
Combined Therapeutics for Atherosclerosis Treatment Using Polymeric Nanovectors

 , , ,
, , ,  , , , , , and
, , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of ATOR-Loaded PLGA/miRNA NPs
2.3. NPs Characterization
2.4. Cargo Entrapment Efficiency and Loading Capacity
2.5. In Vitro Release Kinetics
2.6. Cell Culture
2.7. In Vitro Cytotoxicity
2.8. Cellular Uptake
2.9. Cellular Morphological Changes
2.10. Expression of Pro-Inflammatory Cytokines
2.11. Quantification of Reactive Oxygen Species (ROS) Production
2.12. Oxidized, Low-Density Lipoproteins (Ox-LdL) Internalization
2.13. Evaluation of VCAM and ICAM-1 Expression
2.14. Statistical Analysis
3. Results and Discussion
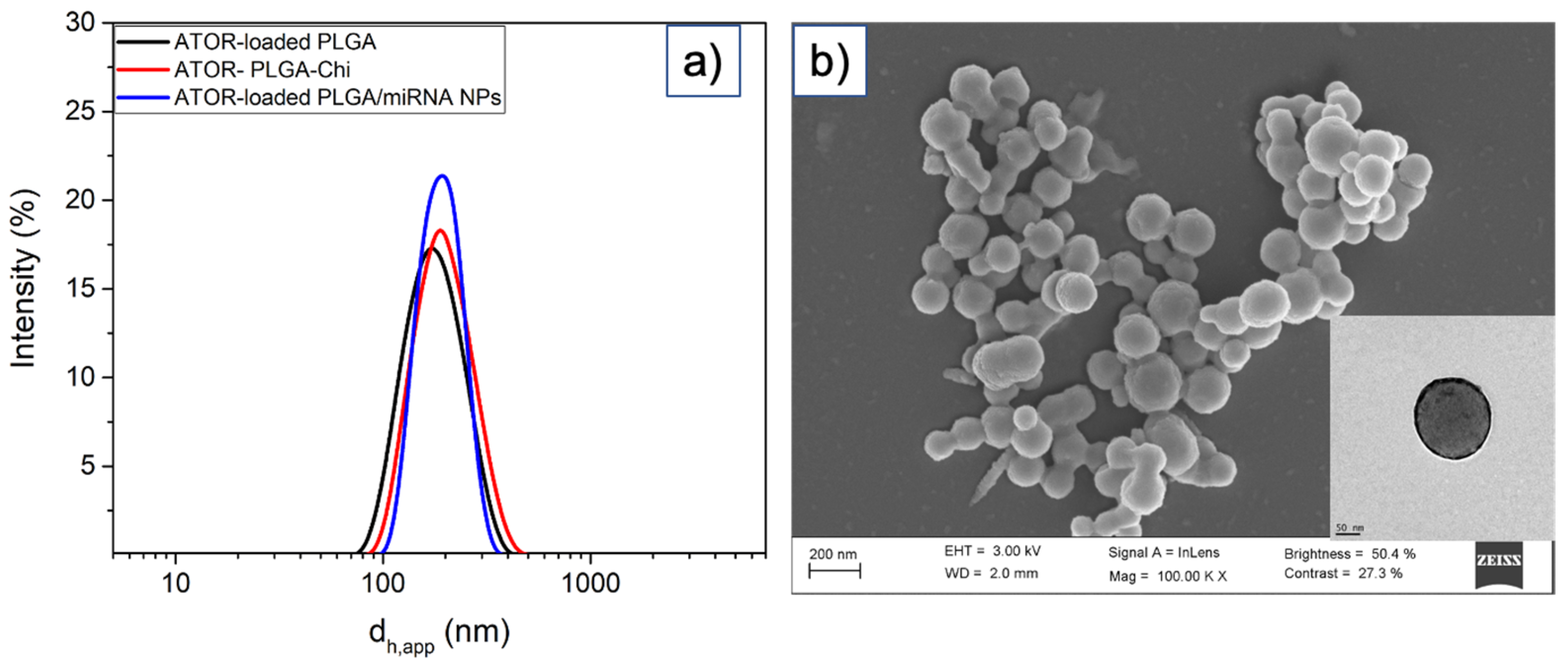
3.1. Synthesis and Characterization of ATOR-Loaded PLGA/miRNA NPs
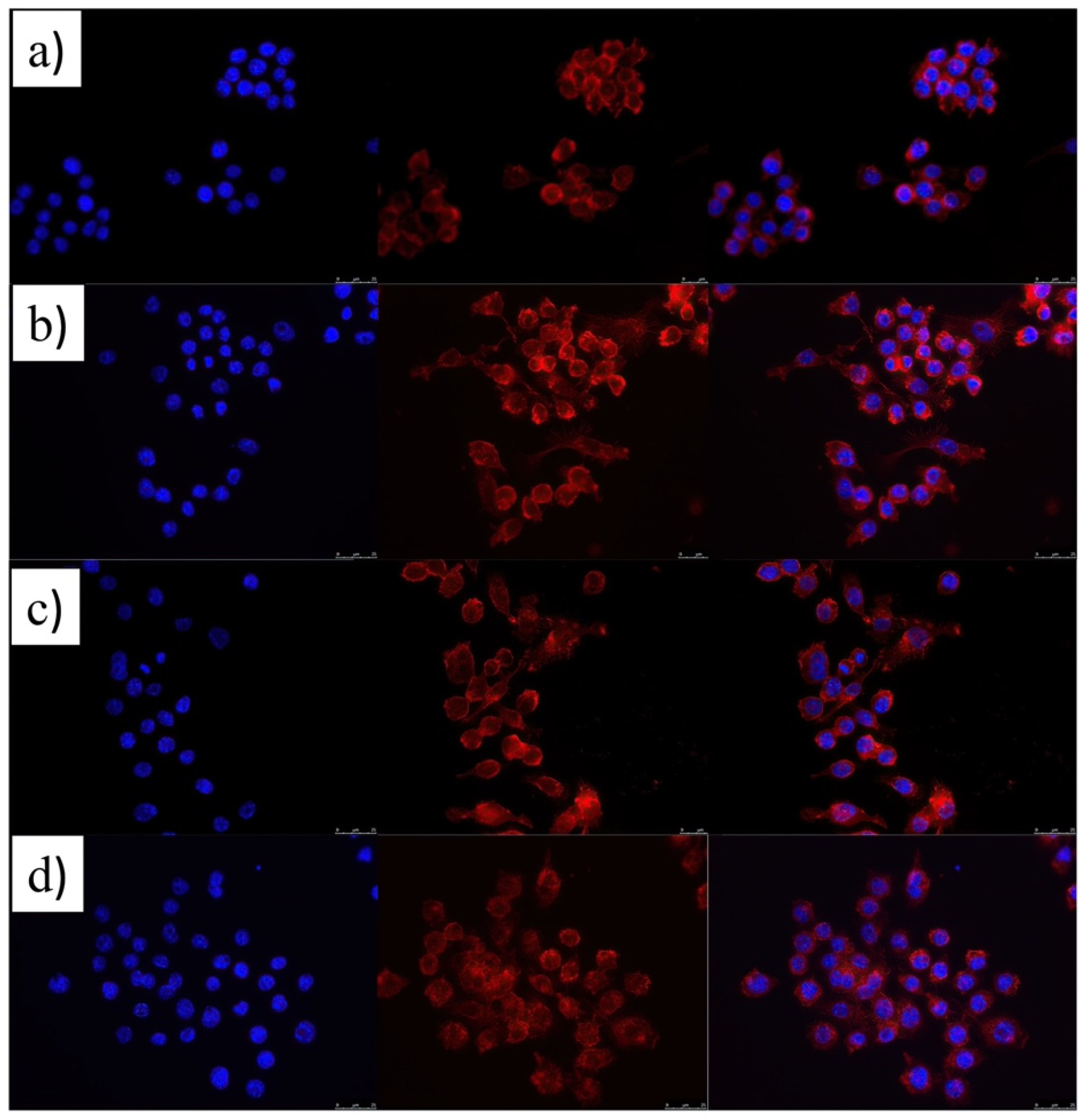
3.2. Cytocompatibility and Cell Uptake ATOR-Loaded PLGA/miRNA NPs
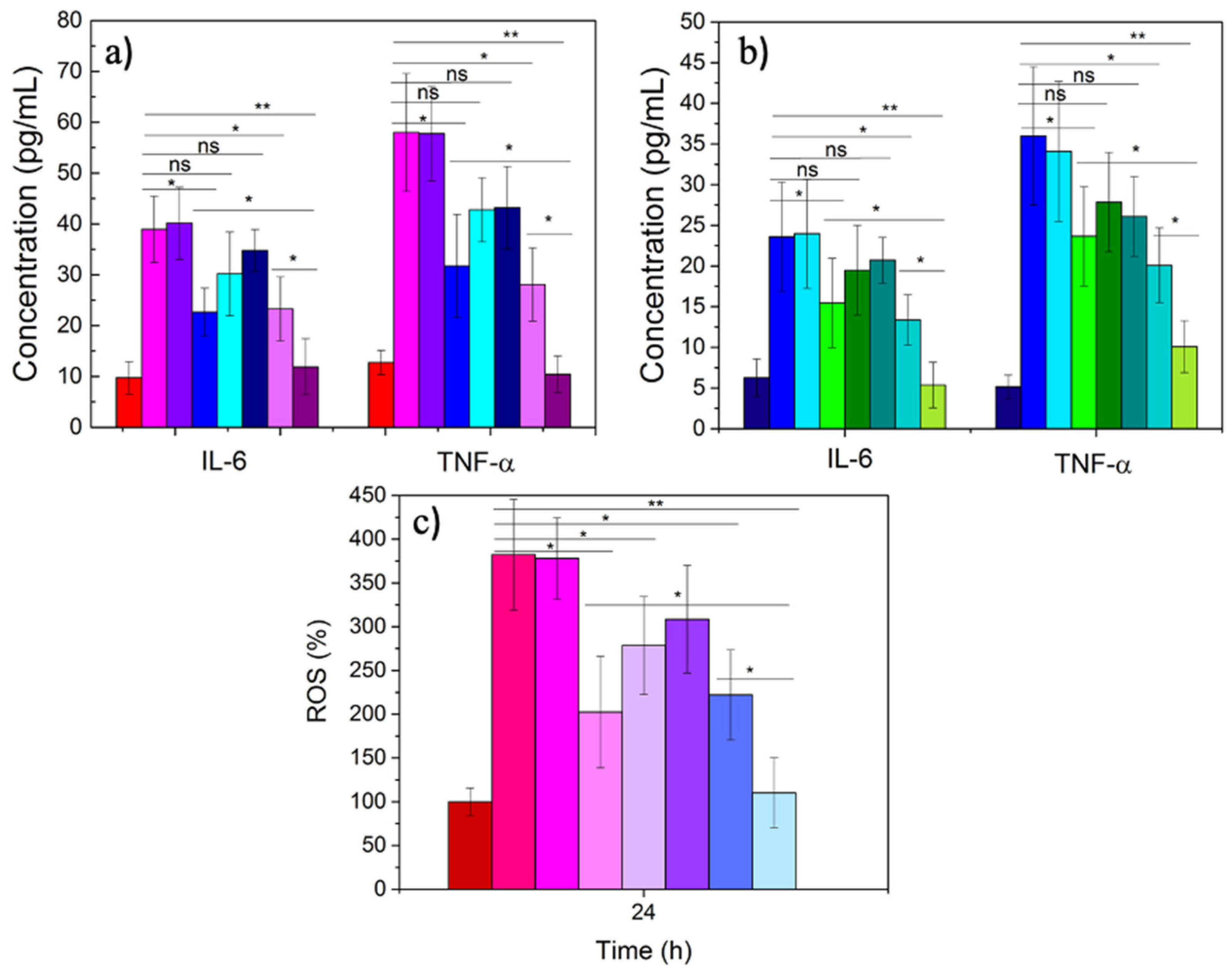
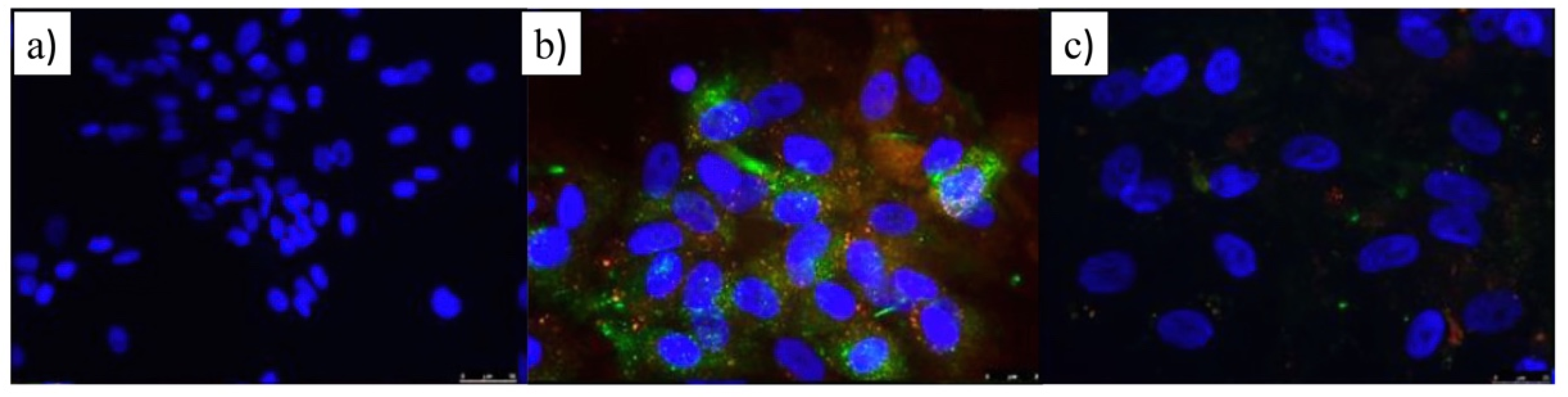
3.3. Therapeutic Activity of ATOR-Loaded PLGA/miRNA NPs
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Ferranti, S.d.; Després, J.-P.; Fullerton, H.J.; Howard, V.J.; et al. Heart Disease and Stroke Statistics—2015 Update. Circulation 2015, 131, e29–e322. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics—2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation, Atherosclerosis, and Coronary Artery Disease. N. J. Engl. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [Green Version]
- Chiu, J.-J.; Chien, S. Effects of Disturbed Flow on Vascular Endothelium: Pathophysiological Basis and Clinical Perspectives. Physiol. Rev. 2011, 91, 327–387. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.B.; Chien, S.; Barakat, A.I.; Nerem, R.M. Endothelial cellular response to altered shear stress. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L529–L533. [Google Scholar] [CrossRef]
- Li, Y.S.; Haga, J.H.; Chien, S. Molecular basis of the effects of shear stress on vascular endothelial cells. J. Biomech. 2005, 38, 1949–1971. [Google Scholar] [CrossRef]
