
Development of In Vitro Evaluation System for Assessing Drug Dissolution Considering Physiological Environment in Nasal Cavity



Abstract
:
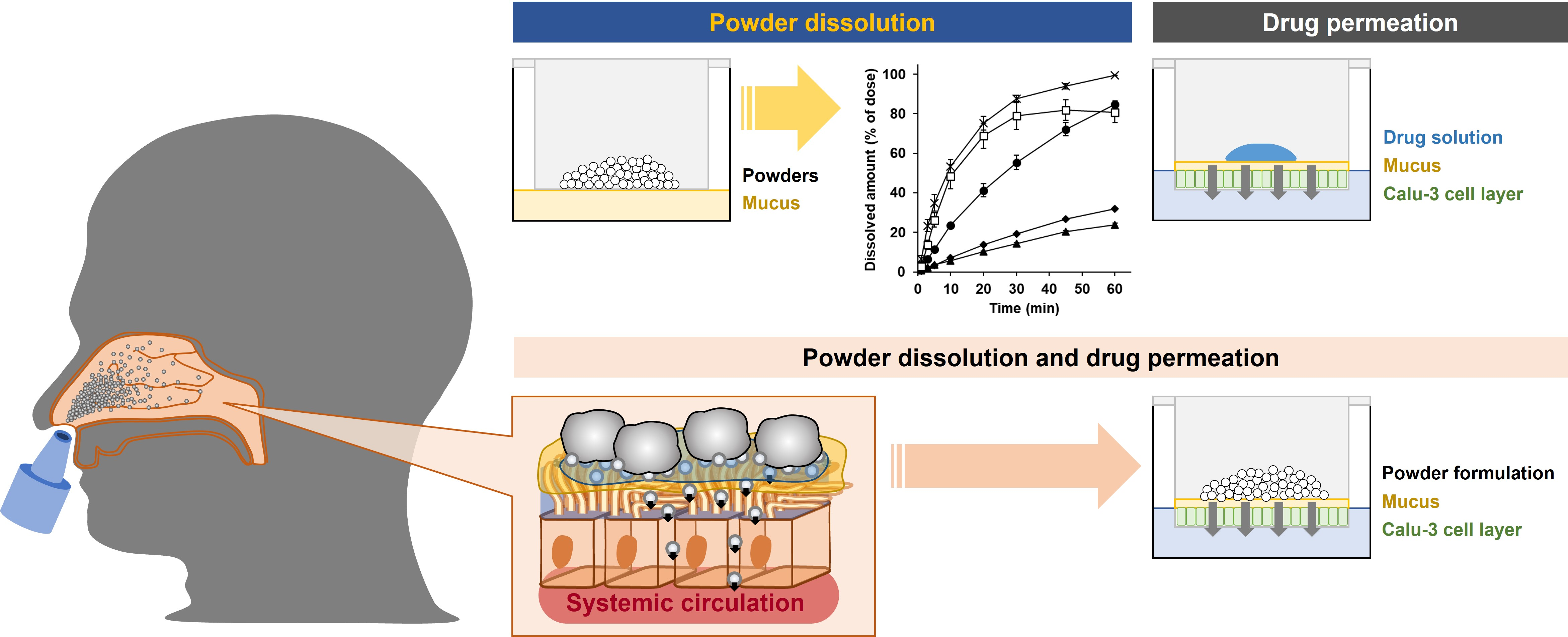
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Drug Dissolution from Powders to Artificial Nasal Fluid (ANF)
2.2.1. Preparation of ANF
2.2.2. Preparation of Powder Dosage Forms
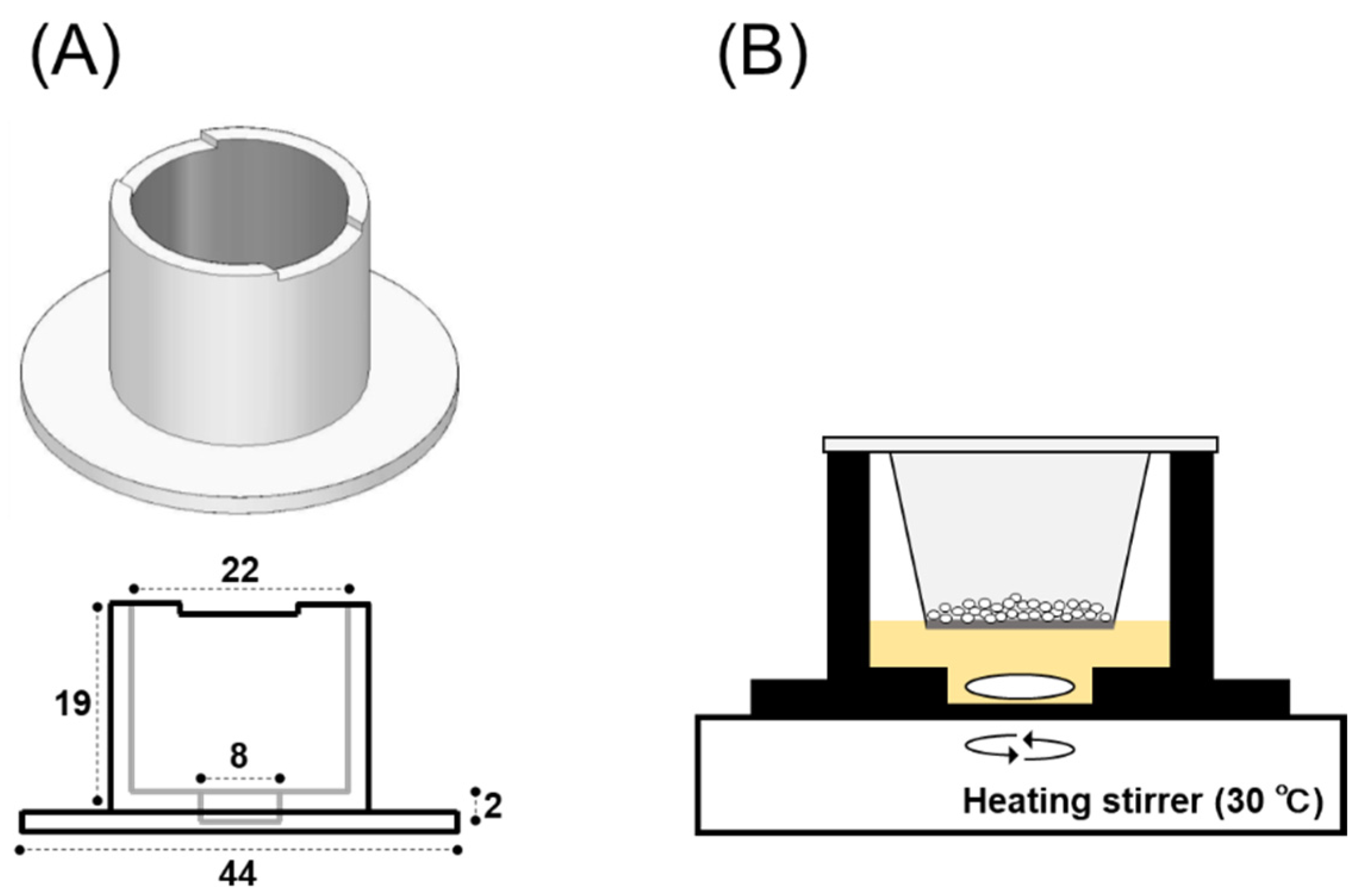
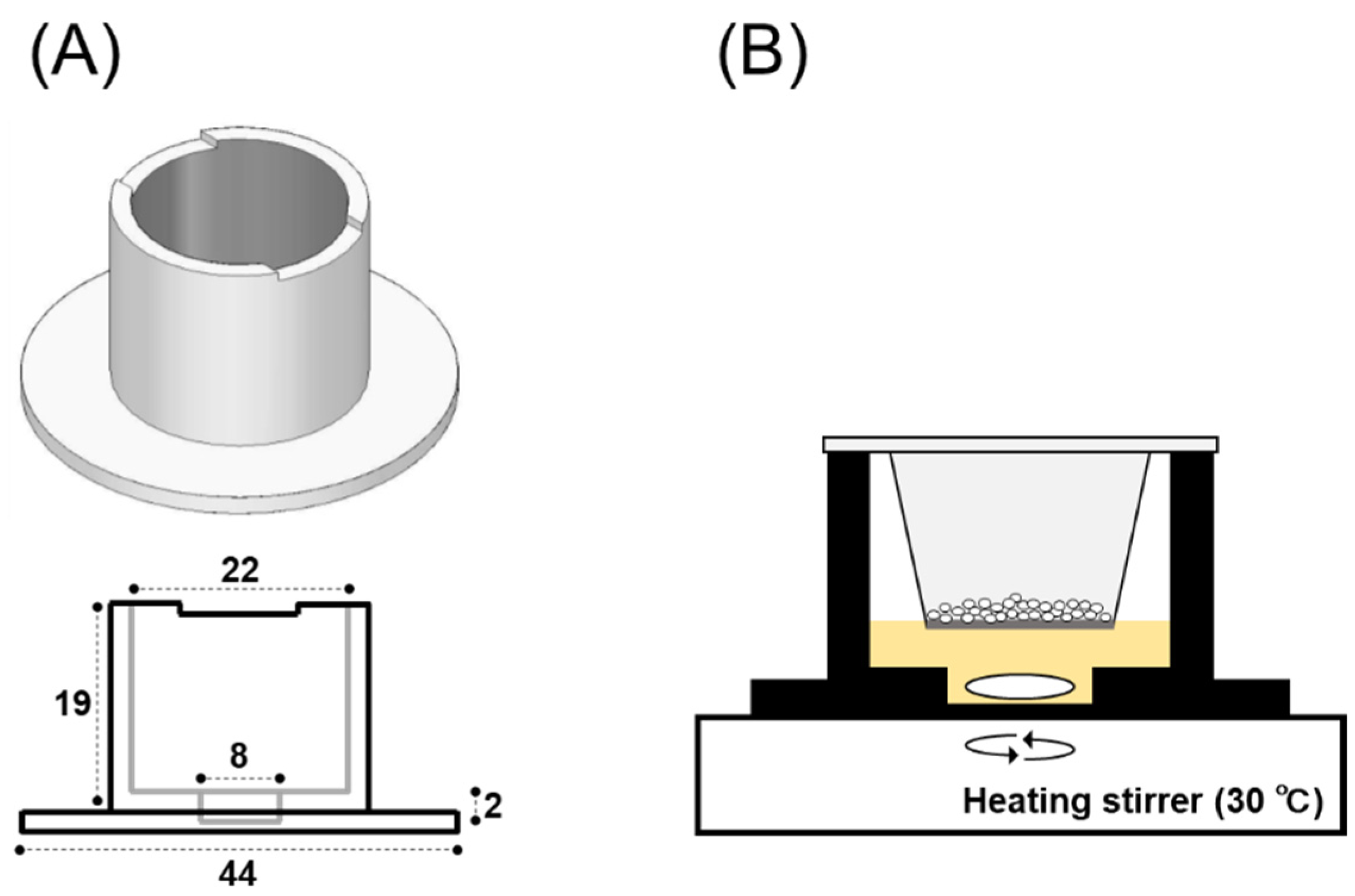
2.2.3. 3D-Printing of a Dissolution Chamber
2.2.4. In Vitro System for Evaluating Powder Dissolution into Nasal Mucus
2.3. Drug Dissolution and Permeation Study for Calu-3 Cell Layers
2.3.1. Culture of Calu-3 Cell Layers
2.3.2. Drug Permeation Study for Powder and Solutions
2.4. Powder Dissolution and Drug Permeation across Calu-3 Cell Layers
2.4.1. Drug Dissolution and Permeation following Powder Application
2.4.2. Permeation Study for Drug Solution
2.5. Sample Treatments
2.6. Statistical Analysis
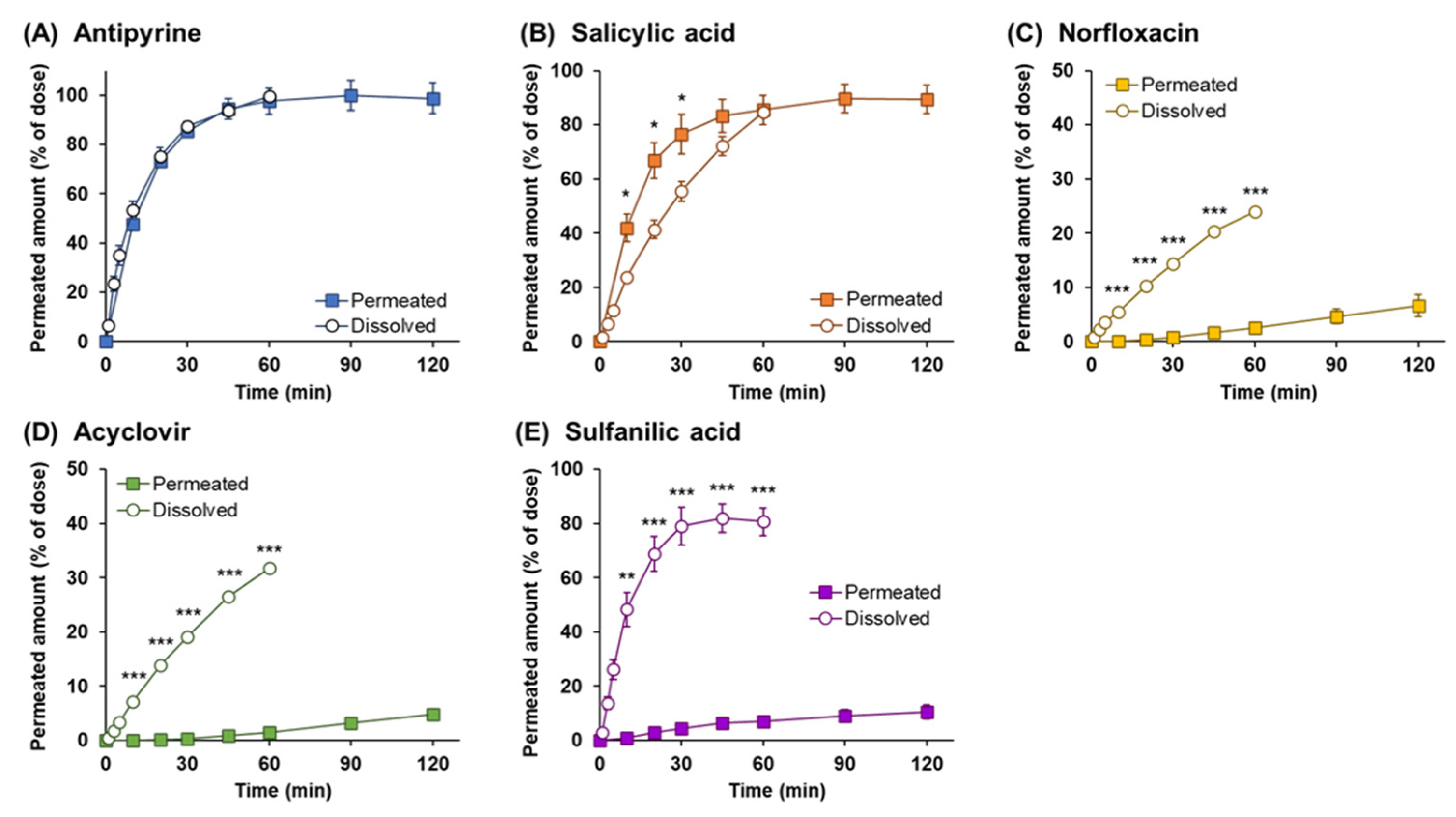
3. Results
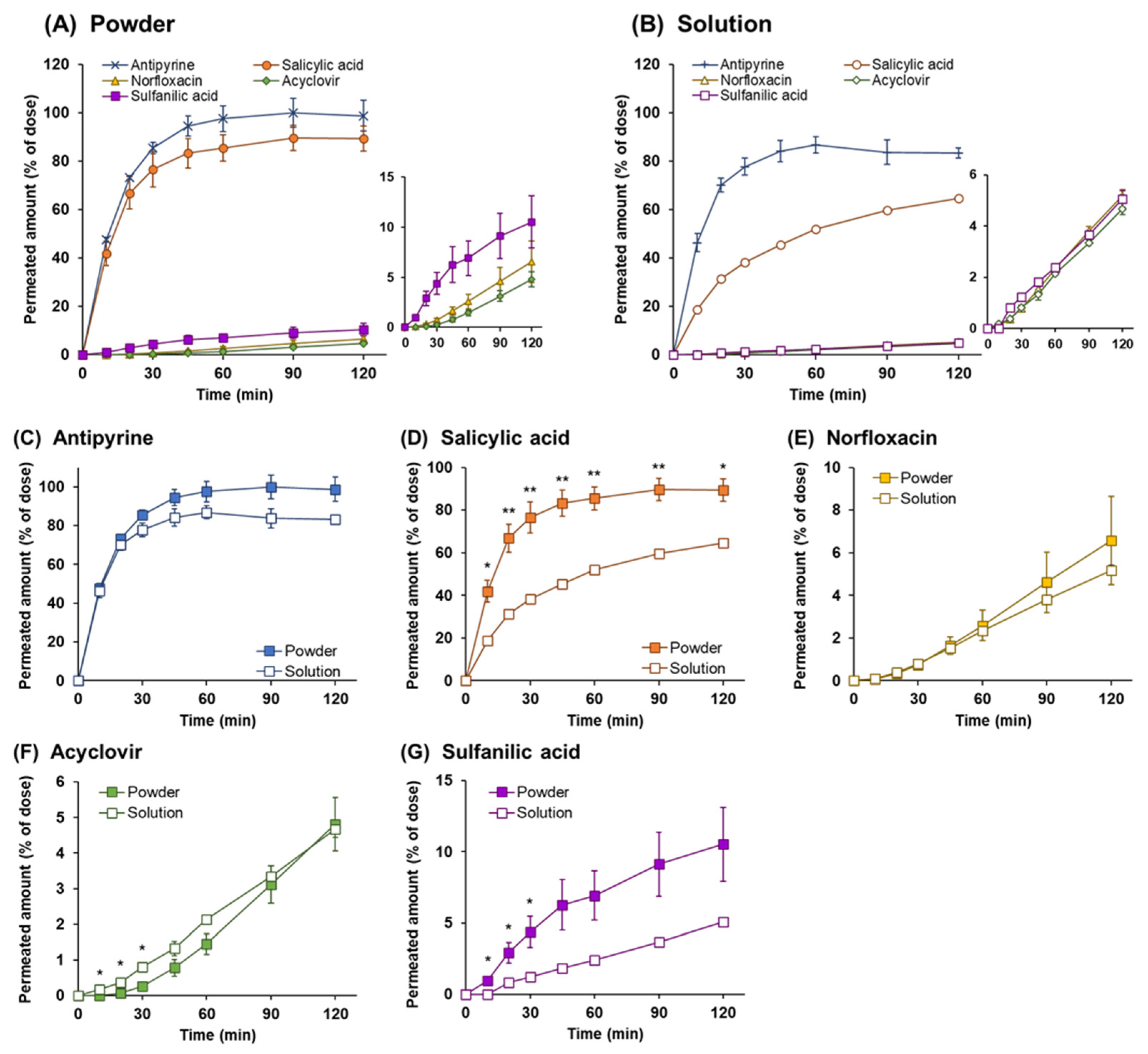
3.1. Drug Permeation Study across Calu-3 Cell Layers
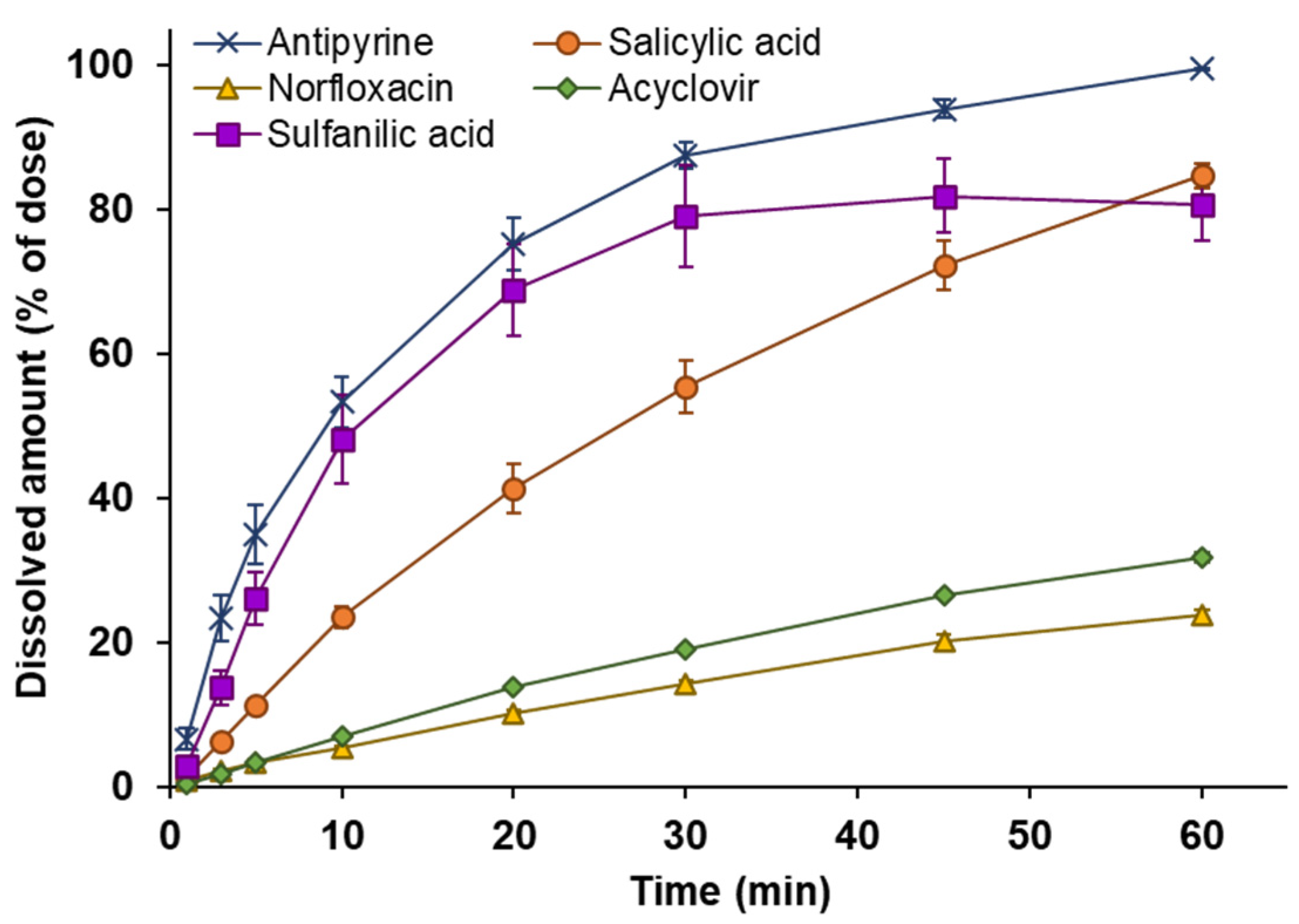
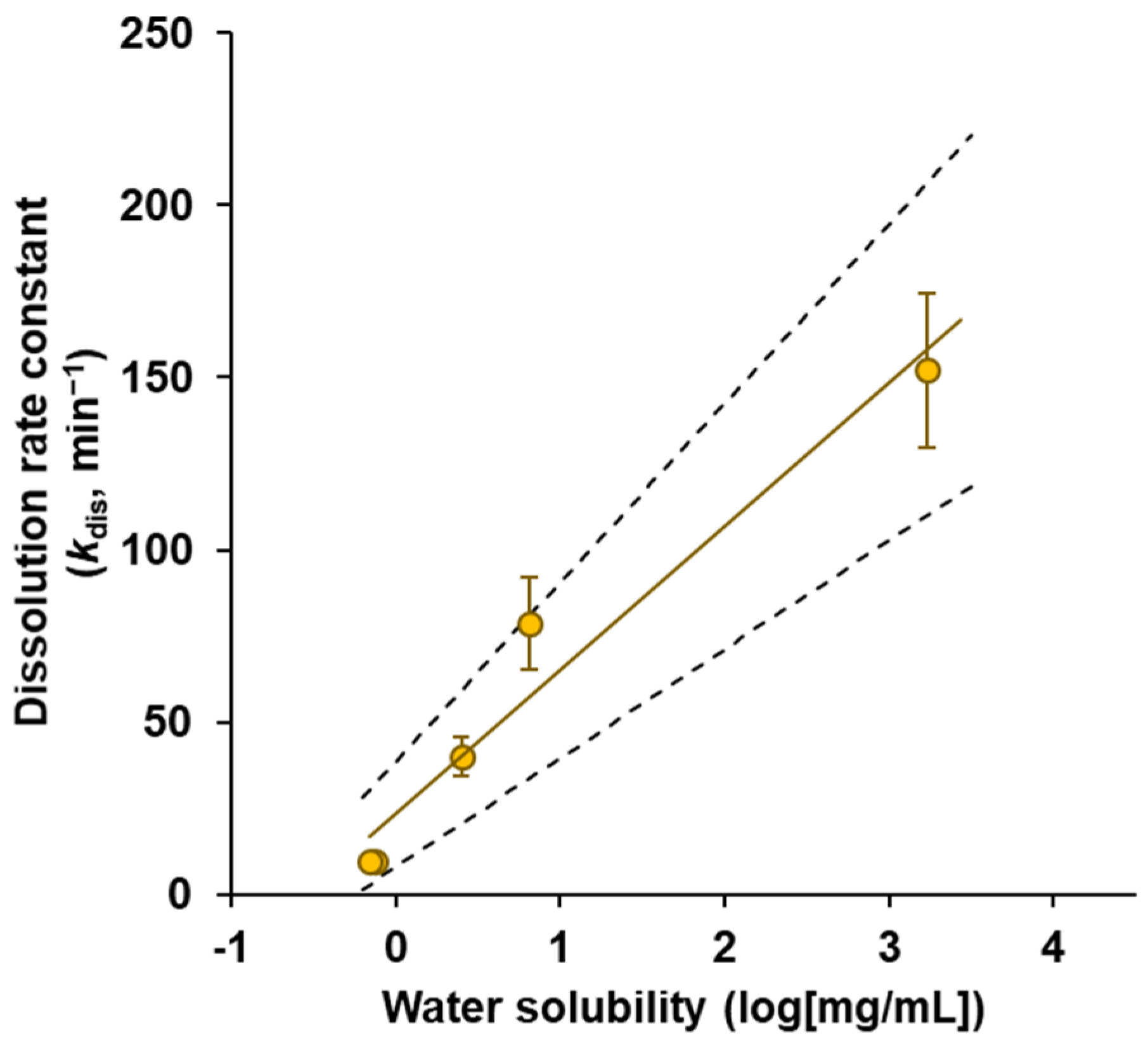
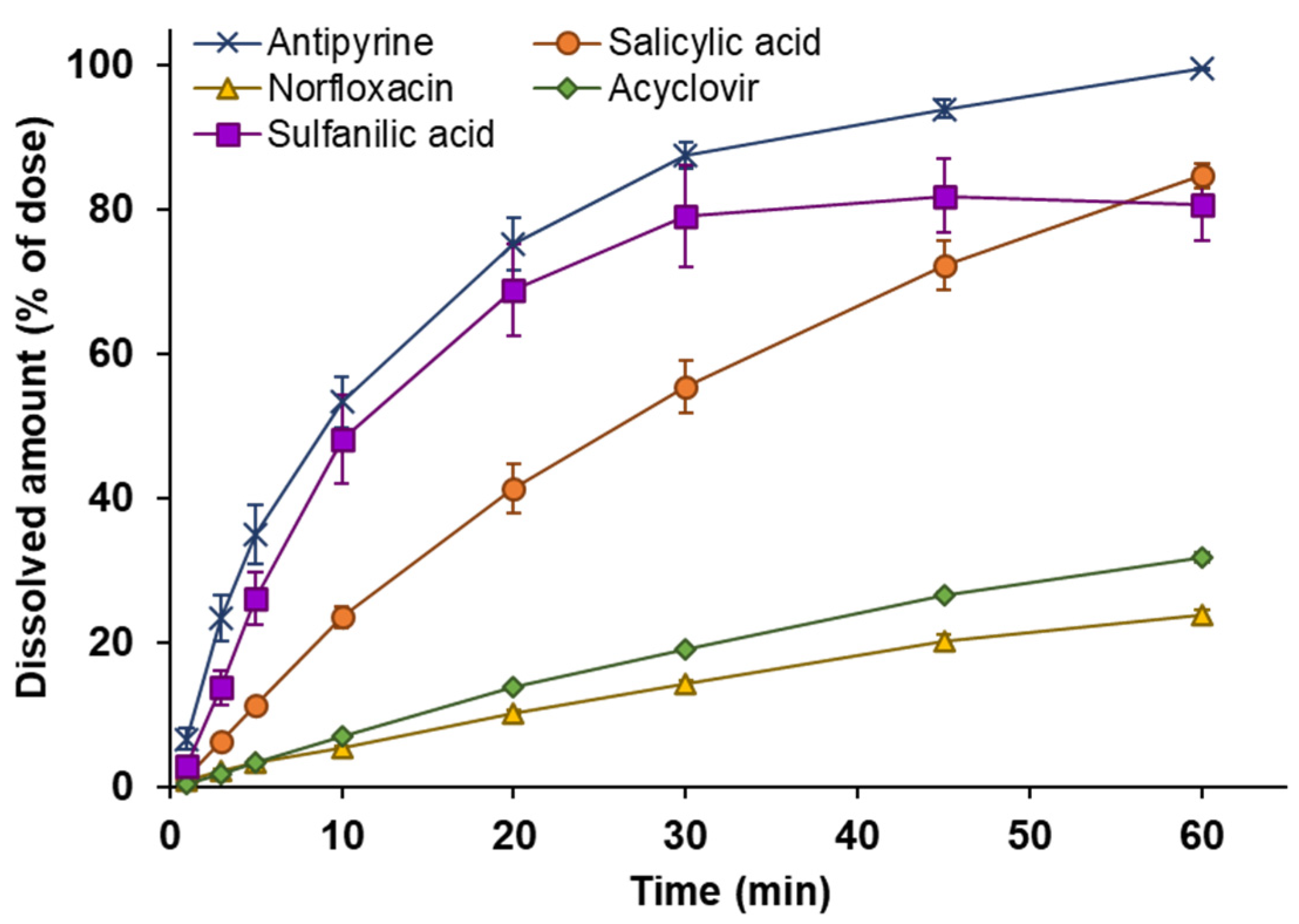
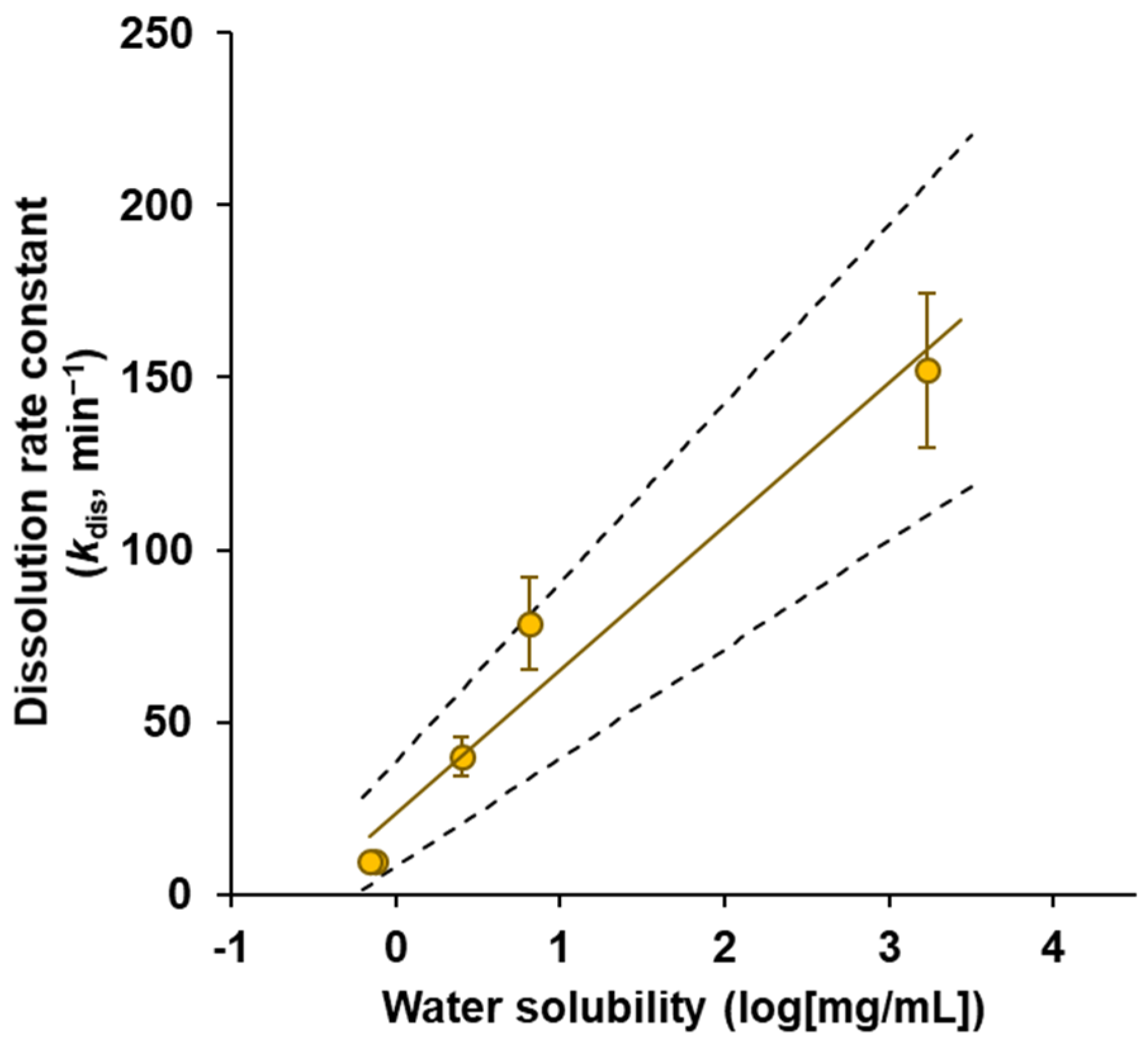
3.2. Powder Dissolution into ANF
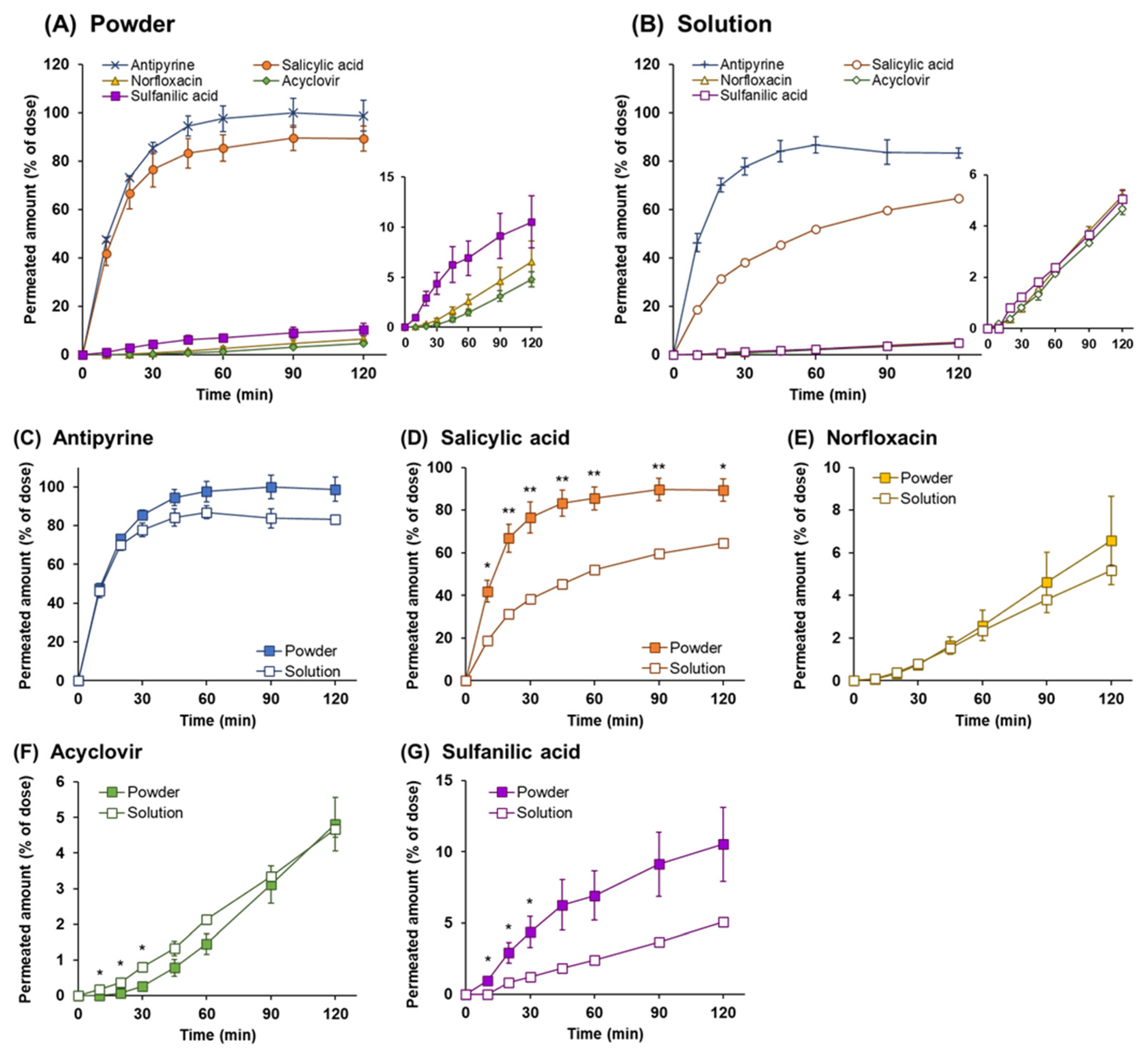
3.3. Powder Dissolution and Drug Permeation across Calu-3 Cell Layers
3.4. Drug Permeation across Calu-3 Cell Layers for Drug Solution
3.5. Comparison of the Drug Permeation Behavior between Powder and Solution Dosage Forms
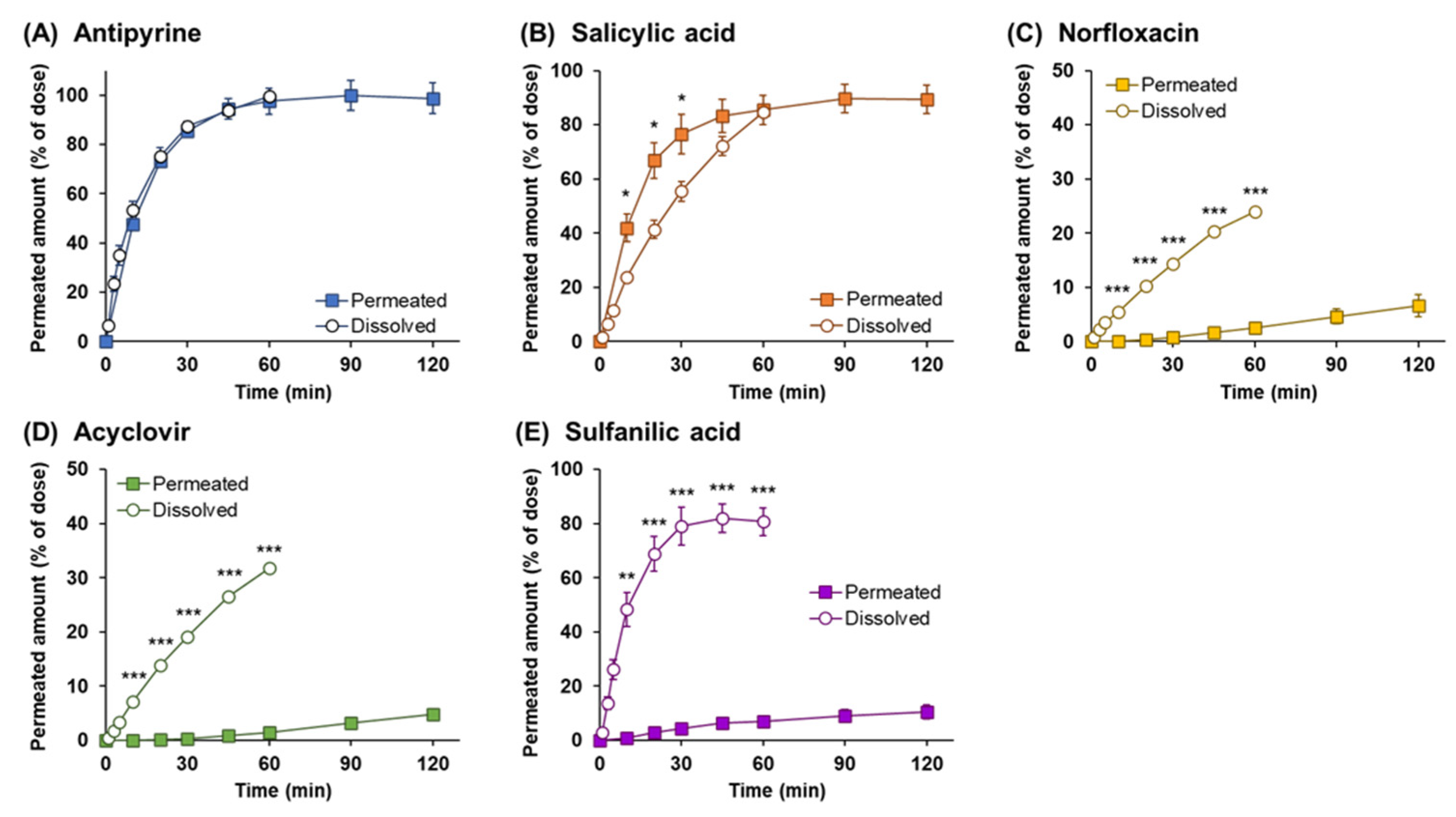
3.6. Analysis of Dissolution and Permeation of Drugs after Powder Application
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Costantino, H.R.; Illum, L.; Brandt, G.; Johnson, P.H.; Quay, S.C. Intranasal Delivery: Physicochemical and Therapeutic Aspects. Int. J. Pharm. 2007, 337, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Borrajo, M.L.; Alonso, M.J. Using Nanotechnology to Deliver Biomolecules from Nose to Brain—Peptides, Proteins, Monoclonal Antibodies and RNA. Drug Deliv. Transl. Res. 2022, 12, 862–880. [Google Scholar] [CrossRef] [PubMed]
