Abstract
(1) Background: This investigation aimed at developing a series of c-Met-targeting cabozantinib-based PROTACs. (2) Methods: Purification of intermediate and target compounds was performed using column chromatography, in vitro antiproliferation activity was measured using a standard MTT assay and a c-Met degradation assay was performed via the immunoblotting technique. (3) Results: Several compounds exhibited antiproliferative activity towards different cell lines of breast cancer (T47D, MDA-MB-231, SKBR3, HCC1954 and MCF7) at the same level as parent cabozantinib and 7-demethyl cabozantinib. Two target conjugates, bearing a VHL-ligand as an E3-ligase binding moiety and glycol-based linkers, exhibited the effective inhibition of c-Met phosphorylation and an ability to decrease the level of c-Met in HCC1954 cells at micromolar concentrations. (4) Conclusions: Two compounds exhibit c-Met inhibition activity in the nanomolar range and can be considered as PROTAC molecules due to their ability to decrease the total level of c-Met in HCC1954 cells. The structures of the offered compounds can be used as starting points for further evaluation of cabozantinib-based PROTACs.
1. Introduction
PROTAC technology has exhibited impressive progress [1] since the first PROTAC molecule was designed in 2001 by C.M. Crews [2] to the point where several candidates entered clinical trials in 2019–2021 [3]. In general, the PROTAC molecule consists of an LP-ligand that connects to the target protein of interest (POI), associated through the linker with LE-ligand, recruiting E3-ligase. Attractive features of this technology are the ability of PROTAC degraders to target proteins without distinct active sites (i.e., “undruggable” proteins) and their catalytic mode of action [4].
The range of targets degraded by PROTAC is constantly growing [5]. Currently, it includes protein kinases [6,7,8] (AKT, BCR-ABL, BTK etc.), epigenetic regulators (BRD4/7/9 [9,10,11,12], TRIM24 [13], Smad3 [14], proteins related to neurodegenerative diseases (Tau [15], mutant huntingtin [16], PSD-95 [17], α-synuclein [18]), nuclear receptors (ERRα [19], AR [20], PARP1 [21], RAR [22]) and anti-apoptotic proteins [23,24,25] (MCL-1, BCL-2, BCL-XL).
Auxiliary proteins named E3 ligases are essential for the ubiquitination of POI. To date, only a few examples (VHL [11,13,19,26], CRBN [9,22,27], MDM2 [10,21,28], IAPs [29,30]) have been confirmed for PROTAC design despite the discovery of more than 700 E3 ligases in humans [31]. Proteins such as VHL can serve not only as E3 ligases but also as POIs [32] due to their ability to interact with HIF1α [33] and facilitate their proteasomal degradation. Based on this consideration, VHL was chosen for the developing of the co-called “homoPROTAC” [32,34]. Nevertheless, expanding the range of potent E3-ligases still remains important for PROTACs’ evolution [35,36,37].
One of the key steps in protein degradation via the ubiquitin–proteasome system (UPS) is the transfer of the polyubiquitin chain to the POI. The linker between LP and LE plays an essential role in this process. All factors: length, flexibility, polarity and presence of functional groups can influence transfer efficiency [38,39,40,41]. The most common motifs for linker design are PEG- or other glycol-based spacers, triazole-, piperidine- and piperazine-bearing fragments. Meanwhile, the choice of linker structure is often carried out empirically by screening an activity of a series of PROTAC molecules.
Despite the fundamental principles of PROTAC action being sufficiently understood, some aspects of PROTAC technology remain unclear. The main issues are related to identifying new possible targets for UPS degradation and to extend the range of E3-ligases. Furthermore, evaluation of PROTACs’ pharmacokinetic and pharmacodynamic characteristics, indicating side targets and determining selectivity, is not trivial. More efforts are needed to fully understand the complete mechanisms of action and utilization of PROTACs [42].
This work is focused on the design and synthesis of cabozantinib-based PROTACs, recruiting CRBN and VHL as E3-ligases and the investigation of their efficiency depending on the linker structure. The choice of cabozantinib as a warhead stemmed from its excellent activity against hepatocellular endothelial growth factor receptor (HEGFR or c-Met), endothelial growth factor receptor (EGFR) and other tumor-associated proteins KIT, RET, AXL, TIE-2 and FLT-3 [43,44]. In addition to the abnormal activation of c-Met along with the progression of glioblastoma, liver [45], colon [46] and pancreatic [47] carcinomas, the selection of c-Met as a target protein for degradation is due to its deep involvement in the activation of several downstream signaling pathways (MAPK, PI3K, SRC, STAT) [48] that play an essential role in cell proliferation and survival.
Cabozantinib itself has been approved by the FDA for progressive metastatic medullary thyroid cancer and advanced renal cell carcinoma [49,50]. However, a wide range of dose-dependent side effects of cabozantinib have occurred in many cases [50,51,52]. Application of cabozantinib-based PROTAC is believed to enhance antitumor action and reduce adverse effects due to the PROTAC catalytic mode of action that could significantly decrease the required drug dose.
2. Materials and Methods
2.1. Chemistry
Agilent DDR2 400 and Varian Mercury 400 Plus spectrometers were used to record the 1H NMR and 13C NMR spectra at 25 °C. The ppm measurement unit was applied for the report of the chemical shifts (δ) in relation to the solution of the compound in DMSO-d6 and CDCl3, while the J values and the internal reference TMS were expressed in Hertz. Only the NMR assignment received the atomic numeration. The Bruker Microflex LT spectrometer was used to record the MALDI spectra. The electron spray ionization (ESI) was applied for the high-resolution mass spectra record on a Bruker Daltonics microOTOF-QII instrument. The Elementar (Vario Micro Cube) apparatus was a tool for the elemental analysis. The Shimadzu Class-VP V6.12SP1 system was used for the HPLC, A: 0.01 M H3PO4 pH 2.6; B: MeCN; all the components were characterized to have purity more than 95%. The Merck Kieselgel 60 (70–230 mesh) was applied for the column chromatography. Commercially available reagents were acquired to perform all the reactions. The purification of the solvents was performed in compliance with the standard procedures. The purchased petroleum ether is appropriate for the fraction of 40–70 °C. Syntheses of VHL-ligand 10 [53], its derivatives 10a,b [54] and conjugates of lenalidomide with caproic acid in forms of N-Boc protected (12a) and free amine (12b) [36], cabozantinib hydroxyl derivative 3 [54] were performed as described earlier.