- Chen, K.D.; Li, Y.S.; Kim, M.; Li, S.; Yuan, S.; Chien, S.; Shyy, J.Y.J. Mechanotransduction in response to shear stress. Roles of receptor tyrosine kinases, integrins, and Shc. J. Biol. Chem. 1999, 274, 18393–18400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.-G.; Ueba, H.; Tanimoto, T.; Lungu, A.O.; Frame, M.D.; Berk, B.C. Ligand-Independent Activation of Vascular Endothelial Growth Factor Receptor 2 by Fluid Shear Stress Regulates Activation of Endothelial Nitric Oxide Synthase. Circ. Res. 2003, 93, 354–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baber, U.; Mehran, R.; Sartori, S.; Schoos, M.M.; Sillesen, H.; Muntendam, P.; Garcia, M.J.; Gregson, J.; Pocock, S.; Falk, E.; et al. Prevalence, impact, and predictive value of detecting subclinical coronary and carotid atherosclerosis in asymptomatic adults: The BioImage study. J. Am. Coll. Cardiol. 2015, 65, 1065–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waksman, R.; Di Mario, C.; Torguson, R.; Ali, Z.A.; Singh, V.; Skinner, W.H.; Artis, A.K.; Cate, T.T.; Powers, E.; Kim, C.; et al. Identification of patients and plaques vulnerable to future coronary events with near-infrared spectroscopy intravascular ultrasound imaging: A prospective, cohort study. Lancet 2019, 394, 1629–1637. [Google Scholar] [CrossRef]
- Piccirillo, F.; Carpenito, M.; Verolino, G.; Chello, C.; Nusca, A.; Lusini, M.; Spadaccio, C.; Nappi, F.; Di Sciascio, G.; Nenna, A. Changes of the coronary arteries and cardiac microvasculature with aging: Implications for translational research and clinical practice. Mech. Ageing Dev. 2019, 184, 111161. [Google Scholar] [CrossRef]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic Effects of Statins on the Cardiovascular System. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef] [Green Version]
- Ou, L.-C.; Zhong, S.; Ou, J.-S.; Tian, J.-W. Application of targeted therapy strategies with nanomedicine delivery for atherosclerosis. Acta Pharmacol. Sin. 2021, 42, 10–17. [Google Scholar] [CrossRef]
- Zhang, J.; Zu, Y.; Dhanasekara, C.S.; Li, J.; Wu, D.; Fan, Z.; Wang, S. Detection and treatment of atherosclerosis using nanoparticles. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1412. [Google Scholar] [CrossRef] [Green Version]
- Flores, A.M.; Ye, J.; Jarr, K.U.; Hosseini-Nassab, N.; Smith, B.R.; Leeper, N.J. Nanoparticle Therapy for Vascular Diseases. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 635–646. [Google Scholar] [CrossRef]
