and Arginine
5. Photo-Oxidation of Excipients
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| OH | hydroxyl radical |
| 1O2 | singlet oxygen, singlet oxygen |
| 2-OH-E+ | 2-hydroxyethidium |
| 3-OH-Kyn | 3-Hydroxykynurenine |
| ABA | p-aminobenzoic acid |
| ABDA | 9,10-anthracenediyl-bis(methylene)dimalonic acid |
| AGEs | advanced glycation endproducts |
| Amplex® Red | N-acetyl-3,7-dihydroxyphenoxazine |
| AMS | 4-acetamido-4‘-maleimidylstilbene-2,2disulfonate |
| APF | 3′-(p-Aminophenyl) fluorescein |
| Arg | arginine |
| ATTA-Eu3+ | [4′-(9-anthryl)-2,2′:6′,2″-terpyridine-6,6″diyl]bis(methylenenitrilo)tetrakis(acetate)-Eu3+ |
| AUC | analytical ultracentrifugation |
| BMPO | 3,4-dihydro-2-methyl-1,1-dimethylethyl ester-2H-pyrrole-2-carboxylic acid-1-oxide |
| CAD | charged aerosol detector |
| CD | circular dichroism |
| CDR | complementary determining region |
| CE | capillary electrophoresis |
| CEX | cation-exchange chromatography, cation-exchange chromatography |
| CID | collision-induced dissociation |
| CML | ε-N-carboxymethyllysine |
| Cys | cysteine |
| cytc | cytochrome c |
| DBZTC | 2-chloro-1,3-dibenzothiazolinecyclohexene |
| DHE | dihydroethidium |
| dimedione | 5,5-dimethyl-1,3-cycloheandione |
| di-Trp | ditryptophan |
| di-Tyr | dityrosine |
| DMPO | 5,5-Dimethyl-1-pyrroline N-oxide |
| DNPH | 2,4-dinitrophenylhydrazine |
| DOPA | 3,4-Dihydroxyphenylalanine |
| DPAX | 9-[2-(3-carboxy-9,10-diphenyl)anthryl]-6-hydroxy-3H-xanthen-3-one |
| DPBF | 1,3-diphenylisobenzofuran |
| DTNB | 5,5’-disulfanediylbis(2-nitrobenzoic acid) |
| DTT | dithiothreitol |
| E+ | ethidium |
| ECD | electron capture dissociation |
| ELISA | enzyme-linked immunosorbent assay |
| ELSD | evaporative light scattering detection |
| EP | endoperoxide |
| EPR | electron paramagnetic resonance spectroscopy |
| ESI | electrospray-ionization |
| ETD | electron-transfer dissociation |
| FA | fatty acid |
| FMA | fluorescence micelle assay |
| FOX | ferrous oxidation−xylenol orange |
| FTIR | Fourier-transform infrared spectroscopy |
| GC | gas chromatography |
| Gly | glycin |
| H2O2 | hydrogen peroxide |
| HDX | Hydrogen-Deuterium exchange |
| HDX-MS | hydrogen deuterium exchange mass spectrometry |
| HIC | hydrophobic-interaction chromatography, hydrophobic-interaction chromatography |
| His | histidine |
| HMW | high molecular weight species |
| HOO· | hydroperoxyl radical |
| HPF | 3′-(p-Hydroxyphenyl) fluorescein |
| HPLC | high performance liquid chromatography |
| HRP | horseradish peroxidase |
| HTPA | 2-hydroxyl terephthalic acid |
| ICG | indocyanine green |
| ICH | International Conference on Harmonization |
| Kyn | kynurenine |
| LC-MS | liquid chromatography with mass spectrometry |
| LOD | limit of detection |
| Lys | lysine |
| MALDI | matrix-assisted laser desorption/ionization |
| Met | methionine |
| MS | mass spectrometry |
| NADH | nicotinamide adenine dinucleotide |
| NBT | nitro blue tetrazolium |
| NFK | N-Formyl kynurenine |
| NIR | near-infrared |
| NMR | nuclear magnetic resonance spectroscopy |
| NPN | N-phenyl-1-naphthylamine |
| O2− | superoxide anions, superoxide anions |
| PEO | poly(ethylene oxide) |
| Phe | phenylalanine |
| POE | polyoxyethylen |
| PPO | poly(propylene oxide) |
| PS | polysorbates |
| PTM | post translational modification |
| R· | organic radicals, carbon-centered radical, carbon-centered radical |
| RO· | alkoxyl radical, alkoxyl radical |
| ROO· | peroxide radical |
| ROOH | organic hydroperoxide, organic hydroperoxide |
| ROS | reactive oxygen species |
| RP | reversed phase, reversed phase |
| SDS-PAGE | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| SEC | size-exclusion chromatography |
| sen | photo-sensitizer |
| SOD | superoxide dismutase |
| SOSG© | singlet oxygen sensor green |
| TBARS | thiobarbituric acid |
| TBT | 2-(2-thienyl)benzothiazoline |
| TEMP | 2,2,6,6-Tetramethylpiperidine |
| TMB | 3,5,3′,5′-tetramethylbenzidine |
| TNB | 5-mercapto-2-nitrobenzoic acid |
| TPA | terephthalic acid |
| TPP | triphenylphosphine |
| TPPO | triphosphine oxide |
| Trp | tryptophan |
| Tyr | tyrosine |
References
- Kesik-Brodacka, M. Progress in biopharmaceutical development. Biotechnol. Appl. Biochem. 2017, 65, 306–322. [Google Scholar] [CrossRef] [Green Version]
- Pattison, D.I.; Rahmanto, A.S.; Davies, M.J. Photo-oxidation of proteins. Photochem. Photobiol. Sci. 2012, 11, 38–53. [Google Scholar] [CrossRef]
- Kerwin, B.A.; Remmele, R.L., Jr. Protect from light: Photodegradation and protein biologics. J. Pharm. Sci. 2007, 96, 1468–1479. [Google Scholar] [CrossRef] [PubMed]
- Tønnesen, H.H. Photostability of Drugs and Drug Formulations; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Mitragotri, S.; Burke, P.A.; Langer, R. Overcoming the challenges in administering biopharmaceuticals: Formulation and delivery strategies. Nat. Rev. Drug Discov. 2014, 13, 655–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Feng, J.; Lin, H.-Y.; Mullapudi, S.; Bishop, E.; Tous, G.I.; Casas-Finet, J.; Hakki, F.; Strouse, A.R.; Schenerman, M.A. Identification of a Single Tryptophan Residue as Critical for Binding Activity in a Humanized Monoclonal Antibody against Respiratory Syncytial Virus. Anal. Chem. 2007, 79, 2797–2805. [Google Scholar] [CrossRef]
- Mason, B.D.; Schöneich, C.; Kerwin, B.A. Effect of pH and Light on Aggregation and Conformation of an IgG1 mAb. Mol. Pharm. 2012, 9, 774–790. [Google Scholar] [CrossRef] [Green Version]
- Qi, P.; Volkin, D.B.; Zhao, H.; Nedved, M.L.; Hughes, R.; Bass, R.; Yi, S.C.; Panek, M.E.; Wang, D.; DalMonte, P.; et al. Characterization of the photodegradation of a human IgG1 monoclonal antibody formulated as a high-concentration liquid dosage form. J. Pharm. Sci. 2009, 98, 3117–3130. [Google Scholar] [CrossRef] [PubMed]
- Amano, M.; Kobayashi, N.; Yabuta, M.; Uchiyama, S.; Fukui, K. Detection of Histidine Oxidation in a Monoclonal Immunoglobulin Gamma (IgG) 1 Antibody. Anal. Chem. 2014, 86, 7536–7543. [Google Scholar] [CrossRef]
- Moussa, E.M.; Panchal, J.P.; Moorthy, B.S.; Blum, J.S.; Joubert, M.K.; Narhi, L.O.; Topp, E.M. Immunogenicity of Therapeutic Protein Aggregates. J. Pharm. Sci. 2016, 105, 417–430. [Google Scholar] [CrossRef] [Green Version]
- Baptista, M.S.; Cadet, J.; Di Mascio, P.; Ghogare, A.A.; Greer, A.; Hamblin, M.R.; Lorente, C.; Nunez, S.C.; Ribeiro, M.S.; Thomas, A.H.; et al. Type I and Type II Photosensitized Oxidation Reactions: Guidelines and Mechanistic Pathways. Photochem. Photobiol. 2017, 93, 912–919. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Polozova, A.; Gruia, F.; Feng, J. Characterization of the Degradation Products of a Color-Changed Monoclonal Antibody: Tryptophan-Derived Chromophores. Anal. Chem. 2014, 86, 6850–6857. [Google Scholar] [CrossRef] [PubMed]
- Silva, E.; Barrias, P.; Lemus, E.F.; Tirapegui, C.; Aspee, A.; Carroll, L.; Davies, M.; López-Alarcón, C. Riboflavin-induced Type 1 photo-oxidation of tryptophan using a high intensity 365 nm light emitting diode. Free Radic. Biol. Med. 2018, 131, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J. Reactive species formed on proteins exposed to singlet oxygen. Photochem. Photobiol. Sci. 2004, 3, 17–25. [Google Scholar] [CrossRef]
- Lemus, E.F.; Mariotti, M.; Hägglund, P.; Leinisch, F.; Fierro, A.; Silva, E.; López-Alarcón, C.; Davies, M.J. Binding of rose bengal to lysozyme modulates photooxidation and cross-linking reactions involving tyrosine and tryptophan. Free Radic. Biol. Med. 2019, 143, 375–386. [Google Scholar] [CrossRef]
- Kanwar, R.; Balasubramanian, D. Structural Studies on Some Dityrosine-Cross-Linked Globular Proteins: Stability Is Weakened, but Activity Is Not Abolished. Biochemistry 2000, 39, 14976–14983. [Google Scholar] [CrossRef] [PubMed]
- Gomyo, T.; Fujimaki, M. Studies on Changes of Protein by Dye Sensitized Photooxidation. Agric. Biol. Chem. Tokyo 1970, 34, 302–309. [Google Scholar] [CrossRef]
- Davies, M.J. Protein oxidation and peroxidation. Biochem. J. 2016, 473, 805–825. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, W.; Schultz-Fademrecht, T.; Blech, M.; Buske, J.; Garidel, P. Investigating photodegradation of antibodies governed by the light dosage. Int. J. Pharm. 2021, 604, 120723. [Google Scholar] [CrossRef]
- Du, C.; Barnett, G.; Borwankar, A.; Lewandowski, A.; Singh, N.; Ghose, S.; Borys, M.; Li, Z.J. Protection of therapeutic antibodies from visible light induced degradation: Use safe light in manufacturing and storage. Eur. J. Pharm. Biopharm. 2018, 127, 37–43. [Google Scholar] [CrossRef]
- Daugaard, S.; Markvart, J.; Bonde, J.P.; Christoffersen, J.; Garde, A.H.; Hansen, M.; Schlunssen, V.; Vestergaard, J.M.; Vistisen, H.T.; Kolstad, H.A. Light Exposure during Days with Night, Outdoor, and Indoor Work. Ann. Work. Expo. Health 2019, 63, 651–665. [Google Scholar] [CrossRef] [PubMed]
- Lingfors, D.; Volotinen, T. Illumination performance and energy saving of a solar fiber optic lighting system. Opt. Express 2013, 21, A642–A655. [Google Scholar] [CrossRef]
- Wallace, A.; Trimble, S.; Valliere-Douglass, J.F.; Allen, M.; Eakin, C.; Balland, A.; Reddy, P.; Treuheit, M.J. Control of Antibody Impurities Induced by Riboflavin in Culture Media During Production. J. Pharm. Sci. 2020, 109, 566–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sackey, S.S.; Vowotor, M.K.; Owusu, A.; Mensah-Amoah, P.; Tatchie, E.T.; Sefa-Ntiri, B.; Hood, C.O.; Atiemo, S.M. Spectroscopic Study of UV Transparency of Some Materials. Environ. Pollut. 2015, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Bertenshaw, D. The standardisation of light and photometry—A historical review. Light. Res. Technol. 2020, 52, 816–848. [Google Scholar] [CrossRef]
- Piechocki, J.T.; Thoma, K. (Eds.) Pharmaceutical Photostability and Stabilization Technology; CRC: Boca Raton, FL, USA, 2007. [Google Scholar]
- Gadgil, R.B. A Study of UV Spectral Transmission Through Different Transparent Media with Spectrophotometer. Indian J. Dermatol. Venereol. Leprol. 1981, 47, 255–258. [Google Scholar]
- Kuhn, H.J.; Braslavsky, S.E.; Schmidt, R. Chemical actinometry (IUPAC Technical Report). Pure Appl. Chem. 2004, 76, 2105–2146. [Google Scholar] [CrossRef]
- Kuhn, H.; Braslavsky, S.; Schmidt, R. Chemical Actinometry. Pure Appl. Chem. 1989, 61, 187–210. [Google Scholar] [CrossRef]
- Roibu, A.; Fransen, S.; Leblebici, M.E.; Meir, G.; Van Gerven, T.; Kuhn, S. An accessible visible-light actinometer for the determination of photon flux and optical pathlength in flow photo microreactors. Sci. Rep. 2018, 8, 5421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolton, J.R.; Stefan, M.I.; Shaw, P.-S.; Lykke, K.R. Determination of the quantum yields of the potassium ferrioxalate and potassium iodide–iodate actinometers and a method for the calibration of radiometer detectors. J. Photochem. Photobiol. A Chem. 2011, 222, 166–169. [Google Scholar] [CrossRef]
- Hatchard, C.G.; Parker, C.A. A new sensitive chemical actinometer—II. Potassium ferrioxalate as a standard chemical actinometer. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1956, 235, 518–536. [Google Scholar] [CrossRef]
- Norton, B. Some Measurement on the Quantum Yield Temperature Coefficient of the Uranyl Oxalate Actinometer at 254 Mu1,2,3. Ohio J. Sci. 1966, 66, 421–425. [Google Scholar]
- Filho, C.A.D.A.; Gomes, D.D.F.; Guedes, J.P.D.M.; Batista, R.M.F.; Santos, B.S. Considerations on the quinine actinometry calibration method used in photostability testing of pharmaceuticals. J. Pharm. Biomed. Anal. 2011, 54, 886–888. [Google Scholar] [CrossRef] [Green Version]
- Christensen, K.L.; Christensen, J.; Frokjaer, S.; Langballe, P.; Hansen, L.L. Influence of temperature and storage time after light exposure on the quinine monohydrochloride chemical actinometric system. Eur. J. Pharm. Sci. 2000, 9, 317–321. [Google Scholar] [CrossRef]
- Baertschi, S.W.; Alsante, K.M.; Tønnesen, H.H. A Critical Assessment of the ICH Guideline on Photostability Testing of New Drug Substances and Products (Q1B): Recommendation for Revision. J. Pharm. Sci. 2010, 99, 2934–2940. [Google Scholar] [CrossRef]
- Buettner, G. The Pecking Order of Free Radicals and Antioxidants: Lipid Peroxidation, α-Tocopherol, and Ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W. Reactions involving singlet oxygen and the superoxide anion. Nature 1976, 262, 420–421. [Google Scholar] [CrossRef]
- Kuznetsova, N.A.; Gretsova, N.S.; Yuzhakova, O.A.; Negrimovskii, V.M.; Kaliya, O.L.; Luk’Yanets, E.A. New Reagents for Determination of the Quantum Efficiency of Singlet Oxygen Generation in Aqueous Media. Russ. J. Gen. Chem. 2001, 71, 36–41. [Google Scholar] [CrossRef]
- Huang, R.; Choe, E.; Min, D. Kinetics for Singlet Oxygen Formation by Riboflavin Photosensitization and the Reaction between Riboflavin and Singlet Oxygen. J. Food Sci. 2006, 69, C726–C732. [Google Scholar] [CrossRef]
- Leem, J.W.; Kim, S.-R.; Choi, K.-H.; Kim, Y.L. Plasmonic photocatalyst-like fluorescent proteins for generating reactive oxygen species. Nano Converg. 2018, 5, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Michaeli, A.; Feitelson, J. Reactivity of Singlet Oxygen Toward Amino Acids and Peptides. Photochem. Photobiol. 1994, 59, 284–289. [Google Scholar] [CrossRef]
- Ogilby, P.R. Singlet oxygen: There is indeed something new under the sun. Chem. Soc. Rev. 2010, 39, 3181–3209. [Google Scholar] [CrossRef] [PubMed]
- Baier, J.; Maier, M.; Engl, R.; Landthaler, M.; Bäumler, W. Time-Resolved Investigations of Singlet Oxygen Luminescence in Water, in Phosphatidylcholine, and in Aqueous Suspensions of Phosphatidylcholine or HT29 Cells. J. Phys. Chem. B 2005, 109, 3041–3046. [Google Scholar] [CrossRef] [PubMed]
- Vittorino, E.; Giancane, G.; Bettini, S.; Valli, L.; Sortino, S. Bichromophoric multilayer films for the light-controlled generation of nitric oxide and singlet oxygen. J. Mater. Chem. 2009, 19, 8253–8258. [Google Scholar] [CrossRef]
- Wilkinson, F.; Helman, W.P.; Ross, A.B. Rate Constants for the Decay and Reactions of the Lowest Electronically Excited Singlet State of Molecular Oxygen in Solution. An Expanded and Revised Compilation. J. Phys. Chem. Ref. Data 1995, 24, 663–677. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, R.; Afshari, E. Collisional Deactivation of O2(1Δg) by Solvent Molecules. Comparative Experiments with 16O2 and 18O2. Ber. Bunsenges. Phys. Chem. 1992, 96, 788–794. [Google Scholar] [CrossRef]
- Carloni, P.; Damiani, E.; Greci, L.; Stipa, P.; Tanfani, F.; Tartaglini, E.; Wozniak, M. On the use of 1,3-diphenylisobenzofuran (DPBF). Reactions with carbon and oxygen centered radicals in model and natural systems. Res. Chem. Intermed. 1993, 19, 395–405. [Google Scholar] [CrossRef]
- Ohyashiki, T.; Nunomura, M.; Katoh, T. Detection of superoxide anion radical in phospholipid liposomal membrane by fluorescence quenching method using 1,3-diphenylisobenzofuran. Biochim. Biophys. Acta Biomembr. 1999, 1421, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Umezawa, N.; Tanaka, K.; Urano, Y.; Kikuchi, K.; Higuchi, T.; Nagano, T. Novel Fluorescent Probes for Singlet Oxygen. Angew. Chem. Int. Ed. 1999, 38, 2899–2901. [Google Scholar] [CrossRef]
- Corey, E.J.; Taylor, W.C. A Study of the Peroxidation of Organic Compounds by Externally Generated Singlet Oxygen Molecules. J. Am. Chem. Soc. 1964, 86, 3881–3882. [Google Scholar] [CrossRef]
- Gollmer, A.; Arnbjerg, J.; Blaikie, F.H.; Pedersen, B.W.; Breitenbach, T.; Daasbjerg, K.; Glasius, M.; Ogilby, P.R. Singlet Oxygen Sensor Green®: Photochemical Behavior in Solution and in a Mammalian Cell. Photochem. Photobiol. 2011, 87, 671–679. [Google Scholar] [CrossRef]
- Tang, C.-Y.; Wu, F.-Y.; Yang, M.-K.; Guo, Y.-M.; Lu, G.-H.; Yang, Y.-H. A Classic Near-Infrared Probe Indocyanine Green for Detecting Singlet Oxygen. Int. J. Mol. Sci. 2016, 17, 219. [Google Scholar] [CrossRef] [PubMed]
- Atchison, J.; Kamila, S.; Nesbitt, H.; Logan, K.A.; Nicholas, D.M.; Fowley, C.; Davis, J.; Callan, B.; McHale, A.P.; Callan, J.F. Iodinated cyanine dyes: A new class of sensitisers for use in NIR activated photodynamic therapy (PDT). Chem. Commun. 2017, 53, 2009–2012. [Google Scholar] [CrossRef]
- Tanaka, K.; Miura, T.; Umezawa, N.; Urano, Y.; Kikuchi, K.; Higuchi, A.T.; Nagano, T. Rational Design of Fluorescein-Based Fluorescence Probes. Mechanism-Based Design of a Maximum Fluorescence Probe for Singlet Oxygen. J. Am. Chem. Soc. 2001, 123, 2530–2536. [Google Scholar] [CrossRef] [PubMed]
- Entradas, T.; Waldron, S.; Volk, M. The detection sensitivity of commonly used singlet oxygen probes in aqueous environments. J. Photochem. Photobiol. B Biol. 2020, 204, 111787. [Google Scholar] [CrossRef] [PubMed]
- Felip-León, C.; Puche, M.; Miravet, J.F.; Galindo, F.; Feliz, M. A spectroscopic study to assess the photogeneration of singlet oxygen by graphene oxide. Mater. Lett. 2019, 251, 45–51. [Google Scholar] [CrossRef]
- Song, B.; Wang, G.; Yuan, J. A new europium chelate-based phosphorescence probe specific for singlet oxygen. Chem. Commun. 2005, 2005, 3553–3555. [Google Scholar] [CrossRef]
- Dong, Y.; Li, G.; Wang, L.; Cao, L.; Li, Y.; Zhao, W. Anti-tumor evaluation of a novel methoxyphenyl substituted chlorin photosensitizer for photodynamic therapy. J. Photochem. Photobiol. B Biol. 2020, 211, 112015. [Google Scholar] [CrossRef]
- Sang, M.; Ma, F.; Xie, J.; Chen, X.-B.; Wang, K.-B.; Qin, X.-C.; Wang, W.-D.; Zhao, J.-Q.; Li, L.-B.; Zhang, J.-P.; et al. High-light induced singlet oxygen formation in cytochrome b6f complex from Bryopsis corticulans as detected by EPR spectroscopy. Biophys. Chem. 2010, 146, 7–12. [Google Scholar] [CrossRef]
- Lagunes, I.; Vázquez-Ortega, F.; Trigos, Á. Singlet Oxygen Detection Using Red Wine Extracts as Photosensitizers. J. Food Sci. 2017, 82, 2051–2055. [Google Scholar] [CrossRef]
- Nardello, V.; Brault, D.; Chavalle, P.; Aubry, J.-M. Measurement of photogenerated singlet oxygen (1O2(1Δg)) in aqueous solution by specific chemical trapping with sodium 1,3-cyclohexadiene-1,4-diethanoate. J. Photochem. Photobiol. B Biol. 1997, 39, 146–155. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, Z.; Zhao, Q.; Chen, X.; Deng, L.; Chen, W.; Jin, Y. Detection of singlet oxygen by chemical trap in ionic liquids. Chem. Phys. Lett. 2019, 739, 136952. [Google Scholar] [CrossRef]
- Lindig, B.A.; Rodgers, M.A.J.; Schaap, A.P. Determination of the lifetime of singlet oxygen in water-d2 using 9,10-anthracenedipropionic acid, a water-soluble probe. J. Am. Chem. Soc. 1980, 102, 5590–5593. [Google Scholar] [CrossRef]
- Tang, W.; Xu, H.; Kopelman, R.; Philbert, M.A. Photodynamic Characterization and In Vitro Application of Methylene Blue-containing Nanoparticle Platforms. Photochem. Photobiol. 2005, 81, 242–249. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, W.; Niu, J.; Chen, Y. Mechanism of Photogenerated Reactive Oxygen Species and Correlation with the Antibacterial Properties of Engineered Metal-Oxide Nanoparticles. ACS Nano 2012, 6, 5164–5173. [Google Scholar] [CrossRef]
- Silva, E.; Ugarte, R.; Andrade, A.; Edwards, A.M. Riboflavin-sensitized photoprocesses of tryptophan. J. Photochem. Photobiol. B Biol. 1994, 23, 43–48. [Google Scholar] [CrossRef]
- Silva, E.; Rückert, V.; Lissi, E.; Abuin, E. Effects of pH and ionic micelles on the riboflavin-sensitized photoprocesses of tryptophan in aqueous solution. J. Photochem. Photobiol. B Biol. 1991, 11, 57–68. [Google Scholar] [CrossRef]
- Wood, P.M. The potential diagram for oxygen at pH 7. Biochem. J. 1988, 253, 287–289. [Google Scholar] [CrossRef] [PubMed]
- Imlay, J.A. Pathways of Oxidative Damage. Annu. Rev. Microbiol. 2003, 57, 395–418. [Google Scholar] [CrossRef]
- Losada-Barreiro, S.; Bravo-Díaz, C. Free radicals and polyphenols: The redox chemistry of neurodegenerative diseases. Eur. J. Med. Chem. 2017, 133, 379–402. [Google Scholar] [CrossRef]
- Kehrer, J.; Robertson, J.; Smith, C. Free Radicals and Reactive Oxygen Species. In Comprehensive Toxicology; Elsevier: Amsterdam, The Netherlands, 2010; pp. 277–307. [Google Scholar] [CrossRef]
- Walling, C. Intermediates in the Reactions of Fenton Type Reagents. Acc. Chem. Res. 1998, 31, 155–157. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M. Biologically relevant metal ion-dependent hydroxyl radical generation. An update. FEBS Lett. 1992, 307, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Koppenol, W.H. The Haber-Weiss cycle—71 years later. Redox Rep. 2002, 7, 59–60. [Google Scholar] [CrossRef]
- Bielski, B.H.J. Reevaluation of the Spectral and Kinetic Properties of Ho2 and O2-Free Radicals. Photochem. Photobiol. 1978, 28, 645–649. [Google Scholar] [CrossRef]
- Vasil’ev, R.; Tsaplev, Y. Light-induced chemiluminescence. Uspekhi Khimii 2006, 75, 1103–1118. [Google Scholar] [CrossRef]
- Yasuta, N.; Takenaka, N.; Takemura, T. Mechanism of Photosensitized Chemiluminescence of 2-Methyl-6-phenylimidazo[1,2-a]pyrazin-3(7H)-one (CLA) in Aqueous Solution. Chem. Lett. 1999, 28, 451–452. [Google Scholar] [CrossRef]
- Lee, J.; Seliger, H.H. Quantum Yields of the Luminol Chemiluminescence Reaction in Aqueous and Aprotic Solvents. Photochem. Photobiol. 1972, 15, 227–237. [Google Scholar] [CrossRef]
- Dorado, A.P.; Llorente, M.A.; Piérola, I.F. Influence of adventitious light scattering on fluorescence anisotropy measurements with front-face excitation. Simulation and correction. J. Photochem. Photobiol. A Chem. 1994, 78, 193–200. [Google Scholar] [CrossRef]
- Wardman, P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: Progress, pitfalls, and prospects. Free Radic. Biol. Med. 2007, 43, 995–1022. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Harrison, D.G. Methods for Detection of Mitochondrial and Cellular Reactive Oxygen Species. Antioxid. Redox Signal. 2014, 20, 372–382. [Google Scholar] [CrossRef] [Green Version]
- Zielonka, J.; Vasquez-Vivar, J.; Kalyanaraman, B. The confounding effects of light, sonication, and Mn(III)TBAP on quantitation of superoxide using hydroethidine. Free Radic. Biol. Med. 2006, 41, 1050–1057. [Google Scholar] [CrossRef]
- Li, H.; Zhang, W.; Fu, Y.; Li, P.; Liu, W.; Chen, J. A novel method for simultaneously screening superoxide anion scavengers and xanthine oxidase inhibitors using hydroethidine as a fluorescent probe coupled with high-performance liquid chromatography-mass spectrometry. Anal. Methods 2020, 12, 255–263. [Google Scholar] [CrossRef]
- Leem, J.W.; Park, J.; Kim, S.-W.; Kim, S.-R.; Choi, S.H.; Choi, K.-H.; Kim, Y.L. Green-Light-Activated Photoreaction via Genetic Hybridization of Far-Red Fluorescent Protein and Silk. Adv. Sci. 2018, 5, 1700863. [Google Scholar] [CrossRef]
- Pagoria, D.; Lee, A.; Geurtsen, W. The effect of camphorquinone (CQ) and CQ-related photosensitizers on the generation of reactive oxygen species and the production of oxidative DNA damage. Biomaterials 2005, 26, 4091–4099. [Google Scholar] [CrossRef]
- Borisenko, G.G.; Martin, I.; Zhao, Q.; Amoscato, A.A.; Tyurina, Y.Y.; Kagan, V.E. Glutathione Propagates Oxidative Stress Triggered by Myeloperoxidase in HL-60 Cells: Evidence for Glutathionyl Radical-Induced Peroxidation of Phospholipids and Cytotoxicity. J. Biol. Chem. 2004, 279, 23453–23462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cline, M.R.; Toscano, J.P. Detection of nitroxyl (HNO) by a prefluorescent probe. J. Phys. Org. Chem. 2011, 24, 993–998. [Google Scholar] [CrossRef]
- Hideg, É.; Barta, C.; Kálai, T.; Vass, I.; Hideg, K.; Asada, K. Detection of Singlet Oxygen and Superoxide with Fluorescent Sensors in Leaves Under Stress by Photoinhibition or UV Radiation. Plant Cell Physiol. 2002, 43, 1154–1164. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Gao, J.; Li, Z.; Zhou, J. Spectrofluorometric Determination of Superoxide Anion Radicals and Superoxide Dismutase Activity Using a 2-(2-Thienyl)Benzothiazoline Probe. Anal. Lett. 2010, 43, 280–288. [Google Scholar] [CrossRef]
- Gao, J.J.; Xu, K.H.; Tang, B.; Yin, L.L.; Yang, G.W.; An, L.G. Selective detection of superoxide anion radicals generated from macrophages by using a novel fluorescent probe. FEBS J. 2007, 274, 1725–1733. [Google Scholar] [CrossRef]
- Butler, J.; Koppenol, W.; Margoliash, E. Kinetics and mechanism of the reduction of ferricytochrome c by the superoxide anion. J. Biol. Chem. 1982, 257, 10747–10750. [Google Scholar] [CrossRef]
- Tarpey, M.M.; Wink, D.A.; Grisham, M.B. Methods for detection of reactive metabolites of oxygen and nitrogen: In vitro and in vivo considerations. Am. J. Physiol. Integr. Comp. Physiol. 2004, 286, R431–R444. [Google Scholar] [CrossRef] [Green Version]
- Greenwald, R.A. Handbook of Methods for Oxygen Radical Research; CRC Press: Boca Raton, FL, USA, 1985. [Google Scholar]
- Mullarkey, C.J.; Edelstein, D.; Brownlee, M. Free radical generation by early glycation products: A mechanism for accelerated atherogenesis in diabetes. Biochem. Biophys. Res. Commun. 1990, 173, 932–939. [Google Scholar] [CrossRef]
- Mossine, V.V.; Linetsky, M.; Glinsky, G.V.; Ortwerth, A.B.J.; Feather, M.S. Superoxide Free Radical Generation by Amadori Compounds: The Role of Acyclic Forms and Metal Ions. Chem. Res. Toxicol. 1999, 12, 230–236. [Google Scholar] [CrossRef]
- Bielski, B.H.J.; Shiue, G.G.; Bajuk, S. Reduction of nitro blue tetrazolium by CO2- and O2- radicals. J. Phys. Chem. 1980, 84, 830–833. [Google Scholar] [CrossRef]
- Choi, H.S.; Kim, J.W.; Cha, Y.-N.; Kim, C. A Quantitative Nitroblue Tetrazolium Assay for Determining Intracellular Superoxide Anion Production in Phagocytic Cells. J. Immunoass. Immunochem. 2006, 27, 31–44. [Google Scholar] [CrossRef]
- Rook, G.A.W.; Steele, J.; Umar, S.; Dockrell, H.M. A simple method for the solubilisation of reduced NBT, and its use as a colorimetric assay for activation of human macrophages by γ-interferon. J. Immunol. Methods 1985, 82, 161–167. [Google Scholar] [CrossRef]
- Rainard, P. A colorimetric microassay for opsonins by reduction of NBT in phagocytosing bovine polymorphs. J. Immunol. Methods 1986, 90, 197–201. [Google Scholar] [CrossRef]
- Ukeda, H.; Shimamura, T.; Tsubouchi, M.; Harada, Y.; Nakai, Y.; Sawamura, M. Spectrophotometric Assay of Superoxide Anion Formed in Maillard Reaction Based on Highly Water-soluble Tetrazolium Salt. Anal. Sci. 2002, 18, 1151–1154. [Google Scholar] [CrossRef] [Green Version]
- Warwar, N.; Mor, A.; Fluhr, R.; Pandian, R.P.; Kuppusamy, P.; Blank, A. Detection and Imaging of Superoxide in Roots by an Electron Spin Resonance Spin-Probe Method. Biophys. J. 2011, 101, 1529–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khramtsov, V.; Berliner, L.J.; Clanton, T.L. NMR spin trapping: Detection of free radical reactions using a phosphorus-containing nitrone spin trap. Magn. Reson. Med. 1999, 42, 228–234. [Google Scholar] [CrossRef]
- Bačić, G.; Spasojević, I.; Šećerov, B.; Mojovic, M. Spin-trapping of oxygen free radicals in chemical and biological systems: New traps, radicals and possibilities. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2008, 69, 1354–1366. [Google Scholar] [CrossRef]
- Konaka, R.; Kasahara, E.; Dunlap, W.C.; Yamamoto, Y.; Chien, K.C.; Inoue, M. Irradiation of titanium dioxide generates both singlet oxygen and superoxide anion. Free Radic. Biol. Med. 1999, 27, 294–300. [Google Scholar] [CrossRef]
- Diaz-Uribe, C.E.; Daza, M.C.; Martínez, F.; Páez-Mozo, E.A.; Guedes, C.L.B.; Di Mauro, E. Visible light superoxide radical anion generation by tetra(4-carboxyphenyl)porphyrin/TiO2: EPR characterization. J. Photochem. Photobiol. A Chem. 2010, 215, 172–178. [Google Scholar] [CrossRef]
- Xia, Q.; Chiang, H.-M.; Yin, J.-J.; Chen, S.; Cai, L.; Yu, H.; Fu, P.P. UVA photoirradiation of benzo[a]pyrene metabolites: Induction of cytotoxicity, reactive oxygen species, and lipid peroxidation. Toxicol. Ind. Health 2013, 31, 898–910. [Google Scholar] [CrossRef]
- Mitchell, D.G.; Rosen, G.M.; Tseitlin, M.; Symmes, B.; Eaton, S.S.; Eaton, G.R. Use of Rapid-Scan EPR to Improve Detection Sensitivity for Spin-Trapped Radicals. Biophys. J. 2013, 105, 338–342. [Google Scholar] [CrossRef] [Green Version]
- Villamena, F.A.; Merle, J.K.; Hadad, C.M.; Zweier, J.L. Superoxide Radical Anion Adduct of 5,5-Dimethyl-1-pyrroline N-Oxide (DMPO). 2. The Thermodynamics of Decay and EPR Spectral Properties. J. Phys. Chem. A 2005, 109, 6089–6098. [Google Scholar] [CrossRef]
- Miller, R.W.; Rapp, U. The oxidation of catechols by reduced flavins and dehydrogenases. An electron spin resonance study of the kinetics and initial products of oxidation. J. Biol. Chem. 1973, 248, 6084–6090. [Google Scholar] [CrossRef]
- Taiwo, F.A. Mechanism of tiron as scavenger of superoxide ions and free electrons. Spectroscopy 2008, 22, 953692. [Google Scholar] [CrossRef]
- Zhu, M.; Lu, J.; Hu, Y.; Liu, Y.; Hu, S.; Zhu, C. Photochemical reactions between 1,4-benzoquinone and O2•−. Environ. Sci. Pollut. 2020, R27, 31289–31299. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R. Free Radical Scavenging Reactions of Tetrahydroxyquinone: A Pulse Radiolysis Study. ChemistrySelect 2016, 1, 1084–1091. [Google Scholar] [CrossRef]
- Daithankar, V.N.; Wang, W.; Trujillo, J.R.; Thorpe, C. Flavin-Linked Erv-Family Sulfhydryl Oxidases Release Superoxide Anion during Catalytic Turnover. Biochemistry 2011, 51, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintero-Saumeth, J.; Rincón, D.A.; Doerr, M.; Daza, M.C. Concerted double proton-transfer electron-transfer between catechol and superoxide radical anion. Phys. Chem. Chem. Phys. 2017, 19, 26179–26190. [Google Scholar] [CrossRef] [PubMed]
- Chin, D.H.; Chiericato, G.; Nanni, E.J.; Sawyer, D.T. Proton-induced disproportionation of superoxide ion in aprotic media. J. Am. Chem. Soc. 1982, 104, 1296–1299. [Google Scholar] [CrossRef]
- Amin, V.; Olson, N. Effect of Temperature on Stability of Hydrogen Peroxide in Milk. J. Dairy Sci. 1967, 50, 1336–1338. [Google Scholar] [CrossRef]
- Noble, R.W.; Gibson, Q.H. The Reaction of Ferrous Horseradish Peroxidase with Hydrogen Peroxide. J. Biol. Chem. 1970, 245, 2409–2413. [Google Scholar] [CrossRef]
- Taylor, R.C.; Cross, P.C. Light Absorption of Aqueous Hydrogen Peroxide Solutions in the Near Ultraviolet Region. J. Am. Chem. Soc. 1949, 71, 2266–2268. [Google Scholar] [CrossRef]
- Kashima-Tanaka, M.; Tsujimoto, Y.; Kawamoto, K.; Senda, N.; Ito, K.; Yamazaki, M. Generation of Free Radicals and/or Active Oxygen by Light or Laser Irradiation of Hydrogen Peroxide or Sodium Hypochlorite. J. Endod. 2003, 29, 141–143. [Google Scholar] [CrossRef]
- Ikai, H.; Nakamura, K.; Shirato, M.; Kanno, T.; Iwasawa, A.; Sasaki, K.; Niwano, Y.; Kohno, M. Photolysis of Hydrogen Peroxide, an Effective Disinfection System via Hydroxyl Radical Formation. Antimicrob. Agents Chemother. 2010, 54, 5086–5091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luna, A.J.; Nascimento, C.A.O.; Chiavone-Filho, O. Photodecomposition of hydrogen peroxide in highly saline aqueous medium. Braz. J. Chem. Eng. 2006, 23, 341–349. [Google Scholar] [CrossRef] [Green Version]
- Baxendale, J.H.; Wilson, J.A. The photolysis of hydrogen peroxide at high light intensities. Trans. Faraday Soc. 1957, 53, 344–356. [Google Scholar] [CrossRef]
- Alfonso-Prieto, M.; Biarnés, X.; Vidossich, P.; Rovira, C. The Molecular Mechanism of the Catalase Reaction. J. Am. Chem. Soc. 2009, 131, 11751–11761. [Google Scholar] [CrossRef] [PubMed]
- Wasylaschuk, W.R.; Harmon, P.A.; Wagner, G.; Harman, A.B.; Templeton, A.C.; Xu, H.; Reed, R.A. Evaluation of Hydroperoxides in Common Pharmaceutical Excipients. J. Pharm. Sci. 2007, 96, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Diwu, Z.; Panchuk-Voloshina, N.; Haugland, R.P. A Stable Nonfluorescent Derivative of Resorufin for the Fluorometric Determination of Trace Hydrogen Peroxide: Applications in Detecting the Activity of Phagocyte NADPH Oxidase and Other Oxidases. Anal. Biochem. 1997, 253, 162–168. [Google Scholar] [CrossRef]
- Gorris, H.; Walt, D.R. Mechanistic Aspects of Horseradish Peroxidase Elucidated through Single-Molecule Studies. J. Am. Chem. Soc. 2009, 131, 6277–6282. [Google Scholar] [CrossRef] [PubMed]
- Nurjayadi, M.; Apriyani, D.; Hasan, U.; Santoso, I.; Kurniadewi, F.; Kartika, I.R.; Agustini, K.; Puspasari, F.; Natalia, D.; Mangunwardoyo, W. Immunogenicity and Specificity of Anti recombinant Protein Fim-C-Salmonella typhimurium Antibody as a Model to Develop Typhoid Vaccine. Procedia Chem. 2016, 18, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Pick, E.; Keisari, Y. A simple colorimetric method for the measurement of hydrogen peroxide produced by cells in culture. J. Immunol. Methods 1980, 38, 161–170. [Google Scholar] [CrossRef]
- Zhao, B.; Summers, F.A.; Mason, R.P. Photooxidation of Amplex red to resorufin: Implications of exposing the Amplex red assay to light. Free Radic. Biol. Med. 2012, 53, 1080–1087. [Google Scholar] [CrossRef] [Green Version]
- Wolff, S.P. [18] Ferrous ion oxidation in presence of ferric ion indicator xylenol orange for measurement of hydroperoxides. Methods Enzymol. 1994, 233, 182–189. [Google Scholar] [CrossRef]
- Jaeger, J.; Sorensen, K.; Wolff, S. Peroxide accumulation in detergents. J. Biochem. Biophys. Methods 1994, 29, 77–81. [Google Scholar] [CrossRef]
- Pinkernell, U.; Effkemann, A.S.; Karst, U. Simultaneous HPLC Determination of Peroxyacetic Acid and Hydrogen Peroxide. Anal. Chem. 1997, 69, 3623–3627. [Google Scholar] [CrossRef]
- Nakamura, T.; Maeda, H. A simple assay for lipid hydroperoxides based on triphenylphosphine oxidation and high-performance liquid chromatography. Lipids 1991, 26, 765–768. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical Review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (⋅OH/⋅O− in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef] [Green Version]
- Stadtman, E.R.; Levine, R.L. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 2003, 25, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, J.M. Lipid peroxidation initiated by superoxide-dependent hydroxyl radicals using complexed iron and hydrogen peroxide. FEBS Lett. 1984, 172, 245–249. [Google Scholar] [CrossRef] [Green Version]
- Hermes-Lima, M.; Willmore, W.G.; Storey, K.B. Quantification of lipid peroxidation in tissue extracts based on Fe(III)xylenol orange complex formation. Free Radic. Biol. Med. 1995, 19, 271–280. [Google Scholar] [CrossRef]
- Gardner, H.W. Oxygen radical chemistry of polyunsaturated fatty acids. Free Radic. Biol. Med. 1989, 7, 65–86. [Google Scholar] [CrossRef]
- Cornwell, O.; Radford, S.E.; Ashcroft, A.E.; Ault, J.R. Comparing Hydrogen Deuterium Exchange and Fast Photochemical Oxidation of Proteins: A Structural Characterisation of Wild-Type and ΔN6 β2-Microglobulin. J. Am. Soc. Mass Spectrom. 2018, 29, 2413–2426. [Google Scholar] [CrossRef] [PubMed]
- Hambly, D.M.; Gross, M.L. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J. Am. Soc. Mass Spectrom. 2005, 16, 2057–2063. [Google Scholar] [CrossRef] [Green Version]
- Aye, T.T.; Low, T.Y.; Sze, S.K. Nanosecond Laser-Induced Photochemical Oxidation Method for Protein Surface Mapping with Mass Spectrometry. Anal. Chem. 2005, 77, 5814–5822. [Google Scholar] [CrossRef]
- Xu, G.; Chance, M.R. Hydroxyl Radical-Mediated Modification of Proteins as Probes for Structural Proteomics. Chem. Rev. 2007, 107, 3514–3543. [Google Scholar] [CrossRef]
- Czapski, G.; Bielski, B.H.J. Absorption spectra of the OH and O- radicals in aqueous solutions. Radiat. Phys. Chem. 1993, 41, 503–505. [Google Scholar] [CrossRef]
- Newton, G.L.; Milligan, J.R. Fluorescence detection of hydroxyl radicals. Radiat. Phys. Chem. 2006, 75, 473–478. [Google Scholar] [CrossRef]
- Jean-M, F.; Xochitl, D.B.; Mika, S. Towards reliable quantification of hydroxyl radicals in the Fenton reaction using chemical probes. RSC Adv. 2018, 8, 5321–5330. [Google Scholar] [CrossRef] [Green Version]
- Lundqvist, M.J.; Eriksson, L.A. Hydroxyl Radical Reactions with Phenol as a Model for Generation of Biologically Reactive Tyrosyl Radicals. J. Phys. Chem. B 2000, 104, 848–855. [Google Scholar] [CrossRef]
- Biondi, R.; Xia, Y.; Rossi, R.; Paolocci, N.; Ambrosio, G.; Zweier, J.L. Detection of Hydroxyl Radicals by d-Phenylalanine Hydroxylation: A Specific Assay for Hydroxyl Radical Generation in Biological Systems. Anal. Biochem. 2001, 290, 138–145. [Google Scholar] [CrossRef]
- Stein, G.; Weiss, J.; Stein, J.W.G. Detection of Free Hydroxyl Radicals by Hydroxylation of Aromatic Compounds. Nature 1950, 166, 1104–1105. [Google Scholar] [CrossRef]
- Freinbichler, W.; Colivicchi, M.A.; Fattori, M.; Ballini, C.; Tipton, K.F.; Linert, W.; Della Corte, L. Validation of a robust and sensitive method for detecting hydroxyl radical formation together with evoked neurotransmitter release in brain microdialysis. J. Neurochem. 2008, 105, 738–749. [Google Scholar] [CrossRef]
- Leichnitz, S.; Heinrich, J.; Kulak, N. A fluorescence assay for the detection of hydrogen peroxide and hydroxyl radicals generated by metallonucleases. Chem. Commun. 2018, 54, 13411–13414. [Google Scholar] [CrossRef]
- Saran, M.; Summer, K.H. Assaying for hydroxyl radicals: Hydroxylated terephthalate is a superior fluorescence marker than hydroxylated benzoate. Free Radic. Res. 1999, 31, 429–436. [Google Scholar] [CrossRef]
- Page, S.E.; Arnold, W.A.; McNeill, K. Terephthalate as a probe for photochemically generated hydroxyl radical. J. Environ. Monit. 2010, 12, 1658–1665. [Google Scholar] [CrossRef]
- Linxiang, L.; Abe, Y.; Nagasawa, Y.; Kudo, R.; Usui, N.; Imai, K.; Mashino, T.; Mochizuki, M.; Miyata, N. An HPLC assay of hydroxyl radicals by the hydroxylation reaction of terephthalic acid. Biomed. Chromatogr. 2004, 18, 470–474. [Google Scholar] [CrossRef]
- Šnyrychová, I.; Hideg, É. The first application of terephthalate fluorescence for highly selective detection of hydroxyl radicals in thylakoid membranes. Funct. Plant Biol. 2007, 34, 1105–1111. [Google Scholar] [CrossRef]
- Price, M.; Reiners, J.J.; Santiago, A.M.; Kessel, D. Monitoring Singlet Oxygen and Hydroxyl Radical Formation with Fluorescent Probes During Photodynamic Therapy. Photochem. Photobiol. 2009, 85, 1177–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setsukinai, K.-I.; Urano, Y.; Kakinuma, K.; Majima, H.J.; Nagano, T. Development of Novel Fluorescence Probes That Can Reliably Detect Reactive Oxygen Species and Distinguish Specific Species. J. Biol. Chem. 2003, 278, 3170–3175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohn, C.; Pedigo, C.; Hylton, S.N.; Simon, S.R.; Schoonen, M.A. Evaluating the use of 3’-(p-Aminophenyl) fluorescein for determining the formation of highly reactive oxygen species in particle suspensions. Geochem. Trans. 2009, 10, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diez, L.; Livertoux, M.-H.; Stark, A.-A.; Wellman-Rousseau, M.; Leroy, P. High-performance liquid chromatographic assay of hydroxyl free radical using salicylic acid hydroxylation during in vitro experiments involving thiols. J. Chromatogr. B Biomed. Sci. Appl. 2001, 763, 185–193. [Google Scholar] [CrossRef]
- Grootveld, M.; Halliwell, B. Aromatic hydroxylation as a potential measure of hydroxyl-radical formation in vivo. Identification of hydroxylated derivatives of salicylate in human body fluids. Biochem. J. 1986, 237, 499–504. [Google Scholar] [CrossRef] [Green Version]
- Freinbichler, W.; Bianchi, L.; Colivicchi, M.A.; Ballini, C.; Tipton, K.F.; Linert, W.; Della Corte, L. The detection of hydroxyl radicals in vivo. J. Inorg. Biochem. 2008, 102, 1329–1333. [Google Scholar] [CrossRef]
- Sel, S.; Nass, N.; Pötzsch, S.; Trau, S.; Simm, A.; Kalinski, T.; Duncker, G.I.; Kruse, F.E.; Auffarth, G.U.; Brömme, H.-J. UVA irradiation of riboflavin generates oxygen-dependent hydroxyl radicals. Redox Rep. 2013, 19, 72–79. [Google Scholar] [CrossRef]
- Biller, J.R.; Tseitlin, M.; Mitchell, D.G.; Yu, Z.; Buchanan, L.A.; Elajaili, H.; Rosen, G.M.; Kao, J.P.Y.; Eaton, S.S.; Eaton, G.R. Improved Sensitivity for Imaging Spin Trapped Hydroxyl Radical at 250 MHz. ChemPhysChem 2014, 16, 528–531. [Google Scholar] [CrossRef]
- Marriott, P.R.; Perkins, M.J.; Griller, D. Spin trapping for hydroxyl in water: A kinetic evaluation of two popular traps. Can. J. Chem. 1980, 58, 803–807. [Google Scholar] [CrossRef]
- Sankuratri, N.; Kotake, Y.; Janzen, E.G. Studies on the stability of oxygen radical spin adducts of a new spin trap: 5-methyl-5-phenylpyrroline-1-oxide (MPPO). Free Radic. Biol. Med. 1996, 21, 889–894. [Google Scholar] [CrossRef]
- Egerton, A.C.; Emte, W.; Minkoff, G.J. Some properties of organic peroxides. Discuss. Faraday Soc. 1951, 10, 278–282. [Google Scholar] [CrossRef]
- Yaremenko, I.; Vil’, V.A.; Demchuk, D.V.; Terent’Ev, A.O. Rearrangements of organic peroxides and related processes. Beilstein J. Org. Chem. 2016, 12, 1647–1748. [Google Scholar] [CrossRef] [Green Version]
- Tommaso, S.D.; Rotureau, P.; Vignes, A.; Alzerreca, M.; Benaissa, W.; Adamo, C. Study of the peroxidation mechanism of diethyl ether (DEE). In Proceedings of the International Symposium on Loss Prevention and Safety Promotion in the Process Industry, Bruges, Belgium, 6–9 June 2010; pp. 327–333. [Google Scholar]
- Davies, M.J. The oxidative environment and protein damage. Biochim. Biophys. Acta Proteins Proteom. 2005, 1703, 93–109. [Google Scholar] [CrossRef]
- Zhu, B.-Z.; Zhao, H.-T.; Kalyanaraman, B.; Liu, J.; Shan, G.-Q.; Du, Y.-G.; Frei, B. Mechanism of metal-independent decomposition of organic hydroperoxides and formation of alkoxyl radicals by halogenated quinones. Proc. Natl. Acad. Sci. USA 2007, 104, 3698–3702. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B.; Gutteridge, J.M. Free Radicals in Biology and Medicine; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Maillard, B.; Ingold, K.U.; Scaiano, J.C. Rate constants for the reactions of free radicals with oxygen in solution. J. Am. Chem. Soc. 1983, 105, 5095–5099. [Google Scholar] [CrossRef]
- Schaich, K.M. Lipid Oxidation: New Perspectives on an Old Reaction. In Bailey’s Industrial Oil and Fat Products; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2020; pp. 1–72. [Google Scholar] [CrossRef]
- Liu, H.; Gu, L.; Zhu, P.; Liu, Z.; Zhou, B. Evaluation on Thermal Hazard of Ter-Butyl Hydroperoxide by Using Accelerating Rate Calorimeter. Procedia Eng. 2012, 45, 574–579. [Google Scholar] [CrossRef] [Green Version]
- Nishiike, T.; Takamura, H.; Matoba, T. Stability of Linoleic Acid Hydroperoxide in the Oil System. J. Cookery Sci. Jpn. 2000, 33, 377–380. [Google Scholar]
- Nishiike, T.; Ichikawa, J.; Kikugawa, N.; Takamura, H.; Matoba, T. Effects of Amino Acids, Sugars, and Ascorbic Acid on the Stability of Linoleic Acid Hydroperoxide in the Water Phase. Biosci. Biotechnol. Biochem. 1999, 63, 1997–2000. [Google Scholar] [CrossRef] [Green Version]
- Pryor, W.A. Oxy-radicals and related species: Their formation, lifetimes, and reactions. Annu. Rev. Physiol. 1986, 48, 657–667. [Google Scholar] [CrossRef]
- Hawkins, C.L.; Davies, M.J. Generation and propagation of radical reactions on proteins. Biochim. Biophys. Acta Bioenergy 2001, 1504, 196–219. [Google Scholar] [CrossRef] [Green Version]
- Schöneich, C. Photo-Degradation of Therapeutic Proteins: Mechanistic Aspects. Pharm. Res. 2020, 37, 45. [Google Scholar] [CrossRef]
- Vlasak, J.; Ionescu, R. Fragmentation of monoclonal antibodies. mAbs 2011, 3, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Tolbert, T.J.; Schöneich, C. Photoinduced Tyrosine Side Chain Fragmentation in IgG4-Fc: Mechanisms and Solvent Isotope Effects. Mol. Pharm. 2018, 16, 258–272. [Google Scholar] [CrossRef]
- Shah, D.D.; Zhang, J.; Maity, H.; Mallela, K.M. Effect of photo-degradation on the structure, stability, aggregation, and function of an IgG1 monoclonal antibody. Int. J. Pharm. 2018, 547, 438–449. [Google Scholar] [CrossRef]
- Cockrell, G.M.; Wolfe, M.S.; Wolfe, J.L.; Schöneich, C. Photoinduced Aggregation of a Model Antibody–Drug Conjugate. Mol. Pharm. 2015, 12, 1784–1797. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Mason, B.D.; Schöneich, C.S.; Carpenter, J.F.; Boone, T.C.; Kerwin, B.A. Light-induced aggregation of type I soluble tumor necrosis factor receptor. J. Pharm. Sci. 2009, 98, 3182–3199. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, M.; Leinisch, F.; Leeming, D.J.; Svensson, B.; Davies, M.J.; Hägglund, P. Mass-Spectrometry-Based Identification of Cross-Links in Proteins Exposed to Photo-Oxidation and Peroxyl Radicals Using 18O Labeling and Optimized Tandem Mass Spectrometry Fragmentation. J. Proteome Res. 2018, 17, 2017–2027. [Google Scholar] [CrossRef] [PubMed]
- Mozziconacci, O.; Kerwin, B.A.; Schoneich, C. Exposure of a monoclonal antibody, IgG1, to UV-light leads to protein dithiohemiacetal and thioether cross-links: A role for thiyl radicals? Chem. Res. Toxicol. 2010, 23, 1310–1312. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, Z.; Cheetham, J.; Ren, D.; Zhou, Z.S. Discovery and Characterization of a Photo-Oxidative Histidine-Histidine Cross-Link in IgG1 Antibody Utilizing 18O-Labeling and Mass Spectrometry. Anal. Chem. 2014, 86, 4940–4948. [Google Scholar] [CrossRef]
- Ehrenshaft, M.; Deterding, L.J.; Mason, R.P. Tripping up Trp: Modification of protein tryptophan residues by reactive oxygen species, modes of detection, and biological consequences. Free Radic. Biol. Med. 2015, 89, 220–228. [Google Scholar] [CrossRef] [Green Version]
- Steinmann, D.; Ji, J.A.; Wang, Y.J.; Schöneich, C. Photodegradation of Human Growth Hormone: A Novel Backbone Cleavage between Glu-88 and Pro-89. Mol. Pharm. 2013, 10, 2693–2706. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Jiménez, J.; Salmerón-García, A.; Cabeza, J.; Vélez, C.; Capitán-Vallvey, L.F.; Navas, N. The Effects of Light-Accelerated Degradation on the Aggregation of Marketed Therapeutic Monoclonal Antibodies Evaluated by Size-Exclusion Chromatography with Diode Array Detection. J. Pharm. Sci. 2016, 105, 1405–1418. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.R.; Zhang, J.; O’Dell, C.; Hsieh, M.-C.; Goldstein, J.; Liu, J.; Srivastava, A. Effect of Polysorbate 80 Quality on Photostability of a Monoclonal Antibody. AAPS Pharm. Sci. Tech. 2012, 13, 422–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeg, S.; Grune, T. Protein Oxidation in Aging: Does It Play a Role in Aging Progression? Antioxid. Redox Signal. 2015, 23, 239–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faraggi, M.; Leopold, J.G. Pulse Radiolysis Studies of Electron-Transfer Reaction in Molecules of Biological Interest: II. The Reduction of Cu(II)-Peptide Complexes. Radiat. Res. 1976, 65, 238. [Google Scholar] [CrossRef]
- Simic, M.G.; Gajewski, E.; Dizdaroglu, M. Kinetics and mechanisms of hydroxyl radical-induced crosslinks between phenylalanine peptides. Radiat. Phys. Chem. 1984, 24, 465–473. [Google Scholar] [CrossRef]
- Kim, H.-J.; Mee, L.K.; Adelstein, S.J.; Taub, I.A.; Carr, S.A.; Reinhold, V.N. Binding Site Specificity of Gamma-Radiation-Induced Crosslinking between Phenylalanine and a Phenylalanine-Containing Tetrapeptide. Radiat. Res. 1984, 100, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Easton, C.; Peters, S. Reactions of α-Substituted Glycine Derivatives with Stannanes in the Presence of Disulfides. Aust. J. Chem. 1994, 47, 859–868. [Google Scholar] [CrossRef]
- Davies, M.J. Protein and Peptide Alkoxyl Radicals Can Give Rise to C-Terminal Decarboxylation and Backbone Cleavage. Arch. Biochem. Biophys. 1996, 336, 163–172. [Google Scholar] [CrossRef]
- Valliere-Douglass, J.; Jones, L.; Shpektor, D.; Kodama, P.; Wallace, A.; Balland, A.; Bailey, R.; Zhang, Y. Separation and Characterization of an IgG2 Antibody Containing a Cyclic Imide in CDR1 of Light Chain by Hydrophobic Interaction Chromatography and Mass Spectrometry. Anal. Chem. 2008, 80, 3168–3174. [Google Scholar] [CrossRef] [PubMed]
- Valliere-Douglass, J.; Wallace, A.; Balland, A. Separation of populations of antibody variants by fine tuning of hydrophobic-interaction chromatography operating conditions. J. Chromatogr. A 2008, 1214, 81–89. [Google Scholar] [CrossRef]
- Xiao, G.; Bondarenko, P.V. Identification and quantification of degradations in the Asp–Asp motifs of a recombinant monoclonal antibody. J. Pharm. Biomed. Anal. 2008, 47, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Cordoba, A.J.; Shyong, B.-J.; Breen, D.; Harris, R.J. Non-enzymatic hinge region fragmentation of antibodies in solution. J. Chromatogr. B 2005, 818, 115–121. [Google Scholar] [CrossRef]
- Liu, H.; Gaza-Bulseco, G.; Chumsae, C. Analysis of reduced monoclonal antibodies using size exclusion chromatography coupled with mass spectrometry. J. Am. Soc. Mass Spectrom. 2009, 20, 2258–2264. [Google Scholar] [CrossRef] [Green Version]
- Hunt, G.; Nashabeh, W. Capillary Electrophoresis Sodium Dodecyl Sulfate Nongel Sieving Analysis of a Therapeutic Recombinant Monoclonal Antibody: A Biotechnology Perspective. Anal. Chem. 1999, 71, 2390–2397. [Google Scholar] [CrossRef] [PubMed]
- Salas-Solano, O.; Tomlinson, B.; Du, S.; Parker, M.; Strahan, A.; Ma, S. Optimization and Validation of a Quantitative Capillary Electrophoresis Sodium Dodecyl Sulfate Method for Quality Control and Stability Monitoring of Monoclonal Antibodies. Anal. Chem. 2006, 78, 6583–6594. [Google Scholar] [CrossRef]
- Ouellette, D.; Alessandri, L.; Piparia, R.; Aikhoje, A.; Chin, A.; Radziejewski, C.; Correia, I. Elevated cleavage of human immunoglobulin gamma molecules containing a lambda light chain mediated by iron and histidine. Anal. Biochem. 2009, 389, 107–117. [Google Scholar] [CrossRef]
- Davagnino, J.; Wong, C.; Shelton, L.; Mankarious, S. Acid hydrolysis of monoclonal antibodies. J. Immunol. Methods 1995, 185, 177–180. [Google Scholar] [CrossRef]
- Smith, M.A.; Easton, M.; Everett, P.; Lewis, G.; Payne, M.; Riveros-Moreno, V.; Allen, G. Specific cleavage of immunoglobulin G by copper ions. Int. J. Pept. Protein Res. 1996, 48, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.E.; Kroon, D.J. Orthoclone OKT3. In Stability and Characterization of Protein and Peptide Drugs; Springer: Boston, MA, USA, 1993; pp. 135–158. [Google Scholar]
- Vlasak, J.; Ionescu, R. Heterogeneity of monoclonal antibodies revealed by charge-sensitive methods. Curr. Pharm. Biotechno. 2008, 9, 468–481. [Google Scholar] [CrossRef]
- Liu, H.; Gaza-Bulseco, G.; Lundell, E. Assessment of antibody fragmentation by reversed-phase liquid chromatography and mass spectrometry. J. Chromatogr. B 2008, 876, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wei, H.; Fu, Y.; Jusuf, S.; Zeng, M.; Ludwig, R.; Krystek, J.S.R.; Chen, G.; Tao, L.; Das, T.K. Isomerization and Oxidation in the Complementarity-Determining Regions of a Monoclonal Antibody: A Study of the Modification–Structure–Function Correlations by Hydrogen–Deuterium Exchange Mass Spectrometry. Anal. Chem. 2016, 88, 2041–2050. [Google Scholar] [CrossRef]
- Perico, N.; Purtell, J.; Dillon, T.M.; Ricci, M.S. Conformational implications of an inversed pH-dependent antibody aggregation. J. Pharm. Sci. 2009, 98, 3031–3042. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.; Bubb, W.A.; Hawkins, C.L.; Davies, M.J. Singlet oxygen-mediated protein oxidation: Evidence for the formation of reactive side chain peroxides on tyrosine residues. Photochem. Photobiol. 2002, 76, 35–46. [Google Scholar] [CrossRef]
- Goosey, J.D.; Zigler, J.S.; Kinoshita, J.H. Cross-Linking of Lens Crystallins in a Photodynamic System: A Process Mediated by Singlet Oxygen. Science 1980, 208, 1278–1280. [Google Scholar] [CrossRef]
- Agarkhed, M.; O’Dell, C.; Hsieh, M.-C.; Zhang, J.; Goldstein, J.; Srivastava, A. Effect of Polysorbate 80 Concentration on Thermal and Photostability of a Monoclonal Antibody. AAPS Pharm. Sci. Tech. 2012, 14, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Sreedhara, A.; Yin, J.; Joyce, M.; Lau, K.; Wecksler, A.T.; Deperalta, G.; Yi, L.; Wang, Y.J.; Kabakoff, B.; Kishore, R.S.K. Effect of ambient light on IgG1 monoclonal antibodies during drug product processing and development. Eur. J. Pharm. Biopharm. 2016, 100, 38–46. [Google Scholar] [CrossRef]
- Lévy, E.; El Banna, N.; Baïlle, D.; Heneman-Masurel, A.; Truchet, S.; Rezaei, H.; Huang, M.-E.; Béringue, V.; Martin, D.; Vernis, L. Causative Links between Protein Aggregation and Oxidative Stress: A Review. Int. J. Mol. Sci. 2019, 20, 3896. [Google Scholar] [CrossRef] [Green Version]
- Gross-Rother, J.; Blech, M.; Preis, E.; Bakowsky, U.; Garidel, P. Particle Detection and Characterization for Biopharmaceutical Applications: Current Principles of Established and Alternative Techniques. Pharmaceutics 2020, 12, 1112. [Google Scholar] [CrossRef]
- Engelsman, J.D.; Garidel, P.; Smulders, R.; Koll, H.; Smith, B.; Bassarab, S.; Seidl, A.; Hainzl, O.; Jiskoot, W. Strategies for the Assessment of Protein Aggregates in Pharmaceutical Biotech Product Development. Pharm. Res. 2010, 28, 920–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Kapp, E.A.; Lothian, A.; Roberts, A.M.; Vasil’Ev, Y.V.; Boughton, B.A.; Barnham, K.J.; Kok, W.M.; Hutton, C.A.; Masters, C.L.; et al. Characterization and Identification of Dityrosine Cross-Linked Peptides Using Tandem Mass Spectrometry. Anal. Chem. 2017, 89, 6136–6145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.-F.; Chen, Y.; Yi, L.; Brantley, T.; Stanley, B.; Sosic, Z.; Zang, L. Discovery and Characterization of Histidine Oxidation Initiated Cross-links in an IgG1 Monoclonal Antibody. Anal. Chem. 2017, 89, 7915–7923. [Google Scholar] [CrossRef] [PubMed]
- Hägglund, P.; Mariotti, M.; Davies, M. Identification and characterization of protein cross-links induced by oxidative reactions. Expert Rev. Proteom. 2018, 15, 665–681. [Google Scholar] [CrossRef]
- Reid, L.O.; Roman, E.A.; Thomas, A.H.; Dántola, M.L. Photooxidation of Tryptophan and Tyrosine Residues in Human Serum Albumin Sensitized by Pterin: A Model for Globular Protein Photodamage in Skin. Biochemistry 2016, 55, 4777–4786. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Carroll, L.; Rasmussen, L.M.; Davies, M.J. Oxidation of protein disulfide bonds by singlet oxygen gives rise to glutathionylated proteins. Redox Biol. 2020, 38, 101822. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.E.; Slaney, T.R.; Wu, L.; Das, T.K.; Kar, S.; Barnett, G.V.; Leone, A.; Tessier, P.M. Unique Impacts of Methionine Oxidation, Tryptophan Oxidation, and Asparagine Deamidation on Antibody Stability and Aggregation. J. Pharm. Sci. 2019, 109, 656–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritzsche, R.; Ihling, C.H.; Götze, M.; Sinz, A. Optimizing the enrichment of cross-linked products for mass spectrometric protein analysis. Rapid Commun. Mass Spectrom. 2012, 26, 653–658. [Google Scholar] [CrossRef]
- Eshan, K.; Subodh, K.M.; Amit, K. Emerging Methods for Structural Analysis of Protein Aggregation. Protein Pept. Lett. 2017, 24, 331–339. [Google Scholar] [CrossRef]
- Munishkina, L.A.; Fink, A.L. Fluorescence as a method to reveal structures and membrane-interactions of amyloidogenic proteins. Biochim. Biophys. Acta Biomembr. 2007, 1768, 1862–1885. [Google Scholar] [CrossRef] [Green Version]
- McAvan, B.S.; Bowsher, L.A.; Powell, T.; O’Hara, J.F.; Spitali, M.; Goodacre, R.; Doig, A.J. Raman Spectroscopy to Monitor Post-Translational Modifications and Degradation in Monoclonal Antibody Therapeutics. Anal. Chem. 2020, 92, 10381–10389. [Google Scholar] [CrossRef] [PubMed]
- Hinterholzer, A.; Stanojlovic, V.; Regl, C.; Huber, C.G.; Cabrele, C.; Schubert, M. Identification and Quantification of Oxidation Products in Full-Length Biotherapeutic Antibodies by NMR Spectroscopy. Anal. Chem. 2020, 92, 9666–9673. [Google Scholar] [CrossRef]
- Staub, A.; Guillarme, D.; Schappler, J.; Veuthey, J.-L.; Rudaz, S. Intact protein analysis in the biopharmaceutical field. J. Pharm. Biomed. Anal. 2011, 55, 810–822. [Google Scholar] [CrossRef] [PubMed]
- Ravi, J.; Hills, A.E.; Cerasoli, E.; Rakowska, P.D.; Ryadnov, M.G. FTIR markers of methionine oxidation for early detection of oxidized protein therapeutics. Eur. Biophys. J. 2011, 40, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Barth, A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta Bioenergy 2007, 1767, 1073–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garidel, P. Single particle characterisation in biologies: From mid-infrared micro-spectroscopy and mapping to spectral imaging. Spectrosc. Eur. 2013, 25, 19–22. [Google Scholar]
- Garidel, P.; Schott, H. Fourier-transform midinfrared spectroscopy for analysis and screening of liquid protein formulations. Part 1: Understanding infrared spectroscopy of proteins. BioProcess Int. 2006, 4, 40–46. [Google Scholar]
- Garidel, P.; Schott, H. Fourier-transform midinfrared spectroscopy for analysis and screening of liquid protein formulations. Part 2: Detailed analysis and applications. Bioprocess Int. 2006, 4, 48–55. [Google Scholar]
- Blaffert, J.; Haeri, H.H.; Blech, M.; Hinderberger, D.; Garidel, P. Spectroscopic methods for assessing the molecular origins of macroscopic solution properties of highly concentrated liquid protein solutions. Anal. Biochem. 2018, 561–562, 70–88. [Google Scholar] [CrossRef]
- Lew, C.; Gallegos-Perez, J.-L.; Fonslow, B.; Lies, M.; Guttman, A. Rapid Level-3 Characterization of Therapeutic Antibodies by Capillary Electrophoresis Electrospray Ionization Mass Spectrometry. J. Chromatogr. Sci. 2015, 53, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Skjoldager, N.; Bang, M.B.; Rykær, M.; Björnberg, O.; Davies, M.; Svensson, B.; Harris, P.; Hägglund, P. The structure of Lactococcus lactis thioredoxin reductase reveals molecular features of photo-oxidative damage. Sci. Rep. 2017, 7, srep46282. [Google Scholar] [CrossRef] [Green Version]
- Haberger, M.; Leiss, M.; Heidenreich, A.-K.; Pester, O.; Hafenmair, G.; Hook, M.; Bonnington, L.; Wegele, H.; Haindl, M.; Reusch, D.; et al. Rapid characterization of biotherapeutic proteins by size-exclusion chromatography coupled to native mass spectrometry. mAbs 2015, 8, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Muneeruddin, K.; Thomas, J.J.; Salinas, P.A.; Kaltashov, I.A. Characterization of Small Protein Aggregates and Oligomers Using Size Exclusion Chromatography with Online Detection by Native Electrospray Ionization Mass Spectrometry. Anal. Chem. 2014, 86, 10692–10699. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, R.A.; Stewart, A.E. Analysis of protein modifications: Recent advances in detection, characterization and mapping. Curr. Opin. Biotechnol. 1994, 5, 85–93. [Google Scholar] [CrossRef]
- Verrastro, I.; Pasha, S.; Jensen, K.T.; Pitt, A.R.; Spickett, C.M. Mass Spectrometry-Based Methods for Identifying Oxidized Proteins in Disease: Advances and Challenges. Biomolecules 2015, 5, 378–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzmann, J.; Hausberger, A.; Rupprechter, A.; Toll, H. Top-down MS for rapid methionine oxidation site assignment in filgrastim. Anal. Bioanal. Chem. 2013, 405, 6667–6674. [Google Scholar] [CrossRef] [Green Version]
- Kükrer, B.; Filipe, V.; Van Duijn, E.; Kasper, P.T.; Vreeken, R.; Heck, A.; Jiskoot, W. Mass Spectrometric Analysis of Intact Human Monoclonal Antibody Aggregates Fractionated by Size-Exclusion Chromatography. Pharm. Res. 2010, 27, 2197–2204. [Google Scholar] [CrossRef] [Green Version]
- Regl, C.; Wohlschlager, T.; Holzmann, J.; Huber, C.G. A Generic HPLC Method for Absolute Quantification of Oxidation in Monoclonal Antibodies and Fc-Fusion Proteins Using UV and MS Detection. Anal. Chem. 2017, 89, 8391–8398. [Google Scholar] [CrossRef]
- Sokolowska, I.; Mo, J.; Dong, J.; Lewis, M.J.; Hu, P. Subunit mass analysis for monitoring antibody oxidation. mAbs 2017, 9, 498–505. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, X.; Liu, Y.-H.; Richardson, D.; Li, H.; Shameem, M.; Yang, X. Simultaneous monitoring of oxidation, deamidation, isomerization, and glycosylation of monoclonal antibodies by liquid chromatography-mass spectrometry method with ultrafast tryptic digestion. mAbs 2016, 8, 1477–1486. [Google Scholar] [CrossRef] [Green Version]
- Hageman, T.; Wei, H.; Kuehne, P.; Fu, J.; Ludwig, R.; Tao, L.; Leone, A.; Zocher, M.; Das, T.K. Impact of Tryptophan Oxidation in Complementarity-Determining Regions of Two Monoclonal Antibodies on Structure-Function Characterized by Hydrogen-Deuterium Exchange Mass Spectrometry and Surface Plasmon Resonance. Pharm. Res. 2018, 36, 24. [Google Scholar] [CrossRef] [Green Version]
- Tinnefeld, V.; Venne, A.S.; Sickmann, A.; Zahedi, R.P. Enrichment of Cross-Linked Peptides Using Charge-Based Fractional Diagonal Chromatography (ChaFRADIC). J. Proteome Res. 2017, 16, 459–469. [Google Scholar] [CrossRef]
- Schmidt, R.; Sinz, A. Improved single-step enrichment methods of cross-linked products for protein structure analysis and protein interaction mapping. Anal. Bioanal. Chem. 2017, 409, 2393–2400. [Google Scholar] [CrossRef]
- Medinas, D.B.; Gozzo, F.C.; Santos, L.F.; Iglesias, A.H.; Augusto, O. A ditryptophan cross-link is responsible for the covalent dimerization of human superoxide dismutase 1 during its bicarbonate-dependent peroxidase activity. Free Radic. Biol. Med. 2010, 49, 1046–1053. [Google Scholar] [CrossRef]
- Degendorfer, G.; Chuang, C.Y.; Mariotti, M.; Hammer, A.; Hoefler, G.; Hägglund, P.; Malle, E.; Wise, S.G.; Davies, M.J. Exposure of tropoelastin to peroxynitrous acid gives high yields of nitrated tyrosine residues, di-tyrosine cross-links and altered protein structure and function. Free Radic. Biol. Med. 2018, 115, 219–231. [Google Scholar] [CrossRef] [Green Version]
- Leinisch, F.; Mariotti, M.; Rykaer, M.; Lopez, C.; Hägglund, P.; Davies, M.J. Peroxyl radical- and photo-oxidation of glucose 6-phosphate dehydrogenase generates cross-links and functional changes via oxidation of tyrosine and tryptophan residues. Free Radic. Biol. Med. 2017, 112, 240–252. [Google Scholar] [CrossRef] [Green Version]
- Leinisch, F.; Mariotti, M.; Hägglund, P.; Davies, M.J. Structural and functional changes in RNAse A originating from tyrosine and histidine cross-linking and oxidation induced by singlet oxygen and peroxyl radicals. Free Radic. Biol. Med. 2018, 126, 73–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creed, D. The Photophysics and Photochemitry of the near UV absorbing Amino acids—I. Tryptophan and its simple derivates. Photochem. Photobiol. 1984, 39, 537–562. [Google Scholar] [CrossRef]
- Ananthanarayanan, V.S.; Bigelow, C.C. Unusual difference spectra of proteins containing tryptophan. I. Studies with model compounds. Biochemistry 1969, 8, 3717–3723. [Google Scholar] [CrossRef] [PubMed]
- Schmid, F.-X. Biological Macromolecules: UV-visible Spectrophotometry. eLS 2001. [Google Scholar] [CrossRef]
- Mujika, J.I.; Uranga, J.; Matxain, J.M. Computational Study on the Attack of.OH Radicals on Aromatic Amino Acids. Chem. A Eur. J. 2013, 19, 6862–6873. [Google Scholar] [CrossRef]
- Davies, M.J.; Fu, S.; Wang, H.; Dean, R.T. Stable markers of oxidant damage to proteins and their application in the study of human disease. Free Radic. Biol. Med. 1999, 27, 1151–1163. [Google Scholar] [CrossRef]
- Fuentes-Lemus, E.; Dorta, E.; Escobar, E.; Aspée, A.; Pino, E.; Abasq, M.L.; Speisky, H.; Silva, E.; Lissi, E.; Davies, M.J.; et al. Oxidation of free, peptide and protein tryptophan residues mediated by AAPH-derived free radicals: Role of alkoxyl and peroxyl radicals. RSC Adv. 2016, 6, 57948–57955. [Google Scholar] [CrossRef]
- Dreaden, T.M.; Chen, J.; Rexroth, S.; Barry, B.A. N-Formylkynurenine as a Marker of High Light Stress in Photosynthesis. J. Biol. Chem. 2011, 286, 22632–22641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walrant, P.; Santus, R. N-Formyl-Kynurenine, A Tryptophan Photooxidation Product, As A Photodynamic Sensitizer. Photochem. Photobiol. 1974, 19, 411–417. [Google Scholar] [CrossRef]
- Kessel, L.; Nielsen, I.B.; Bochenkova, A.V.; Bravaya, K.B.; Andersen, L.H. Gas-Phase Spectroscopy of Protonated 3-OH Kynurenine and Argpyrimidine. Comparison of Experimental Results to Theoretical Modeling. J. Phys. Chem. A 2007, 111, 10537–10543. [Google Scholar] [CrossRef]
- Korlimbinis, A.; Hains, P.G.; Truscott, R.J.W.; Aquilina, J.A. 3-Hydroxykynurenine Oxidizes α-Crystallin: Potential Role in Cataractogenesis. Biochemistry 2006, 45, 1852–1860. [Google Scholar] [CrossRef]
- Goldstein, L.E.; Leopold, M.C.; Huang, X.; Atwood, C.S.; Saunders, A.J.; Hartshorn, M.; Lim, J.T.; Faget, K.Y.; Muffat, J.A.; Scarpa, R.C.; et al. 3-Hydroxykynurenine and 3-Hydroxyanthranilic Acid Generate Hydrogen Peroxide and Promote α-Crystallin Cross-Linking by Metal Ion Reduction. Biochemistry 2000, 39, 7266–7275. [Google Scholar] [CrossRef]
- Parker, N.R.; Jamie, J.; Davies, M.J.; Truscott, R.J. Protein-bound kynurenine is a photosensitizer of oxidative damage. Free Radic. Biol. Med. 2004, 37, 1479–1489. [Google Scholar] [CrossRef] [PubMed]
- Haywood, J.; Mozziconacci, O.; Allegre, K.M.; Kerwin, B.A.; Schöneich, C. Light-Induced Conversion of Trp to Gly and Gly Hydroperoxide in IgG1. Mol. Pharm. 2013, 10, 1146–1150. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; Fu, S.; Dean, R.T. Protein hydroperoxides can give rise to reactive free radicals. Biochem. J. 1995, 305, 643–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakowicz, J. Principles of Fluorescence Spectroscopy; Springer: Boston, MA, USA, 2006; Volume 1. [Google Scholar]
- Dalsgaard, T.K.; Otzen, D.; Nielsen, J.H.; Larsen, L.B. Changes in Structures of Milk Proteins upon Photo-oxidation. J. Agric. Food Chem. 2007, 55, 10968–10976. [Google Scholar] [CrossRef]
- Paz, A.; Roth, E.; Ashani, Y.; Xu, Y.; Shnyrov, V.L.; Sussman, J.L.; Silman, I.; Weiner, L. Structural and functional characterization of the interaction of the photosensitizing probe methylene blue with Torpedo californica acetylcholinesterase. Protein Sci. 2012, 21, 1138–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simat, T.J.; Steinhart, H. Oxidation of Free Tryptophan and Tryptophan Residues in Peptides and Proteins. J. Agric. Food Chem. 1998, 46, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wang, S.; Liu, J.; Raghani, A. Determination of tryptophan oxidation of monoclonal antibody by reversed phase high performance liquid chromatography. J. Chromatogr. A 2007, 1156, 174–182. [Google Scholar] [CrossRef]
- Fukunaga, Y.; Katsuragi, Y.; Izumi, T.; Sakiyama, F. Fluorescence Characteristics of Kynurenine and N′-Formylkynurenine, Their Use as Reporters of the Environment of Tryptophan 62 in Hen Egg-White Lysozyme1. J. Biochem. 1982, 92, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Domingues, M.R.M.; Domingues, P.; Reis, A.; Fonseca, C.; Amado, F.M.L.; Ferrer-Correia, A.J.V. Identification of oxidation products and free radicals of tryptophan by mass spectrometry. J. Am. Soc. Mass Spectrom. 2003, 14, 406–416. [Google Scholar] [CrossRef] [Green Version]
- Perdivara, I.; Deterding, L.J.; Przybylski, M.; Tomer, K.B. Mass spectrometric identification of oxidative modifications of tryptophan residues in proteins: Chemical artifact or post-translational modification? J. Am. Soc. Mass Spectr. 2010, 21, 1114–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staniszewska, M.; Nagaraj, R.H. Detection of kynurenine modifications in proteins using a monoclonal antibody. J. Immunol. Methods 2007, 324, 63–73. [Google Scholar] [CrossRef]
- Ehrenshaft, M.; Silva, S.D.O.; Perdivara, I.; Bilski, P.; Sik, R.H.; Chignell, C.F.; Tomer, K.B.; Mason, R.P. Immunological detection of N-formylkynurenine in oxidized proteins. Free Radic. Biol. Med. 2009, 46, 1260–1266. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, C.L.; Morgan, P.E.; Davies, M.J. Quantification of protein modification by oxidants. Free Radic. Biol. Med. 2009, 46, 965–988. [Google Scholar] [CrossRef]
- Chen, R.F. Measurements of absolute values in biochemical fluorescence spectroscopy. J. Res. Natl. Bur. Stand. Sect. A Phys. Chem. 1972, 76A, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Pavon, J.A.; Xiao, L.; Li, X.; Zhao, J.; Aldredge, D.; Dank, E.; Fridman, A.; Liu, Y.-H. Selective Tryptophan Oxidation of Monoclonal Antibodies: Oxidative Stress and Modeling Prediction. Anal. Chem. 2019, 91, 2192–2200. [Google Scholar] [CrossRef]
- Dunn, J.D.; Reid, G.E.; Bruening, M.L. Techniques for phosphopeptide enrichment prior to analysis by mass spectrometry. Mass Spectrom. Rev. 2009, 29, 29–54. [Google Scholar] [CrossRef]
- Gundry, R.L.; White, M.Y.; Murray, C.I.; Kane, L.A.; Fu, Q.; Stanley, B.A.; Van Eyk, J.E. Preparation of Proteins and Peptides for Mass Spectrometry Analysis in a Bottom-Up Proteomics Workflow. Curr. Protoc. Mol. Biol. 2010, 10, 1025. [Google Scholar] [CrossRef] [Green Version]
- Yates, J.R.; Ruse, C.I.; Nakorchevsky, A. Proteomics by Mass Spectrometry: Approaches, Advances, and Applications. Annu. Rev. Biomed. Eng. 2009, 11, 49–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrenshaft, M.; Bonini, M.G.; Feng, L.; Chignell, C.F.; Mason, R.P. Partial Colocalization of Oxidized, N-formylkynurenine-containing Proteins in Mitochondria and Golgi of Keratinocytes. Photochem. Photobiol. 2010, 86, 752–756. [Google Scholar] [CrossRef] [PubMed]
- Ehrenshaft, M.; Roberts, J.E.; Mason, R.P. Hypericin-mediated photooxidative damage of α-crystallin in human lens epithelial cells. Free Radic. Biol. Med. 2013, 60, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Sherin, P.S.; Zelentsova, E.A.; Sormacheva, E.D.; Yanshole, V.V.; Duzhak, T.G.; Tsentalovich, Y.P. Aggregation of α-crystallins in kynurenic acid-sensitized UVA photolysis under anaerobic conditions. Phys. Chem. Chem. Phys. 2015, 18, 8827–8839. [Google Scholar] [CrossRef]
- Carroll, L.; Pattison, D.I.; Davies, J.B.; Anderson, R.F.; Lopez, C.; Davies, M. Formation and detection of oxidant-generated tryptophan dimers in peptides and proteins. Free Radic. Biol. Med. 2017, 113, 132–142. [Google Scholar] [CrossRef]
- Aeschbach, R.; Amadoò, R.; Neukom, H. Formation of dityrosine cross-links in proteins by oxidation of tyrosine residues. Biochim. Biophys. Acta Protein Struct. 1976, 439, 292–301. [Google Scholar] [CrossRef]
- Giulivi, C.; Traaseth, N.J.; Davies, K.J.A. Tyrosine oxidation products: Analysis and biological relevance. Amino Acids 2003, 25, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, S.S.; Fasman, G.D. Ultraviolet Irradiation Effects in Poly-L-tyrosine and Model Compounds. Identification of Bityrosine as a Photoproduct. Biochemistry 1967, 6, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Malencik, D.A.; Sprouse, J.F.; Swanson, C.A.; Anderson, S.R. Dityrosine: Preparation, Isolation, and Analysis. Anal. Biochem. 1996, 242, 202–213. [Google Scholar] [CrossRef]
- Davies, M.J.; Truscott, R.J. Photo-oxidation of proteins and its role in cataractogenesis. J. Photochem. Photobiol. B Biol. 2001, 63, 114–125. [Google Scholar] [CrossRef]
- Mahmoud, S.F.; Bialkowski, S. Detection of Dityrosine in Apoferritin. Appl. Spectrosc. 1995, 49, 1677–1681. [Google Scholar] [CrossRef]
- Giulivi, C.; Davies, K.J. Dityrosine: A marker for oxidatively modified proteins and selective proteolysis. Methods Enzymol. 1994, 233, 363–371. [Google Scholar] [CrossRef]
- Huggins, T.G.; Wells-Knecht, M.C.; Detorie, N.; Baynes, J.W.; Thorpe, S.R. Formation of o-tyrosine and dityrosine in proteins during radiolytic and metal-catalyzed oxidation. J. Biol. Chem. 1993, 268, 12341–12347. [Google Scholar] [CrossRef]
- Herold, S. Nitrotyrosine, dityrosine, and nitrotryptophan formation from metmyoglobin, hydrogen peroxide, and nitrite. Free Radic. Biol. Med. 2004, 36, 565–579. [Google Scholar] [CrossRef]
- Malencik, D.A.; Anderson, S.R. Dityrosine as a product of oxidative stress and fluorescent probe. Amino Acids 2003, 25, 233–247. [Google Scholar] [CrossRef]
- Atwood, C.S.; Perry, G.; Zeng, H.; Kato, Y.; Jones, W.D.; Ling, K.-Q.; Huang, X.; Moir, R.D.; Wang, D.; Sayre, L.M.; et al. Copper mediates dityrosine cross-linking of Alzheimer’s amyloid-beta. Biochemistry 2004, 43, 560–568. [Google Scholar] [CrossRef]
- Trnka, M.; Baker, P.R.; Robinson, P.J.J.; Burlingame, A.L.; Chalkley, R.J. Matching Cross-linked Peptide Spectra: Only as Good as the Worse Identification. Mol. Cell. Proteom. 2014, 13, 420–434. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Maruyama, W.; Naoi, M.; Hashizume, Y.; Osawa, T. Immunohistochemical detection of dityrosine in lipofuscin pigments in the aged human brain. FEBS Lett. 1998, 439, 231–234. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Wub, X.; Naitoc, M.; Nomurac, H.; Kitamotoa, N.; Osawa, T. Immunochemical Detection of Protein Dityrosine in Atherosclerotic Lesion of Apo-E-Deficient Mice Using a Novel Monoclonal Antibody. Biochem. Biophys. Res. Commun. 2000, 275, 11–15. [Google Scholar] [CrossRef]
- Paz, M.A.; Fluckiger, R.; Boak, A.; Kagan, H.M.; Gallop, P.M. Specific detection of quinoproteins by redox-cycling staining. J. Biol. Chem. 1991, 266, 689–692. [Google Scholar] [CrossRef]
- Waite, J.H. Determination of (catecholato)borate complexes using difference spectrophotometry. Anal. Chem. 1984, 56, 1935–1939. [Google Scholar] [CrossRef]
- Armstrong, S.G.; Dean, R.T. A sensitive fluorometric assay for protein-bound DOPA and related products of radical-mediated protein oxidation. Redox Rep. 1995, 1, 291–298. [Google Scholar] [CrossRef]
- Ke, Z.; Huang, Q. Effect of protein structure and/or conformation on the dityrosine cross-linking induced by haem-hydrogen peroxide. Biochim. Biophys. Acta 2016, 1860, 2232–2238. [Google Scholar] [CrossRef]
- Kumarathasan, P.; Vincent, R. New approach to the simultaneous analysis of catecholamines and tyrosines in biological fluids. J. Chromatogr. A 2002, 987, 349–358. [Google Scholar] [CrossRef]
- Dyer, J.; Bringans, S.; Bryson, W. Characterisation of photo-oxidation products within photoyellowed wool proteins: Tryptophan and tyrosine derived chromophores. Photochem. Photobiol. Sci. 2006, 5, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Pattison, D.I.; Dean, R.T.; Davies, M.J. Oxidation of DNA, proteins and lipids by DOPA, protein-bound DOPA, and related catechol(amine)s. Toxicology 2002, 177, 23–37. [Google Scholar] [CrossRef]
- Eslami, M.; Zare, H.R.; Namazian, M. Thermodynamic Parameters of Electrochemical Oxidation of l-DOPA: Experimental and Theoretical Studies. J. Phys. Chem. B 2012, 116, 12552–12557. [Google Scholar] [CrossRef]
- McPherson, P.A.; Türemen, B.T. 3,4-Dihydroxy-l-phenylalanine as a biomarker of oxidative damage in proteins: Improved detection using cloud-point extraction and HPLC. Biochem. Biophys. Res. Commun. 2014, 452, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Maidt, M.L.; Yu, Z.; Sang, H.; Markesbery, W.R.; Floyd, R.A. Electrochemical Analysis of Protein Nitrotyrosine and Dityrosine in the Alzheimer Brain Indicates Region-Specific Accumulation. J. Neurosci. 1998, 18, 8126–8132. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; Gilbert, B.C. ChemInform Abstract: Free Radical Reactions: Fragmentation and Rearrangements in Aqueous Solution. ChemInform 1992, 23, 285. [Google Scholar] [CrossRef]
- Dizdaroglu, M.; Simic, M. Radiation-induced conversion of phenylalanine to tyrosines. Radiat. Res. 1980, 83, 437. [Google Scholar]
- Orhan, H.; Vermeulen, N.P.; Tump, C.; Zappey, H.; Meerman, J.H. Simultaneous determination of tyrosine, phenylalanine and deoxyguanosine oxidation products by liquid chromatography-tandem mass spectrometry as non-invasive biomarkers for oxidative damage. J. Chromatogr. B 2004, 799, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, O.H.; Montalvo, R. Radiolysis of Phenylalanine and Tyrosine and Aqueous Solution. Radiat. Res. 1969, 40, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Martín-Tornero, E.; Sierra-Tadeo, F.J.; Durán-Merás, I.; Espinosa-Mansilla, A. Phenylalanine Photoinduced Fluorescence and Characterization of the Photoproducts by LC-MS. J. Fluoresc. 2019, 29, 1445–1455. [Google Scholar] [CrossRef]
- Balakrishnan, G.; Barnett, G.V.; Kar, S.R.; Das, T.K. Detection and Identification of the Vibrational Markers for the Quantification of Methionine Oxidation in Therapeutic Proteins. Anal. Chem. 2018, 90, 6959–6966. [Google Scholar] [CrossRef]
- Mo, J.; Yan, Q.; So, C.K.; Soden, T.; Lewis, M.J.; Hu, P. Understanding the Impact of Methionine Oxidation on the Biological Functions of IgG1 Antibodies Using Hydrogen/Deuterium Exchange Mass Spectrometry. Anal. Chem. 2016, 88, 9495–9502. [Google Scholar] [CrossRef]
- Pan, H.; Chen, K.; Chu, L.; Kinderman, F.; Apostol, I.; Huang, G. Methionine oxidation in human IgG2 Fc decreases binding affinities to protein A and FcRn. Protein Sci. 2009, 18, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassi, L.; Cabrele, C. Susceptibility of protein therapeutics to spontaneous chemical modifications by oxidation, cyclization, and elimination reactions. Amino Acids 2019, 51, 1409–1431. [Google Scholar] [CrossRef] [Green Version]
- Sen, K.I.; Hepler, R.; Nanda, H. Detection and Measurement of Methionine Oxidation in Proteins. Curr. Protoc. Protein Sci. 2017, 87, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Schöneich, C. Methionine oxidation by reactive oxygen species: Reaction mechanisms and relevance to Alzheimer’s disease. Biochim. Biophys. Acta 2005, 1703, 111–119. [Google Scholar] [CrossRef]
- Saunders, C.C.; Stites, W.E. An electrophoretic mobility shift assay for methionine sulfoxide in proteins. Anal. Biochem. 2012, 421, 767–769. [Google Scholar] [CrossRef] [Green Version]
- Loew, C.; Knoblich, C.; Fichtl, J.; Alt, N.; Diepold, K.; Bulau, P.; Goldbach, P.; Adler, M.; Mahler, H.-C.; Grauschopf, U. Analytical Protein A Chromatography as a Quantitative Tool for the Screening of Methionine Oxidation in Monoclonal Antibodies. J. Pharm. Sci. 2012, 101, 4248–4257. [Google Scholar] [CrossRef]
- Pavon, J.A.; Li, X.; Chico, S.; Kishnani, U.; Soundararajan, S.; Cheung, J.; Li, H.; Richardson, D.; Shameem, M.; Yang, X. Analysis of monoclonal antibody oxidation by simple mixed mode chromatography. J. Chromatogr. A 2016, 1431, 154–165. [Google Scholar] [CrossRef]
- Lundblad, R.L. Chemical Reagents for Protein Modification; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- Mo, J.; Tymiak, A.A.; Chen, G. Structural mass spectrometry in biologics discovery: Advances and future trends. Drug Discov. Today 2012, 17, 1323–1330. [Google Scholar] [CrossRef]
- Guan, Z.; Yates, N.A.; Bakhtiar, R. Detection and characterization of methionine oxidation in peptides by collision-induced dissociation and electron capture dissociation. J. Am. Soc. Mass Spectrom. 2003, 14, 605–613. [Google Scholar] [CrossRef] [Green Version]
- Spraggins, J.M.; Rizzo, D.G.; Moore, J.L.; Rose, K.L.; Hammer, N.D.; Skaar, E.P.; Caprioli, R.M. MALDI FTICR IMS of Intact Proteins: Using Mass Accuracy to Link Protein Images with Proteomics Data. J. Am. Soc. Mass Spectrom. 2015, 26, 974–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oien, D.B.; Canello, T.; Gabizon, R.; Gasset, M.; Lundquist, B.L.; Burns, J.M.; Moskovitz, J. Detection of oxidized methionine in selected proteins, cellular extracts and blood serums by novel anti-methionine sulfoxide antibodies. Arch. Biochem. Biophys. 2009, 485, 35–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehr, N.B.; Levine, R.L. Wanted and wanting: Antibody against methionine sulfoxide. Free Radic. Biol. Med. 2012, 53, 1222–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hada, S.; Kim, N.A.; Lim, D.G.; Lim, J.Y.; Kim, K.H.; Adhikary, P.; Jeong, S.H. Evaluation of antioxidants in protein formulation against oxidative stress using various biophysical methods. Int. J. Biol. Macromol. 2016, 82, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Knepp, V.M.; Whatley, J.L.; Muchnik, A.; Calderwood, T.S. Identification of antioxidants for prevention of peroxide-mediated oxidation of recombinant human ciliary neurotrophic factor and recombinant human nerve growth factor. PDA J. Pharm. Sci. Technol. 1996, 50, 163–171. [Google Scholar] [PubMed]
- Dion, M.Z.; Leiske, D.; Sharma, V.K.; De Zafra, C.L.Z.; Salisbury, C.M. Mitigation of Oxidation in Therapeutic Antibody Formulations: A Biochemical Efficacy and Safety Evaluation of N-Acetyl-Tryptophan and L-Methionine. Pharm. Res. 2018, 35, 222. [Google Scholar] [CrossRef]
- Lam, X.M.; Yang, J.Y.; Cleland, J.L. Antioxidants for Prevention of Methionine Oxidation in Recombinant Monoclonal Antibody HER2. J. Pharm. Sci. 1997, 86, 1250–1255. [Google Scholar] [CrossRef]
- Hermeling, S.; Crommelin, D.J.A.; Schellekens, H.; Jiskoot, W. Structure-Immunogenicity Relationships of Therapeutic Proteins. Pharm. Res. 2004, 21, 897–903. [Google Scholar] [CrossRef]
- Devarie-Baez, N.O.; Lopez, E.I.S.; Furdui, C.M. Biological chemistry and functionality of protein sulfenic acids and related thiol modifications. Free Radic. Res. 2015, 50, 172–194. [Google Scholar] [CrossRef] [Green Version]
- Hoyle, C.E.; Bowman, C.N. Thiol-ene click chemistry. Angew. Chem. Int. Ed. 2010, 49, 1540–1573. [Google Scholar] [CrossRef]
- Choi, H.; Kim, M.; Jang, J.; Hong, S. Visible-Light-Induced Cysteine-Specific Bioconjugation: Biocompatible Thiol-Ene Click Chemistry. Angew. Chem. Int. Ed. 2020, 59, 22514–22522. [Google Scholar] [CrossRef] [PubMed]
- Keylor, M.; Park, J.E.; Wallentin, C.J.; Stephenson, C. Photocatalytic initiation of thiol–ene reactions: Synthesis of thiomorpholin-3-ones. Tetrahedron 2014, 70, 4264–4269. [Google Scholar] [CrossRef]
- Tyson, E.L.; Ament, M.S.; Yoon, T.P. Transition Metal Photoredox Catalysis of Radical Thiol-Ene Reactions. J. Org. Chem. 2012, 78, 2046–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikalov, S.; Khramtsov, V.; Zimmer, G. Determination of rate constants of the reactions of thiols with superoxide radical by electron paramagnetic resonance: Critical remarks on spectrophotometric approaches. Arch. Biochem. Biophys. 1996, 326, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Hampton, M.B. Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 2008, 45, 549–561. [Google Scholar] [CrossRef]
- Poole, L.; Claiborne, A. The non-flavin redox center of the streptococcal NADH peroxidase. II. Evidence for a stabilized cysteine-sulfenic acid. J Biol Chem. 1989, 264, 12330–12338. [Google Scholar] [CrossRef]
- Riener, C.K.; Kada, G.; Gruber, H.J. Quick measurement of protein sulfhydryls with Ellman’s reagent and with 4,4′-dithiodipyridine. Anal. Bioanal. Chem. 2002, 373, 266–276. [Google Scholar] [CrossRef]
- Habeeb, A. [37] Reaction of protein sulfhydryl groups with Ellman’s reagent. Methods Enzymol. 1972, 25, 457–464. [Google Scholar] [CrossRef]
- Alcock, L.J.; Perkins, M.V.; Chalker, J.M. Chemical methods for mapping cysteine oxidation. Chem. Soc. Rev. 2017, 47, 231–268. [Google Scholar] [CrossRef] [Green Version]
- Grassetti, D.R.; Murray, J.F. Determination of sulfhydryl groups with 2,2′- or 4,4′-dithiodipyridine. Radiolysis of Phenylalanine and Tyrosine and Aqueous Solution. Biochem. Biophys. 1967, 119, 41–49. [Google Scholar] [CrossRef]
- Robotham, A.C.; Kelly, J.F. Detection and quantification of free sulfhydryls in monoclonal antibodies using maleimide labeling and mass spectrometry. mAbs 2019, 11, 757–766. [Google Scholar] [CrossRef]
- Wu, C.W.; Yarbrough, L.R. N-(1-pyrene)maleimide: A fluorescent cross-linking reagent. Biochemistry 1976, 15, 2863–2868. [Google Scholar] [CrossRef]
- Hoff, S.; Larsen, F.H.; Andersen, M.L.; Lund, M.N. Quantification of protein thiols using ThioGlo 1 fluorescent derivatives and HPLC separation. Analyst 2013, 138, 2096–2103. [Google Scholar] [CrossRef]
- Ercal, N.; Yang, P.; Aykin, N. Determination of biological thiols by high-performance liquid chromatography following derivatization by ThioGlo maleimide reagents. J. Chromatogr. B Biomed. Sci. Appl. 2001, 753, 287–292. [Google Scholar] [CrossRef]
- Getz, E.B.; Xiao, M.; Chakrabarty, T.; Cooke, R.; Selvin, P.R. A comparison between the sulfhydryl reductants tris(2-carboxyethyl)phosphine and dithiothreitol for use in protein biochemistry. Anal. Biochem. 1999, 273, 73–80. [Google Scholar] [CrossRef]
- Jakob, U.; Muse, W.; Eser, M.; Bardwell, J.C.A. Chaperone Activity with a Redox Switch. Cell 1999, 96, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-R.; Kwon, K.-S.; Kim, S.-R.; Rhee, S.G. Reversible Inactivation of Protein-tyrosine Phosphatase 1B in A431 Cells Stimulated with Epidermal Growth Factor. J. Biol. Chem. 1998, 273, 15366–15372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, P.L.; Chen, S.-F.; Huang, S.Y. Mass spectrometry-based strategies for protein disulfide bond identification. Rev. Anal. Chem. 2013, 32, 257–268. [Google Scholar] [CrossRef]
- Sharma, D.; Rajarathnam, K. 13C NMR chemical shifts can predict disulfide bond formation. J. Biomol. NMR 2000, 18, 165–171. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, A.A.; Haebel, P.W.; Törrönen, A.; Rybin, V.; Baker, E.; Metcalf, P. Crystal structure of the protein disulfide bond isomerase, DsbC, from Escherichia coli. Nat. Genet. 2000, 7, 196–199. [Google Scholar] [CrossRef]
- Poole, L.B.; Klomsiri, C.; Knaggs, S.A.; Furdui, C.M.; Nelson, K.J.; Thomas, M.J.; Fetrow, J.S.; Daniel, L.W.; King, S.B. Fluorescent and Affinity-Based Tools to Detect Cysteine Sulfenic Acid Formation in Proteins. Bioconjugate Chem. 2007, 18, 2004–2017. [Google Scholar] [CrossRef] [Green Version]
- Poole, L.B.; Zeng, B.-B.; Knaggs, S.; Yakubu, M.; King, S.B. Synthesis of Chemical Probes to Map Sulfenic Acid Modifications on Proteins. Bioconjugate Chem. 2005, 16, 1624–1628. [Google Scholar] [CrossRef] [PubMed]
- Charles, R.L.; Schröder, E.; May, G.; Free, P.; Gaffney, P.; Wait, R.; Begum, S.; Heads, R.J.; Eaton, P. Protein Sulfenation as a Redox Sensor. Mol. Cell. Proteom. 2007, 6, 1473–1484. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.H.; Carroll, K.S. Profiling protein thiol oxidation in tumor cells using sulfenic acid-specific antibodies. Proc. Natl. Acad. Sci. USA 2009, 106, 16163–16168. [Google Scholar] [CrossRef] [Green Version]
- Saurin, A.; Neubert, H.; Brennan, J.P.; Eaton, P. Widespread sulfenic acid formation in tissues in response to hydrogen peroxide. Proc. Natl. Acad. Sci. USA 2004, 101, 17982–17987. [Google Scholar] [CrossRef] [Green Version]
- Eaton, P. Protein thiol oxidation in health and disease: Techniques for measuring disulfides and related modifications in complex protein mixtures. Free Radic. Biol. Med. 2006, 40, 1889–1899. [Google Scholar] [CrossRef]
- Winther, J.R.; Thorpe, C. Quantification of thiols and disulfides. Biochim. Biophys. Acta Gen. Subj. 2013, 1840, 838–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Kwon, K.-S.; Rhee, S.G. Probing cellular protein targets of H2O2 with fluorescein-conjugated iodoacetamide and antibodies to fluorescein. FEBS Lett. 1998, 440, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-R.; Yoon, H.W.; Kwon, K.-S.; Lee, S.-R.; Rhee, S.G. Identification of Proteins Containing Cysteine Residues That Are Sensitive to Oxidation by Hydrogen Peroxide at Neutral pH. Anal. Biochem. 2000, 283, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.L.; Davies, M.J. Detection, identification, and quantification of oxidative protein modifications. J. Biol. Chem. 2019, 294, 19683–19708. [Google Scholar] [CrossRef] [Green Version]
- Von Ossowski, L.; Tossavainen, H.; von Ossowski, I.; Cai, C.; Aitio, O.; Fredriksson, K.; Permi, P.; Annila, A.; Keinänen, K. Peptide Binding and NMR Analysis of the Interaction between SAP97 PDZ2 and GluR-A: Potential Involvement of a Disulfide Bond. Biochemistry 2006, 45, 5567–5575. [Google Scholar] [CrossRef]
- Walewska, A.; Skalicky, J.J.; Davis, D.R.; Zhang, M.-M.; Vera, E.L.; Watkins, M.; Han, T.S.; Yoshikami, D.; Olivera, B.M.; Bulaj, G. NMR-Based Mapping of Disulfide Bridges in Cysteine-Rich Peptides: Application to the μ-Conotoxin SxIIIA. J. Am. Chem. Soc. 2008, 130, 14280–14286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, H.R.; Poole, L.B. Novel Application of 7-Chloro-4-nitrobenzo-2-oxa-1,3-diazole To Identify Cysteine Sulfenic Acid in the AhpC Component of Alkyl Hydroperoxide Reductase. Biochemistry 1997, 36, 15013–15018. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, D.M.; Levine, R.L.; Finkel, T. Detection and Affinity Purification of Oxidant-Sensitive Proteins Using Biotinylated Glutathione Ethyl Ester. Methods Enzymol. 2002, 353, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Kast, J. Chapter Fifteen—Biotin Switch Assays for Quantitation of Reversible Cysteine Oxidation. Methods Enzymol. 2017, 585, 269–284. [Google Scholar] [PubMed]
- Charron, G.; Zhang, M.; Yount, J.; Wilson, J.; Raghavan, A.S.; Shamir, E.; Hang, H.C. Robust Fluorescent Detection of Protein Fatty-Acylation with Chemical Reporters. J. Am. Chem. Soc. 2009, 131, 4967–4975. [Google Scholar] [CrossRef]
- Fauq, A.H.; Kache, R.; Khan, M.A.; Vega, I.E. Synthesis of Acid-Cleavable Light Isotope-Coded Affinity Tags (ICAT-L) for Potential Use in Proteomic Expression Profiling Analysis. Bioconjugate Chem. 2005, 17, 248–254. [Google Scholar] [CrossRef]
- Szychowski, J.; Mahdavi, A.; Hodas, J.J.L.; Bagert, J.D.; Ngo, J.T.; Landgraf, P.; Dieterich, D.C.; Schuman, E.M.; Tirrell, D.A. Cleavable Biotin Probes for Labeling of Biomolecules via Azide−Alkyne Cycloaddition. J. Am. Chem. Soc. 2010, 132, 18351–18360. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.-Y.; Grammel, M.; Raghavan, A.S.; Charron, G.; Hang, H.C. Comparative Analysis of Cleavable Azobenzene-Based Affinity Tags for Bioorthogonal Chemical Proteomics. Chem. Biol. 2010, 17, 1212–1222. [Google Scholar] [CrossRef] [Green Version]
- Park, K.D.; Liu, R.; Kohn, H. Useful tools for biomolecule isolation, detection, and identification: Acylhydrazone-based cleavable linkers. Chem. Biol. 2009, 16, 763–772. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Bautista, R.; Yu, K.; Zapata, G.A.; Mulkerrin, M.G.; Chamow, S.M. Influence of histidine on the stability and physical properties of a fully human antibody in aqueous and solid forms. Pharm. Res. 2003, 20, 1952–1960. [Google Scholar] [CrossRef]
- Zbacnik, T.J.; Holcomb, R.E.; Katayama, D.S.; Murphy, B.M.; Payne, R.W.; Coccaro, R.C.; Evans, G.J.; Matsuura, J.E.; Henry, C.S.; Manning, M.C. Role of Buffers in Protein Formulations. J. Pharm. Sci. 2017, 106, 713–733. [Google Scholar] [CrossRef]
- Lei, M.; Quan, C.; Wang, Y.J.; Kao, Y.-H.; Schöneich, C. Light-Induced Covalent Buffer Adducts to Histidine in a Model Protein. Pharm. Res. 2018, 35, 67. [Google Scholar] [CrossRef]
- Lei, M.; Quan, C.; Wang, J.Y.; Kao, Y.-H.; Schöneich, C. Light-Induced Histidine Adducts to an IgG1 Molecule Via Oxidized Histidine Residue and the Potential Impact of Polysorbate-20 Concentration. Pharm. Res. 2021, 38, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Huvaere, K.; Skibsted, L.H. Light-Induced Oxidation of Tryptophan and Histidine. Reactivity of Aromatic N-Heterocycles toward Triplet-Excited Flavins. J. Am. Chem. Soc. 2009, 131, 8049–8060. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lu, W.; Fang, Y.; Liu, J. Evolution of oxidation dynamics of histidine: Non-reactivity in the gas phase, peroxides in hydrated clusters, and pH dependence in solution. Phys. Chem. Chem. Phys. 2014, 16, 22179–22191. [Google Scholar] [CrossRef] [PubMed]
- Agon, V.V.; Bubb, W.A.; Wright, A.; Hawkins, C.L.; Davies, M.J. Sensitizer-mediated photooxidation of histidine residues: Evidence for the formation of reactive side-chain peroxides. Free Radic. Biol. Med. 2006, 40, 698–710. [Google Scholar] [CrossRef]
- Miyahara, Y.; Shintani, K.; Hayashihara-Kakuhou, K.; Zukawa, T.; Morita, Y.; Nakazawa, T.; Yoshida, T.; Ohkubo, T.; Uchiyama, S. Effect of UVC Irradiation on the Oxidation of Histidine in Monoclonal Antibodies. Sci. Rep. 2020, 10, 6333–6339. [Google Scholar] [CrossRef]
- Dean, R.T.; Wolff, S.P.; McElligott, M.A. Histidine and Proline are Important Sites of Free Radical Damage to Proteins. Free Radic. Res. Commun. 1989, 7, 97–103. [Google Scholar] [CrossRef]
- Stroop, S.D.; Conca, D.M.; Lundgard, R.P.; Renz, M.E.; Peabody, L.M.; Leigh, S.D. Photosensitizers Form in Histidine Buffer and Mediate the Photodegradation of a Monoclonal Antibody. J. Pharm. Sci. 2011, 100, 5142–5155. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Kawakishi, S. 2-Oxo-histidine as a novel biological marker for oxidatively modified proteins. FEBS Lett. 1993, 332, 208–210. [Google Scholar] [CrossRef] [Green Version]
- Uchida, K.; Kawakishi, S. Identification of oxidized histidine generated at the active site of Cu,Zn-superoxide dismutase exposed to H2O2. Selective generation of 2-oxo-histidine at the histidine 118. J. Biol. Chem. 1994, 269, 2405–2410. [Google Scholar] [CrossRef]
- Srikanth, R.; Wilson, J.; Vachet, R.W. Correct identification of oxidized histidine residues using electron-transfer dissociation. J. Mass Spectrom. 2009, 44, 755–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridgewater, J.D.; Srikanth, R.; Lim, J.; Vachet, R.W. The effect of histidine oxidation on the dissociation patterns of peptide ions. J. Am. Soc. Mass Spectrom. 2007, 18, 553–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.-R.; Spikes, J.; Smith, C.; Kopeček, J. Photodynamic cross-linking of proteins: IV. Nature of the His–His bond(s) formed in the rose bengal-photosensitized cross-linking of N-benzoyl-L-histidine. J. Photochem. Photobiol. Chem. 2000, 130, 1–6. [Google Scholar] [CrossRef]
- Uchida, K. Histidine and lysine as targets of oxidative modification. Amino Acids 2003, 25, 249–257. [Google Scholar] [CrossRef]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Fields, R.; Dixon, H.B.F. Micro method for determination of reactive carbonyl groups in proteins and peptides, using 2,4-dinitrophenylhydrazine. Biochem. J. 1971, 121, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Akagawa, M.; Suyama, K.; Uchida, K. Fluorescent detection of alpha-aminoadipic and gamma-glutamic semialdehydes in oxidized proteins. Free Radic. Biol. Med. 2009, 46, 701–706. [Google Scholar] [CrossRef]
- Estévez, M.; Ollilainen, V.; Heinonen, M. Analysis of Protein Oxidation Markers α-Aminoadipic and γ-Glutamic Semialdehydes in Food Proteins Using Liquid Chromatography (LC)−Electrospray Ionization (ESI)−Multistage Tandem Mass Spectrometry (MS). J. Agric. Food Chem. 2009, 57, 3901–3910. [Google Scholar] [CrossRef]
- Mirzaei, H.; Regnier, F. Affinity Chromatographic Selection of Carbonylated Proteins Followed by Identification of Oxidation Sites Using Tandem Mass Spectrometry. Anal. Chem. 2005, 77, 2386–2392. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, H.; Regnier, F. Identification and quantification of protein carbonylation using light and heavy isotope labeled Girard’s P reagent. J. Chromatogr. A 2006, 1134, 122–133. [Google Scholar] [CrossRef]
- Temple, A.; Yen, T.-Y.; Gronert, S. Identification of specific protein carbonylation sites in model oxidations of human serum albumin. J. Am. Soc. Mass Spectrom. 2006, 17, 1172–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, J.D.; Bisson, W.H.; Maier, C.S. A targeted mass spectrometry-based approach for the identification and characterization of proteins containing α-aminoadipic and γ-glutamic semialdehyde residues. Anal. Bioanal. Chem. 2010, 398, 2905–2914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Q.; Moore, B.; Beardsley, R.L. Discovery and characterization of hydroxylysine in recombinant monoclonal antibodies. mAbs 2015, 8, 371–378. [Google Scholar] [CrossRef] [Green Version]
- Ayala, A.; Cutler, R.G. Comparison of 5-hydroxy-2-amino valeric acid with carbonyl group content as a marker of oxidized protein in human and mouse liver tissues. Free Radic. Biol. Med. 1996, 21, 551–558. [Google Scholar] [CrossRef]
- Baynes, B.M.; Wang, D.I.C.; Trout, B.L. Role of Arginine in the Stabilization of Proteins against Aggregation. Biochemistry 2005, 44, 4919–4925. [Google Scholar] [CrossRef]
- Papeschi, G.; Monici, M.; Pinzauti, S. pH effect on dye sensitized photooxidation of aminoacids and albumins. Med. Biol. Environ. 1982, 10, 245–250. [Google Scholar]
- Pietzsch, J. Measurement of 5-Hydroxy-2-aminovaleric Acid as a Specific Marker of Iron-Mediated Oxidation of Proline and Arginine Side-Chain Residues of Low-Density Lipoprotein Apolipoprotein B-100. Biochem. Biophys. Res. Commun. 2000, 270, 852–857. [Google Scholar] [CrossRef]
- Subelzu, N.; Schöneich, C. Near UV and Visible Light Induce Iron-Dependent Photodegradation Reactions in Pharmaceutical Buffers: Mechanistic and Product Studies. Mol. Pharm. 2020, 17, 4163–4179. [Google Scholar] [CrossRef]
- Abrahamson, H.B.; Rezvani, A.B.; Brushmiller, J. Photochemical and spectroscopic studies of complexes, of iron(III) with citric acid and other carboxylic acids. Inorg. Chim. Acta 1994, 226, 117–127. [Google Scholar] [CrossRef]
- Valliere-Douglass, J.F.; Connell-Crowley, L.; Jensen, R.; Schnier, P.D.; Trilisky, E.; Leith, M.; Follstad, B.D.; Kerr, J.; Lewis, N.; Vunnum, S.; et al. Photochemical degradation of citrate buffers leads to covalent acetonation of recombinant protein therapeutics. Protein Sci. 2010, 19, 2152–2163. [Google Scholar] [CrossRef] [Green Version]
- Prajapati, I.; Peters, B.-H.; Larson, N.R.; Wei, Y.; Choudhary, S.; Kalonia, C.; Hudak, S.; Esfandiary, R.; Middaugh, C.R.; Schöneich, C. Cis/Trans Isomerization of Unsaturated Fatty Acids in Polysorbate 80 During Light Exposure of a Monoclonal Antibody–Containing Formulation. J. Pharm. Sci. 2019, 109, 603–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, A.; Koulov, A.; Huwyler, J.; Mahler, H.-C.; Jahn, M. Stabilizing Polysorbate 20 and 80 Against Oxidative Degradation. J. Pharm. Sci. 2020, 109, 1924–1932. [Google Scholar] [CrossRef] [PubMed]
- Ha, E.; Wang, W.; Wang, Y.J. Peroxide formation in polysorbate 80 and protein stability. J. Pharm. Sci. 2002, 91, 2252–2264. [Google Scholar] [CrossRef]
- Dwivedi, M.; Blech, M.; Presser, I.; Garidel, P. Polysorbate degradation in biotherapeutic formulations: Identification and discussion of current root causes. Int. J. Pharm. 2018, 552, 422–436. [Google Scholar] [CrossRef]
- Yao, J.; Dokuru, D.K.; Noestheden, M.; Park, S.S.; Kerwin, B.A.; Jona, J.; Ostovic, D.; Reid, D.L. A Quantitative Kinetic Study of Polysorbate Autoxidation: The Role of Unsaturated Fatty Acid Ester Substituents. Pharm. Res. 2009, 26, 2303–2313. [Google Scholar] [CrossRef] [PubMed]
- Larson, N.R.; Wei, Y.; Prajapati, I.; Chakraborty, A.; Peters, B.; Kalonia, C.; Hudak, S.; Choudhary, S.; Esfandiary, R.; Dhar, P.; et al. Comparison of Polysorbate 80 Hydrolysis and Oxidation on the Aggregation of a Monoclonal Antibody. J. Pharm. Sci. 2019, 109, 633–639. [Google Scholar] [CrossRef] [Green Version]
- Evers, D.-H.; Schultz-Fademrecht, T.; Garidel, P.; Buske, J. Development and validation of a selective marker-based quantification of polysorbate 20 in biopharmaceutical formulations using UPLC QDa detection. J. Chromatogr. B 2020, 1157, 122287. [Google Scholar] [CrossRef] [PubMed]
- Lam, X.M.; Lai, W.G.; Chan, E.K.; Ling, V.; Hsu, C.C. Site-Specific Tryptophan Oxidation Induced by Autocatalytic Reaction of Polysorbate 20 in Protein Formulation. Pharm. Res. 2011, 28, 2543–2555. [Google Scholar] [CrossRef]
- Brito, R.; Vaz, W. Determination of the critical micelle concentration of surfactants using the fluorescent probe N-phenyl-1-naphthylamine. Anal. Biochem. 1986, 152, 250–255. [Google Scholar] [CrossRef]
- Lippold, S.; Koshari, S.H.; Kopf, R.; Schuller, R.; Buckel, T.; Zarraga, I.E.; Koehn, H. Impact of mono- and poly-ester fractions on polysorbate quantitation using mixed-mode HPLC-CAD/ELSD and the fluorescence micelle assay. J. Pharm. Biomed. Anal. 2017, 132, 24–34. [Google Scholar] [CrossRef]
- Martos, A.; Koch, W.; Jiskoot, W.; Wuchner, K.; Winter, G.; Friess, W.; Hawe, A. Trends on Analytical Characterization of Polysorbates and Their Degradation Products in Biopharmaceutical Formulations. J. Pharm. Sci. 2017, 106, 1722–1735. [Google Scholar] [CrossRef]
- Dahotre, S.; Tomlinson, A.; Lin, B.; Yadav, S. Novel markers to track oxidative polysorbate degradation in pharmaceutical formulations. J. Pharm. Biomed. Anal. 2018, 157, 201–207. [Google Scholar] [CrossRef]
- Klein, M.; Preston, J. Polysorbate 80 (Tween® 80) HPLC-UV Analysis on a YarraTM 1.8 µm SEC-X150 Aqueous GFC/SEC Column; Application Note from Phenomenex; Phenomenex: Torrance, CA, USA, 2016. [Google Scholar]
- Lobback, C.; Backensfeld, T.; Funke, A.; Weitschies, W. Quantitative determination of nonionic surfactants with CAD. Chromatogr. Tech. 2007, 11, 18–20. [Google Scholar]
- Nayak, V.S.; Tan, Z.; Ihnat, P.M.; Russell, R.J.; Grace, M.J. Evaporative Light Scattering Detection Based HPLC Method for the Determination of Polysorbate 80 in Therapeutic Protein Formulations. J. Chromatogr. Sci. 2011, 50, 21–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kou, D.; Manius, G.; Tian, H.; Chokshi, H.P. Photo-Oxidation of Therapeutic Protein Formulations. Polym. Surfactants 2017, 327–339. [Google Scholar] [CrossRef]
- Fekete, S.; Ganzler, K.; Fekete, J. Fast and sensitive determination of Polysorbate 80 in solutions containing proteins. J. Pharm. Biomed. Anal. 2010, 52, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Kranz, W.; Wuchner, K.; Corradini, E.; Berger, M.; Hawe, A. Factors Influencing Polysorbate’s Sensitivity against Enzymatic Hydrolysis and Oxidative Degradation. J. Pharm. Sci. 2019, 108, 2022–2032. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hewitt, D.; Lentz, Y.K.; Ji, J.A.; Zhang, T.Y.; Zhang, K. Characterization and Stability Study of Polysorbate 20 in Therapeutic Monoclonal Antibody Formulation by Multidimensional Ultrahigh-Performance Liquid Chromatography–Charged Aerosol Detection–Mass Spectrometry. Anal. Chem. 2014, 86, 5150–5157. [Google Scholar] [CrossRef]
- Ayorinde, F.O.; Gelain, S.V.; Johnson, J.H., Jr.; Wan, L.W. Analysis of some commercial polysorbate formulations using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2000, 14, 2116–2124. [Google Scholar] [CrossRef]
- Frison-Norrie, S.; Sporns, P. Investigating the Molecular Heterogeneity of Polysorbate Emulsifiers by MALDI-TOF MS. J. Agric. Food Chem. 2001, 49, 3335–3340. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yadav, S.; Demeule, B.; Wang, Y.J.; Mozziconacci, O.; Schӧneich, C. Degradation Mechanisms of Polysorbate 20 Differentiated by 18O-labeling and Mass Spectrometry. Pharm. Res. 2016, 34, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Hvattum, E.; Yip, W.L.; Grace, D.; Dyrstad, K. Characterization of polysorbate 80 with liquid chromatography mass spectrometry and nuclear magnetic resonance spectroscopy: Specific determination of oxidation products of thermally oxidized polysorbate 80. J. Pharm. Biomed. Anal. 2012, 62, 7–16. [Google Scholar] [CrossRef]
- Kishore, R.S.; Pappenberger, A.; Dauphin, I.B.; Ross, A.; Buergi, B.; Staempfli, A.; Mahler, H.-C. Degradation of Polysorbates 20 and 80: Studies on Thermal Autoxidation and Hydrolysis. J. Pharm. Sci. 2011, 100, 721–731. [Google Scholar] [CrossRef]
- Kishore, R.S.K.; Kiese, S.; Fischer, S.; Pappenberger, A.; Grauschopf, U.; Mahler, H.-C. The Degradation of Polysorbates 20 and 80 and its Potential Impact on the Stability of Biotherapeutics. Pharm. Res. 2011, 28, 1194–1210. [Google Scholar] [CrossRef]
- Poppe, L.; Knutson, N.; Cao, S.; Wikstrom, M. In Situ Quantification of Polysorbate in Pharmaceutical Samples of Therapeutic Proteins by Hydrodynamic Profiling by NMR Spectroscopy. Anal. Chem. 2019, 91, 7807–7811. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, A.; Meng, Y.; Ning, T.; Yang, H.; Ding, L.; Xiao, X.; Li, X. NMR Method for Accurate Quantification of Polysorbate 80 Copolymer Composition. Anal. Chem. 2015, 87, 9810–9816. [Google Scholar] [CrossRef]
- Donbrow, M.; Azaz, E.; Pillersdorf, A. Autoxidation of Polysorbates. J. Pharm. Sci. 1978, 67, 1676–1681. [Google Scholar] [CrossRef]
- Esue, O.; Sharma, V.K. Compositions and Methods for Stabilizing Protein-Containing Formulations. U.S. Patent 9.254,321, 9 February 2016. [Google Scholar]
- Bodratti, A.M.; Alexandridis, P. Formulation of Poloxamers for Drug Delivery. J. Funct. Biomater. 2018, 9, 11. [Google Scholar] [CrossRef] [Green Version]
- Costa, L.; Camino, G.; Luda, M.P.; Cameron, G.; Qureshi, M. The thermal oxidation of poly(propylene oxide) and its complexes with LiBr and LiI. Polym. Degrad. Stab. 1996, 53, 301–310. [Google Scholar] [CrossRef]
- Gallet, G.; Carroccio, S.; Rizzarelli, P.; Karlsson, S. Thermal degradation of poly(ethylene oxide–propylene oxide–ethylene oxide) triblock copolymer: Comparative study by SEC/NMR, SEC/MALDI-TOF-MS and SPME/GC-MS. Polymer 2002, 43, 1081–1094. [Google Scholar] [CrossRef]
- Wang, T.; Markham, A.; Thomas, S.J.; Wang, N.; Huang, L.; Clemens, M.; Rajagopalan, N. Solution Stability of Poloxamer 188 Under Stress Conditions. J. Pharm. Sci. 2018, 108, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Thompson, M.J.; Wang, Q.; Tsai, E.W. Quantitation of poloxamers in pharmaceutical formulations using size exclusion chromatography and colorimetric methods. J. Pharm. Biomed. Anal. 2004, 35, 1127–1142. [Google Scholar] [CrossRef]
- Chung, H.H.; Zhou, C.; Khor, H.K.; Qiu, J. Direct determination of residual Pluronic F-68 in in-process samples from monoclonal antibody preparations by high performance liquid chromatography. J. Chromatogr. A 2011, 1218, 2106–2113. [Google Scholar] [CrossRef]
- Liu, J.-Y.; Li, Y.-Z.; Chang, W.-B. Measurement of the peroxidation of Brji-35 in aqueous solution by hemin and horseradish peroxidase catalyzed fluorogenic reaction. Anal. Bioanal. Chem. 1999, 365, 448–451. [Google Scholar] [CrossRef]
- Mensink, M.A.; Frijlink, H.W.; Maarschalk, K.v.d.V.; Hinrichs, W.L. How sugars protect proteins in the solid state and during drying (review): Mechanisms of stabilization in relation to stress conditions. Eur. J. Pharm. Biopharm. 2017, 114, 288–295. [Google Scholar] [CrossRef]
- Singh, S.K. Sucrose and Trehalose in Therapeutic Protein Formulations; Springer International Publishing: Cham, Switzerland, 2018; pp. 63–95. [Google Scholar] [CrossRef]
- Chang, L.; Shepherd, D.; Sun, J.; Ouellette, D.; Grant, K.L.; Tang, X.; Pikal, M.J. Mechanism of protein stabilization by sugars during freeze-drying and storage: Native structure preservation, specific interaction, and/or immobilization in a glassy matrix? J. Pharm. Sci. 2005, 94, 1427–1444. [Google Scholar] [CrossRef]
- Ohtake, S.; Kita, Y.; Arakawa, T. Interactions of formulation excipients with proteins in solution and in the dried state. Adv. Drug Deliv. Rev. 2011, 63, 1053–1073. [Google Scholar] [CrossRef] [PubMed]
- Gervasi, V.; Agnol, R.D.; Cullen, S.; McCoy, T.; Vucen, S.; Crean, A. Parenteral protein formulations: An overview of approved products within the European Union. Eur. J. Pharm. Biopharm. 2018, 131, 8–24. [Google Scholar] [CrossRef]
- André, P.; Villain, F. Free radical scavenging properties of mannitol and its role as a constituent of hyaluronic acid fillers: A literature review. Int. J. Cosmet. Sci. 2017, 39, 355–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Li, W.-M.; Wang, W. Trehalose: Protector of antioxidant enzymes or reactive oxygen species scavenger under heat stress? Environ. Exp. Bot. 2008, 63, 378–384. [Google Scholar] [CrossRef]
- Matros, A.; Peshev, D.; Peukert, M.; Mock, H.-P.; Van Den Ende, W. Sugars as hydroxyl radical scavengers: Proof-of-concept by studying the fate of sucralose in Arabidopsis. Plant J. 2015, 82, 822–839. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Jensen, R.G.; Bohnert, H.J. Mannitol Protects against Oxidation by Hydroxyl Radicals. Plant Physiol. 1997, 115, 527–532. [Google Scholar] [CrossRef] [Green Version]
- Takagi, Y.; Kashiwagi, A.; Tanaka, Y.; Asahina, T.; Kikkawa, R.; Shigeta, Y. Significance of fructose-induced protein oxidation and formation of advanced glycation end product. J. Diabetes Complicat. 1995, 9, 87–91. [Google Scholar] [CrossRef]
- Fuentealba, D.; Friguet, B.; Silva, E. Advanced Glycation Endproducts Induce Photocrosslinking and Oxidation of Bovine Lens Proteins Through Type-I Mechanism. Photochem. Photobiol. 2009, 85, 185–194. [Google Scholar] [CrossRef]
- Wondrak, G.T.; Jacobson, E.L.; Jacobson, M.K. Photosensitization of DNA damage by glycated proteins. Photochem. Photobiol. Sci. 2002, 1, 355–363. [Google Scholar] [CrossRef]
- Gkogkolou, P.; Böhm, M. Advanced glycation end products: Key players in skin aging? Dermatoendocrinology 2012, 4, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, T.; Cai, W.; Peppa, M.; Dardaine, V.; Baliga, B.S.; Uribarri, J.; Vlassara, H. Advanced glycoxidation end products in commonly consumed foods. J. Am. Diet. Assoc. 2004, 104, 1287–1291. [Google Scholar] [CrossRef] [PubMed]
- Menini, S.; Iacobini, C.; de Latouliere, L.; Manni, I.; Ionta, V.; Fantauzzi, C.B.; Pesce, C.; Cappello, P.; Novelli, F.; Piaggio, G.; et al. The advanced glycation end-product Nϵ-carboxymethyllysine promotes progression of pancreatic cancer: Implications for diabetes-associated risk and its prevention. J. Pathol. 2018, 245, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, H.; Motomiya, Y.; Miura, K.; Morisawa, M.; Yoshimura, Y.; Maruyama, I. Immunochemical assay of hemoglobin with N(epsilon)-(carboxymethyl)lysine at lysine 66 of the beta chain. Clin. Chem. 2001, 47, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Séro, L.; Sanguinet, L.; Blanchard, P.; Dang, B.T.; Morel, S.; Richomme, P.; Seraphin, D.; Derbré, S. Tuning a 96-Well Microtiter Plate Fluorescence-Based Assay to Identify AGE Inhibitors in Crude Plant Extracts. Molecules 2013, 18, 14320–14339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagisawa, K.; Makita, Z.; Shiroshita, K.; Ueda, T.; Fusegawa, T.; Kuwajima, S.; Takeuchi, M.; Koike, T. Specific fluorescence assay for advanced glycation end products in blood and urine of diabetic patients. Metabolism 1998, 47, 1348–1353. [Google Scholar] [CrossRef]
- Henle, T.; Deppisch, R.; Beck, W.; Hergesell, O.; Hänsch, G.M.; Ritz, E. Advanced glycated end-products (AGE) during haemodialysis treatment: Discrepant results with different methodologies reflecting the heterogeneity of AGE compounds. Nephrol. Dial. Transplant. 1999, 14, 1968–1975. [Google Scholar] [CrossRef] [Green Version]
- Soboleva, A.; Vikhnina, M.; Grishina, T.; Frolov, A. Probing Protein Glycation by Chromatography and Mass Spectrometry: Analysis of Glycation Adducts. Int. J. Mol. Sci. 2017, 18, 2557. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Thornalley, P.; Dawczynski, J.; Franke, S.; Strobel, J.; Stein, G.; Haik, G.M. Methylglyoxal-Derived Hydroimidazolone Advanced Glycation End-Products of Human Lens Proteins. Investig. Opthalmol. Vis. Sci. 2003, 44, 5287–5292. [Google Scholar] [CrossRef] [PubMed]
- Scheijen, J.L.; van de Waarenburg, M.P.; Stehouwer, C.D.; Schalkwijk, C.G. Measurement of pentosidine in human plasma protein by a single-column high-performance liquid chromatography method with fluorescence detection. J. Chromatogr. B 2009, 877, 610–614. [Google Scholar] [CrossRef]
- Nomi, Y.; Annaka, H.; Sato, S.; Ueta, E.; Ohkura, T.; Yamamoto, K.; Homma, S.; Suzuki, E.; Otsuka, Y. Simultaneous Quantitation of Advanced Glycation End Products in Soy Sauce and Beer by Liquid Chromatography-Tandem Mass Spectrometry without Ion-Pair Reagents and Derivatization. J. Agric. Food Chem. 2016, 64, 8397–8405. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Battah, S.; Ahmed, N.; Karachalias, N.; Agalou, S.; Babaei-Jadidi, R.; Dawnay, A. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem. J. 2003, 375, 581–592. [Google Scholar] [CrossRef]
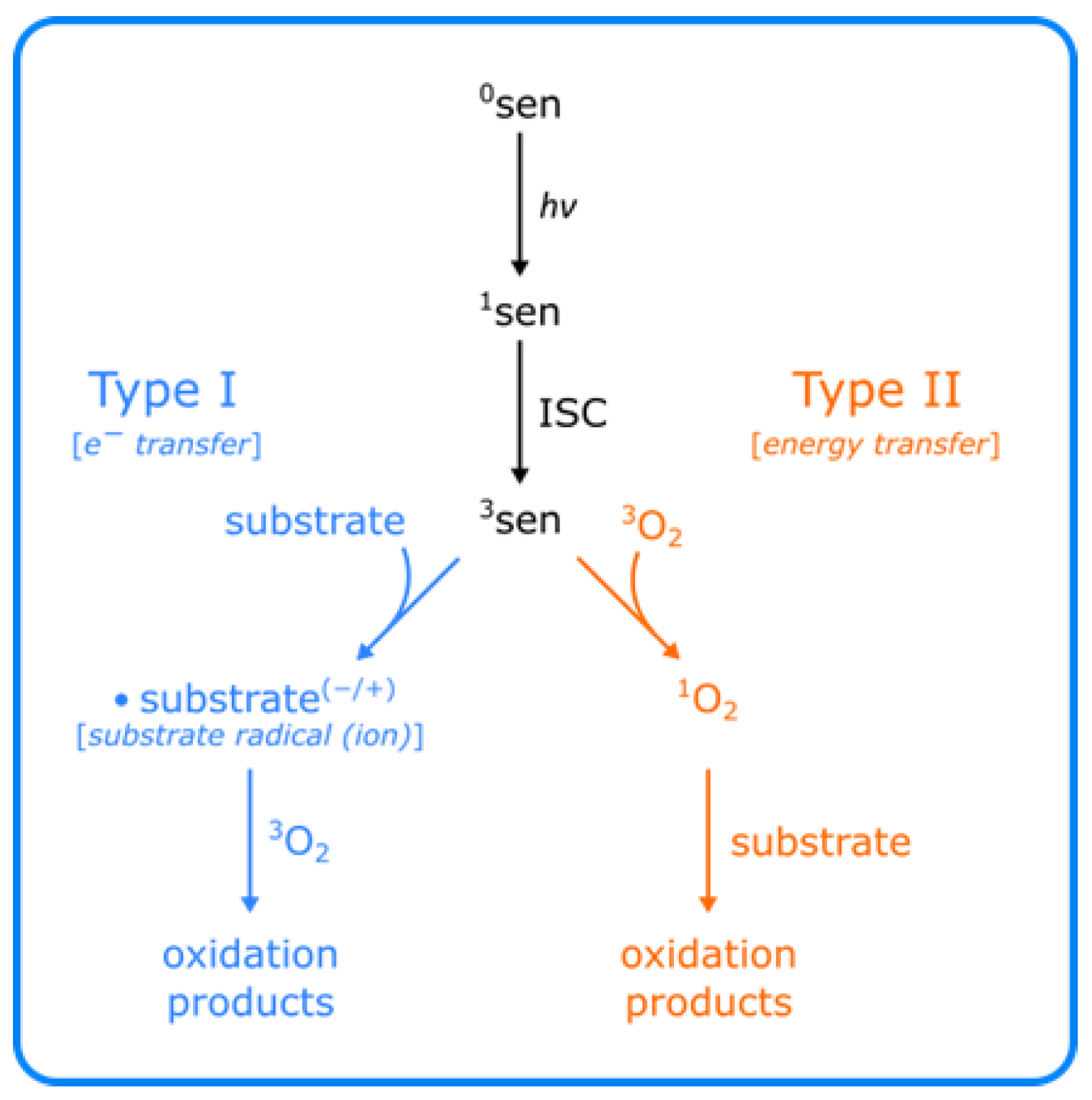


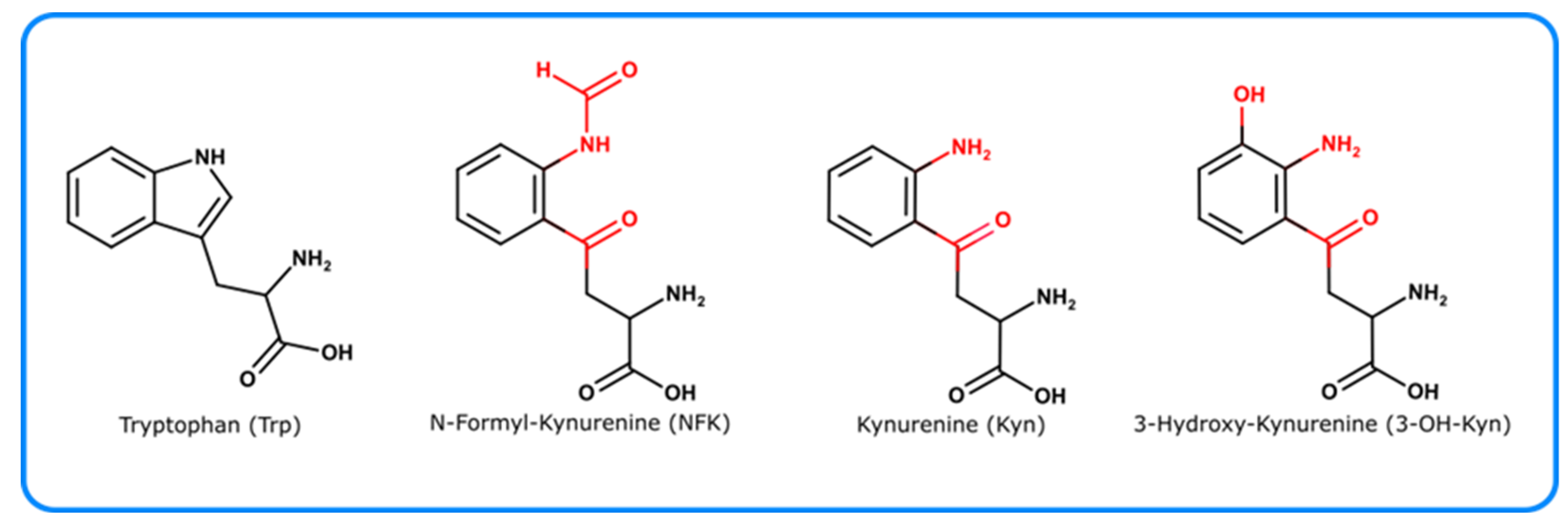


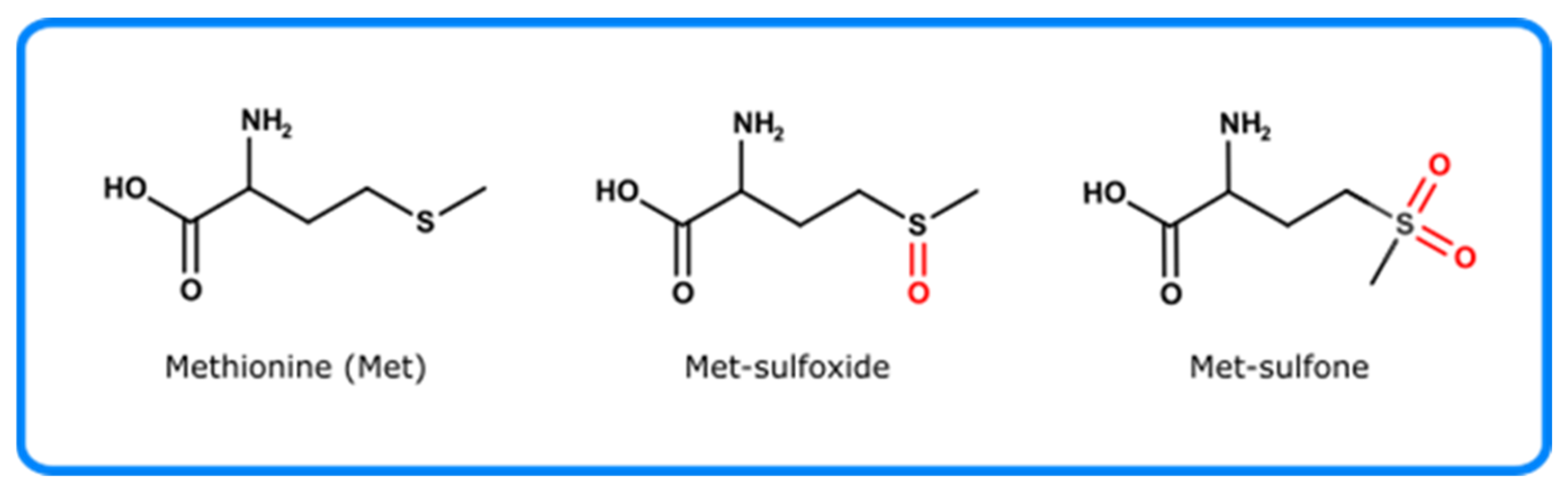
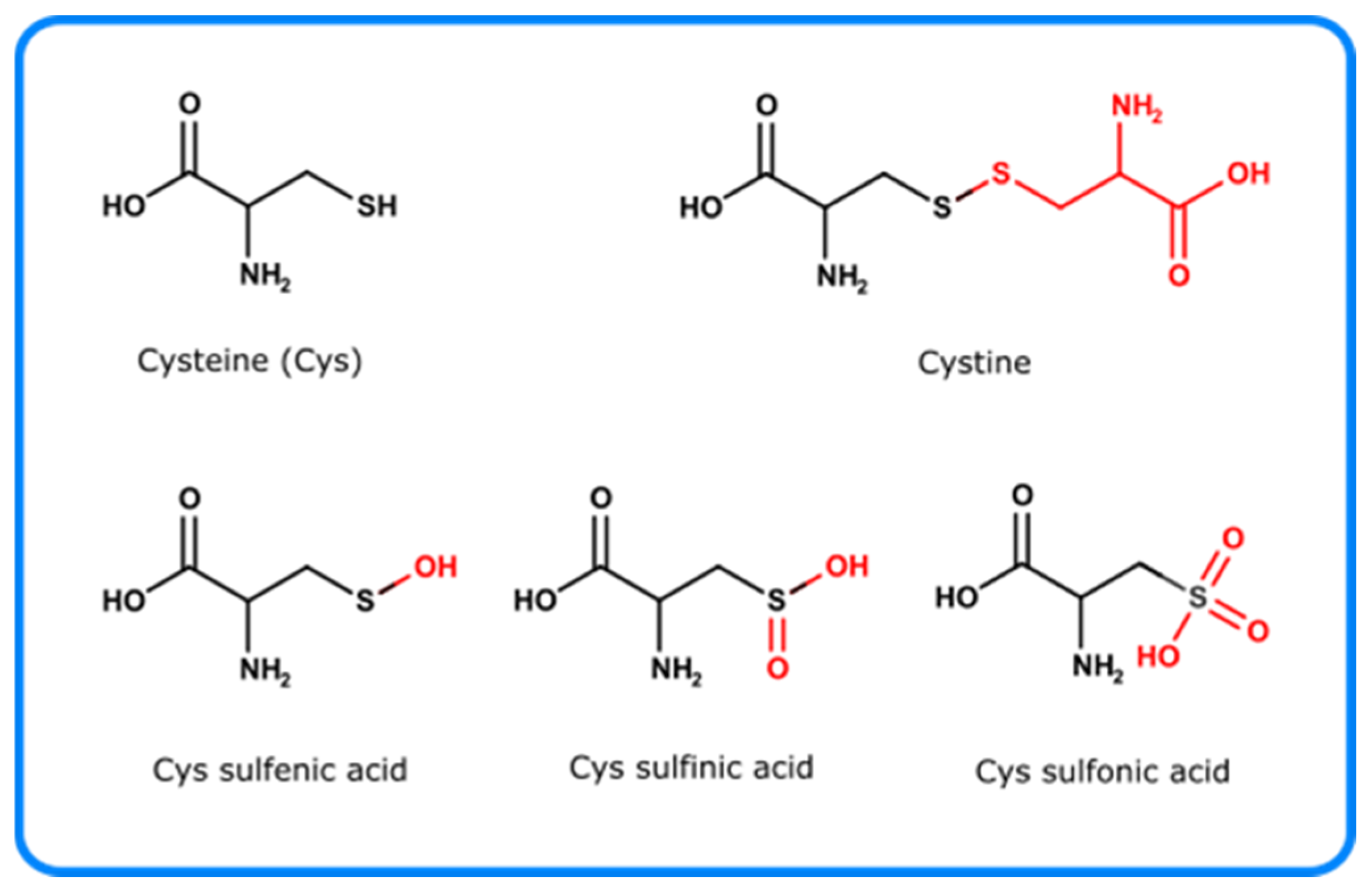


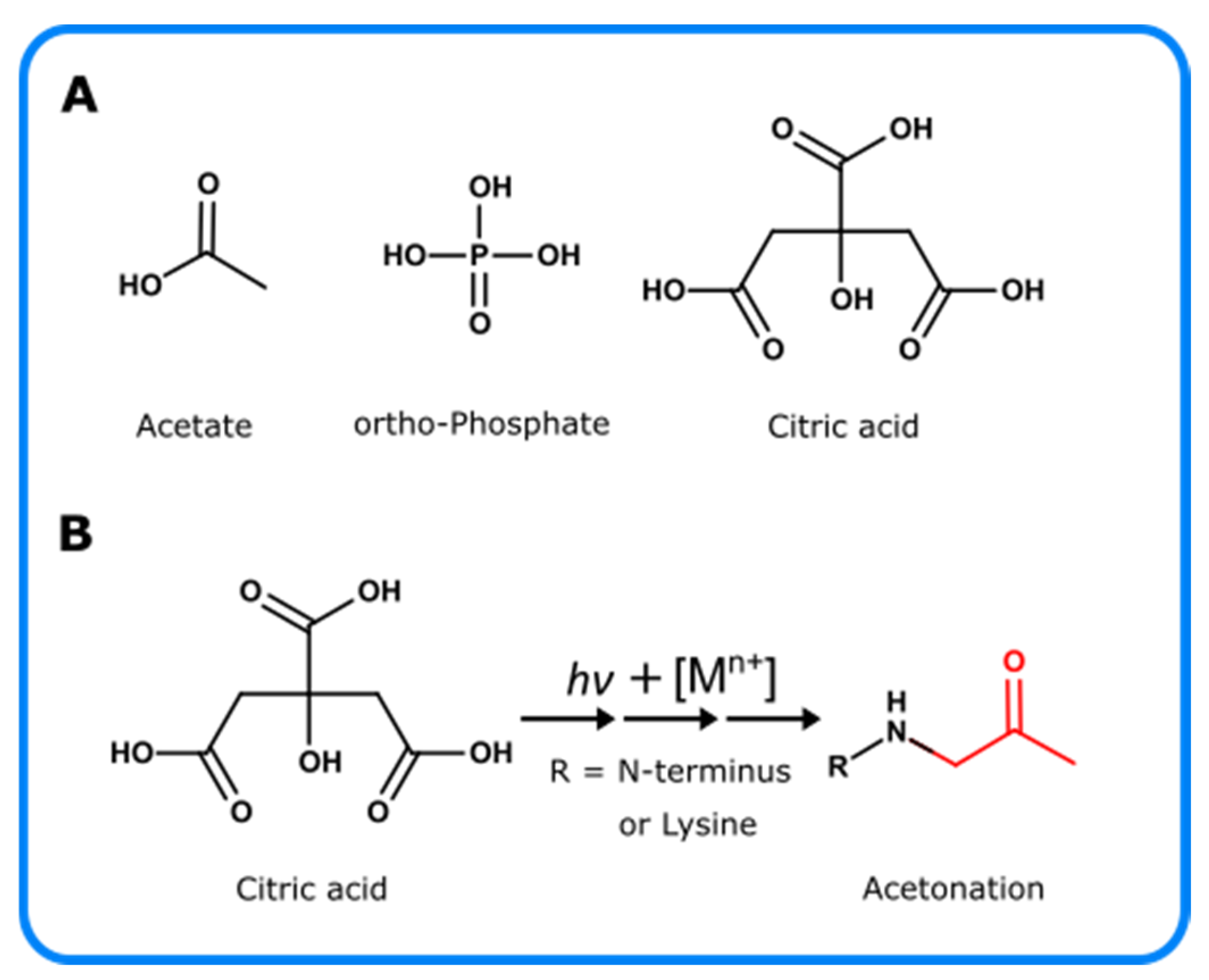
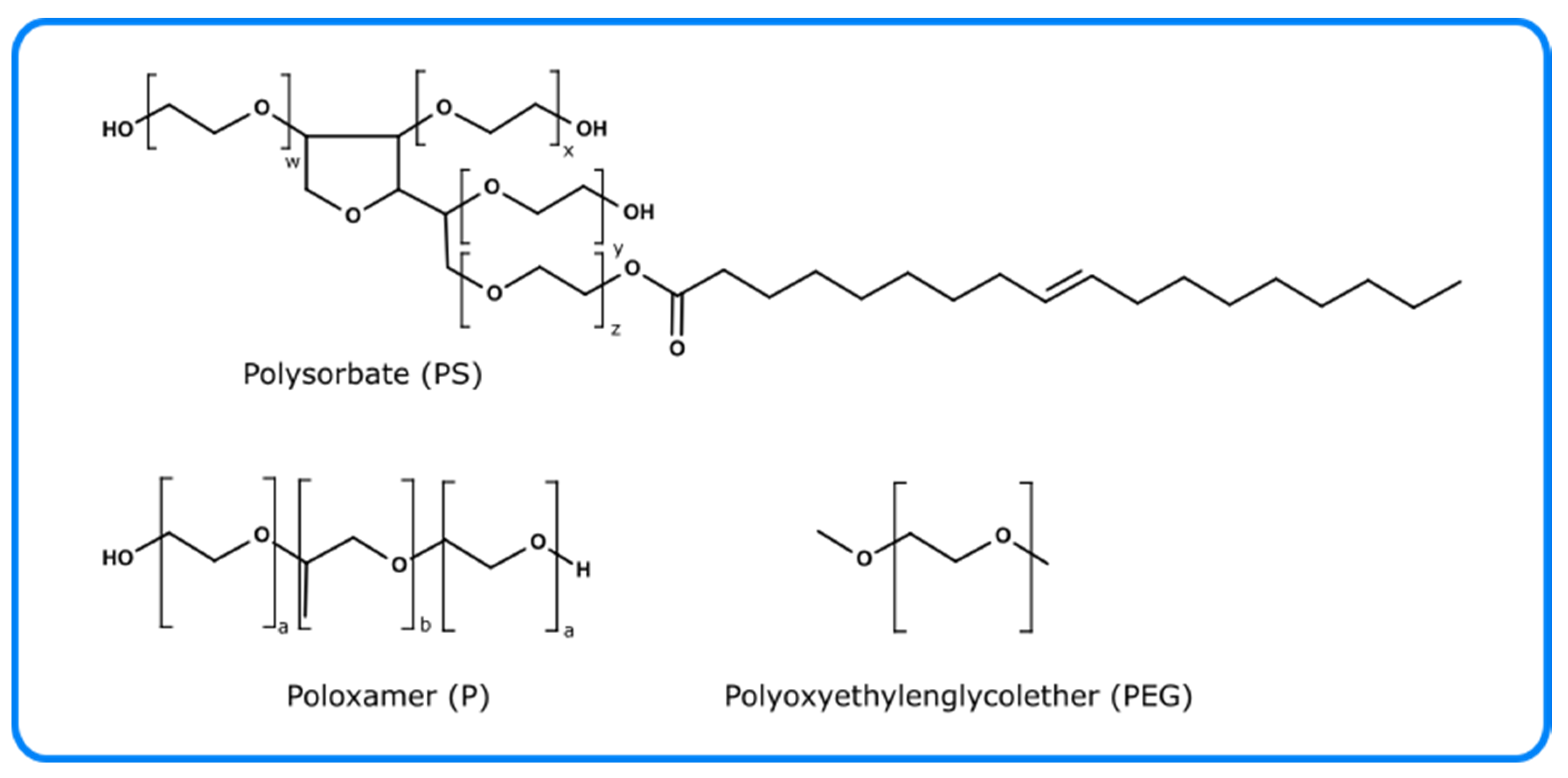
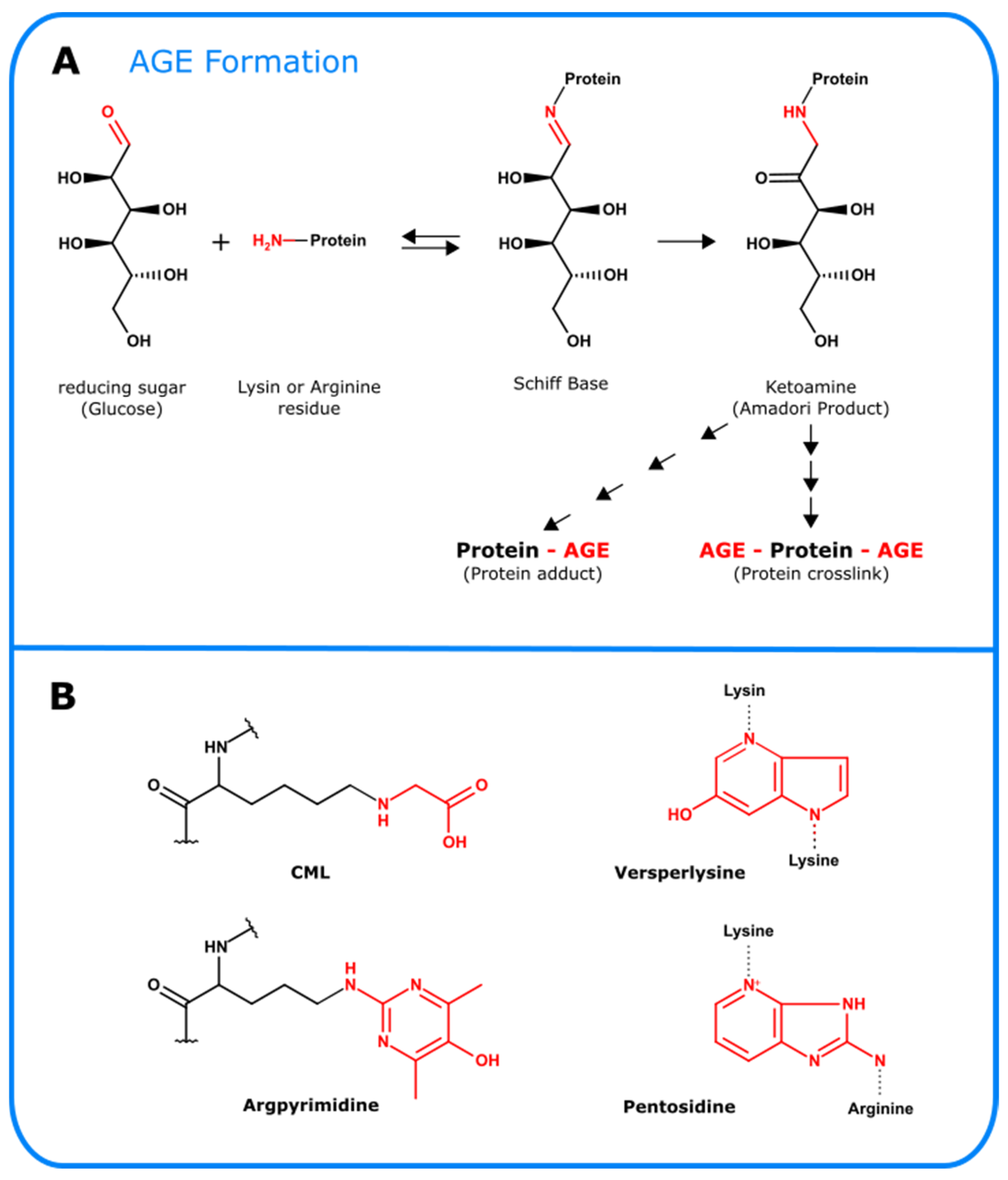

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
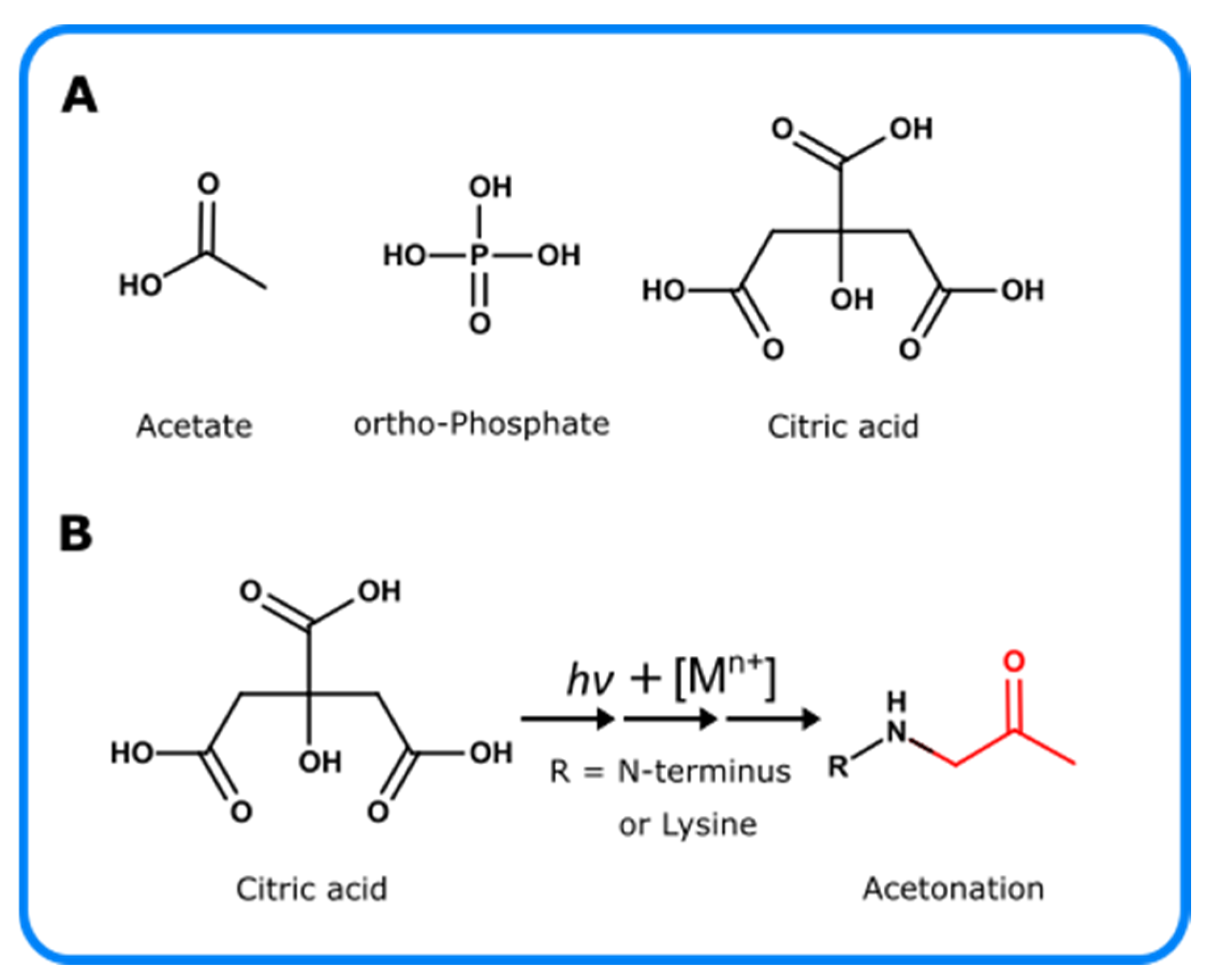{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reactive Oxygen Species | Analytical Technique | Probes/Spin Traps | Reference |
|---|---|---|---|
| 1O2 | Infrared spectroscopy | - | Ogilby et al. (2010), Baier et al. (2005), Vittorino et al. (2009) [43,44,45] |
| Fluorescence spectroscopy | DPAX, SOSG©, ATTA-Eu3+ | Umezawa et al. (1999), Corey et al. (1964), Gollmer et al. (2011), Dong et al. (2020) [50,51,52,59] | |
| Infrared spectroscopy | ICG | Tang et al. (2016) [53] | |
| UV-VIS spectroscopy | DPBF, ABDA | Entradas et al. (2020) [56] | |
| EPR | TEMP | Sang et al. (2010) [60] |
| Reactive Oxygen Species | Analytical Technique | Probes/Spin Traps | Reference |
|---|---|---|---|
| ·O2− | UV-Vis spectroscopy | - | Bielski et al. (1978) [76] |
| Chemiluminescence | Luminol, Lucigenin | Vasil‘ev et al. (2006), Yasuuta et al. (1999), Lee et al. (2008) [77,78,79] | |
| Fluorescence spectroscopy | DHE, mitoSOX©, TEMPO-9-Ac, HO-1889NH, TBT, DBZTC | Dikalov et al. (2014), Leem et al. (2018), Gao et al. (2007) [82,85,91] | |
| Colorimetric assay | Cytc, NBT | Mullarkey et al. (1990), Mossine et al. (1999), Bielski et al. (1980) [92,93,94,95,96,97] | |
| EPR, | DMPO; BMPO | Warwar et al. (2011), Khramtsov et al. (1999), Konaka et al. (1999), Diaz-Uribe et al. (2010), Xia et al. (2015) [102,103,105,106,107] | |
| NMR | |||
| HPLC | - | Taiwo et al. (1990) [111] |
| Reactive Oxygen Species | Analytical Technique | Probes | Reference |
|---|---|---|---|
| H2O2 | UV-Vis spectroscopy | FOX Assay HRP | Noble et al. (1970), Taylor et al. (1949), Pick et al. (1980) [118,119,129] |
| Fluorescence spectroscopy | Amplex Red® | Zhou et al. (1997), Nurjayadi et al. (2016) [126,128] | |
| HPLC | TPP | Pinckernell et al. (1997) [133] |
| Reactive Oxygen Species | Analytical Technique | Probes | Reference |
|---|---|---|---|
| ·OH | Fluorescence spectroscopy | TPA, APF, HPF | Saran et al. (1999), Proce et al. (2009), Freinbichler et al. (2008), Setsukinai et al. (2003), Cohn et al. (2009) [150,152,156,157,158] |
| HPLC | salicylic acid, phenylalanine | Diez et al. (2001), Freinbichler et al. (2008) [159,161] | |
| EPR | DMPO, BMPO | Sel et al. (2014), Biller et al. (2015) [162,163] |
| Amino Acid Residue | Oxidized Amino Acid Residues | Analytical Technique | Reference |
|---|---|---|---|
| Trp | Loss of Trp residue | Fluorescence spectroscopy | Davies et al. (1999), Lakowicz et al. (2006), Dalsgarrd et al. (2007), Paz et al. (2012) [260,270,271,272] |
| NFK, Kyn, 5-OH-Trp | HPLC-RP | Simat et al. (1998), Yang et al. (2007) [273,274] | |
| NFK, Kyn | Fluorescence spectroscopy | Fakunaga et al. (1982), Ehrenshaft et al. (2015) [188,275] | |
| NFK, Kyn, 3-OH-Trp | Raman spectroscopy with principal component analysis | McAvan et al. (2020) [229] | |
| CD spectroscopy with principal component analysis | McAvan et al. (2020) [229] | ||
| NFK, Kyn, 3-OH-Kyn | ESI-LCMS | Domingues et al. (2003), Perdivara et al. (2010) [276,277] | |
| NFK, Kyn | NMR | Hinterholzer et al. (2020) [230] | |
| Antiserum | Staniszewska et al. (2007), Ehrenshaft el al (2009) [278,279] | ||
| Di-Trp | UV and Fluorescence spectroscopy | Hawkins et al. (2009) [280] | |
| 18O-labeling MS spectroscopy | Medinas et al. (2010), Mariotti et al. (2018) [185,252] |
| Amino Acid Residue | Oxidized Amino Acid Residues | Analytical Technique | Reference |
|---|---|---|---|
| Tyr | Di-Tyr | UV spectroscopy | Hawkins et al. (2009) [280] |
| Fluorescence spectroscopy | Malencik et al. (1996) [293] | ||
| Affinity & RP-HPLC with fluorescence detection | Malencik et al. (1996), Davies et al. (2001), Mahmoud et al. (1995), Malencik et al. (2003) [293,294,295,299] | ||
| Di-Tyr, Tyr-Trp, Tyr-Lys, Tyr-His | ESI-MS/MS (and with 18O labeling with trypsin) | Mukherjee et al. (2017), Atwood et al. (2004), Trnka et al. (2014), Mariotti et al. (2018), Degendorfer et al. (2018), Leinisch et al. (2018) [185,220,253,254,255,300,301] | |
| Di-Tyr | Immunodetection | Kato et al. (1998), Kato et al. (2000) [302,303] | |
| DOPA | SDS-Page | Paz et al. (1991) [304] | |
| DOPA, o-Tyr, m-Tyr | Fluorescence spectroscopy | Creed et al. (1984), Kerwin et al. (2007) [3,256] | |
| DOPA | UV- spectroscopy | Waite et al. (1984) [305] | |
| HPLC with fluorescence detection | Armstrong et al. (1995) [306] | ||
| HPLC with electrochemical detection | Armstrong et al. (1995), Ke et al. (2016), Kumarathasan et al. (2003) [306,307,308] |
| Amino Acid Residue | Oxidized Amino Acid Residues | Analytical Technique | Reference |
|---|---|---|---|
| Phe | o-Tyr, m-Tyr | see Tyr | Orhan et al. (2004), Wheeler et al. (1969), Martin- Tornero et al. (2019) [316,317,318] |
| Amino Acid Residue | Oxidized Amino Acid Residues | Analytical Technique | Reference |
|---|---|---|---|
| Met | Met-sulfoxide | Electrophoretic assay | Schöneich et al. (2005), Saunders et al. (2012) [324,325] |
| Affinity HPLC (Protein A) | Mo et al. (2016), Loew et al. (2012) [320,326] | ||
| Met-oxidation | Raman and FTIR | Balakrishnan et al. (2018) [319] | |
| LC/MS (ECD; CID and FTICR-MS) | Balakrishnan et al. (2018), Mo et al. (2012), Sen et al. (2017), Guan et al. (2003) [319,323,329,330] | ||
| NMR | Hinterholzer et al. (2020) [230] | ||
| Immunological detection | Oien et al. (2009), Wehr et al. (2012) [332,333] |
| Amino Acid Residue | Oxidized Amino Acid Residues | Analytical Technique | Reference |
|---|---|---|---|
| Cys | Loss of Cys | UV-Vis-spectroscopy with TNB | Poole et al. (1989) [346] |
| Fluorescence spectroscopy | Wu et al. (1976), Hoff et al. (2013), Ercal et al. (2001) [352,353,354] | ||
| Disulfides | Reduction with DTT, 2-Mercaptoethanol then thiol detection methods | Getz et al. (1999) [355] | |
| SDS-Page (with, e.g., AMS, 3H-IAM) | Alcock et al. (2018), Jakob et al. (1999), Lee et al. (1998) [349,356,357] | ||
| LC/MS | Tsai et al. (2013) [358] | ||
| NMR | Sharma et al. (2000) [359] | ||
| X-ray crystallography | McCarthy et al. (2000) [360] | ||
| Sulfenic acid | LC-MS with 1,3-diketone such as dimedione or biotin tags | Poole et al. (2007), Poole et al. (2005), Charles et al. (2007) [361,362,363] | |
| Immunological detection | Seo et al. (2009) [364] |
| Amino Acid Residue | Oxidized Amino Acid Residues | Analytical Technique | Reference |
|---|---|---|---|
| His | 2-Oxo-His | RP-HPLC-ECD/CID-MS | Uchida et al. (1993), Uchida et al. (1994), Srikanth et al. (2009), Bridgewater et al. (2007) [391,392,393,394] |
| His-His, His-Cys, His-Lys, His-Arg | 18O labelling MS approach | Shen et al. (2000), Leinisch et al. (2018), Xu et al. (20179; Liu et al. (2014) [187,221,255,395] |
| Amino Acid Residue | Oxidized Amino Acid Residues | Analytical Technique | Reference |
|---|---|---|---|
| Lys | α-aminoadipic | UV/Vis spectroscopy with TBARS/DNPH | Ohkawa et al. (1979), Fields et al. (1971) [397,398] |
| HPLC with fluorometric detection and labeling with ABA | Akagawa et al. (2009) [399] | ||
| LC-ESI-MS (with labeling with ABA) | Chavez et al. (2010), Estévez et al. (2009) [400,404] | ||
| 5-Hydroxylysine | LC-MS/MS | Xie et al. (2016) [405] |
| Amino Acid Residue | Oxidized Amino Acid Residues | Analytical Technique | Reference |
|---|---|---|---|
| Arg | glutamic semi-aldehyd | See α-aminoadipic | |
| 5-hydroxy-2-aminovaleric acid can | GC-MS | Ayala et al. (1996), [406] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hipper, E.; Blech, M.; Hinderberger, D.; Garidel, P.; Kaiser, W. Photo-Oxidation of Therapeutic Protein Formulations: From Radical Formation to Analytical Techniques. Pharmaceutics 2022, 14, 72. https://doi.org/10.3390/pharmaceutics14010072
Hipper E, Blech M, Hinderberger D, Garidel P, Kaiser W. Photo-Oxidation of Therapeutic Protein Formulations: From Radical Formation to Analytical Techniques. Pharmaceutics. 2022; 14(1):72. https://doi.org/10.3390/pharmaceutics14010072
Chicago/Turabian StyleHipper, Elena, Michaela Blech, Dariush Hinderberger, Patrick Garidel, and Wolfgang Kaiser. 2022. "Photo-Oxidation of Therapeutic Protein Formulations: From Radical Formation to Analytical Techniques" Pharmaceutics 14, no. 1: 72. https://doi.org/10.3390/pharmaceutics14010072