2.2. Biological Research
2.2.1. Cell Culture Screening and Assessment of the Antiproliferative Effects
The A-431 (skin), A549 (lung), HeLa (cervical), T47D, MDA-MB-231, SKBR3, HCC1954 and MCF7 (breast) cancer cells were acquired from the ATCC center and incubated in normal media (Gibco, New York, NY, USA) recommended by the manufacturer. The MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) (Applichem) assay with modifications [55] was used to assess the cell growth as described earlier in [56]. The seeding of the tumor cells was performed in 24-well plates (TPP) using 900 μL of the appropriate medium. DMSO (AppliChem, Darmstadt, Germany) was applied for the compound dissolution to a concentration of 10 mM immediately before the experiments. Subsequently, the obtained solutions were diluted in the DMEM medium to the necessary doses. The solutions of the investigated compounds with various concentrations in 100 μL of the DMEM were added a day following the seeding, and the cell cultures were incubated for 3 days. After the cell growth with the compounds, the DMEM was ablated, and the MTT dye as part of the medium was added to the end concentration of 0.2 mg/mL to every well for the incubation during 2 h. All the wells were supplemented with DMSO in the amount of 350 μL for solvation of the MTT formazan purple crystals. A MultiScan reader (ThermoFisher, Waltham, MA, USA) was applied for the measurement of the solutions’ absorbance at a wavelength of 571 nanometers. The cell survival was evaluated after subtraction of the blank value (the absorbance in each well without cells) from all wells. All of the tests were performed 3 times. The investigation of the dose–response curves was performed with the usage of regression analysis and sigmoidal curves (Log(concentration) vs. normalized absorbance). GraphPad Prism was applied for the establishment of the half-maximal inhibitory concentrations (IC50) (Table 1).

Table 1.
Antiproliferative activity of target compounds 15a–d, 16a,b, 17a–d against cancer cells (IC50 values 1, µM); the dose–response curves are presented in Figures S1–S5.
2.2.2. Immunoblotting
To prepare samples for immunoblotting analysis, the cells were seeded on 100 mm dishes (Corning, New York, NY, USA). For experiments with HCC1954 breast cancer cells, the compounds 15a and 15c were added in fresh medium after 24 h of seeding. For the cell extracts’ preparation, cell cultures were washed in phosphate-buffered saline (PBS) 2 times and incubated for 10 min on ice in the lysis buffer. The aforementioned buffer had a pH of 7.5 and contained Tris-HCl (50 mM), NaCl (150 mM), Igepal CA-630 (0.5%), DTT (1 mM), EDTA (1 mM), sodium orthovanadate (0.1 mM), PMSF (1 mM) and pepstatin, leupeptin, aprotinin (1 µg/mL each) as described earlier [57]. The Bradford method [58] was used to define the level of the protein. The cell extracts were divided in SDS-PAGE (10%) at reducing conditions, transmitted to a nitrocellulose membrane (GE HealthCare, Chicago, IL, USA) and treated following a regular methodology. To prevent nonspecific absorption, the membranes were processed with 5% nonfat milk solution in TBS buffer at a pH of 7.5. The aforesaid buffer contained Tris (20 mM), NaCl (500 mM) and Tween-20 (0.1%). Thereafter, the membranes were incubated with primary antibodies during the night at a temperature of 4 °C. c-Met and phospho-c-Met (Tyr1234/1235, Tyr1003) antibodies were acquired from Cell Signaling Technology; the antibodies versus α-tubulin (Cell Signaling Technology, Danvers, MA, USA) were applied for a loading control. Goat anti-rabbit IgGs (Jackson ImmunoResearch, West Grove, PA, USA) associated with horseradish peroxidase were involved in immunoblotting in the quality of secondary antibodies. The detection of the signals was performed with the usage of the ECL reagent as characterized by Mruk and Cheng [59] and an ImageQuant LAS4000 system (GE HealthCare, Chicago, IL, USA). Densitometry for the tested proteins/α-tubulin ratio was carried out using ImageJ software.
3. Results and Discussion
3.1. Chemistry
All synthesized conjugates included cabozantinib as a warhead, whereas CRBN-ligand lenalidomide (an analogue of thalidomide, a derivative of naturally occurring glutamic acid) or VHL-ligand (S,R,S)-AHPC were exploited as E3-ligase ligands. The mentioned molecules were joined with linkers of various lengths and structures. Simple aliphatic spacers, glycol-based spacers and linkers bearing a triazole core were applied in conjugate synthesis. The general structures of target conjugates are represented in Figure 1.
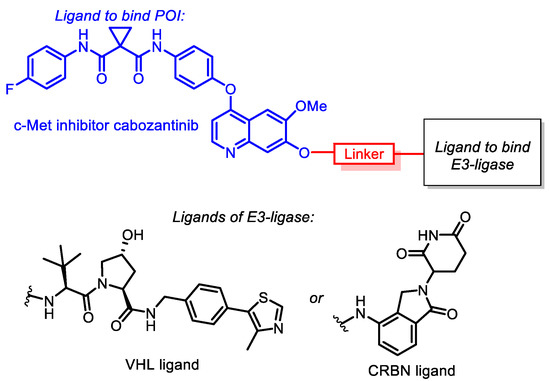
Figure 1.
General structures of cabozantinib-based PROTACs.
Since the binding affinity of cabozantinib after conjugation should remain at the same level as for the intact molecule, it was important to determine the possible conjugation site. Previously [60], it was reported that structural analogues of cabozantinib functionalized on the position “7” of the quinazoline core demonstrated almost the same activity as parent compounds towards c-Met.
The synthetic approach to 7-demethylated cabozantinib congener 3, a common building block for all target molecules, was based on a previously published protocol [61] and included 11 steps. 4-Hydroxy-3-methoxybenzaldehyde (1) and cyclopropane-1,1-dicarboxylic acid (2) were used as starting materials (Scheme 1).