- Koga, J.; Matoba, T.; Egashira, K. Anti-inflammatory Nanoparticle for Prevention of Atherosclerotic Vascular Diseases. J. Atheroscler. Thromb. 2016, 23, 757–765. [Google Scholar] [CrossRef] [Green Version]
- Katsuki, S.; Matoba, T.; Koga, J.-I.; Nakano, K.; Egashira, K. Anti-inflammatory Nanomedicine for Cardiovascular Disease. Front. Cardiovasc. Med. 2017, 4, 87. [Google Scholar] [CrossRef] [Green Version]
- Nakhlband, A.; Eskandani, M.; Omidi, Y.; Saeedi, N.; Ghaffari, S.; Barar, J.; Garjani, A. Combating atherosclerosis with targeted nanomedicines: Recent advances and future prospective. BioImpacts BI 2018, 8, 59–75. [Google Scholar] [CrossRef]
- DiStasio, N.; Lehoux, S.; Khademhosseini, A.; Tabrizian, M. The Multifaceted Uses and Therapeutic Advantages of Nanoparticles for Atherosclerosis Research. Materials 2018, 11, 754. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Qin, B.; Xia, G.; Choi, S.H. FDA’s Poly (Lactic-Co-Glycolic Acid) Research Program and Regulatory Outcomes. AAPS J. 2021, 23, 92. [Google Scholar] [CrossRef]
- Park, K.; Skidmore, S.; Hadar, J.; Garner, J.; Park, H.; Otte, A.; Soh, B.K.; Yoon, G.; Yu, D.; Yun, Y.; et al. Injectable, long-acting PLGA formulations: Analyzing PLGA and understanding microparticle formation. J. Control. Release 2019, 304, 125–134. [Google Scholar] [CrossRef]
- Nenna, A.; Nappi, F.; Larobina, D.; Verghi, E.; Chello, M.; Ambrosio, L. Polymers and Nanoparticles for Statin Delivery: Current Use and Future Perspectives in Cardiovascular Disease. Polymers 2021, 13, 711. [Google Scholar] [CrossRef]
- Katsuki, S.; Matoba, T.; Nakashiro, S.; Sato, K.; Koga, J.-I.; Nakano, K.; Nakano, Y.; Egusa, S.; Sunagawa, K.; Egashira, K. Nanoparticle-Mediated Delivery of Pitavastatin Inhibits Atherosclerotic Plaque Destabilization/Rupture in Mice by Regulating the Recruitment of Inflammatory Monocytes. Circulation 2014, 129, 896–906. [Google Scholar] [CrossRef]
- Nagaoka, K.; Matoba, T.; Mao, Y.; Nakano, Y.; Ikeda, G.; Egusa, S.; Tokutome, M.; Nagahama, R.; Nakano, K.; Sunagawa, K.; et al. A New Therapeutic Modality for Acute Myocardial Infarction: Nanoparticle-Mediated Delivery of Pitavastatin Induces Cardioprotection from Ischemia-Reperfusion Injury via Activation of PI3K/Akt Pathway and Anti-Inflammation in a Rat Model. PLoS ONE 2015, 10, e0132451. [Google Scholar] [CrossRef]
