
Metabolic Impact of Anticancer Drugs Pd2Spermine and Cisplatin on the Brain of Healthy Mice

 ,
,  , ,
, ,  , and
, and 
Abstract
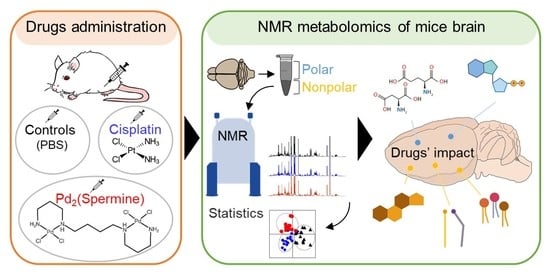
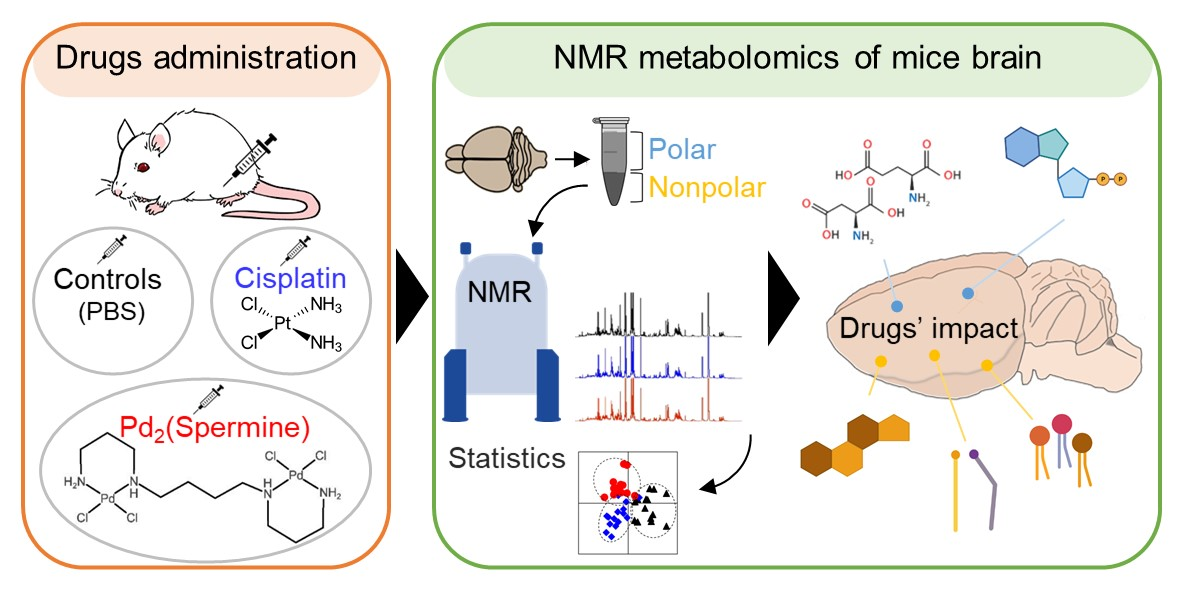
:
1. Introduction
2. Materials and Methods
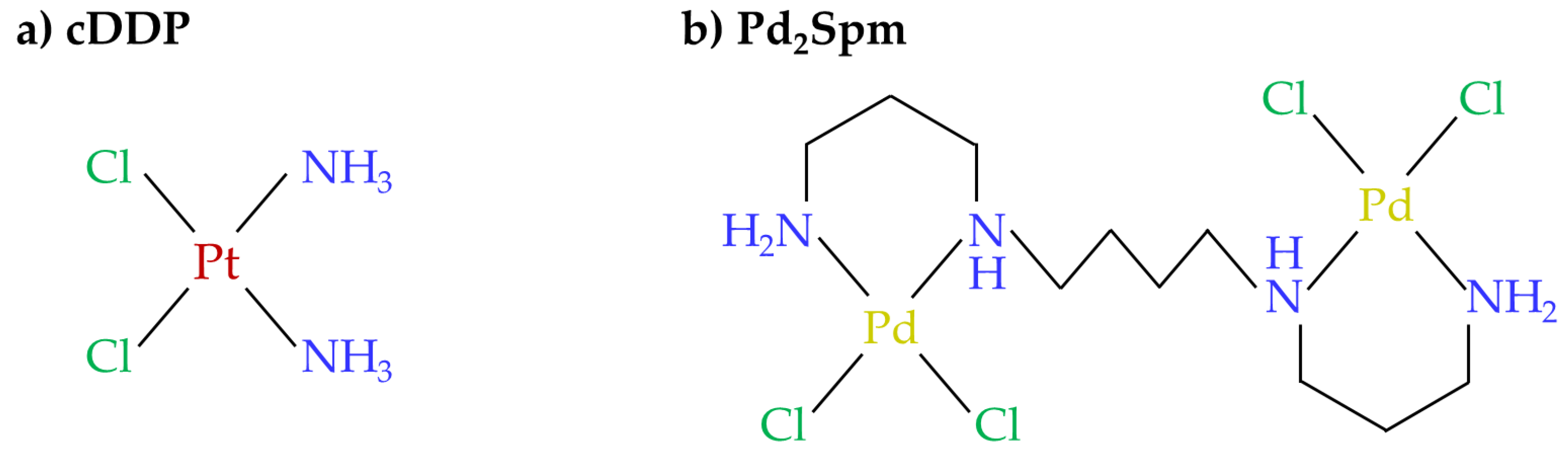
2.1. Chemicals and Solutions
2.2. Ethical Considerations
2.3. Animals Procedures
2.4. Preparation of Brain Extracts
2.5. NMR Spectroscopy
2.6. Data Processing and Statistical Analysis
3. Results
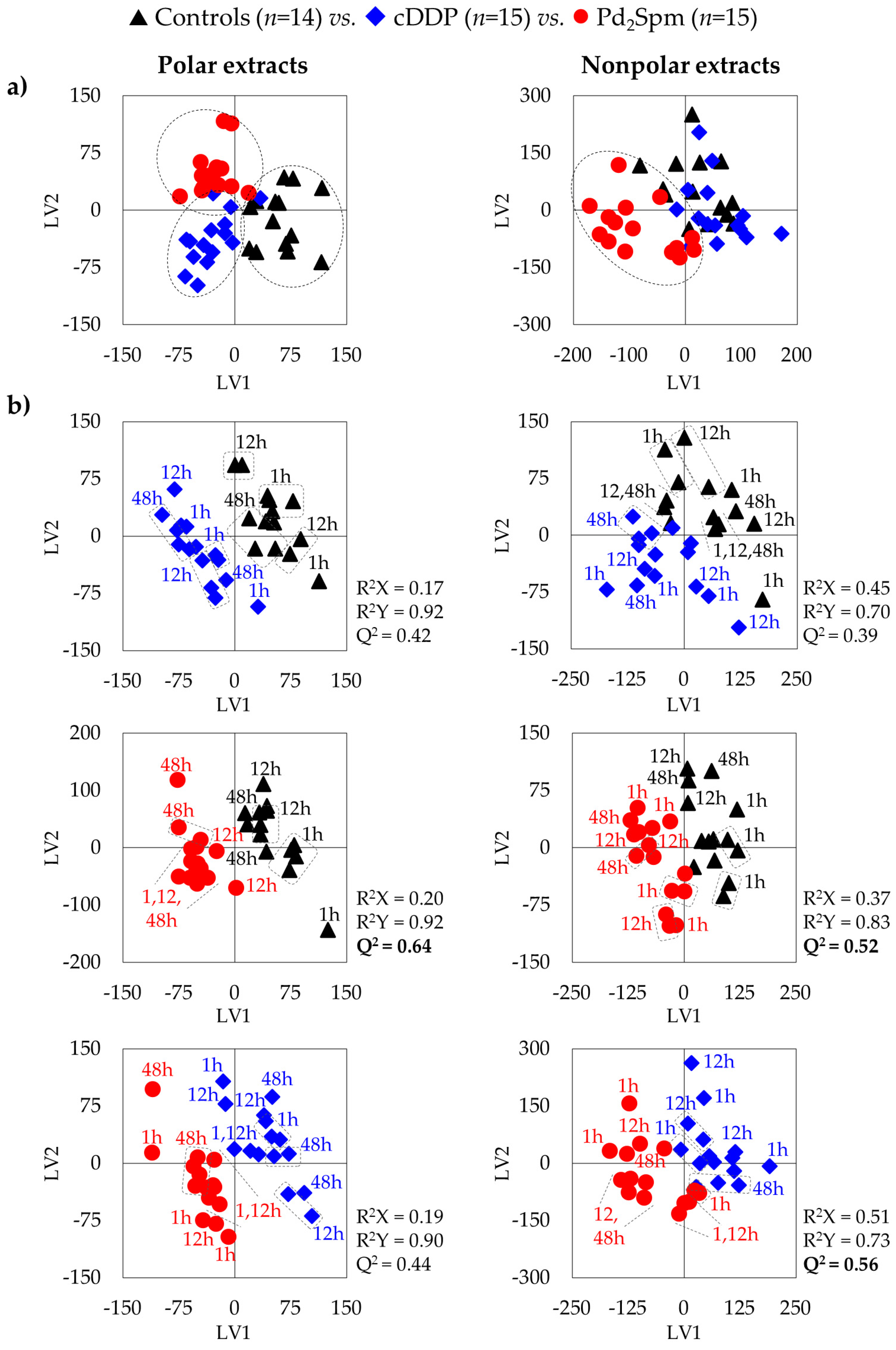
3.1. Impact of Pd2Spm on Mice Brain, Compared to cDDP: Polar Metabolome
3.2. Impact of Pd2Spm on Mice Brain, Compared to cDDP: Nonpolar Metabolome
4. Discussion
4.1. Amino Acids Metabolism
4.2. Nucleotides’ Metabolism
4.3. Choline Compounds and Lipid Metabolism
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosenberg, B.; Van Camp, L.; Krigas, T. Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, B.H.; Vancamp, L.; Trosko, J.E.; Mansour, V.H. Platinum compounds: A new class of potent antitumour agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Amptoulach, S.; Tsavaris, N. Neurotoxicity caused by the treatment with platinum analogues. Chemother. Res. Pract. 2011, 2011, 843019. [Google Scholar] [CrossRef] [PubMed]
- Aldossary, S.A. Review on Pharmacology of Cisplatin: Clinical Use, Toxicity and Mechanism of Resistance of Cisplatin. Biomed. Pharmacol. J. 2019, 12, 7–15. [Google Scholar] [CrossRef]