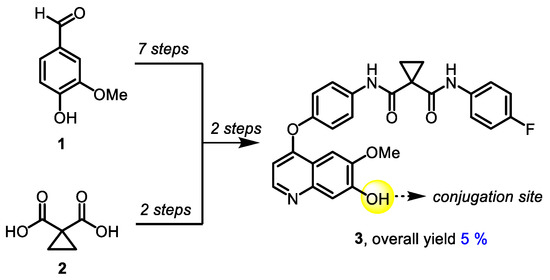
Scheme 1.
Synthesis of 7-demethylated cabozantinib 3.
Starting from 7-demethylated cabozantinib analogue 3, a series of carboxyl-containing derivatives were synthesized via two approaches. The first route (Scheme 2, pathway A) was based on the alkylation of 3 with esters of halogenated carboxylic acids with the subsequent hydrolysis of the ester group (Scheme 2). Various bases (Cs2CO3, K2CO3, Na2CO3, DBU, NaH), solvents (1,4-dioxane, DMF, THF, toluene, acetone) and temperatures were applied, and the highest yields were achieved using NaH in DMF at 65 °C. The treatment of 3 with methyl esters of 5-bromovaleric and 11-bromoundecanoic acids in the presence of NaH in DMF resulted in 7-O-alkylated cabozantinib derivatives 4a,b (67% and 63%, respectively). When methyl 2-bromoacetate and methyl 3-bromopropionate were used for the alkylation of 3, the yields of isolated products dramatically dropped; therefore only two derivatives were entered in the next step. Cleavage of the ester group in 4a,b by alkaline treatment led to appropriate acids 5a,b in good to high yields. A similar synthetic approach (Scheme 2, pathway B) was applied to obtain carboxylic derivatives containing ethylene glycol (7a) as well as a propane-1,3-diol-based linker (7b).
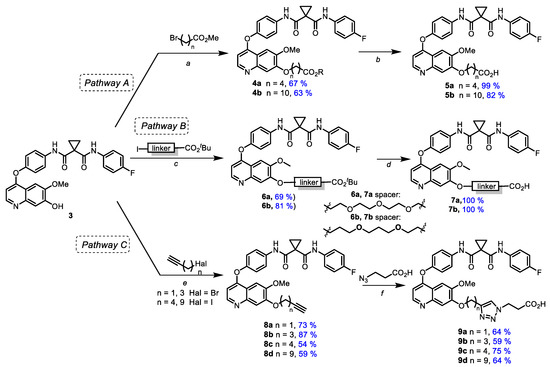
Scheme 2.
Synthesis of ω-carboxyl bearing cabozantinib derivatives. Reagents and conditions: (a) NaH, DMF, 0 °C → 65 °C; (b) NaOH, MeOH-H2O (2:1), 60 °C; (c) Cs2CO3, DMF, 65 °C; (d) TFA, rt; (e) Cs2CO3, DMF, rt; (f) TBTA, CuSO4*5H2O, AscNa, DMF-H2O (1:1), 55 °C.
Another pathway (Scheme 2, pathway C) included the alkylation of 3 with ω-haloalkynes containing a terminal triple bond and subsequent azide-alkyne cycloaddition with 3-azidopropionic acid followed by the formation of carboxylic cabozantinib derivatives 9a–d containing triazole linkers. Alkynes with various meanings of the chain length were used to define the impact of triazole moiety location on PROTACs’ activity (Scheme 2).
Next, synthetic blocks, containing moieties-targeted E3-ligases CRBN and VHL, were synthesized (Scheme 3). For this purpose peptidomimetic ligand 10 for VHL, obtained by previously published methods, and commercially available lenalidomide (11) for CRBN [53,62] were treated with ω-N-Boc protected carboxylic acids [36,54] 13 and 14 (Scheme 3) that led to protected amines 10a, 11a and 12a with good to high yields.
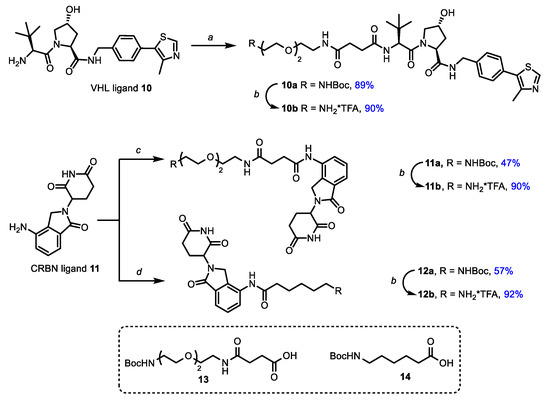
Scheme 3.
Synthesis of ligands for E3-ligases. Reagents and conditions: (a) 13, DIPEA, HATU, DCM, rt; (b) TFA, DCM, rt; (c) 13, HATU, DIPEA, DMF, rt; (d) 14, HATU, DIPEA, DMF, rt.
At the next step, protecting groups were removed by a trifluoroacetic acid treatment and corresponding amines 10b, 11b and 12b were coupled with carboxylic cabozantinib derivatives 5a,b, 7a,b and 9a–d to obtain target PROTACs 15a–d, 16a,b and 17a–d (Figure 2). The final step was carried out under a standard peptide coupling procedure with HATU in the presence of DIPEA in DMF (Figure 2).
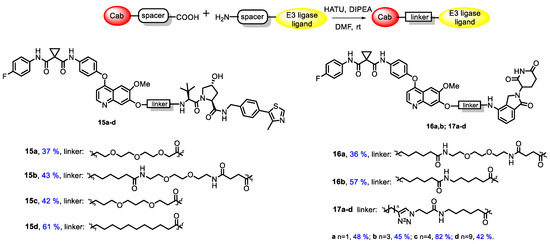
Figure 2.
Synthesis and structures of target conjugates.
3.2. Biology
3.2.1. Cancer Cell Line Screening and Antiproliferative Evaluation
The first aim of the biological part of the study was to find the proper cell model for testing of obtained conjugates. The published data on c-Met expression in cancer cell lines are quite contradictory. The c-Met expression described in several papers varies considerably. The levels of c-Met expression depend on the method of analysis and characteristics of antibodies used by the researchers. Moreover, sample preparation techniques can affect the identified c-Met expression level [63,64,65,66,67,68]. Considering the above, we screened cancer cell lines to check the level of c-Met expression. The different cell lines of breast cancer (T47D, MDA-MB-231, SKBR3, HCC1954 and MCF7), skin cancer (A-431), lung cancer (A549) and cervical cancer (HeLa) were cultured without obtained compounds and then subjected to immunoblotting with antibodies against c-Met, as characterized in the Materials and Methods section. As demonstrated in Figure 3, the c-Met expression level varied considerably between cells. First, no c-Met expression was observed in MCF7, SKBR3 and T47D breast cancer cells. A rather low c-Met level was revealed in MDA-MB-231 breast tumor cell cultures; nevertheless, 140 and 170 kDa c-Met proteins were clearly visible in these cells, as can be seen in Figure 3.