- Mao, Y.; Koga, J.I.; Tokutome, M.; Matoba, T.; Ikeda, G.; Nakano, K.; Egashira, K. Nanoparticle-Mediated Delivery of Pitavastatin to Monocytes/Macrophages Inhibits Left Ventricular Remodeling After Acute Myocardial Infarction by Inhibiting Monocyte-Mediated Inflammation. Int. Heart J. 2017, 58, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Huang, H.; Huang, L.; Du, L.; Sun, Y.; Duan, Y. Prevention of Oxidized Low Density Lipoprotein-Induced Endothelial Cell Injury by DA-PLGA-PEG-cRGD Nanoparticles Combined with Ultrasound. Int. J. Mol. Sci. 2017, 18, 815. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, R.; Ii, M.; Masuda, M.; Tabata, Y.; Hoshiga, M.; Ishizaka, N.; Asahi, M. Cardiac Regeneration by Statin-Polymer Nanoparticle-Loaded Adipose-Derived Stem Cell Therapy in Myocardial Infarction. Stem Cells Transl. Med. 2019, 8, 1055–1067. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Rana, T.M. Therapeutic targeting of microRNAs: Current status and future challenges. Nat. Rev. Drug Discov. 2014, 13, 622–638. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Calin, G.A.; Lopez-Berestein, G.; Sood, A.K. miRNA Deregulation in Cancer Cells and the Tumor Microenvironment. Cancer Discov. 2016, 6, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zhang, J.; Wang, Y.; Chen, M. Hyaluronic acid-coated PEI-PLGA nanoparticles mediated co-delivery of doxorubicin and miR-542-3p for triple negative breast cancer therapy. Nanomedicine 2016, 12, 411–420. [Google Scholar] [CrossRef]
- Xie, Y.; Murray-Stewart, T.; Wang, Y.; Yu, F.; Li, J.; Marton, L.J.; Casero, R.A., Jr.; Oupický, D. Self-immolative nanoparticles for simultaneous delivery of microRNA and targeting of polyamine metabolism in combination cancer therapy. J. Control. Release 2017, 246, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Sun, Y.; Shen, M.; Song, K.; Yin, X.; Di, W.; Duan, Y. Enhanced Chemotherapeutic Efficacy of Paclitaxel Nanoparticles Co-delivered with MicroRNA-7 by Inhibiting Paclitaxel-Induced EGFR/ERK pathway Activation for Ovarian Cancer Therapy. ACS Appl. Mater. Interfaces 2018, 10, 7821–7831. [Google Scholar] [CrossRef]
- Weitz-Schmidt, G.; Welzenbach, K.; Brinkmann, V.; Kamata, T.; Kallen, J.; Bruns, C.; Cottens, S.; Takada, Y.; Hommel, U. Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nat. Med. 2001, 7, 687–692. [Google Scholar] [CrossRef]
- Saraiva, C.; Talhada, D.; Rai, A.; Ferreira, R.; Ferreira, L.; Bernardino, L.; Ruscher, K. MicroRNA-124-loaded nanoparticles increase survival and neuronal differentiation of neural stem cells in vitro but do not contribute to stroke outcome in vivo. PLoS ONE 2018, 13, e0193609. [Google Scholar] [CrossRef]
- Sun, Y.; Li, Q.; Gui, H.; Xu, D.-P.; Yang, Y.-L.; Su, D.-F.; Liu, X. MicroRNA-124 mediates the cholinergic anti-inflammatory action through inhibiting the production of pro-inflammatory cytokines. Cell Res. 2013, 23, 1270–1283. [Google Scholar] [CrossRef] [Green Version]
- Polo, E.; Puertas, S.; Moros, M.; Batalla, P.; Guisán, J.M.; de la Fuente, J.M.; Grazú, V. Tips for the Functionalization of Nanoparticles with Antibodies. In Immobilization of Enzymes and Cells, 3rd ed.; Guisan, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 149–163. [Google Scholar] [CrossRef]
- Valdepérez, D.; Del Pino, P.; Sánchez, L.; Parak, W.J.; Pelaz, B. Highly active antibody-modified magnetic polyelectrolyte capsules. J. Colloid Interface Sci. 2016, 474, 1–8. [Google Scholar] [CrossRef]
- Sorensen, P.S.; Lycke, J.; Erälinna, J.P.; Edland, A.; Wu, X.; Frederiksen, J.L.; Oturai, A.; Malmeström, C.; Stenager, E.; Sellebjerg, F.; et al. Simvastatin as add-on therapy to interferon β-1a for relapsing-remitting multiple sclerosis (SIMCOMBIN study): A placebo-controlled randomised phase 4 trial. Lancet Neurol. 2011, 10, 691–701. [Google Scholar] [CrossRef]
- Palaga, T.; Buranaruk, C.; Rengpipat, S.; Fauq, A.H.; Golde, T.E.; Kaufmann, S.H.; Osborne, B.A. Notch signaling is activated by TLR stimulation and regulates macrophage functions. Eur. J. Immunol. 2008, 38, 174–183. [Google Scholar] [CrossRef]
- Shimizu, K.; Aikawa, M.; Takayama, K.; Libby, P.; Mitchell, R.N. Direct anti-inflammatory mechanisms contribute to attenuation of experimental allograft arteriosclerosis by statins. Circulation 2003, 108, 2113–2120. [Google Scholar] [CrossRef] [Green Version]