- Samaridou, E.; Alonso, M.J. Nose-to-Brain Peptide Delivery–The Potential of Nanotechnology. Bioorganic Med. Chem. 2018, 26, 2888–2905. [Google Scholar] [CrossRef]
- Soleimanizadeh, A.; Dinter, H.; Schindowski, K. Central Nervous System Delivery of Antibodies and Their Single-domain Antibodies and Variable Fragment Derivatives with Focus on Intranasal Nose to Brain Administration. Antibodies 2021, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Fortuna, A.; Alves, G.; Serralheiro, A.; Sousa, J.; Falcão, A. Intranasal Delivery of Systemic-Acting Drugs: Small-Molecules and Biomacromolecules. Eur. J. Pharm. Biopharm. 2014, 88, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xiong, G.; Tsang, W.C.; Schätzlein, A.G.; Uchegbu, I.F. Special Section on Drug Delivery Technologies-Minireview Nose-to-Brain Delivery. J. Pharmacol. Exp. Ther. 2019, 370, 593–601. [Google Scholar] [CrossRef] [Green Version]
- Bourganis, V.; Kammona, O.; Alexopoulos, A.; Kiparissides, C. Recent Advances in Carrier Mediated Nose-to-Brain Delivery of Pharmaceutics. Eur. J. Pharm. Biopharm. 2018, 128, 337–362. [Google Scholar] [CrossRef]
- Agrawal, M.; Saraf, S.; Saraf, S.; Antimisiaris, S.G.; Chougule, M.B.; Shoyele, S.A.; Alexander, A. Nose-to-Brain Drug Delivery: An Update on Clinical Challenges and Progress towards Approval of Anti-Alzheimer Drugs. J. Control. Release 2018, 281, 139–177. [Google Scholar] [CrossRef]
- Hussain, A.; Hirai, S.; Bawarshi, R. Nasal Absorption of Propranolol from Different Dosage Forms by Rats and Dogs. J. Pharm. Sci. 1980, 69, 1411–1413. [Google Scholar] [CrossRef]
- Shinichiro, H.; Takatsuka, Y.; Tai, M.; Hiroyuki, M. Absorption of Drugs from the Nasal Mucosa of Rat. Int. J. Pharm. 1981, 7, 317–325. [Google Scholar] [CrossRef]
- Bahamondez-Canas, T.F.; Cui, Z. Intranasal Immunization with Dry Powder Vaccines. Eur. J. Pharm. Biopharm. 2018, 122, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Al-Salama, Z.T.; Scott, L.J. Sumatriptan Nasal Powder: A Review in Acute Treatment of Migraine. Drugs 2016, 76, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Pozzoli, M.; Rogueda, P.; Zhu, B.; Smith, T.; Young, P.M.; Traini, D.; Sonvico, F. Dry Powder Nasal Drug Delivery: Challenges, Opportunities and a Study of the Commercial Teijin Puvlizer Rhinocort Device and Formulation. Drug Dev. Ind. Pharm. 2016, 42, 1660–1668. [Google Scholar] [CrossRef] [PubMed]
- Mohamad, S.A.; Badawi, A.M.; Mansour, H.F. Insulin Fast-Dissolving Film for Intranasal Delivery via Olfactory Region, a Promising Approach for the Treatment of Anosmia in COVID-19 Patients: Design, in-Vitro Characterization and Clinical Evaluation. Int. J. Pharm. 2021, 601, 120600. [Google Scholar] [CrossRef] [PubMed]
- Laffleur, F. Nasal Adhesive Patches-Approach for Topical Application for Dry Nasal Syndrome. Int. J. Biol. Macromol. 2018, 111, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Inoue, D.; Yamashita, A.; To, H. Formulation and In Vitro Characterization of a Vacuum-Dried Drug–Polymer Thin Film for Intranasal Application. Polymers 2022, 14, 2954. [Google Scholar] [CrossRef]
- Beule, A.G. Physiology and Pathophysiology of Respiratory Mucosa of the Nose and the Paranasal Sinuses. GMS Curr. Top. Otorhinolaryngol. Head Neck Surg. 2010, 9, Doc07. [Google Scholar] [CrossRef]
- Widdicombe, J.G. Airway Liquid: A Barrier to Drug Diffusion? Eur. Respir. J. 1997, 10, 2194–2197. [Google Scholar] [CrossRef] [Green Version]
- Marttin, E.; Schipper, N.G.M.; Coos Verhoef, J.; Merkus, F.W.H.M. Nasal Mucociliary Clearance as a Factor in Nasal Drug Delivery. Adv. Drug Deliv. Rev. 1998, 29, 13–38. [Google Scholar] [CrossRef]
- Inoue, D.; Tanaka, A.; Kimura, S.; Kiriyama, A.; Katsumi, H.; Yamamoto, A.; Ogawara, K.I.; Kimura, T.; Higaki, K.; Yutani, R.; et al. The Relationship between in Vivo Nasal Drug Clearance and in Vitro Nasal Mucociliary Clearance: Application to the Prediction of Nasal Drug Absorption. Eur. J. Pharm. Sci. 2018, 117, 21–26. [Google Scholar] [CrossRef]
- Inoue, D.; Kimura, S.; Kiriyama, A.; Katsumi, H.; Yamamoto, A.; Ogawara, K.I.; Higaki, K.; Tanaka, A.; Yutani, R.; Sakane, T.; et al. Quantitative Estimation of the Effect of Nasal Mucociliary Function on in Vivo Absorption of Norfloxacin after Intranasal Administration to Rats. Mol. Pharm. 2018, 15, 4462–4469. [Google Scholar] [CrossRef] [PubMed]
- Tiozzo Fasiolo, L.; Manniello, M.D.; Tratta, E.; Buttini, F.; Rossi, A.; Sonvico, F.; Bortolotti, F.; Russo, P.; Colombo, G. Opportunity and Challenges of Nasal Powders: Drug Formulation and Delivery. Eur. J. Pharm. Sci. 2018, 113, 2–17. [Google Scholar] [CrossRef]
- Buttini, F.; Colombo, P.; Rossi, A.; Sonvico, F.; Colombo, G. Particles and Powders: Tools of Innovation for Non-Invasive Drug Administration. J. Control. Release 2012, 161, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Ambrus, R.; Gieszinger, P.; Gáspár, R.; Sztojkov-Ivanov, A.; Ducza, E.; Márki, Á.; Janáky, T.; Tömösi, F.; Kecskeméti, G.; Szabó-Révész, P.; et al. Investigation of the Absorption of Nanosized Lamotrigine Containing Nasal Powder via the Nasal Cavity. Molecules 2020, 25, 1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiozzo Fasiolo, L.; Manniello, M.D.; Bortolotti, F.; Buttini, F.; Rossi, A.; Sonvico, F.; Colombo, P.; Valsami, G.; Colombo, G.; Russo, P. Anti-Inflammatory Flurbiprofen Nasal Powders for Nose-to-Brain Delivery in Alzheimer’s Disease. J. Drug Target. 2019, 27, 984–994. [Google Scholar] [CrossRef]
- Vasa, D.M.; Buckner, I.S.; Cavanaugh, J.E.; Wildfong, P.L.D. Improved Flux of Levodopa via Direct Deposition of Solid Microparticles on Nasal Tissue. AAPS PharmSciTech 2017, 18, 904–912. [Google Scholar] [CrossRef]
- Torikai, Y.; Sasaki, Y.; Sasaki, K.; Kyuno, A.; Haruta, S.; Tanimoto, A. Evaluation of Systemic and Mucosal Immune Responses Induced by a Nasal Powder Delivery System in Conjunction with an OVA Antigen in Cynomolgus Monkeys. J. Pharm. Sci. 2021, 110, 2038–2046. [Google Scholar] [CrossRef]
- Thakkar, S.G.; Warnken, Z.N.; Alzhrani, R.F.; Valdes, S.A.; Aldayel, A.M.; Xu, H.; Williams, R.O.; Cui, Z. Intranasal Immunization with Aluminum Salt-Adjuvanted Dry Powder Vaccine. J. Control. Release 2018, 292, 111–118. [Google Scholar] [CrossRef]
- Salade, L.; Wauthoz, N.; Vermeersch, M.; Amighi, K.; Goole, J. Chitosan-Coated Liposome Dry-Powder Formulations Loaded with Ghrelin for Nose-to-Brain Delivery. Eur. J. Pharm. Biopharm. 2018, 129, 257–266. [Google Scholar] [CrossRef]