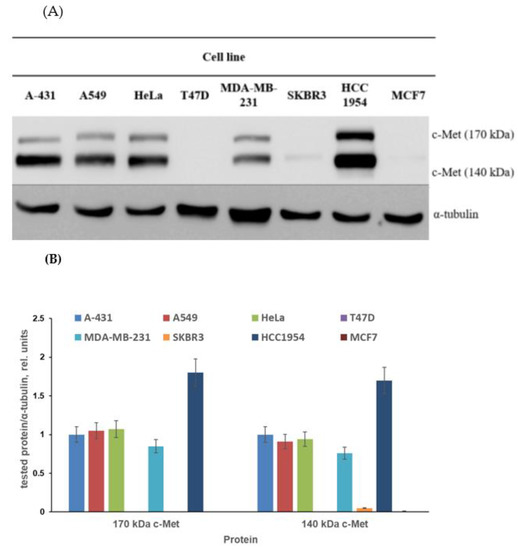
Figure 3.
The c-Met expression level in selected cell lines. (A) Representative immunoblotting images; (B) densitometry for immunoblotting data (n = 3) was carried out using ImageJ software (Wayne Rasband, NIH) with the protocol provided by The University of Queensland and the recommendations from the work [69].
The c-Met expression level evaluated in A-431, A549 and HeLa cells was moderate. Finally, the highest level of c-Met was detected in HCC1954 breast cancer cells. Thus, for further experiments, we used cell lines with different levels of c-Met expression (Table 1).
The antiproliferative activity of target compounds against selected cancer cell lines is shown in Table 1. Compounds 17a–d bearing a triazole ring in the spacer between cabozantinib (LP) and lenalidomide (LE) demonstrated low activity towards all tested cell lines. The length of the spacer and position of triazole did not play any role in antiproliferative activity. Derivatives 16a,b, in which LP and LE were bound by using a combination of ethylene or propylene glycol fragments, were slightly superior to triazole-based analogues 17a–d; however, they had low antiproliferative potency towards HCC1954 cells. In contrast, conjugates 15a–c were found to exhibit the most promising activity, especially towards the HCC1954 cell line with the highest c-Met expression. It may be supposed that the introduction of VHL as the LE-end of PROTAC conjugates can generally enhance the antitumor potency of compounds. However, compound 15d, bearing an aliphatic nonpolar linker, was less potent in comparison with other compounds series 15 as well as with reference molecules 3 and cabozantinib. The high activity of cabozantinib in T47D cancer cells draws attention. Interestingly, these cells do not express known cabozantinib targets, including c-Met. The cabozantinib activity against T47D cells described in our work coincides with that described in [70]. We believe that the search for new targets of cabozantinib in c-Met/VEGFRs/KIT-negative cells is a promising task. Mimicking the activity of cabozantinib, compounds 3, 15a, and 16a,b were also active against c-Met-negative T47D breast cancer cells.
3.2.2. Assessment of c-Met Inhibition
Compounds 15a and 15c significantly inhibited the growth of c-Met-positive HCC1954 cells and had notably lower antiproliferative effects on the c-Met-negative MCF7 cells. These compounds were chosen for in-depth evaluation as c-Met targeting agents. HCC1954 cells were treated with compounds 15a and 15c at concentrations ranging from 14 to 10,000 nM, and then the levels of c-Met protein and its phosphorylated forms were determined by immunoblotting (Figure 4).
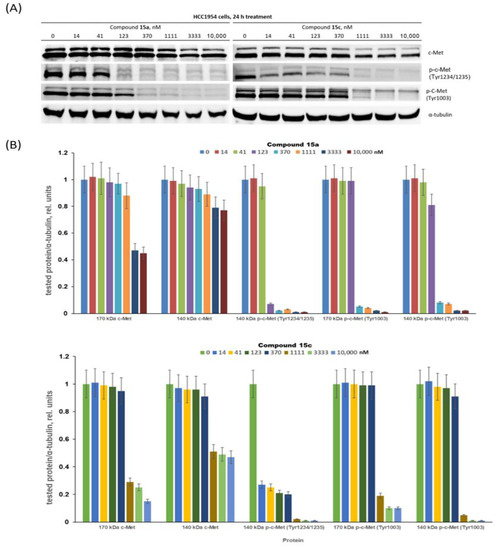
Figure 4.
Decreasing c-Met phosphorylation and expression with conjugates 15a and 15c. (A) Representative immunoblotting images; (B) densitometry for immunoblotting data (n = 3) was carried out using ImageJ software (Wayne Rasband, NIH) with the protocol provided by The University of Queensland and the recommendations from the work [69].
First, we proved that the cabozantinib part of the PROTAC molecules 15a and 15c can still interact with the c-Met kinase. The c-Met intracellular sequence consists of a juxtamembrane domain, a tyrosine kinase domain and a C-terminal multifunctional docking site [71,72]. The juxtamembrane domain contains amino acid residues including Tyr1003, which interacts with casitas B-lineage lymphoma (c-Cbl) and leads to ubiquitin-dependent c-Met degradation [73,74]. This signalling process is a mechanism of a negative regulatory loop, which modulates the c-Met activation status. Pascal Peschard and colleagues found that mutation of the c-Cbl tyrosine kinase binding domain on the c-Met receptor converts it into a transforming protein [73]. The authors replaced the juxtamembrane tyrosine of c-Met by phenylalanine. The mutant c-Met had transforming activity in fibroblast and epithelial cells. The tyrosine kinase c-Met domain, upon phosphorylation of Tyr1234 and Tyr1235, undergoes a conformational change resulting in increased kinase activity. Although both compounds demonstrate a similar effect on the Tyr1003 phosphorylation residue at juxtamembrane c-Met domain, it appears at 123 nM and above (Figure 4), conjugate 15c can block phosphorylation Tyr1234/1235 (belonging to tyrosine kinase c-Met domain) already at 14 nM, whereas similar effects for 15a were observed at concentrations higher than 100 nM. We believe that the simultaneous blocking of tyrosine phosphorylation in two receptor domains is a very important characteristic of the lead PROTAC molecules. Treatment of HCC1954 cells with both conjugates 15a and 15c resulted in a decrease in the levels of c-Met in a dose-dependent manner. Figure 4 shows that compounds 15a and 15c at nanomolar concentrations had a weak effect on the level of c-Met, while low micromolar concentrations led to near complete blocking of the accumulation of p-c-Met in HCC1954 breast cancer cells. Altogether, PROTAC conjugates 15a and 15c, exhibiting antiproliferative effects and suppressing c-Met kinase at low concentrations, are promising leaders for further development.
4. Conclusions
We characterized the design, synthesis and biological assessment of a series of cabozantinib-based PROTACs, recruiting CRBN and VHL as E3-ligases. The obtained compounds exhibited antiproliferative effects against a number of tumor cell lines. Compounds 17a–d, bearing a triazole linker, demonstrated low antiproliferative activity towards all tested cell lines. No correlation was observed between the distance of triazole and cabozantinib fragments and antiproliferative activity. Compounds 15a–d demonstrated more pronounced antiproliferative activity towards the panel of selected cell lines. However, compound 15d, bearing a nonpolar linker with 11 atoms as well as compound 15b with a polar but longer spacer, were less active than compounds with glycol- and propanediol-based linkers 15a and 15c. They caused a 50% suppression of HCC1954 cell proliferation at a dose of approximately 6 μM and 7 μM, respectively, meaning they provide a new perspective for further studies considering c-Met as the main target of the synthesized conjugates. Both compounds at nanomolar concentrations reduced the level of the phosphorylated form of c-Met protein and were able to block the c-Met accumulation in tumor cells at higher concentrations, indicating the high potency of novel anticancer cabozantinib-based PROTACs. Further research may be aimed at the optimization of the PROTAC linker structure and the search for other PROTAC targets in the cancer cells (MET, RET, KIT, FLT1, FLT3, FLT4, TIE2, AXL, etc.). The obtained data clearly indicate the complex relationship between the biological activity of the conjugate and the length, flexibility and polarity of the linker between LP and LE. Apparently, the glycol-based linkers, bearing 10–12 atoms, are the most suitable in the case of the described molecules. Moreover, the mechanisms of action of the leader conjugates, and particularly an assessment of the antitumor efficacy of these agents in vivo, will be established.
Supplementary Materials
The following supporting data can be downloaded at https://www.mdpi.com/article/10.3390/pharmaceutics14122829/s1: synthetic procedures, description and copies of NMR spectra, and dose–response curves. Figure S1. Antiproliferative activity of target compounds 15a–d, 16a,b, 17a–d against A-431 cells; the A-431 cells were incubated with compounds for 3 days and the cell viability was assessed by the MTT test. Figure S2. Antiproliferative activity of target compounds 15a–d, 16a,b, 17a–d against T47D cells. Figure S3. Antiproliferative activity of target compounds 15a–d, 16a,b, 17a–d against MCF7 cells. Figure S4. Antiproliferative activity of target compounds 15a–d, 16a,b, 17a–d against HCC1954 cells. Figure S5. Antiproliferative activity of target compounds 15a–d, 16a,b, 17a–d against SKBR3 cells. Reference [75] is cited in the supplementary materials.
Author Contributions
Conceptualization, E.S.S., A.Y.F., A.S.T. and A.E.S.; data curation, D.V.A., D.I.S., D.V.S. and F.B.B.; formal analysis, Y.D.R.; funding acquisition, E.S.S.; investigation, A.A.S., D.V.A., A.S.T., A.M.S., D.I.S., D.V.S. and F.B.B.; methodology, A.S.T. and A.M.S.; project administration, E.S.S., A.Y.F. and A.E.S.; supervision, E.S.S. and A.E.S.; visualization, D.I.S. and D.V.S.; writing—original draft, E.S.S. and A.M.S.; writing—review and editing, A.Y.F., A.S.T. and A.E.S. All authors have read and agreed to the published version of the manuscript.
Funding
This investigation was supported by Russian Science Foundation, grant number 22-23-00736 (https://www.rscf.ru/project/22-23-00736/ (accessed on 1 December 2022)).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The densitometry protocol for immunoblotting “Using ImageJ to quantify blots” was provided by The University of Queensland.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Garber, K. The PROTAC Gold Rush. Nat. Biotechnol. 2022, 40, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric Molecules That Target Proteins to the Skp1–Cullin–F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Benowitz, A.B.; Jones, K.L.; Harling, J.D. The Therapeutic Potential of PROTACs. Expert Opin. Ther. Pat. 2021, 31, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, X.; Lv, D.; Yuan, Y.; Zheng, G.; Zhou, D. Assays and Technologies for Developing Proteolysis Targeting Chimera Degraders. Future Med. Chem. 2020, 12, 1155–1179. [Google Scholar] [CrossRef]
- Zou, Y.; Ma, D.; Wang, Y. The PROTAC Technology in Drug Development. Cell Biochem. Funct. 2019, 37, 21–30. [Google Scholar] [CrossRef]
- Smith, B.E.; Wang, S.L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC Substrate Specificity Dictated by Orientation of Recruited E3 Ligase. Nat. Commun. 2019, 10, 131. [Google Scholar] [CrossRef]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great Opportunities for Academia and Industry. Signal Transduct. Target Ther. 2019, 4, 64. [Google Scholar] [CrossRef]
- Remillard, D.; Buckley, D.L.; Paulk, J.; Brien, G.L.; Sonnett, M.; Seo, H.-S.; Dastjerdi, S.; Wühr, M.; Dhe-Paganon, S.; Armstrong, S.A.; et al. Degradation of the BAF Complex Factor BRD9 by Heterobifunctional Ligands. Angew. Chem. Int. Ed. 2017, 56, 5738–5743. [Google Scholar] [CrossRef]
- Hines, J.; Lartigue, S.; Dong, H.; Qian, Y.; Crews, C.M. MDM2-Recruiting PROTAC Offers Superior, Synergistic Antiproliferative Activity via Simultaneous Degradation of BRD4 and Stabilization of P53. Cancer Res. 2019, 79, 251–262. [Google Scholar] [CrossRef]
- Zoppi, V.; Hughes, S.J.; Maniaci, C.; Testa, A.; Gmaschitz, T.; Wieshofer, C.; Koegl, M.; Riching, K.M.; Daniels, D.L.; Spallarossa, A.; et al. Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel–Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7. J. Med. Chem. 2019, 62, 699–726. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Wang, W.-L.; Yang, Y.-Y.; Hu, X.-T.; Wang, Q.-W.; Zuo, W.-Q.; Xu, Y.; Feng, Q.; Wang, N.-Y. Identification of a Selective BRD4 PROTAC with Potent Antiproliferative Effects in AR-Positive Prostate Cancer Based on a Dual BET/PLK1 Inhibitor. Eur. J. Med. Chem. 2022, 227, 113922. [Google Scholar] [CrossRef] [PubMed]
- Gechijian, L.N.; Buckley, D.L.; Lawlor, M.A.; Reyes, J.M.; Paulk, J.; Ott, C.J.; Winter, G.E.; Erb, M.A.; Scott, T.G.; Xu, M.; et al. Functional TRIM24 Degrader via Conjugation of Ineffectual Bromodomain and VHL Ligands. Nat. Chem. Biol. 2018, 14, 405–412. [Google Scholar] [CrossRef]
- Wang, X.; Feng, S.; Fan, J.; Li, X.; Wen, Q.; Luo, N. New Strategy for Renal Fibrosis: Targeting Smad3 Proteins for Ubiquitination and Degradation. Biochem. Pharmacol. 2016, 116, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.C.; Nandi, G.; Donovan, K.A.; Cai, Q.; Berry, B.C.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; Ferguson, F.M.; Haggarty, S.J. Discovery and Optimization of Tau Targeted Protein Degraders Enabled by Patient Induced Pluripotent Stem Cells-Derived Neuronal Models of Tauopathy. Front. Cell. Neurosci. 2022, 16, 801179. [Google Scholar] [CrossRef] [PubMed]
- Tomoshige, S.; Nomura, S.; Ohgane, K.; Hashimoto, Y.; Ishikawa, M. Discovery of Small Molecules That Induce the Degradation of Huntingtin. Angew. Chem. Int. Ed. 2017, 56, 11530–11533. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Jin, W.Y.; Lu, J.; Wang, J.; Wang, Y.T. Rapid and Reversible Knockdown of Endogenous Proteins by Peptide-Directed Lysosomal Degradation. Nat. Neurosci. 2014, 17, 471–480. [Google Scholar] [CrossRef]
- Kargbo, R.B. PROTAC Compounds Targeting α-Synuclein Protein for Treating Neurogenerative Disorders: Alzheimer’s and Parkinson’s Diseases. ACS Med. Chem. Lett. 2020, 11, 1086–1087. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Mares, A.; Smith, I.E.D.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in Vivo Protein Knockdown by Small-Molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef]
- Itoh, Y.; Kitaguchi, R.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Design, Synthesis and Biological Evaluation of Nuclear Receptor-Degradation Inducers. Bioorg. Med. Chem. 2011, 19, 6768–6778. [Google Scholar] [CrossRef]
- Zhao, Q.; Lan, T.; Su, S.; Rao, Y. Induction of Apoptosis in MDA-MB-231 Breast Cancer Cells by a PARP1-Targeting PROTAC Small Molecule. Chem. Commun. 2019, 55, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Han, L.; Mao, S.; Xu, P.; Xu, X.; Zhao, R.; Wu, Z.; Zhong, K.; Yu, G.; Wang, X. Discovery of A031 as Effective Proteolysis Targeting Chimera (PROTAC) Androgen Receptor (AR) Degrader for the Treatment of Prostate Cancer. Eur. J. Med. Chem. 2021, 216, 113307. [Google Scholar] [CrossRef]
- Papatzimas, J.W.; Gorobets, E.; Maity, R.; Muniyat, M.I.; MacCallum, J.L.; Neri, P.; Bahlis, N.J.; Derksen, D.J. From Inhibition to Degradation: Targeting the Antiapoptotic Protein Myeloid Cell Leukemia 1 (MCL1). J. Med. Chem. 2019, 62, 5522–5540. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Thummuri, D.; He, Y.; Liu, X.; Zhang, P.; Zhou, D.; Zheng, G. Utilizing PROTAC Technology to Address the On-Target Platelet Toxicity Associated with Inhibition of BCL-XL. Chem. Commun. 2019, 55, 14765–14768. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; He, N.; Guo, Z.; Niu, C.; Song, T.; Guo, Y.; Cao, K.; Wang, A.; Zhu, J.; Zhang, X.; et al. Proteolysis Targeting Chimeras for the Selective Degradation of Mcl-1/Bcl-2 Derived from Nonselective Target Binding Ligands. J. Med. Chem. 2019, 62, 8152–8163. [Google Scholar] [CrossRef] [PubMed]
- Zengerle, M.; Chan, K.-H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide Conjugation as a Strategy for in Vivo Target Protein Degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef]
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted Intracellular Protein Degradation Induced by a Small Molecule: En Route to Chemical Proteomics. Bioorg. Med. Chem. Lett. 2008, 18, 5904–5908. [Google Scholar] [CrossRef]
- Itoh, Y.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Protein Knockdown Using Methyl Bestatin—Ligand Hybrid Molecules: Design and Synthesis of Inducers of Ubiquitination-Mediated Degradation of Cellular Retinoic Acid-Binding Proteins. J. Am. Chem. Soc. 2010, 132, 5820–5826. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, Y.; Shi, L.; Yang, S.; Chang, J.; Zhong, Y.; Li, Q.; Xing, D. Recent Advances in IAP-Based PROTACs (SNIPERs) as Potential Therapeutic Agents. J. Enzym. Inhib. Med. Chem. 2022, 37, 1437–1453. [Google Scholar] [CrossRef]
- Humphreys, L.M.; Smith, P.; Chen, Z.; Fouad, S.; D’Angiolella, V. The Role of E3 Ubiquitin Ligases in the Development and Progression of Glioblastoma. Cell Death Differ. 2021, 28, 522–537. [Google Scholar] [CrossRef] [PubMed]
- Maniaci, C.; Hughes, S.J.; Testa, A.; Chen, W.; Lamont, D.J.; Rocha, S.; Alessi, D.R.; Romeo, R.; Ciulli, A. Homo-PROTACs: Bivalent Small-Molecule Dimerizers of the VHL E3 Ubiquitin Ligase to Induce Self-Degradation. Nat. Commun. 2017, 8, 830. [Google Scholar] [CrossRef] [PubMed]
- Tyers, M.; Rottapel, R. VHL: A Very Hip Ligase. Proc. Natl. Acad. Sci. USA 1999, 96, 12230–12232. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Li, T.; Miao, Z.; Wang, P.; Sheng, C.; Zhuang, C. Homobivalent, Trivalent, and Covalent PROTACs: Emerging Strategies for Protein Degradation. J. Med. Chem. 2022, 65, 8798–8827. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Liu, T.; Jiao, Q.; Ji, J.; Tao, M.; Liu, Y.; You, Q.; Jiang, Z. Discovery of a Keap1-Dependent Peptide PROTAC to Knockdown Tau by Ubiquitination-Proteasome Degradation Pathway. Eur. J. Med. Chem. 2018, 146, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wu, Z.; Chen, P.; Zhang, J.; Wang, T.; Zhou, J.; Zhang, H. Discovery of a New Class of PROTAC BRD4 Degraders Based on a Dihydroquinazolinone Derivative and Lenalidomide/Pomalidomide. Bioorg. Med. Chem. 2020, 28, 115228. [Google Scholar] [CrossRef] [PubMed]
- Kannt, A.; Đikić, I. Expanding the Arsenal of E3 Ubiquitin Ligases for Proximity-Induced Protein Degradation. Cell Chem. Biol. 2021, 28, 1014–1031. [Google Scholar] [CrossRef]
- Cyrus, K.; Wehenkel, M.; Choi, E.-Y.; Han, H.-J.; Lee, H.; Swanson, H.; Kim, K.-B. Impact of Linker Length on the Activity of PROTACs. Mol. BioSyst. 2011, 7, 359–364. [Google Scholar] [CrossRef]
- Troup, R.I.; Fallan, C.; Baud, M.G.J. Current Strategies for the Design of PROTAC Linkers: A Critical Review. Explor. Target. Anti Tumor Ther. 2020, 1, 273–312. [Google Scholar] [CrossRef]
- Bemis, T.A.; La Clair, J.J.; Burkart, M.D. Unraveling the Role of Linker Design in Proteolysis Targeting Chimeras. J. Med. Chem. 2021, 64, 8042–8052. [Google Scholar] [CrossRef]
- Klein, V.G.; Bond, A.G.; Craigon, C.; Lokey, R.S.; Ciulli, A. Amide-to-Ester Substitution as a Strategy for Optimizing PROTAC Permeability and Cellular Activity. J. Med. Chem. 2021, 64, 18082–18101. [Google Scholar] [CrossRef] [PubMed]
- Goracci, L.; Desantis, J.; Valeri, A.; Castellani, B.; Eleuteri, M.; Cruciani, G. Understanding the Metabolism of Proteolysis Targeting Chimeras (PROTACs): The Next Step toward Pharmaceutical Applications. J. Med. Chem. 2020, 63, 11615–11638. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a Novel MET and VEGFR2 Inhibitor, Simultaneously Suppresses Metastasis, Angiogenesis, and Tumor Growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef]
- Cochin, V.; Gross-Goupil, M.; Ravaud, A.; Godbert, Y.; Le Moulec, S. Cabozantinib: Modalités d’action, Efficacité et Indications. Bull. Cancer 2017, 104, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Bouattour, M.; Raymond, E.; Qin, S.; Cheng, A.-L.; Stammberger, U.; Locatelli, G.; Faivre, S. Recent Developments of c-Met as a Therapeutic Target in Hepatocellular Carcinoma. Hepatology 2018, 67, 1132–1149. [Google Scholar] [CrossRef]
- Liu, Y. Prognostic Value of c-Met in Colorectal Cancer: A Meta-Analysis. World J. Gastroenterol. 2015, 21, 3706. [Google Scholar] [CrossRef] [PubMed]
- Pothula, S.P.; Xu, Z.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Targeting HGF/c-Met Axis in Pancreatic Cancer. Int. J. Mol. Sci. 2020, 21, 9170. [Google Scholar] [CrossRef]
- Scagliotti, G.V.; Novello, S.; von Pawel, J. The Emerging Role of MET/HGF Inhibitors in Oncology. Cancer Treat. Rev. 2013, 39, 793–801. [Google Scholar] [CrossRef]
- Lacy, S.A.; Miles, D.R.; Nguyen, L.T. Clinical Pharmacokinetics and Pharmacodynamics of Cabozantinib. Clin. Pharmacokinet. 2017, 56, 477–491. [Google Scholar] [CrossRef]
- Markowitz, J.N.; Fancher, K.M. Cabozantinib: A Multitargeted Oral Tyrosine Kinase Inhibitor. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2018, 38, 357–369. [Google Scholar] [CrossRef]
- Schmidinger, M.; Danesi, R. Management of Adverse Events Associated with Cabozantinib Therapy in Renal Cell Carcinoma. Oncologist 2018, 23, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Lien, V.T.; Pettersen, S.; Haugen, M.H.; Olberg, D.E.; Mælandsmo, G.M.; Klaveness, J. Design, Synthesis and Biological Evaluation of 6-substituted Quinolines Derived from Cabozantinib as c-Met Inhibitors. Arch. Pharm. 2019, 352, 1900101. [Google Scholar] [CrossRef] [PubMed]
- Steinebach, C.; Kehm, H.; Lindner, S.; Vu, L.P.; Köpff, S.; López Mármol, Á.; Weiler, C.; Wagner, K.G.; Reichenzeller, M.; Krönke, J.; et al. PROTAC-Mediated Crosstalk between E3 Ligases. Chem. Commun. 2019, 55, 1821–1824. [Google Scholar] [CrossRef]
- Zheng, G.; Zhou, D.; Zhang, X.; Wang, Y.; Chang, J. Compounds That Induce Degradation of Anti-Apoptotic bcl-2 Family Proteins and the Uses Thereof. Patent WO2017184995, 26 October 2017. [Google Scholar]
- Iselt, M.; Holtei, W.; Hilgard, P. The Tetrazolium Dye Assay for Rapid in Vitro Assessment of Cytotoxicity. Arzneimittelforschung 1989, 39, 747–749. [Google Scholar]
- Volkova, Y.A.; Antonov, Y.S.; Komkov, A.V.; Scherbakov, A.M.; Shashkov, A.S.; Menchikov, L.G.; Chernoburova, E.I.; Zavarzin, I.V. Access to Steroidal Pyridazines via Modified Thiohydrazides. RSC Adv. 2016, 6, 42863–42868. [Google Scholar] [CrossRef]
- Scherbakov, A.M.; Lobanova, Y.S.; Shatskaya, V.A.; Onopchenko, O.V.; Gershtein, E.S.; Krasil’nikov, M.A. Activation of Mitogenic Pathways and Sensitization to Estrogen-Induced Apoptosis: Two Independent Characteristics of Tamoxifen-Resistant Breast Cancer Cells? Breast Cancer Res. Treat. 2006, 100, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Mruk, D.D.; Cheng, C.Y. Enhanced Chemiluminescence (ECL) for Routine Immunoblotting. Spermatogenesis 2011, 1, 121–122. [Google Scholar] [CrossRef]
- Chen, T.; Zhuo, L.-S.; Liu, P.-F.; Fang, W.-R.; Li, Y.-M.; Huang, W. Discovery of 1,6-Naphthyridinone-Based MET Kinase Inhibitor Bearing Quinoline Moiety as Promising Antitumor Drug Candidate. Eur. J. Med. Chem. 2020, 192, 112174. [Google Scholar] [CrossRef]
- Forsyth, T.P.; Mac, M.B.; Leahy, J.W.; Nuss, J.M.; Xu, W. c-Met Modulators and Methods of Use. U.S. Patent 2007054928-A1, 26 September 2003. [Google Scholar]
- Gore, V.G.; Shukla, V.K.; Bhandari, S.S.; Hasbe, S. Process for the Preparation of Lenalidomide. Patent US8946265B2, 3 February 2015. [Google Scholar]
- Sawada, K.; Radjabi, A.R.; Shinomiya, N.; Kistner, E.; Kenny, H.; Becker, A.R.; Turkyilmaz, M.A.; Salgia, R.; Yamada, S.D.; Vande Woude, G.F.; et al. c-Met Overexpression Is a Prognostic Factor in Ovarian Cancer and an Effective Target for Inhibition of Peritoneal Dissemination and Invasion. Cancer Res. 2007, 67, 1670–1679. [Google Scholar] [CrossRef]
- Tang, Z.; Du, R.; Jiang, S.; Wu, C.; Barkauskas, D.S.; Richey, J.; Molter, J.; Lam, M.; Flask, C.; Gerson, S.; et al. Dual MET–EGFR Combinatorial Inhibition against T790M-EGFR-Mediated Erlotinib-Resistant Lung Cancer. Br. J. Cancer 2008, 99, 911–922. [Google Scholar] [CrossRef]
- Mueller, K.L.; Yang, Z.-Q.; Haddad, R.; Ethier, S.P.; Boerner, J.L. EGFR/Met Association Regulates EGFR TKI Resistance in Breast Cancer. J. Mol. Signal. 2010, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread Potential for Growth-Factor-Driven Resistance to Anticancer Kinase Inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.-J.; Wu, Y.; Hou, W.-H.; Wang, Y.-X.; Yuan, Q.-Y.; Wang, H.-J.; Yu, M. A Novel Bispecific c-Met/PD-1 Antibody with Therapeutic Potential in Solid Cancer. Oncotarget 2017, 8, 29067–29079. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, Q.; Zhang, C.; Huang, Z.; Wang, T.; Wang, X.; Wang, X.; Xu, G.; Liu, Y.; Yang, S.; et al. Discovery of a Highly Potent, Selective and Novel CDK9 Inhibitor as an Anticancer Drug Candidate. Bioorg. Med. Chem. Lett. 2017, 27, 3231–3237. [Google Scholar] [CrossRef]
- Taylor, S.C.; Berkelman, T.; Yadav, G.; Hammond, M. A Defined Methodology for Reliable Quantification of Western Blot Data. Mol. Biotechnol. 2013, 55, 217–226. [Google Scholar] [CrossRef]
- Yang, L.; Li, Y.; Bhattacharya, A.; Zhang, Y. A Recombinant Human Protein Targeting HER2 Overcomes Drug Resistance in HER2-Positive Breast Cancer. Sci. Transl. Med. 2019, 11, eaav1620. [Google Scholar] [CrossRef]
- Organ, S.L.; Tsao, M.-S. An Overview of the c-Met Signaling Pathway. Ther. Adv. Med. Oncol. 2011, 3, S7–S19. [Google Scholar] [CrossRef]
- Yao, H.-P.; Tong, X.-M.; Wang, M.-H. Oncogenic Mechanism-Based Pharmaceutical Validation of Therapeutics Targeting MET Receptor Tyrosine Kinase. Ther. Adv. Med. Oncol. 2021, 13, 175883592110069. [Google Scholar] [CrossRef]
- Peschard, P.; Fournier, T.M.; Lamorte, L.; Naujokas, M.A.; Band, H.; Langdon, W.Y.; Park, M. Mutation of the C-Cbl TKB Domain Binding Site on the Met Receptor Tyrosine Kinase Converts It into a Transforming Protein. Mol. Cell 2001, 8, 995–1004. [Google Scholar] [CrossRef]
- Petrelli, A.; Gilestro, G.F.; Lanzardo, S.; Comoglio, P.M.; Migone, N.; Giordano, S. The Endophilin–CIN85–Cbl Complex Mediates Ligand-Dependent Downregulation of c-Met. Nature 2002, 416, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Bannen, L.C.; Chan, D.S.-M.; Forsyth, T.P.; Khoury, R.G.; Leahy, J.W.; Mac, M.B.; Mann, L.W.; Nuss, J.M.; Parks, J.J.; Wang, Y.; et al. c-Met Modulators and Methods of Use. Patent US7579473B2, 25 August 2009. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).