- Avan, A.; Postma, T.J.; Ceresa, C.; Avan, A.; Cavaletti, G.; Giovannetti, E.; Peters, G.J. Platinum-induced neurotoxicity and preventive strategies: Past, present, and future. Oncologist 2015, 20, 411–432. [Google Scholar] [CrossRef] [Green Version]
- Kanat, O.; Ertas, H.; Caner, B. Platinum-induced neurotoxicity: A review of possible mechanisms. World J. Clin. Oncol. 2017, 8, 329–335. [Google Scholar] [CrossRef]
- Staff, N.P.; Cavaletti, G.; Islam, B.; Lustberg, M.; Psimaras, D.; Tamburin, S. Platinum-induced peripheral neurotoxicity: From pathogenesis to treatment. J. Peripher. Nerv. Syst. 2019, 24, S26–S39. [Google Scholar] [CrossRef]
- Pellacani, C.; Eleftheriou, G. Neurotoxicity of antineoplastic drugs: Mechanisms, susceptibility, and neuroprotective strategies. Adv. Med. Sci. 2020, 65, 265–285. [Google Scholar] [CrossRef]
- Santos, N.A.G.D.; Ferreira, R.S.; Santos, A.C.D. Overview of cisplatin-induced neurotoxicity and ototoxicity, and the protective agents. Food Chem. Toxicol. 2020, 136, 111079. [Google Scholar] [CrossRef]
- Gregg, R.W.; Molepo, J.M.; Monpetit, V.J.; Mikael, N.Z.; Redmond, D.; Gadia, M.; Stewart, D.J. Cisplatin neurotoxicity: The relationship between dosage, time, and platinum concentration in neurologic tissues, and morphologic evidence of toxicity. J. Clin. Oncol. 1992, 10, 795–803. [Google Scholar] [CrossRef]