- He, J.; Zhang, G.; Zhang, Q.; Chen, J.; Zhang, Y.; An, X.; Wang, P.; Xie, S.; Fang, F.; Zheng, J.; et al. Evaluation of Inhaled Recombinant Human Insulin Dry Powders: Pharmacokinetics, Pharmacodynamics and 14-Day Inhalation. J. Pharm. Pharmacol. 2019, 71, 176–184. [Google Scholar] [CrossRef]
- Inoue, D.; Furubayashi, T.; Tanaka, A.; Sakane, T.; Sugano, K. Quantitative Estimation of Drug Permeation through Nasal Mucosa Using in vitro Membrane Permeability across Calu-3 Cell Layers for Predicting in Vivo Bioavailability after Intranasal Administration to Rats. Eur. J. Pharm. Biopharm. 2020, 149, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Benet, L.Z.; Broccatelli, F.; Oprea, T.I. BDDCS Applied to over 900 Drugs. AAPS J. 2011, 13, 519–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, E.A.; Swenberg, J.A.; Fields, S.; Popp, J.A. Comparative Morphometry of the Nasal Cavity in Rats and Mice. J. Anat 1982, 135, 83–88. [Google Scholar] [PubMed]
- Schriever, V.A.; Hummel, T.; Lundström, J.N.; Freiherr, J. Size of Nostril Opening as a Measure of Intranasal Volume. Physiol. Behav. 2013, 110–111, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, J.; Leiacker, R.; Rettinger, G.; Keck, T. Nasal Mucosal Temperature during Respiration. Clin. Otolaryngol. Allied. Sci. 2002, 27, 135–139. [Google Scholar] [CrossRef]
- Tanaka, A.; Furubayashi, T.; Tomisaki, M.; Kawakami, M.; Kimura, S.; Inoue, D.; Kusamori, K.; Katsumi, H.; Sakane, T.; Yamamoto, A. Nasal Drug Absorption from Powder Formulations: The Effect of Three Types of Hydroxypropyl Cellulose (HPC). Eur. J. Pharm. Sci. 2017, 96, 284–289. [Google Scholar] [CrossRef]
- Chatton, J.Y.; Roch-Ramel, F. Nonionic Diffusion of Salicylic Acid through MDCK Cell Monolayers. J. Pharmacol. Exp. Ther. 1992, 261, 1071–1079. [Google Scholar]
- Neuhoff, S.; Ungell, A.L.; Zamora, I.; Artursson, P. PH-Dependent Passive and Active Transport of Acidic Drugs across Caco-2 Cell Monolayers. Eur. J. Pharm. Sci. 2005, 25, 211–220. [Google Scholar] [CrossRef]
- McMartin, C.; Hutchinson, L.E.F.; Hyde, R.; Peters, G.E. Analysis of Structural Requirements for the Absorption of Drugs and Macromolecules from the Nasal Cavity. J. Pharm. Sci. 1987, 76, 535–540. [Google Scholar] [CrossRef]
- Hosoya, K.-I.; Kubo, H.; Natsume, H.; Sugibayashi, K.; Morimoto, Y.; Yamashita, S. The Structural Barrier of Absorptive Mucosae: Site Difference of the Permeability of Fluorescein Isothiocyanate-Labelled Dextran in Rabbits. Biopharm. Drug Dispos. 1993, 14, 685–695. [Google Scholar] [CrossRef]
- Song, Y.; Wang, Y.; Thakur, R.; Meidan, V.M.; Michniak, B. Mucosal Drug Delivery: Membranes, Methodologies, and Applications. Crit. Rev. Ther. Drug Carrier Syst. 2004, 21, 195–256. [Google Scholar] [CrossRef] [PubMed]
- Marttin, E.; Verhoef, J.C.; Merkus, F.W.H.M. Efficacy, Safety and Mechanism of Cyclodextrins as Absorption Enhancers in Nasal Delivery of Peptide and Protein Drugs. J. Drug Target. 1998, 6, 17–36. [Google Scholar] [CrossRef] [PubMed]
- Ugwoke, M.I.; Verbeke, N.; Kinget, R. The Biopharmaceutical Aspects of Nasal Mucoadhesive Drug Delivery. J. Pharm. Pharmacol. 2010, 53, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Casettari, L.; Illum, L. Chitosan in Nasal Delivery Systems for Therapeutic Drugs. J. Control. Release 2014, 190, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.S.; Illum, L. Absorption Enhancers for Nasal Drug Delivery. Clin. Pharmacokinet. 2003, 42, 1107–1128. [Google Scholar] [CrossRef]
- Ghadiri, M.; Young, P.; Traini, D. Strategies to Enhance Drug Absorption via Nasal and Pulmonary Routes. Pharmaceutics 2019, 11, 113. [Google Scholar] [CrossRef] [Green Version]
- Alhalaweh, A.; Andersson, S.; Velaga, S.P. Preparation of Zolmitriptan-Chitosan Microparticles by Spray Drying for Nasal Delivery. Eur. J. Pharm. Sci. 2009, 38, 206–214. [Google Scholar] [CrossRef]
- Bartos, C.; Varga, P.; Szabó-Révész, P.; Ambrus, R. Physico-Chemical and In Vitro Characterization of Chitosan-Based Microspheres Intended for Nasal Administration. Pharmaceutics 2021, 13, 608. [Google Scholar] [CrossRef]
- Luppi, B.; Bigucci, F.; Abruzzo, A.; Corace, G.; Cerchiara, T.; Zecchi, V. Freeze-Dried Chitosan/Pectin Nasal Inserts for Antipsychotic Drug Delivery. Eur. J. Pharm. Biopharm. 2010, 75, 381–387. [Google Scholar] [CrossRef]
- Rautiola, D.; Updyke, J.L.; Nelson, K.M.; Siegel, R.A. Diazepam Prodrug Stabilizes Human Aminopeptidase B during Lyophilization. Mol. Pharm. 2020, 17, 453–460. [Google Scholar] [CrossRef]
- Papakyriakopoulou, P.; Manta, K.; Kostantini, C.; Kikionis, S.; Banella, S.; Ioannou, E.; Christodoulou, E.; Rekkas, D.M.; Dallas, P.; Vertzoni, M.; et al. Nasal Powders of Quercetin-β-Cyclodextrin Derivatives Complexes with Mannitol/Lecithin Microparticles for Nose-to-Brain Delivery: In Vitro and Ex Vivo Evaluation. Int. J. Pharm. 2021, 607, 121016. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Du, L.; Chen, X.; Ge, P.; Wang, Y.; Fu, Y.; Sun, H.; Jiang, Q.; Jin, Y. Nasal Delivery of Analgesic Ketorolac Tromethamine Thermo- and Ion-Sensitive in Situ Hydrogels. Int. J. Pharm. 2015, 489, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Tas, C.; Ozkan, C.K.; Savaser, A.; Ozkan, Y.; Tasdemir, U.; Altunay, H. Nasal Absorption of Metoclopramide from Different Carbopol® 981 Based Formulations: In Vitro, Ex Vivo and in Vivo Evaluation. Eur. J. Pharm. Biopharm. 2006, 64, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Reno, F.E.; Normand, P.; McInally, K.; Silo, S.; Stotland, P.; Triest, M.; Carballo, D.; Piché, C. A Novel Nasal Powder Formulation of Glucagon: Toxicology Studies in Animal Models. BMC Pharmacol. Toxicol. 2015, 16, 29. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Water Solubility | LogP | BCS Class | |
|---|---|---|---|---|
| (mg/mL) | (log[mg/mL]) | |||
| Antipyrine | 1700 | 3.23 | 0.23 | 1 |
| Salicylic acid | 2.51 | 0.40 | 2.26 | 1 |
| Norfloxacin | 0.75 | −0.12 | −1.03 | 4 |
| Acyclovir | 0.7 | −0.15 | −1.95 | 3 |
| Sulfanilic acid | 6.5 | 0.81 | −0.9 | 3 |
| Rat | Human | |||
|---|---|---|---|---|
| Nasal cavity | Length | (mm) | 91 ± 0.3 a | 100–140 c |
| Volume | (mm3) | 256.7 ± 4.1 a | 20,000 c | |
| Surface area of epithelium | Squamous | (mm2) | 44.2 ± 5.2 a | - |
| Respiratory | (mm2) | 623.1 ± 14.0 a | - | |
| Olfactory | (mm2) | 675.2 ± 43.0 a | 200–400 c | |
| Total | (mm2) | 1343.5 ± 55.0 a | 16,000 c | |
| Thickness of mucus layer | Periciliary layer | (μm) | 5–10 b | 5 d |
| Surface layer | (μm) | 1–10 b | 10–15 d |
| Drug | Calu-3 Permeability papp (×10−5 cm/s) | ||
|---|---|---|---|
| Antipyrine | 3.88 | ± | 0.08 |
| Salicylic acid | 1.43 | ± | 0.02 |
| Norfloxacin | 0.081 | ± | 0.001 |
| Acyclovir | 0.067 | ± | 0.002 |
| Sulfanilic acid | 0.044 | ± | 0.004 |
| Drug | Dissolution Rate Constant kdis (min−1) | ||
|---|---|---|---|
| Antipyrine | 152.12 | ± | 22.53 |
| Salicylic acid | 40.10 | ± | 5.59 |
| Norfloxacin | 9.89 | ± | 1.62 |
| Acyclovir | 9.76 | ± | 0.29 |
| Sulfanilic acid | 78.84 | ± | 13.37 |
| Drug | TEER (% of Initial) | |||||
|---|---|---|---|---|---|---|
| Solution | Powder | |||||
| Antipyrine | 91.04 | ± | 1.63 | 94.82 | ± | 1.12 |
| Salicylic acid | 82.24 | ± | 3.78 | 75.30 | ± | 2.76 |
| Norfloxacin | 88.32 | ± | 1.95 | 84.37 | ± | 3.71 |
| Acyclovir | 83.35 | ± | 1.81 | 94.98 | ± | 1.80 |
| Sulfanilic acid | 93.87 | ± | 7.72 | 84.74 | ± | 2.46 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inoue, D.; Yamashita, A.; To, H. Development of In Vitro Evaluation System for Assessing Drug Dissolution Considering Physiological Environment in Nasal Cavity. Pharmaceutics 2022, 14, 2350. https://doi.org/10.3390/pharmaceutics14112350
Inoue D, Yamashita A, To H. Development of In Vitro Evaluation System for Assessing Drug Dissolution Considering Physiological Environment in Nasal Cavity. Pharmaceutics. 2022; 14(11):2350. https://doi.org/10.3390/pharmaceutics14112350
Chicago/Turabian StyleInoue, Daisuke, Ayari Yamashita, and Hideto To. 2022. "Development of In Vitro Evaluation System for Assessing Drug Dissolution Considering Physiological Environment in Nasal Cavity" Pharmaceutics 14, no. 11: 2350. https://doi.org/10.3390/pharmaceutics14112350
APA StyleInoue, D., Yamashita, A., & To, H. (2022). Development of In Vitro Evaluation System for Assessing Drug Dissolution Considering Physiological Environment in Nasal Cavity. Pharmaceutics, 14(11), 2350. https://doi.org/10.3390/pharmaceutics14112350