- Topete, A.; Melgar, D.; Alatorre-Meda, M.; Iglesias, P.; Argibay, B.; Vidawati, S.; Barbosa, S.; Costoya, J.A.; Taboada, P.; Mosquera, V. NIR-light active hybrid nanoparticles for combined imaging and bimodal therapy of cancerous cells. J. Mater. Chem. B 2014, 2, 6967–6977. [Google Scholar] [CrossRef]
- Zhao, Y.; Gao, H.; He, J.; Jiang, C.; Lu, J.; Zang, W.; Yang, H.; Liu, J. Co-delivery of LOX-1 siRNA and statin to endothelial cells and macrophages in the atherosclerotic lesions by a dual-targeting core-shell nanoplatform: A dual cell therapy to regress plaques. J. Control. Release 2018, 283, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Tao, W.; Zhang, D.; Wu, C.; Song, B.; Wang, S.; Wang, T.; Hu, M.; Liu, X.; Wang, Y.; et al. The studies of PLGA nanoparticles loading atorvastatin calcium for oral administration in vitro and in vivo. Asian J. Pharm. Sci. 2017, 12, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Aroa, S.; Swaminathan, S.K.; Kirtane, A.; Srivastava, S.K.; Bhardwaj, A.; Singh, S.; Panyam, J.; Singh, A.P. Synthesis, characterization, and evaluation of poly (D, L-lactide-co-glycolide)-based nanoformulation of miRNA-150: Potential implications for pancreatic cancer therapy. Int. J. Nanomed. 2014, 9, 2933–2942. [Google Scholar] [CrossRef]
- Sun, Y.; Zheng, Y.; Ran, H.; Zhou, Y.; Shen, H.; Chen, Y.; Chen, H.; Krupka, T.M.; Li, A.; Li, P.; et al. Superparamagnetic PLGA-iron oxide microcapsules for dual-modality US/MR imaging and high intensity focused US breast cancer ablation. Biomaterials 2012, 33, 5854–5864. [Google Scholar] [CrossRef]
- Forrest, M.L.; Won, C.Y.; Malick, A.W.; Kwon, G.S. In vitro release of the mTOR inhibitor rapamycin from poly(ethylene glycol)-b-poly(epsilon-caprolactone) micelles. J. Control. Release 2006, 110, 370–377. [Google Scholar] [CrossRef]
- Whitfield, C.J.; Zhang, M.; Winterwerber, P.; Wu, Y.; Ng, D.Y.W.; Weil, T. Functional DNA–Polymer Conjugates. Chem. Rev. 2021, 121, 11030–11084. [Google Scholar] [CrossRef]
- Santos-Carballal, B.; Aaldering, L.J.; Ritzefeld, M.; Pereira, S.; Sewald, N.; Moerschbacher, B.M.; Götte, M.; Goycoolea, F.M. Physicochemical and biological characterization of chitosan-microRNA nanocomplexes for gene delivery to MCF-7 breast cancer cells. Sci. Rep. 2015, 5, 13567. [Google Scholar] [CrossRef]