- Vojtek, M.; Gonçalves-Monteiro, S.; Pinto, E.; Kalivodová, S.; Almeida, A.; Marques, M.P.M.; Batista de Carvalho, A.L.M.; Martins, C.B.; Mota-Filipe, H.; Ferreira, I.M.P.L.V.O.; et al. Preclinical Pharmacokinetics and Biodistribution of Anticancer Dinuclear Palladium(II)-Spermine Complex (Pd2Spm) in Mice. Pharmaceuticals 2021, 14, 173. [Google Scholar] [CrossRef] [PubMed]
- Shabani, M.; Larizadeh, M.H.; Parsania, S.; Hajali, V.; Shojaei, A. Evaluation of destructive effects of exposure to cisplatin during developmental stage: No profound evidence for sex differences in impaired motor and memory performance. Int. J. Neurosci. 2012, 122, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Owoeye, O.; Adedara, I.A.; Farombi, E.O. Pretreatment with taurine prevented brain injury and exploratory behaviour associated with administration of anticancer drug cisplatin in rats. Biomed. Pharmacother. 2018, 102, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.; Reyes, T.M.; Heijnen, C.J.; Kavelaars, A. Cisplatin treatment induces attention deficits and impairs synaptic integrity in the prefrontal cortex in mice. Sci. Rep. 2018, 8, 17400. [Google Scholar] [CrossRef] [PubMed]
- Waseem, M.; Parvez, S. Mitochondrial dysfunction mediated cisplatin induced toxicity: Modulatory role of curcumin. Food Chem. Toxicol. 2013, 53, 334–342. [Google Scholar] [CrossRef]
- Zanotto-Filho, A.; Braganhol, E.; Edelweiss, M.I.; Behr, G.A.; Zanin, R.; Schröder, R.; Simões-Pires, A.; Battastini, A.M.; Moreira, J.C. The curry spice curcumin selectively inhibits cancer cells growth in vitro and in preclinical model of glioblastoma. J. Nutr. Biochem. 2012, 23, 591–601. [Google Scholar] [CrossRef]
- Cankara, F.N.; Günaydın, C.; Çelik, Z.B.; Şahin, Y.; Pekgöz, Ş.; Erzurumlu, Y.; Gülle, K. Agomelatine confers neuroprotection against cisplatin-induced hippocampal neurotoxicity. Metab. Brain Dis. 2021, 36, 339–349. [Google Scholar] [CrossRef]
- Zhou, W.; Kavelaars, A.; Heijnen, C.J. Metformin Prevents Cisplatin-Induced Cognitive Impairment and Brain Damage in Mice. PLoS ONE 2016, 11, e0151890. [Google Scholar] [CrossRef]
- Alam, M.N.; Huq, F. Comprehensive Review on Tumour Active Palladium Compounds and Structure-Activity Relationships. Coord. Chem. Rev. 2016, 316, 36–67. [Google Scholar] [CrossRef]
- Simpson, P.V.; Desai, N.M.; Casari, I.; Massi, M.; Falasca, M. Metal-based antitumor compounds: Beyond cisplatin. Future Med. Chem. 2019, 11, 119–135. [Google Scholar] [CrossRef]
- Fiuza, S.M.; Holy, J.; Batista de Carvalho, L.A.E.; Marques, M.P.M. Biologic activity of a dinuclear pd(II)-spermine complex toward human breast cancer. Chem. Biol. Drug Des. 2011, 77, 477–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batista de Carvalho, A.L.M.; Pilling, M.; Gardner, P.; Doherty, J.; Cinque, G.; Wehbe, K.; Kelley, C.; Batista de Carvalho, L.A.E.; Marques, M.P.M. Chemotherapeutic response to cisplatin-like drugs in human breast cancer cells probed by vibrational microspectroscopy. