
Development of Heterocyclic PPAR Ligands for Potential Therapeutic Applications



Abstract
:1. Introduction
2. Structure of PPAR
3. Types and Expressions of PPARs
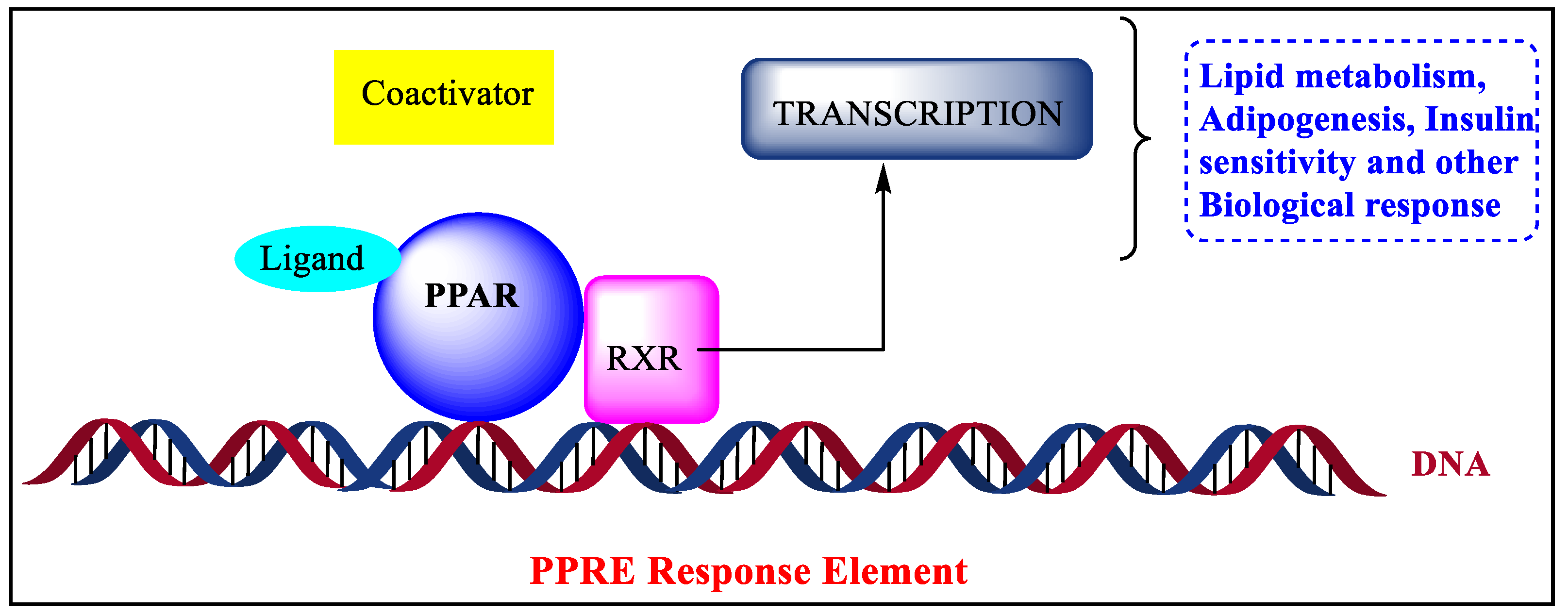
4. Functions of PPARs
5. Recent Developments in the Medicinal Chemistry of PPARs
5.1. Thiazolidinediones
5.2. Oxazole and Oxadiazole
5.3. Benzoimidazole
5.4. Thiazole
5.5. Indole
5.6. Furan
5.7. Benzopyran
5.8. Bavachinin (BVC)
5.9. Miscellaneous
6. Conclusions
7. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
List of Abbreviations
| 5-ASA | 5-Aminosalicylic acid |
| ACCs | Acetyl-CoA carboxylase |
| ADAM | 6-(5-heptyl-1,2-oxazol-3-yl)hexanoic acid |
| BVC | Bavachinin |
| CPT1 | Carnitine palmityl transferase 1 |
| FF | Fenofibrate |
| hBM-MSCs | human bone marrow-mesenchymal stem cells |
| LBP | Ligand-binding pocket |
| LDL-C | Low-density lipoprotein cholesterin |
| MDS | Molecular design suite |
| NHR assay | Nuclear hormone assay |
| OFAI | Oxohexadecenoic acids (7E)-9-oxohexadec-7-enoic |
| OFAII | (10E)-9 -oxohexadec-10-enoic acid |
| OGTT | Oral glucose tolerance test |
| PB | 2-Prenylated benzopyrans |
| PPAR | Peroxisome proliferator-activated receptors |
| PPREs | Peroxisome proliferator hormone response elements |
| RXR | Retinoid X receptor |
| SAR | Structural activity relationship |
| T2DM | Type 2 diabetes mellitus |
| TC | Total cholesterol |
| TG | Triglycerides TR-FRET: the time-resolved fluorescence resonance energy transfer |
| TZD | Thiazolidinedione |
References
- Lee, S.J.; Samala, M.; Woo, S.Y.; Hahn, D.; Kim, D.; Kadayat, T.M.; Jung, K.; Kim, J.; Kim, D.S.; Kwon, S.; et al. Enantioselective Synthesis of a Novel Thiazoline Core as a Potent Peroxisome Proliferator-Activated Receptor δ Agonist. ACS Omega 2018, 3, 1970–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadayat, T.M.; Lee, G.; Jung, K.; Hwang, H.J.; Joo, J.; Hahn, D.; Hwang, H.; Park, K.G.; Cho, S.J.; Kim, K.H.; et al. Synthesis of a unique dimethyl thiazoline containing intermediate of novel peroxisome proliferator-activated receptors(PPAR)δ agonists. Tetrahedron Lett. 2018, 59, 4384–4386. [Google Scholar] [CrossRef]
- Miyamoto, T.; Kaneko, A.; Kakizawa, T.; Yajima, H.; Kamijo, K.; Sekine, R.; Hiramatsu, K.; Nishii, Y.; Hashimoto, T.; Hashizume, K. Inhibition of Peroxisome Proliferator Signaling Pathways by Thyroid Hormone Receptor Competitive Binding To The Response Element*. J. Biol. Chem. 1997, 272, 7752–7758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Li, J.; Kennedy, L.J.; Tao, S.; Hernández, A.S.; Lai, Z.; Chen, S.; Wong, H.; Zhu, J.; Trehan, A.; et al. Discovery and Preclinical Evaluation of BMS-711939, an Oxybenzylglycine Based PPARα Selective Agonist. ACS Med. Chem. Lett. 2016, 7, 590–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Cho, J.H.; Lee, S.; Lee, W.; Chang, S.C.; Chung, H.Y.; Moon, H.R.; Lee, J. Neuroprotective effects of MHY908, a PPAR α/γ dual agonist, in an MPTP-induced Parkinson’s disease model. Brain Res. 2019, 1704, 47–58. [Google Scholar] [CrossRef]
- Mirza, R.; Sharma, B. A selective peroxisome proliferator-activated receptor-γ agonist benefited propionic acid-induced autism-like behavioral phenotypes in rats by attenuation of neuroinflammation and oxidative stress. Chem.-Biol. Interact. 2019, 311, 108758. [Google Scholar] [CrossRef]
- Locci, A.; Pinna, G. Stimulation of Peroxisome Proliferator-Activated Receptor-α by N-Palmitoylethanolamine Engages Allopregnanolone Biosynthesis to Modulate Emotional Behavior. Biol. Psychiatry 2019, 85, 1036–1045. [Google Scholar] [CrossRef]
- Ohura, A.; Itoh, T.; Ishida, H.; Saito, A.; Yamamoto, K. Three-Component Regioselective Synthesis of Tetrahydrofuro [2,3-d]oxazoles and Their Efficient Conversion to Oxazoles. Asian J. Org. Chem. 2017, 6, 673–676. [Google Scholar] [CrossRef]
- Li, Y.; Chen, H.; Ke, Z.; Huang, J.; Huang, L.; Yang, B.; Fan, S.; Huang, C. Identification of isotschimgine as a novel farnesoid X receptor agonist with potency for the treatment of obesity in mice. Biochem. Biophys. Res. Commun. 2020, 521, 639–645. [Google Scholar] [CrossRef]
- Fan, S.; Tong, T.; Fang, L.; Wu, J.; Li, E.; Kang, H.; Lv, X.; Wang, X. A facile one-pot synthesis of 2-o-cyanoaryl oxazole derivatives mediated by CuCN. Tetrahedron Lett. 2018, 59, 1409–1413. [Google Scholar] [CrossRef]
- Giampietro, L.; Gallorini, M.; de Filippis, B.; Amoroso, R.; Cataldi, A.; di Giacomo, V. PPAR-γ agonist GL516 reduces oxidative stress and apoptosis occurrence in a rat astrocyte cell line. Neurochem. Int. 2019, 126, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, N.S.; Kumar, N.; Duseja, A. Peroxisome Proliferator-Activated Receptors and Their Agonists in Nonalcoholic Fatty Liver Disease. J. Clin. Exp. Hepatol. 2019, 9, 731–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shioi, R.; Okazaki, S.; Noguchi-Yachide, T.; Ishikawa, M.; Makishima, M.; Hashimoto, Y.; Yamaguchi, T. Switching subtype-selectivity: Fragment replacement strategy affords novel class of peroxisome proliferator-activated receptor α/δ (PPARα/δ) dual agonists. Bioorgan. Med. Chem. Lett. 2017, 27, 3131–3134. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.Z.; Althagafi, I.I.; Shamshad, H. Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. Eur. J. Med. Chem. 2019, 166, 502–513. [Google Scholar] [CrossRef]
- Takada, I.; Makishima, M. Peroxisome proliferator-activated receptor agonists and antagonists: A patent review (2014-present). Expert Opin. Ther. Pat. 2020, 30, 1–13. [Google Scholar] [CrossRef]
- Zoete, V.; Grosdidier, A.; Michielin, O. Peroxisome proliferator-activated receptor structures: Ligand specificity, molecular switch and interactions with regulators. Biochim. Et Biophys. Acta—Mol. Cell Biol. Lipids 2007, 1771, 915–925. [Google Scholar] [CrossRef]
- Christofides, A.; Konstantinidou, E.; Jani, C.; Boussiotis, V.A. The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metab. Clin. Exp. 2021, 114, 154338. [Google Scholar] [CrossRef]
- D’Aniello, E.; Fellous, T.; Iannotti, F.A.; Gentile, A.; Allarà, M.; Balestrieri, F.; Gray, R.; Amodeo, P.; Vitale, R.M.; di Marzo, V. Identification and characterization of phytocannabinoids as novel dual PPARα/γ agonists by a computational and in vitro experimental approach. Biochim. Biophys. Acta—Gen. Subj. 2019, 1863, 586–597. [Google Scholar] [CrossRef]
- Dou, X.Z.; Nath, D.; Shin, Y.; Ma, J.X.; Duerfeldt, A.S. Structure-guided evolution of a 2-phenyl-4-carboxyquinoline chemotype into PPARα selective agonists: New leads for oculovascular conditions. Bioorgan. Med. Chem. Lett. 2018, 28, 2717–2722. [Google Scholar] [CrossRef]
- Cam, M.E.; Hazar-Yavuz, A.N.; Yildiz, S.; Keles, R.; Ertas, B.; Kabasakal, L. Dapagliflozin attenuates depressive-like behavior of male rats in the forced swim test. Eur. Neuropsychopharmacol. 2019, 29, S262–S263. [Google Scholar] [CrossRef]
- Feng, X.Y.; Jia, W.Q.; Liu, X.; Jing, Z.; Liu, Y.Y.; Xu, W.R.; Cheng, X.C. Identification of novel PPARα/γ dual agonists by pharmacophore screening, docking analysis, ADMET prediction, and molecular dynamics simulations. Comput. Biol. Chem. 2019, 78, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as metabolic regulators in the liver: Lessons from liver-specific PPAR-null mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamichane, S.; Lamichane, B.D.; Kwon, S.M. Pivotal roles of peroxisome proliferator-activated receptors (PPARs) and their signal cascade for cellular and whole-body energy homeostasis. Int. J. Mol. Sci. 2018, 19, 949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagi, S.; Gupta, P.; Saini, A.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Popeijus, H.E. Peroxisome Proliferator-Activated Receptor Alpha (PPAR-Alpha). In Encyclopedia of Signaling Molecules; Springer: New York, NY, USA, 2016; pp. 1–6. [Google Scholar] [CrossRef]
- Popeijus, H.E.; van Otterdijk, S.D.; van der Krieken, S.E.; Konings, M.; Serbonij, K.; Plat, J.; Mensink, R.P. Fatty acid chain length and saturation influences PPARα transcriptional activation and repression in HepG2 cells. Mol. Nutr. Food Res. 2014, 58, 2342–2349. [Google Scholar] [CrossRef]
- Wirth, M.; Benson, G.; Schwarz, C.; Obe, T.K.; Stekovic, S.; Madeo, F.; Oel, A.F. O1-12-03 Spermidine Supplementation To Improve Episodic Memory In Aging Adults At Risk Of Dementia. Alzheimer’s Dement. 2018, 14, 250. [Google Scholar] [CrossRef]
- Kim, D.S.; Lee, J.; Londhe, A.M.; Kadayat, T.M.; Joo, J.; Hwang, H.; Kim, K.H.; Pae, A.N.; Chin, J.; Cho, S.J.; et al. Synthesis and evaluation of an orally available “Y”-shaped biaryl peroxisome proliferator-activated receptor δ agonist. Bioorgan. Med. Chem. 2018, 26, 4382–4389. [Google Scholar] [CrossRef]
- Rohini, A.; Agrawal, N.; Kumar, H.; Nath, V.; Kumar, V. Norbixin, an apocarotenoid derivative activates PPARγ in cardiometabolic syndrome: Validation by in silico and in vivo experimental assessment. Life Sci. 2018, 209, 69–77. [Google Scholar] [CrossRef]
- Grygiel-Górniak, B. Peroxisome Proliferator-Activated Receptors and Their Ligands: Nutritional and Clinical Implications-a Review. 2014. Available online: http://www.nutritionj.com/content/13/1/17 (accessed on 15 July 2022).
- Govindarajulu, M.Y. P3-040: Novel Ppar-Γ Agonist Improves Pathologies And Memory Deficits In In Vitro And In Vivo Models Of Alzheimer’s Disease. Alzheimer’s Dementia 2018, 14, 1079. [Google Scholar] [CrossRef]
- Li, Y.; Xu, L.; Zeng, K.; Xu, Z.; Suo, D.; Peng, L.; Ren, T.; Sun, Z.; Yang, W.; Jin, X.; et al. Propane-2-sulfonic acid octadec-9-enyl-amide, a novel PPARα/γ dual agonist, protects against ischemia-induced brain damage in mice by inhibiting inflammatory responses. Brain Behav. Immun. 2017, 66, 289–301. [Google Scholar] [CrossRef]
- Stebbins, K.J.; Broadhead, A.R.; Cabrera, G.; Correa, L.D.; Messmer, D.; Bundey, R.; Baccei, C.; Bravo, Y.; Chen, A.; Stock, N.S.; et al. In vitro and in vivo pharmacology of NXT629, a novel, and selective PPARα antagonist. Eur. J. Pharmacol. 2017, 809, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Galambos, M.R.; Bowlus, C.L.; Swain, M.G.; Doerffel, Y.; Steinberg, A.S.; Varga, M.; Choi, Y.-J.; Martin, R.L.; Chera, H.; et al. Proof of efficacy for Seladelpar, a selective PPAR-δ agonist, in patients with primary biliary cholangitis non-responsive to ursodeoxycholic acid: Results of an international Phase 2 randomized controlled clinical study. J. Hepatol. 2017, 66, S357. [Google Scholar] [CrossRef]
- Wang, Y.; Guan, X.; Gao, C.L.; Ruan, W.; Zhao, S.; Kai, G.; Li, F.; Pang, T. Medioresinol as a novel PGC-1α activator prevents pyroptosis of endothelial cells in ischemic stroke through PPARα-GOT1 axis. Pharmacol. Res. 2021, 169, 105640. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Zeng, Z.; Zhang, W.; Yan, Z.; Chen, J.; Yu, L.; Yang, Q.; Li, Y.; Yu, H.; Chen, J.; et al. Design, synthesis, and biological evaluation of novel sulindac derivatives as partial agonists of PPARγ with potential anti-diabetic efficacy. Eur. J. Med. Chem. 2021, 222, 113542. [Google Scholar] [CrossRef]
- Zhou, Y.; Guo, Y.; Zhu, Y.; Sun, Y.; Li, W.; Li, Z.; Wei, L. Dual PPARγ/ɑ agonist oroxyloside suppresses cell cycle progression by glycolipid metabolism switch-mediated increase of reactive oxygen species levels. Free Radic. Biol. Med. 2021, 167, 205–217. [Google Scholar] [CrossRef]
- Decara, J.; Rivera, P.; López-Gambero, A.J.; Serrano, A.; Pavón, F.J.; Baixeras, E.; de Fonseca, F.R.; Suárez, J. Peroxisome Proliferator-Activated Receptors: Experimental Targeting for the Treatment of Inflammatory Bowel Diseases. Front. Pharmacol. 2020, 11, 730. [Google Scholar] [CrossRef]
- Ferré, P. The Biology of Peroxisome Proliferator-Activated Receptors Relationship With Lipid Metabolism and Insulin Sensitivity. Diabetes 2004, 53, S43–S50. [Google Scholar] [CrossRef] [Green Version]
- Tinto, F.; Archambault, A.S.; Dumais, É.; Rakotoarivelo, V.; Kostrzewa, M.; Martin, C.; Plante, P.L.; Desjardins, Y.; Simard, M.; Pouliot, R.; et al. Synthesis and molecular targets of N-13-hydroxy-octadienoyl-ethanolamine, a novel endogenous bioactive 15-lipoxygenase-derived metabolite of N-linoleoyl-ethanolamine found in the skin and saliva. Biochim. Biophys. Acta—Mol. Cell Biol. Lipids 2021, 1866, 158954. [Google Scholar] [CrossRef]
- DeFronzo, R.A. Chiglitazar: A novel pan-PPAR agonist. Sci. Bull. 2021, 66, 1497–1498. [Google Scholar] [CrossRef]
- Dharavath, R.N.; Arora, S.; Kondepudi, K.K.; Bishnoi, M.; Chopra, K. Saroglitazar, a novel dual PPAR-α/γ agonist, reverses high fat-low protein diet-induced metabolic and cognitive aberrations in C57BL/6J male mice. Life Sci. 2021, 271, 119191. [Google Scholar] [CrossRef]
- Rzemieniec, J.; Litwa, E.; Wnuk, A.; Lason, W.; Kajta, M. Bazedoxifene and raloxifene protect neocortical neurons undergoing hypoxia via targeting ERα and PPAR-γ. Mol. Cell. Endocrinol. 2018, 461, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Usui-Ouchi, A.; Ouchi, Y.; Ebihara, N. The peroxisome proliferator-activated receptor pan-agonist bezafibrate suppresses microvascular inflammatory responses of retinal endothelial cells and vascular endothelial growth factor production in retinal pigmented epithelial cells. Int. Immunopharmacol. 2017, 52, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Takei, K.; Nakagawa, Y.; Wang, Y.; Han, S.I.; Satoh, A.; Sekiya, M.; Matsuzaka, T.; Shimano, H. Effects of K-877, a novel selective PPARα modulator, on small intestine contribute to the amelioration of hyperlipidemia in low-density lipoprotein receptor knockout mice. J. Pharmacol. Sci. 2017, 133, 214–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piemontese, L.; Cerchia, C.; Laghezza, A.; Ziccardi, P.; Sblano, S.; Tortorella, P.; Iacobazzi, V.; Infantino, V.; Convertini, P.; Piaz, F.D.; et al. New diphenylmethane derivatives as peroxisome proliferator-activated receptor alpha/gamma dual agonists endowed with anti-proliferative effects and mitochondrial activity. Eur. J. Med. Chem. 2017, 127, 379–397. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Pascau, L.; Britti, E.; Calap-Quintana, P.; Dong, Y.N.; Vergara, C.; Delaspre, F.; Medina-Carbonero, M.; Tamarit, J.; Pallardó, F.v.; Gonzalez-Cabo, P.; et al. PPAR gamma agonist leriglitazone improves frataxin-loss impairments in cellular and animal models of Friedreich Ataxia. Neurobiol. Dis. 2021, 148, 105162. [Google Scholar] [CrossRef]
- Ji, L.; Song, W.; Fang, H.; Li, W.; Geng, J.; Wang, Y.; Guo, L.; Cai, H.; Yang, T.; Li, H.; et al. Efficacy and safety of chiglitazar, a novel peroxisome proliferator-activated receptor pan-agonist, in patients with type 2 diabetes: A randomized, double-blind, placebo-controlled, phase 3 trial (CMAP). Sci. Bull. 2021, 66, 1571–1580. [Google Scholar] [CrossRef]
- Barak, Y.; Liao, D.; He, W.; Ong, E.S.; Nelson, M.C.; Olefsky, J.M.; Boland, R.; Evans, R.M. Effects of peroxisome proliferator-activated receptor δ on placentation, adiposity, and colorectal cancer. Proc. Natl. Acad. Sci. USA 2002, 99(1), 303–308. [Google Scholar] [CrossRef] [Green Version]
- Boyer-Diaz, Z.; Aristu-Zabalza, P.; Andrés-Rozas, M.; Robert, C.; Ortega-Ribera, M.; Fernández-Iglesias, A.; Broqua, P.; Junien, J.L.; Wettstein, G.; Bosch, J.; et al. Pan-PPAR agonist lanifibranor improves portal hypertension and hepatic fibrosis in experimental advanced chronic liver disease. J. Hepatol. 2021, 74, 1188–1199. [Google Scholar] [CrossRef]
- Abdel-Razek, E.A.N.; Abo-Youssef, A.M.; Azouz, A.A. Benzbromarone mitigates cisplatin nephrotoxicity involving enhanced peroxisome proliferator-activated receptor-alpha (PPAR-α) expression. Life Sci. 2020, 243, 117272. [Google Scholar] [CrossRef]
- Tutunchi, H.; Ostadrahimi, A.; Saghafi-Asl, M.; Hosseinzadeh-Attar, M.J.; Shakeri, A.; Asghari-Jafarabadi, M.; Roshanravan, N.; Farrin, N.; Naemi, M.; Hasankhani, M. Oleoylethanolamide supplementation in obese patients newly diagnosed with non-alcoholic fatty liver disease: Effects on metabolic parameters, anthropometric indices, and expression of PPAR-α, UCP1, and UCP2 genes. Pharmacol. Res. 2020, 156, 104770. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, D.H.; Heck, B.E.; Shaffer, M.; Yoo, K.H.; Hur, J. PPAR-δ agonist affects adipo-chondrogenic differentiation of human mesenchymal stem cells through the expression of PPAR-γ. Regen. Ther. 2020, 15, 103–111. [Google Scholar] [CrossRef]
- Horikawa, T.; Kawanami, T.; Hamaguchi, Y.; Tanaka, Y.; Kita, S.; Ryorin, R.; Takashi, Y.; Takahashi, H.; Tanabe, M.; Yanase, T.; et al. Pemafibrate, a PPAR alpha agonist, attenuates neointima formation after vascular injury in mice fed normal chow and a high-fat diet. Heliyon 2020, 6, e05431. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, M.B.; Miranda-Perez, E.; Verjan, J.C.G.; Barrera, M.d.F.; Perez-Ramos, J.; Alarcon-Aguilar, F.J. Potential of the chlorogenic acid as multitarget agent: Insulin-secretagogue and PPAR α/γ dual agonist. Biomed. Pharmacother. 2017, 94, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, A.; Giammarco, M.L.; de Nuccio, C.; Ajmone-Cat, M.A.; Visentin, S.; de Simone, R.; Minghetti, L. Docosahexaenoic acid promotes oligodendrocyte differentiation via PPAR-γ signaling and prevents tumor necrosis factor-α-dependent maturational arrest. Biochim. Biophys. Acta—Mol. Cell Biol. Lipids 2017, 1862, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Song, G.J.; Nam, Y.; Jo, M.; Jung, M.; Koo, J.Y.; Cho, W.; Koh, M.; Park, S.B.; Suk, K. A novel small-molecule agonist of PPAR-γ potentiates an anti-inflammatory M2 glial phenotype. Neuropharmacology 2016, 109, 159–169. [Google Scholar] [CrossRef]
- Okazaki, S.; Shioi, R.; Noguchi-Yachide, T.; Ishikawa, M.; Makishima, M.; Hashimoto, Y.; Yamaguchi, T. Structure-activity relationship studies of non-carboxylic acid peroxisome proliferator-activated receptor α/δ (PPARα/δ) dual agonists. Bioorgan. Med. Chem. 2016, 24, 5455–5461. [Google Scholar] [CrossRef]
- Kinarivala, N.; Suh, J.H.; Botros, M.; Webb, P.; Trippier, P.C. Pharmacophore elucidation of phosphoiodyn A—Potent and selective peroxisome proliferator-activated receptor β/δ agonists with neuroprotective activity. Bioorgan. Med. Chem. Lett. 2016, 26, 1889–1893. [Google Scholar] [CrossRef]
- Lund, J.; Stensrud, C.; Rajender; Bohov, P.; Thoresen, G.H.; Berge, R.K.; Wright, M.; Kamal, A.; Rustan, A.C.; Miller, A.D.; et al. The molecular structure of thioether fatty acids influences PPAR-dependent regulation of lipid metabolism. Bioorgan. Med. Chem. 2016, 24, 1191–1203. [Google Scholar] [CrossRef]
- Agarwal, S.; Yadav, A.; Chaturvedi, R.K. Peroxisome proliferator-activated receptors (PPARs) as therapeutic target in neurodegenerative disorders. Biochem. Biophys. Res. Commun. 2017, 483, 1166–1177. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Goel, H.; Kotwani, A. Implications of the endogenous PPAR-gamma ligand, 15-deoxy-delta-12, 14-prostaglandin J2, in diabetic retinopathy. Life Sci. 2016, 153, 93–99. [Google Scholar] [CrossRef]
- Ishibashi, S.; Yamashita, S.; Arai, H.; Araki, E.; Yokote, K.; Suganami, H.; Fruchart, J.C.; Kodama, T. Effects of K-877, a novel selective PPARα modulator (SPPARMα), in dyslipidaemic patients: A randomized, double-blind, active- and placebo-controlled, phase 2 trial. Atherosclerosis 2016, 249, 36–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, M.; Foulquier, S.; Dupuis, F.; Flament, S.; Grimaud, L.; Henrion, D.; Lartaud, I.; Monard, G.; Grillier-Vuissoz, I.; Boisbrun, M. Synthesis and evaluation of new designed multiple ligands directed towards both peroxisome proliferator-activated receptor-γ and angiotensin II type 1 receptor. Eur. J. Med. Chem. 2018, 158, 334–352. [Google Scholar] [CrossRef] [PubMed]
- Jacintho, J.D.; Baccei, C.S.; Bravo, Y.; Broadhead, A.; Chen, A.; Correa, L.; Fischer, K.; Laffitte, B.; Lee, C.; Lorrain, D.S.; et al. Discovery of potent and selective PPARα/δ dual antagonists and initial biological studies. Bioorgan. Med. Chem. Lett. 2019, 29, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Jia, D.; Wu, X.; Shi, K.; Ren, C.; Dou, Y.; Guo, M.; Wang, J.; Ma, M.; Wu, Z.; et al. A novel metformin derivative showed improvement of lipid metabolism in obese rats with type 2 diabetes. Clin. Exp. Pharmacol. Physiol. 2020, 47, 1382–1392. [Google Scholar] [CrossRef]
- Lin, L.; Metherel, A.H.; di Miceli, M.; Liu, Z.; Sahin, C.; Fioramonti, X.; Cummins, C.L.; Layé, S.; Bazinet, R.P. Tetracosahexaenoylethanolamide, a novel N-acylethanolamide, is elevated in ischemia and increases neuronal output. J. Lipid Res. 2020, 61, 1480–1490. [Google Scholar] [CrossRef]
- Laghezza, A.; Piemontese, L.; Tortorella, P.; Loiodice, F. An update about the crucial role of stereochemistry on the effects of Peroxisome Proliferator-Activated Receptor ligands. Eur. J. Med. Chem. 2019, 176, 326–342. [Google Scholar] [CrossRef]
- Wada, Y.; Maekawa, M.; Ohnishi, T.; Balan, S.; Matsuoka, S.; Iwamoto, K.; Iwayama, Y.; Ohba, H.; Watanabe, A.; Hisano, Y.; et al. Peroxisome proliferator-activated receptor α as a novel therapeutic target for schizophrenia. EBioMedicine 2020, 62, 103130. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, Z.; Hu, L.; Deng, L.; Ren, Q.; Zhang, L. ZLY032, the first-in-class dual FFA1/PPARδ agonist, improves glucolipid metabolism and alleviates hepatic fibrosis. Pharmacol. Res. 2020, 159, 105035. [Google Scholar] [CrossRef]
- Lim, J.J.; Dutta, M.; Dempsey, J.L.; Lehmler, H.J.; MacDonald, J.; Bammler, T.; Walker, C.; Kavanagh, T.J.; Gu, H.; Mani, S.; et al. Neonatal exposure to BPA, BDE-99, and PCB produces persistent changes in hepatic transcriptome associated with gut dysbiosis in adult mouse livers. Toxicol. Sci. 2021, 184, 83–103. [Google Scholar] [CrossRef]
- Eeda, V.; Wu, D.; Lim, H.Y.; Wang, W. Design, synthesis, and evaluation of potent novel peroxisome proliferator-activated receptor γ indole partial agonists. Bioorgan. Med. Chem. Lett. 2019, 29, 126664. [Google Scholar] [CrossRef]
- Judkins, R.; Benthem, L. Sulfinic Acid and Sulfonic Acid Compounds for Use in Modulating Peroxisome Proliferator-Activated Receptors. WO2021161218, 19 August 2021. Available online: https://patents.google.com/patent/WO2021161218A1 (accessed on 15 July 2022).
- Imig, J.D.; Khan, M.A.H.; Proschak, E.; Blocher, R. Diabetes and Metabolic Syndrome Treatment with a Novel Dual Modulator of Soluble Epoxide Hydrolase and Peroxisome Proliferator-Activated Receptors. U.S. Patent 20210238125, 8 May 2021. [Google Scholar]
- Yu, D.D.; Forman, B.M.; Li, H. Peroxisome Proliferator-Activated Receptor Agonists. U.S. Patent 20190077774A1, 14 March 2019. [Google Scholar]
- Evans, R.M.; Downes, M. PPAR Agonists and Methods of Use Thereof. U.S. Patent 20180305403A1, 4 February 2020. [Google Scholar]
- Antkaly, T. PPAR-γ Activators and Their Therapeutic Usages. U.S. Patent 2020190000790A1, 3 January 2019. [Google Scholar]
- Pahan, K.; Roy, A. Brain PPARα Ligands. U.S. Patent 20190060269A1, 28 February 2019. [Google Scholar]
- Uji-Shi, T.K.; Uji-Shi, T.G.; Uji-Shi, H.T.; Uji-Shi, H.C.; Ichip, N.; Nakata, K. PPARα Activator, Pharmaceutical Composition, Food and Drink, Food Additive, Supplement and Method of Manufacturing the Same. U.S. Patent 20180015062A1, 2017. Available online: https://patents.google.com/patent/US20180015062A1 (accessed on 15 July 2022).
- Legu, B.; Senaiar, R. PPAR Agonist, Compounds, Pharmaceutical Composition, and Method of Use Thereof. WO2017180818A1, 2017. Available online: https://patents.google.com/patent/WO2017180818A1 (accessed on 15 July 2022).
- Baiga, T.; Downes, M.; Evans, R.; Kluge, A.; Laga, B.; Miura, M.; Panograhi, S.K. PPAR Agonist, Compounds, Pharmaceutical Composition, and Method of Use Thereof. US20170304255A1, 26 October 2017. Available online: https://patents.google.com/patent/US20170304255A1 (accessed on 15 July 2022).
- Knape, T.; Knethen, A.V.; Parnham, M.J.; Schubert-Zsilavecz, M.; Wurglics, M.; Flesch, D. Comoetative PPAR-α Antagonista. U.S. Patent 20170210711A1, 27 July 2017. [Google Scholar]
- Darwish, K.M.; Ismail, S.; Samia, M.; Mohamed, S.G.; Mohamed, A.H. Design, synthesis, and biological evaluation of novel thiazolidinediones as PPARγ/FFAR1 dual agonists. Eur. J. Med. Chem. 2016, 109, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Chhajed, S.S.; Chaskar, S.; Kshirsagar, S.K.; Haldar, G.M.A.; Mahapatra, D.K. Rational design and synthesis of some PPAR-γ agonists: Substituted benzylideneamino-benzylidene-thiazolidine-2,4-diones. Comput. Biol. Chem. 2017, 67, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, K.R.A.; Fadaly, W.A.A.; Kamel, G.M.; Elshaier, Y.A.M.M.; El-Magd, M.A. Design, synthesis, modeling studies and biological evaluation of thiazolidine derivatives containing pyrazole core as potential anti-diabetic PPAR-γ agonists and anti-inflammatory COX-2 selective inhibitors. Bioorgan. Chem. 2019, 82, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.R.; Bhatia, R.; Chawla, P. Synthesis, biological evaluation and molecular docking studies of novel 3,5-disubstituted 2,4-thiazolidinediones derivatives. Bioorgan. Chem. 2019, 89, 102993. [Google Scholar] [CrossRef]
- Dai, L.; Feng, Z.; Zha, R.; Cheng, K.; Wen, X.; Sun, H.; Yuan, H. Discovery of Novel Peroxisome Proliferator-Activated Receptor α (PPARα) Agonists by Virtual Screening and Biological Evaluation. J. Chem. Inf. Model. 2020, 60, 1717–1727. [Google Scholar] [CrossRef]
- Shakour, N.; Sahebkar, A.; Karimi, G.; Paseban, M.; Tasbandi, A.; Mosaffa, F.; Tayarani-Najaran, Z.; Ghodsi, R.; Hadizadeh, F. Design, synthesis and biological evaluation of novel 5-(imidazolyl-methyl) thiazolidinediones as antidiabetic agents. Bioorgan. Chem. 2021, 115, 105162. [Google Scholar] [CrossRef]
- Okazaki, S.; Noguchi-Yachide, T.; Sakai, T.; Ishikawa, M.; Makishima, M.; Hashimoto, Y.; Yamaguchi, T. Discovery of N-(1-(3-(4-phenoxyphenyl)-1,2,4-oxadiazol-5-yl)ethyl)acetamides as novel acetyl-CoA carboxylase 2 (ACC2) inhibitors with peroxisome proliferator-activated receptor α/δ (PPARα/δ) dual agonistic activity. Bioorgan. Med. Chem. 2016, 24, 5258–5269. [Google Scholar] [CrossRef]
- Arnesen, H.; Haj-Yasein, N.N.; Tungen, J.E.; Soedling, H.; Matthews, J.; Paulsen, S.M.; Nebb, H.I.; Sylte, I.; Hansen, T.V.; Sæther, T. Molecular modeling, synthesis, and biological evaluations of a 3,5-disubstituted isoxazole fatty acid analog as a PPARα-selective agonist. Bioorgan. Med. Chem. 2019, 27, 4059–4068. [Google Scholar] [CrossRef]
- Jiang, Z.; Liu, X.; Yuan, Z.; He, H.; Wang, J.; Zhang, X.; Gong, Z.; Hou, L.; Shen, L.; Guo, F.; et al. Discovery of a Novel Selective Dual Peroxisome Proliferator-Activated Receptor α/δAgonist for the Treatment of Primary Biliary Cirrhosis. ACS Med. Chem. Lett. 2019, 10, 1068–1073. [Google Scholar] [CrossRef]
- Kaur, P.; Bhat, Z.R.; Bhat, S.; Kumar, R.; Kumar, R.; Tikoo, K.; Gupta, J.; Khurana, N.; Kaur, J.; Khatik, G.L. Synthesis and evaluation of new 1,2,4-oxadiazole based trans- acrylic acid derivatives as potential PPAR-alpha/gamma dual agonist. Bioorgan. Chem. 2020, 100, 103867. [Google Scholar] [CrossRef]
- Obermoser, V.; Mauersberger, R.; Schuster, D.; Czifersky, M.; Lipova, M.; Siegl, M.; Kintscher, U.; Gust, R. Importance of 5/6-aryl substitution on the pharmacological profile of 4ʹ-((2-propyl-1H-benzo[d]imidazol-1-yl)methyl)-[1,1ʹ-biphenyl]-2-carboxylic acid derived PPARγ agonists. Eur. J. Med. Chem. 2017, 126, 590–603. [Google Scholar] [CrossRef] [PubMed]
- Shinozuka, T.; Tsukada, T.; Fujii, K.; Tokumaru, E.; Shimada, K.; Onishi, Y.; Matsui, Y.; Wakimoto, S.; Kuroha, M.; Ogata, T.; et al. Discovery of DS-6930, a potent selective PPARγ modulator. Part I: Lead Identif. Bioorgan. Med. Chem. 2018, 26, 5079–5098. [Google Scholar] [CrossRef]
- Yamamoto, K.; Tamura, T.; Nakamura, R.; Hosoe, S.; Matsubara, M.; Nagata, K.; Kodaira, H.; Uemori, T.; Takahashi, Y.; Suzuki, M.; et al. Development of a novel class of peroxisome proliferator-activated receptor (PPAR) gamma ligands as an anticancer agent with a unique binding mode based on a non-thiazolidinedione scaffold. Bioorgan. Med. Chem. 2019, 27, 115122. [Google Scholar] [CrossRef]
- An, S.; Kim, G.; Kim, H.J.; Ahn, S.; Kim, H.Y.; Ko, H.; Hyun, Y.E.; Nguyen, M.; Jeong, J.; Liu, Z.; et al. Discovery and Structure-Activity Relationships of Novel Template, Truncated 1′-Homologated Adenosine Derivatives as Pure Dual PPARγ/δModulators. J. Med. Chem. 2020, 63, 16012–16027. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xu, Y.; Cai, Z.; Wang, X.; Ren, Q.; Zhou, Z.; Xie, R. Discovery of novel dual PPARα/δ agonists based on benzimidazole scaffold for the treatment of non-alcoholic fatty liver disease. Bioorgan. Chem. 2020, 99, 103803. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Lian, Y.; Tang, J.; Chen, F.; Gao, H.; Zhou, Z.; Hou, N.; Yi, W. Identification of the anti-fungal drug fenticonazole nitrate as a novel PPARγ-modulating ligand with good therapeutic index: Structure-based screening and biological validation. Pharmacol. Res. 2021, 173, 105860. [Google Scholar] [CrossRef]
- Bhalla, J.; Bari, S.S.; Chaudhary, G.R.; Kumar, A.; Rathee, A.; Sharma, R.; Bhalla, A. Stereoselective synthesis and in-silico evaluation of C4-benzimidazolyloxyphenyl substituted trans-β-lactam derivatives as promising novel PPARγ activators. Synth. Commun. 2021, 51, 3758–3767. [Google Scholar] [CrossRef]
- Kharbanda, C.; Alam, M.S.; Hamid, H.; Javed, K.; Bano, S.; Ali, Y.; Dhulap, A.; Alam, P.; Pasha, M.A.Q. Novel Piperine Derivatives with Antidiabetic Effect as PPAR-γ Agonists. Chem. Biol. Drug Des. 2016, 88, 354–362. [Google Scholar] [CrossRef]
- Li, Z.; Chen, Y.; Zhou, Z.; Deng, L.; Xu, Y.; Hu, L.; Liu, B.; Zhang, L. Discovery of first-in-class thiazole-based dual FFA1/PPARδ agonists as potential anti-diabetic agents. Eur. J. Med. Chem. 2019, 164, 352–365. [Google Scholar] [CrossRef]
- Ammazzalorso, A.; de Lellis, L.; Florio, R.; Laghezza, A.; de Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Tortorella, P.; Veschi, S.; et al. Synthesis of novel benzothiazole amides: Evaluation of PPAR activity and anti-proliferative effects in paraganglioma, pancreatic and colorectal cancer cell lines. Bioorgan. Med. Chem. Lett. 2019, 29, 2302–2306. [Google Scholar] [CrossRef]
- Mourad, A.A.E.; Mourad, M.A.E. Enhancing insulin sensitivity by dual PPARγ partial agonist, β-catenin inhibitor: Design, synthesis of new αphthalimido-o-toluoyl2-aminothiazole hybrids. Life Sci. 2020, 259, 118270. [Google Scholar] [CrossRef] [PubMed]
- Patchipala, S.B.; Pasupuleti, V.R.; Audipudi, A.V.; babu Bollikolla, H. Synthesis, in-vivo anti-diabetic & anticancer activities and molecular modeling studies of tetrahydrobenzo[d]thiazole tethered nicotinohydrazide derivatives. Arab. J. Chem. 2022, 15, 103546. [Google Scholar] [CrossRef]
- Fu, Y.; Ma, J.; Shi, X.; Song, X.Y.; Yang, Y.; Xiao, S.; Li, J.; Gu, W.J.; Huang, Z.; Zhang, J.; et al. A novel pyrazole-containing indolizine derivative suppresses NF-κB activation and protects against TNBS-induced colitis via a PPAR-γ-dependent pathway. Biochem. Pharmacol. 2017, 135, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Boubia, B.; Poupardin, O.; Barth, M.; Binet, J.; Peralba, P.; Mounier, L.; Jacquier, E.; Gauthier, E.; Lepais, V.; Chatar, M.; et al. Design Synthesis, and Evaluation of a Novel Series of Indole Sulfonamide Peroxisome Proliferator-Activated Receptor (PPAR) α/γ/δ Triple Activators: Discovery of Lanifibranor, a New Antifibrotic Clinical Candidate. J. Med. Chem. 2018, 61, 2246–2265. [Google Scholar] [CrossRef]
- El-Zahabi, M.A.; Elbendary, E.R.; Bamanie, F.H.; Radwan, M.F.; Ghareib, S.A.; Eissa, I.H. Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of phthalimide-sulfonylurea hybrids as PPARγ and SUR agonists. Bioorgan. Chem. 2019, 91, 103115. [Google Scholar] [CrossRef]
- Liu, J.; Su, X.; Li, H.; Fan, L.; Li, Y.; Tang, X.; Yan, J.; Chen, X.; Chen, F.; Liu, J.; et al. Design, synthesis, and evaluation of novel L-phenyl glycine derivatives as potential PPARγ lead compounds. Bioorgan. Med. Chem. 2018, 26, 4153–4167. [Google Scholar] [CrossRef]
- Ding, X.; Kang, D.; Sun, L.; Zhan, P.; Liu, X. Combination of 2D and 3D-QSAR studies on DAPY and DANA derivatives as potent HIV-1 NNRTIs. J. Mol. Struct. 2022, 1249, 131603. [Google Scholar] [CrossRef]
- Liu, K.; Zhao, X.; Qi, X.; Hou, D.L.; Li, H.b.; Gu, Y.H.; Xu, Q.L. Design, synthesis, and biological evaluation of a novel dual peroxisome proliferator-activated receptor alpha/delta agonist for the treatment of diabetic kidney disease through anti-inflammatory mechanisms. Eur. J. Med. Chem. 2021, 218, 1802–1812. [Google Scholar] [CrossRef]
- Niu, H.; Wang, W.; Li, J.; Lei, Y.; Zhao, Y.; Yang, W.; Zhao, C.; Lin, B.; Song, S.; Wang, S. A novel structural class of coumarin-chalcone fibrates as PPARα/γ agonists with potent antioxidant activities: Design, synthesis, biological evaluation, and molecular docking studies. Eur. J. Med. Chem. 2017, 138, 212–220. [Google Scholar] [CrossRef]
- Bermejo, A.; Collado, A.; Barrachina, I.; Marqués, P.; el Aouad, N.; Franck, X.; Garibotto, F.; Dacquet, C.; Caignard, D.H.; Suvire, F.D.; et al. Polycerasoidol, a Natural Prenylated Benzopyran with a Dual PPARα/PPARγAgonist Activity and Anti-inflammatory Effect. J. Nat. Prod. 2019, 82, 1802–1812. [Google Scholar] [CrossRef]
- Ahn, S.; Kim, J.; An, S.; Pyo, J.J.; Jung, D.; Lee, J.; Hwang, S.Y.; Gong, J.; Shin, I.; Kim, H.P.; et al. 2-Phenyl-8-(1-phenyl allyl)-chromenone compounds have a pan-PPAR modulator pharmacophore. Bioorgan. Med. Chem. 2019, 27, 2948–2958. [Google Scholar] [CrossRef] [PubMed]
- Vila, L.; Cabedo, N.; Villarroel-Vicente, C.; García, A.; Bernabeu, Á.; Hennuyer, N.; Staels, B.; Franck, X.; Figadère, B.; Sanz, M.J.; et al. Synthesis and biological studies of “Polycerasoidol” and “trans-δ-Tocotrienolic acid” derivatives as PPARα and/or PPARγ agonists. Bioorgan. Med. Chem. 2022, 53, 116532. [Google Scholar] [CrossRef] [PubMed]
- Du, G.; Zhao, Y.; Feng, L.; Yang, Z.; Shi, J.; Huang, C.; Li, B.; Guo, F.; Zhu, W.; Li, Y. Design, Synthesis, and Structure-Activity Relationships of Bavachinin Analogues as Peroxisome Proliferator-Activated Receptor γ Agonists. ChemMedChem 2017, 12, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Du, G.; Zhao, Y.; Zhang, L.; Li, B.; Zhu, W.; Huang, C.; Li, Y.; Guo, F. Bavachinin analogs as agonists of pan-peroxisome proliferator-activated receptors. Med. Chem. Res. 2018, 27, 1851–1862. [Google Scholar] [CrossRef]
- Ahn, S.; Lee, M.; An, S.; Hyun, S.; Hwang, J.; Lee, J.; Noh, M. 2-Formyl-komarovicine promotes adiponectin production in human mesenchymal stem cells through PPARγ partial agonism. Bioorgan. Med. Chem. 2018, 26, 1069–1075. [Google Scholar] [CrossRef]
- Dixit, V.A.; Rathi, P.C.; Bhagat, S.; Gohlke, H.; Petersen, R.K.; Kristiansen, K.; Chakraborti, A.K.; Bharatam, P.V. Design and synthesis of novel Y-shaped barbituric acid derivatives as PPARγ activators. Eur. J. Med. Chem. 2016, 108, 423–435. [Google Scholar] [CrossRef]
- Frkic, R.L.; He, Y.; Rodriguez, B.B.; Chang, M.R.; Kuruvilla, D.; Ciesla, A.; Abell, A.D.; Kamenecka, T.M.; Griffin, P.R.; Bruning, J.B. Structure-Activity Relationship of 2,4-Dichloro-N-(3,5-dichloro-4-(quinolin-3-yloxy)phenyl)benzenesulfonamide (INT131) Analogs for PPARγ-Targeted Antidiabetics. J. Med. Chem. 2017, 60, 4584–4593. [Google Scholar] [CrossRef]
- Gan, L.; Gan, Z.; Dan, Y.; Li, Y.; Zhang, P.; Chen, S.; Ye, Z.; Pan, T.; Wan, C.; Hu, X.; et al. Tetrazanbigen Derivatives as Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) Partial Agonists: Design, Synthesis, Structure-Activity Relationship, and Anticancer Activities. J. Med. Chem. 2021, 64, 1018–1036. [Google Scholar] [CrossRef]
- Zhao, S.; Kanno, Y.; Li, W.; Wakatabi, H.; Sasaki, T.; Koike, K.; Nemoto, K.; Li, H. Picrasidine N Is a Subtype-Selective PPARβ/δ Agonist. J. Nat. Prod. 2016, 79, 879–885. [Google Scholar] [CrossRef]
- Ibrahim, M.K.; Eissa, I.H.; Alesawy, M.S.; Metwaly, A.M.; Radwan, M.M.; ElSohly, M.A. Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of quinazolin-4(3H)-one derivatives as potential PPARγ and SUR agonists. Bioorgan. Med. Chem. 2017, 25, 4723–4744. [Google Scholar] [CrossRef]
- Yu, D.D.; van Citters, G.; Li, H.; Stoltz, B.M.; Forman, B.M. Discovery of novel modulators for the PPARα (peroxisome proliferator-activated receptor α): Potential therapies for nonalcoholic fatty liver disease. Bioorgan. Med. Chem. 2021, 41, 116193. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.P.; Reji, A.; Bhavimani, G.; Prabitha, P.; Durai, P.; Yuvaraj, S.; Shashank, A.; Krishna, K.L.; Kumar, B.R.P. Rational Design, Synthesis and Evaluation of Novel Rodanine Derivatives for Antihyperglycemic Activity. Polycycl. Aromat. Compd. 2020, 42, 1794–1805. [Google Scholar] [CrossRef]
- Hu, X.; Wan, C.; Gan, Z.; Liu, R.; Chen, Y.; Wang, J.; Gan, L.; Chen, Y.; Li, Y.; He, B.; et al. TNBG-5602, a novel derivative of quinoxaline, inhibits liver cancer growth via upregulating peroxisome proliferator-activated receptor γ in vitro and in vivo. J. Pharm. Pharmacol. 2019, 71, 1684–1694. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.D.; Xu, Y.H.; Liu, J.P.; Wang, B.; Shi, Y.H.; Wang, W.; Wang, X.P.; Sun, M.; Xu, X.Y.; Bian, X.L. 1,3-Benzodioxole-based fibrate derivatives as potential hypolipidemic and hepatoprotective agents. Bioorgan. Med. Chem. Lett. 2021, 43, 127898. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ren, Q.; Zhou, Z.; Cai, Z.; Wang, B.; Han, J.; Zhang, L. Discovery of the first-in-class dual PPARδ/γ partial agonist for the treatment of metabolic syndrome. Eur. J. Med. Chem. 2021, 225, 113807. [Google Scholar] [CrossRef]
- Wagenaar, L.J.; Kuck, E.M.; Hoekstra, J.B.L. Drug therapy (invited) Troglitazone. Is it all over? Neth. J. Med. 1999, 55, 4–12. [Google Scholar] [CrossRef]
- Darwish, K.M.; Salama, I.; Mostafa, S.; Gomaa, M.S.; Khafagy, E.-S.; Helal, M.A. Synthesis, biological evaluation, and molecular docking investigation of benzhydrol- and indole-based dual PPAR-γ/FFAR1 agonists. Bioorgan. Med. Chem. Lett. 2018, 28, 1595–1602. [Google Scholar] [CrossRef]
- Szychowski, K.A.; Leja, M.L.; Kaminskyy, D.V.; Kryshchyshyn, A.P.; Binduga, U.E.; Pinyazhko, O.R.; Lesyk, R.B.; Tobiasz, J.; Gmiński, J. Anticancer properties of 4-thiazolidinone derivatives depend on peroxisome proliferator-activated receptor gamma (PPARγ). Eur. J. Med. Chem. 2017, 141, 162–168. [Google Scholar] [CrossRef]
- Bansal, G.; Singh, S.; Monga, V.; Thanikachalam, P.V.; Chawla, P. Synthesis and biological evaluation of thiazolidine-2,4-dione-pyrazole conjugates as antidiabetic, anti-inflammatory, and antioxidant agents. Bioorgan. Chem. 2019, 92, 103271. [Google Scholar] [CrossRef]
- El-Hussieny, M.; El-Sayed, N.F.; Ewies, E.F.; Ibrahim, N.M.; Mahran, M.R.H.; Fouad, M.A. Synthesis, molecular docking and biological evaluation of 2-(thiophen-2-yl)-1H-indoles as potent HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorgan. Chem. 2020, 95, 103521. [Google Scholar] [CrossRef]


























































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr No | Date | Patent Number | Description | Ref. |
|---|---|---|---|---|
| 1. | 19 August 2021 | WO2021161218 | Sulfinic acid and sulfonic acid compounds for use in modulating peroxisome proliferator-activated receptors | [73] |
| 2. | 23 December 2020 | US20210238125 | Diabetes and Metabolic Syndrome Treatment with a Novel Dual Modulator of Soluble Epoxide Hydrolase and Peroxisome Proliferator-Activated Receptors | [74] |
| 3. | 18 July 2018 | US20190077774A1 | Peroxisome proliferator-activated receptor agonists | [75] |
| 4. | 28 July 2018 | US20180305403A1 | PPAR agonists and methods of use thereof | [76] |
| 5. | 01 July 2018 | US20190000790A1 | PPAR-γ activators and their therapeutical usage | [77] |
| 6. | 13 December 2017 | US20190060269A1 | Brain-Derived PPARα Ligands | [78] |
| 7. | 07 April 2017 | US20180015062A1 | PPAR-α Activator, Pharmaceutical Composition, Food and Drink, Food Additive, Supplement, and Method of Manufacturing the Same | [79] |
| 8. | 13 April 2017 | WO2017180818A1 | PPAR agonists, compounds, pharmaceutical compositions, and methods of use thereof | [80] |
| 9. | 07 April 2017 | US20170304255A1 | PPAR agonists, compounds, pharmaceutical compositions, and methods of use thereof | [81] |
| 10. | 30 January 2017 | US20170210711A1 | Competitive PPAR-α antagonists | [82] |
| Heterocycles | Most Potent Substitution | Activity | Reference |
|---|---|---|---|
| Thiazolidinediones | Allyl derivative (1a) | EC50 of −4.95 μM in the human PPAR-γ transactivation assay | [83] |
| Amino derivative (3b) | 1.7 times more than reference compounds in glucose uptake assay | [84] | |
| Chloro derivative (8a) | 63.15%, PPAR-γ transactivation | [85] | |
| Dichloro derivative (9a) | (−11.6930) Docking score | [86] | |
| Nitro and carboxylic acid derivative (10d) | (−17.44) Docking score | [87] | |
| Flouro derivative (12e) | Reduced 0.09-fold the expression gene of PPARγ in cultured adipocytes compared to the control group | [88] | |
| Oxadiazole | No substitution (13d) | EC50 = −0.15 for PPARα EC50 = −0.29 for PPARδ | [89] |
| ADAM (14) | 2.5-fold greater efficacy in activating PPARα | [90] | |
| Bromo derivative (15a) | Potency and selectivity towards PPARα/δ receptors with PPARα/δ/γ EC50, EC50 γ/α ratio, and EC50 γ/δ ratio value was 8/5/2939 nM, 367, 588 respectively | [91] | |
| Flouro derivative (16d) | PPARα −0.06 ± 0.0005, PPARγ −0.07 ± 0.0006 | [92] | |
| Bemzoimidazole | Chloro derivative | EC50 = −0.19 ± 0.01 | [93] |
| Flouro and chloro derivative | −68 ± 28j | [94] | |
| (20c) | −10.6 ± 0.4 | [95] | |
| Chloro and iodo derivative | Ki = 1023 µM for PPARγ and Ki = 0.106 µM for PPARδ | [96] | |
| Chloro derivative (22b) | PPARα 307 PPARγ 2052 PPARδ 214 | [97] | |
| (23) | Kd value = −2.8 ± 0.8 nM | [98] | |
| Methoxy derivative (24a) | [99] | ||
| Thiazole | - | - | [100] |
| (26) | EC50 > 10 µM | [101] | |
| Phenyl derivative (27b) | PPARα Imax% 22 ± 16 | [102] | |
| p-halogenated phenyl substituted thiazole derivative (28f) | Increased PPARγ activity by almost 4- to 5-fold while rosiglitazone exhibited approximately 10-fold activation | [103] | |
| fluoro and nitro derivative (29b) | - | [104] | |
| indole | 2,4 dimethoxy derivative 30d | - | [105] |
| Chloro and phenyl derivative (31a) | PPARα/δ/γ profile at potency and efficacy level.(31a, Emax = 50%) | [106] | |
| Cyclohexyl derivative | 24.43% Reduction in blood glucose level | [107] | |
| Furan | phenyl derivative 33h | hPPARα (LBD)-GAL4 EC50 (µM) −7.31 hPPARγ (LBD)-GAL4 EC50 (µM) −2.97 hPPARδ (LBD)-GAL4 EC50 (µM) −1.98 | [108] |
| Triflouro carbon phenyl derivative (34a) | 16.2-fold and 8.4-fold more PPAR-α/δ agonistic activity than PPARγ | [109] | |
| Triflouro carbon phenyl derivative (35a) | High potency toward PPAR- α/δ (0.26 ± 0.08 µM 0.50 ± 0.10 µM) and higher selectivity against PPARγ (4.22 ± 0.18 M) than that of GFT505 | [110] | |
| Benzopyran | Nitro derivative (36b) | EC50 −0.91 µM | [111] |
| Butane derivative (37c) | hPPARα and γ with % values of 91% and 88% using a transactivation assay | [112] | |
| Di-hydroxyl deritive (38b) | Ki value 1.41 μM | [113] | |
| 39 | hPPARα with high selectivity (123 % and 38% for α and γ, respectively). | [114] | |
| Bavachinin | (40) | PPARα agonistic activity EC50 −0.43 | [115] |
| 41 | 21 (EC50 = −22.28 μM) | [116] | |
| Miscellaneous | 42 | - | [117] |
| Cyanophenyl (43a) | IC50 6.5 µM time-resolved FRET technique | [118] | |
| 44 | EC50 (nM) = −170 ± 10 | [119] | |
| 45a | HepG2 and A549 cell growth with IC50 values of −0.54 and −0.47 μM, respectively, CCK-8 assay | [120] | |
| 46 | - | [121] | |
| Methoxy and cyclohexyl derivative | IC50 values −0.350 fluorescence polarization assay | [122] | |
| 50a | [123] | ||
| o-Tolyl (51a1–5) | glucose uptake activity in vivo evaluation, with −37.05 ± 0.44 | [124] | |
| 52 | - | [125] | |
| 53a | - | [126] | |
| Ethylcyclopropane derivative (54a) | - | [127] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Virendra, S.A.; Kumar, A.; Chawla, P.A.; Mamidi, N. Development of Heterocyclic PPAR Ligands for Potential Therapeutic Applications. Pharmaceutics 2022, 14, 2139. https://doi.org/10.3390/pharmaceutics14102139
Virendra SA, Kumar A, Chawla PA, Mamidi N. Development of Heterocyclic PPAR Ligands for Potential Therapeutic Applications. Pharmaceutics. 2022; 14(10):2139. https://doi.org/10.3390/pharmaceutics14102139
Chicago/Turabian StyleVirendra, Sharma Arvind, Ankur Kumar, Pooja A. Chawla, and Narsimha Mamidi. 2022. "Development of Heterocyclic PPAR Ligands for Potential Therapeutic Applications" Pharmaceutics 14, no. 10: 2139. https://doi.org/10.3390/pharmaceutics14102139