- Ubrich, N.; Bouillot, P.; Pellerin, C.; Hoffman, M.; Maincent, P. Preparation and characterization of propranolol hydrochloride nanoparticles: A comparative study. J. Control. Release 2004, 97, 291–300. [Google Scholar] [CrossRef]
- Lee, Y.; Park, S.Y.; Mok, H.; Park, T.G. Synthesis, Characterization, Antitumor Activity of Pluronic Mimicking Copolymer Micelles Conjugated with Doxorubicin via Acid-Cleavable Linkage. Bioconjugate Chem. 2008, 19, 525–531. [Google Scholar] [CrossRef]
- Gaschignard, J.; Levy, C.; Chrabieh, M.; Boisson, B.; Bost-Bru, C.; Dauger, S.; Dubos, F.; Durand, P.; Gaudelus, J.; Gendrel, D.; et al. Invasive pneumococcal disease in children can reveal a primary immunodeficiency. Clin. Infect. Dis. 2014, 59, 244–251. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Luo, T.; Mei, Y.; Liu, H.; Dong, J.; Fang, Y.; Peng, J.; Guo, Y. Simvastatin alters M1/M2 polarization of murine BV2 microglia via Notch signaling. J. Neuroimmunol. 2018, 316, 56–64. [Google Scholar] [CrossRef]
- Duivenvoorden, R.; Tang, J.; Cormode, D.P.; Mieszawska, A.J.; Izquierdo-Garcia, D.; Ozcan, C.; Otten, M.J.; Zaidi, N.; Lobatto, M.E.; van Rijs, S.M.; et al. A statin-loaded reconstituted high-density lipoprotein nanoparticle inhibits atherosclerotic plaque inflammation. Nat. Commun. 2014, 5, 3065. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Wei, W.; Zhang, J.; Nie, W.; Yuan, L.; Huang, Y.; Zuo, L.; Huang, L.; Xi, X.; Xie, H.Y. A self-driven bioinspired nanovehicle by leukocyte membrane-hitchhiking for early detection and treatment of atherosclerosis. Biomaterials 2020, 250, 119963. [Google Scholar] [CrossRef]
- Gao, C.; Huang, Q.; Liu, C.; Kwong, C.H.T.; Yue, L.; Wan, J.-B.; Lee, S.M.Y.; Wang, R. Treatment of atherosclerosis by macrophage-biomimetic nanoparticles via targeted pharmacotherapy and sequestration of proinflammatory cytokines. Nat. Commun. 2020, 11, 2622. [Google Scholar] [CrossRef]
- Hossain, E.; Ota, A.; Karnan, S.; Takahashi, M.; Mannan, S.B.; Konishi, H.; Hosokawa, Y. Lipopolysaccharide augments the uptake of oxidized LDL by up-regulating lectin-like oxidized LDL receptor-1 in macrophages. Mol. Cell. Biochem. 2015, 400, 29–40. [Google Scholar] [CrossRef]
