

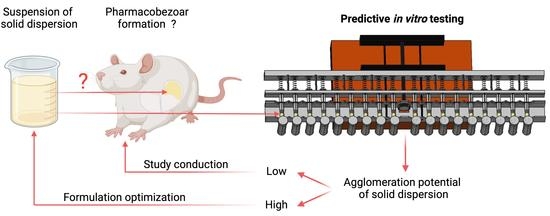
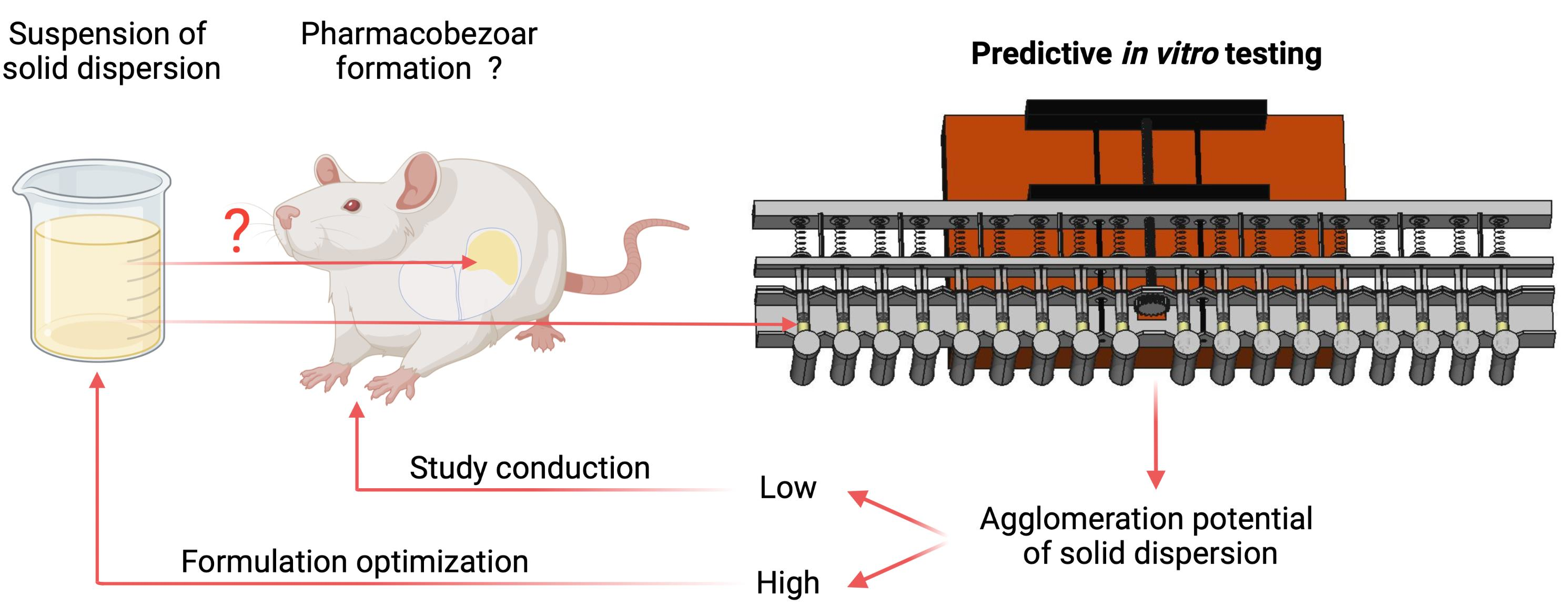
An In Vitro Model to Investigate the Potential of Solid Dispersions to Form Pharmacobezoars
 , ,
, , 
Abstract
:
1. Introduction
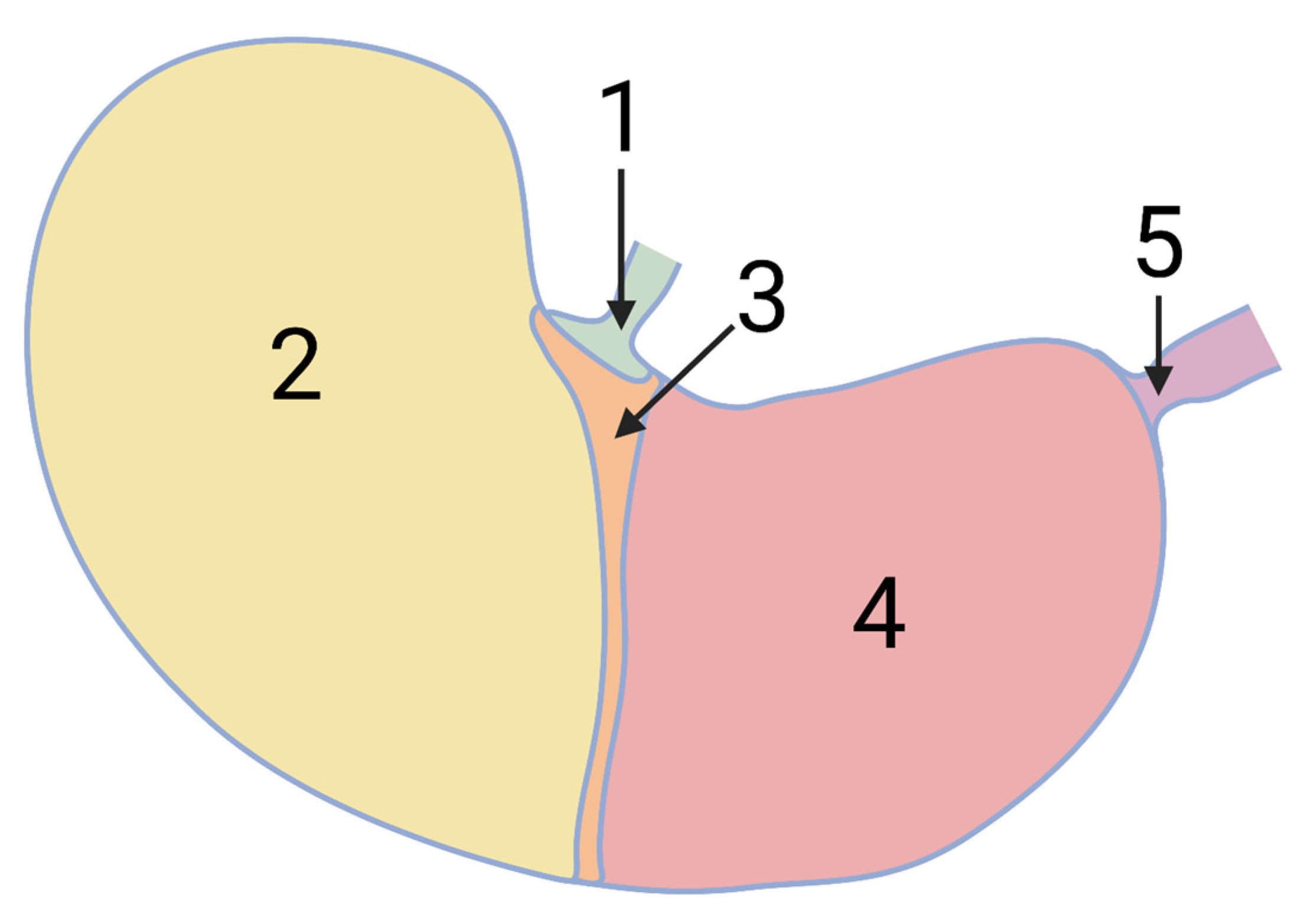
2. Materials and Methods
2.1. In Vitro Model
2.1.1. Mechanical Model Description
2.1.2. 3D Printed Parts
2.1.3. Volume Pattern in Syringe Cavities
2.2. Tested Spray-Dried Formulations
2.3. Preparation of Suspensions for In Vitro Testing
2.4. Conduction of In Vitro Experiments
In Vitro Agglomerates
3. Results and Discussion
3.1. In Vitro Model Development
3.2. Experiments on In Vitro Agglomeration Potential of Formulation 1 and Formulation 2
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- ICH. ICH M3 (R2)—Non-Clinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorisation for Pharmaceuticals; ICH: Geneva, Switzerland, 2009. [Google Scholar]
- Butreddy, A. Hydroxypropyl methylcellulose acetate succinate as an exceptional polymer for amorphous solid dispersion formulations: A review from bench to clinic. Eur. J. Pharm. Biopharm. 2022, 177, 289–307. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, N.; Ueno, K.; Igarashi, T.; Kitagawa, H.; Fujita, T.; Ichikawa, N.; Kondo, Y.; Isoda, M. Studies of hydroxypropylmethylcellulose acetate succinate on fertility in rats. J. Toxicol. Sci. 1985, 10, 187–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshi, N.; Ueno, K.; Yano, H.; Hirashima, K.; Kitagawa, H. General pharmacological studies of hydroxypropylmethylcellulose acetate succinate in experimental animals. J. Toxicol. Sci. 1985, 10 (Suppl. 2), 129–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshi, N.; Yano, H.; Hirashima, K.; Kitagawa, H.; Fukuda, Y. Toxicological studies of hydroxypropylmethylcellulose acetate succinate. J. Toxicol. Sci. 1985, 10, 147–185. [Google Scholar] [CrossRef]
- Gierke, H.; Pfrommer, T.; Schaefer, K.; Weitschies, W.; Nolte, T. Pharmacobezoar Formation from HPMC-AS containing Spray dried Formulations in Nonclinical Safety Studies in Rats. Toxicol. Pathol. 2022. [Google Scholar]
- McConnell, E.L.; Basit, A.W.; Murdan, S. Measurements of rat and mouse gastrointestinal pH, fluid and lymphoid tissue, and implications for in-vivo experiments. J. Pharm. Pharmacol. 2008, 60, 63–70. [Google Scholar] [CrossRef]
- Lentle, R.G.; Janssen, P.W.M.; Goh, K.; Chambers, P.; Hulls, C. Quantification of the effects of the volume and viscosity of gastric contents on antral and fundic activity in the rat stomach maintained ex vivo. Dig. Dis. Sci. 2010, 55, 3349–3360. [Google Scholar] [CrossRef] [PubMed]
- Gärtner, K. The forestomach of rats and mice, an effective device supporting digestive metabolism in muridae (Review). J. Exp. Anim. Sci. 2002, 42, 1–20. [Google Scholar] [CrossRef]
- Vdoviaková, K.; Petrovová, E.; Maloveská, M.; Krešáková, L.; Teleky, J.; Elias, M.Z.J.; Petrášová, D. Surgical anatomy of the gastrointestinal tract and its vasculature in the laboratory rat. Gastroenterol. Res. Pract. 2016. [Google Scholar] [CrossRef] [Green Version]
- Raybould, H.E.; Tache, Y. Cholecystokinin inhibits gastric motility and emptying via a capsaicin-sensitive vagal pathway in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 1988, 255, G242–G246. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Cao, J.; Oleson, S.; Powley, T.L.; Liu, Z.; Ieee, M. Contrast Enhanced Magnetic Resonance Imaging of Gastric Emptying and Motility in Rats. IEEE 2017, 64, 2546–2554. [Google Scholar]
- Smith, H.W. Observations on the Flora of the Alimentary Tract of Animals and Factors Affecting Its Composition. J. Pathol. Bacteriol. 1965, 89, 95–122. [Google Scholar] [CrossRef]
- Ward, F.W.; Coates, M.E. Gastrointestinal pH measurement in rats: Influence of the microbial flora, diet and fasting. Lab. Anim. 1987, 21, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Zecevic, D.E.; Meier, R.; Daniels, R.; Wagner, K.G. Site specific solubility improvement using solid dispersions of HPMC-AS/HPC SSL-Mixtures. Eur. J. Pharm. Biopharm. 2014, 87, 264–270. [Google Scholar] [CrossRef]
- Chen, L.; Wu, X.; Chen, X.D. Comparison between the digestive behaviors of a new in vitro rat soft stomach model with that of the in vivo experimentation on living rats-Motility and morphological influences. J. Food Eng. 2013, 117, 183–192. [Google Scholar] [CrossRef]
- Wu, P.; Liao, Z.; Luo, T.; Chen, L.; Chen, X.D. Enhancement of Digestibility of Casein Powder and Raw Rice Particles in an Improved Dynamic Rat Stomach Model Through an Additional Rolling Mechanism. J. Food Sci. 2017, 82, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Wu, P.; Chen, X.D. Quantitative visualization study on the physical movement and gastric emptying of diced carrot particle in a transparent rat stomach-duodenum model. Int. J. Food Eng. 2022, 18, 461–478. [Google Scholar] [CrossRef]
- Jang, S.-F.; Goins, B.A.; Phillips, W.T.; Santoyo, C.; Rice-Ficht, A.; McConville, J.T. Size discrimination in rat and mouse gastric emptying. Biopharm. Drug Dispos. 2013, 29, 13–19. [Google Scholar] [CrossRef]
- Lichtenberger, L.M.; Szabo, S.; Reynolds, E.S. Gastric emptying in the rat is inhibited by the duodenal ulcerogens, cysteamine and propionitrile. Gastroenterology 1977, 73, 1072–1076. [Google Scholar] [CrossRef]
- Tomlin, J.; Brown, N.; Ellis, A.; Carlsson, A.; Bogentoft, C.; Read, N.W. The effect of liquid fibre on gastric emptying in the rat and humans and the distribution of small intestinal contents in the rat. Gut 1993, 34, 1177–1181. [Google Scholar] [CrossRef] [Green Version]
- Reilly, J.A.; Forst, C.F.; Quigley, E.M.M.; Rikkers, L.F. Gastric emptying of liquids and solids in the portal hypertensive rat. Dig. Dis. Sci. 1990, 35, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Stellar, E.; Hill, J.H. The rat’s rate of drinking as a function of water deprivation. J. Comp. Physiol. Psychol. 1952, 45, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Fuller, L.M. The pharmacology of drinking behavior. Pharmacol. Ther. 1984, 24, 179–206. [Google Scholar] [CrossRef] [PubMed]
- Peck, J.W.; Blass, W.M. Localization of thirst and antidiuretic osmoreceptors by intracranial injections in rats. Am. J. Physiol. 1975, 228, 1501–1509. [Google Scholar] [CrossRef] [PubMed]
- Grignard, E.; Taylor, R.; McAllister, M.; Box, K.; Fotaki, N. Considerations for the development of in vitro dissolution tests to reduce or replace preclinical oral absorption studies. Eur. J. Pharm. Sci. 2017, 99, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Jensen, T.L.; Kiersgaard, M.K.; Sørensen, D.B.; Mikkelsen, L.F. Fasting of mice: A review. Lab. Anim. 2013, 47, 225–240. [Google Scholar] [CrossRef] [Green Version]
- Gad, S.C.; Spainhour, C.B.; Shoemake, C.; Pallman, D.R.S.; Stricker-Krongrad, A.; Downing, P.A.; Seals, R.E.; Eagle, L.A.; Polhamus, K.; Daly, J. Tolerable Levels of Nonclinical Vehicles and Formulations Used in Studies by Multiple Routes in Multiple Species with Notes on Methods to Improve Utility. Int. J. Toxicol. 2016, 35, 95–178. [Google Scholar] [CrossRef]
- Li, P.; Zhao, L. Developing early formulations: Practice and perspective. Int. J. Pharm. 2007, 341, 1–19. [Google Scholar] [CrossRef]
- Liu, H.H.; Surawanvijit, S.; Rallo, R.; Orkoulas, G.; Cohen, Y. Analysis of nanoparticle agglomeration in aqueous suspensions via constant-number Monte Carlo simulation. Environ. Sci. Technol. 2011, 45, 9284–9292. [Google Scholar] [CrossRef]
- Pestel, S.; Martin, H.J.; Maier, G.M.; Guth, B. Effect of commonly used vehicles on gastrointestinal, renal, and liver function in rats. J. Pharmacol. Toxicol. Methods 2006, 54, 200–214. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCE | BI 1026706 | BI 1026891 |
|---|---|---|
| pKa (Acid/Base) | 8.6 (Acid) | 4.4 (Base) |
| Water solubility pH 7.4 (mg/mL) | 0.02 | 0.002 |
| Intrinsic dissolution rate (µg/cm2*min) | <50 | <20 |
| LogP | 3.2 | 3.8 |
| Solid dispersion | Formulation 1 | Formulation 2 |
| Drugload (%) | 70 | 80 |
| Spray-drying solvent | Methanol | Acetone/H2O 80/20 (%, m/m) |
| Bulk density (g/cm3) | 0.33 | 0.37 |
| Particle density (g/cm3) | 1.45 | 1.51 |
| D50 (µm) | 6.61 | 5.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gierke, H.; Schaefer, K.; Gerlich, L.; Willmann, A.-C.; Bialetzki, V.; Boeck, G.; Pfrommer, T.; Nolte, T.; Weitschies, W. An In Vitro Model to Investigate the Potential of Solid Dispersions to Form Pharmacobezoars. Pharmaceutics 2022, 14, 2608. https://doi.org/10.3390/pharmaceutics14122608
Gierke H, Schaefer K, Gerlich L, Willmann A-C, Bialetzki V, Boeck G, Pfrommer T, Nolte T, Weitschies W. An In Vitro Model to Investigate the Potential of Solid Dispersions to Form Pharmacobezoars. Pharmaceutics. 2022; 14(12):2608. https://doi.org/10.3390/pharmaceutics14122608
Chicago/Turabian StyleGierke, Hannes, Kerstin Schaefer, Lukas Gerlich, Ann-Cathrin Willmann, Verena Bialetzki, Georg Boeck, Teresa Pfrommer, Thomas Nolte, and Werner Weitschies. 2022. "An In Vitro Model to Investigate the Potential of Solid Dispersions to Form Pharmacobezoars" Pharmaceutics 14, no. 12: 2608. https://doi.org/10.3390/pharmaceutics14122608
APA StyleGierke, H., Schaefer, K., Gerlich, L., Willmann, A. -C., Bialetzki, V., Boeck, G., Pfrommer, T., Nolte, T., & Weitschies, W. (2022). An In Vitro Model to Investigate the Potential of Solid Dispersions to Form Pharmacobezoars. Pharmaceutics, 14(12), 2608. https://doi.org/10.3390/pharmaceutics14122608