- La Heij, E.; Kuijpers, R.W.; Baarsma, S.G.; Kijlstra, A.; van der Weiden, M.; Mooy, C.M. Adhesion molecules in iris biopsy specimens from patients with uveitis. Br. J. Ophthalmol. 1998, 82, 432–437. [Google Scholar] [CrossRef]
- Kjaergaard, A.G.; Dige, A.; Krog, J.; Tønnesen, E.; Wogensen, L. Soluble adhesion molecules correlate with surface expression in an in vitro model of endothelial activation. Basic Clin. Pharmacol. Toxicol. 2013, 113, 273–279. [Google Scholar] [CrossRef]
- Brooks, A.R.; Lelkes, P.I.; Rubanyi, G.M. Gene expression profiling of human aortic endothelial cells exposed to disturbed flow and steady laminar flow. Physiol. Genom. 2002, 9, 27–41. [Google Scholar] [CrossRef] [Green Version]
- Cybulsky, M.I.; Gimbrone, M.A., Jr. Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science 1991, 251, 788–791. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leal, B.H.; Velasco, B.; Cambón, A.; Pardo, A.; Fernandez-Vega, J.; Arellano, L.; Al-Modlej, A.; Mosquera, V.X.; Bouzas, A.; Prieto, G.; et al. Combined Therapeutics for Atherosclerosis Treatment Using Polymeric Nanovectors. Pharmaceutics 2022, 14, 258. https://doi.org/10.3390/pharmaceutics14020258
Leal BH, Velasco B, Cambón A, Pardo A, Fernandez-Vega J, Arellano L, Al-Modlej A, Mosquera VX, Bouzas A, Prieto G, et al. Combined Therapeutics for Atherosclerosis Treatment Using Polymeric Nanovectors. Pharmaceutics. 2022; 14(2):258. https://doi.org/10.3390/pharmaceutics14020258
Chicago/Turabian StyleLeal, Baltazar Hiram, Brenda Velasco, Adriana Cambón, Alberto Pardo, Javier Fernandez-Vega, Lilia Arellano, Abeer Al-Modlej, Víctor X. Mosquera, Alberto Bouzas, Gerardo Prieto, and et al. 2022. "Combined Therapeutics for Atherosclerosis Treatment Using Polymeric Nanovectors" Pharmaceutics 14, no. 2: 258. https://doi.org/10.3390/pharmaceutics14020258
APA StyleLeal, B. H., Velasco, B., Cambón, A., Pardo, A., Fernandez-Vega, J., Arellano, L., Al-Modlej, A., Mosquera, V. X., Bouzas, A., Prieto, G., Barbosa, S., & Taboada, P. (2022). Combined Therapeutics for Atherosclerosis Treatment Using Polymeric Nanovectors. Pharmaceutics, 14(2), 258. https://doi.org/10.3390/pharmaceutics14020258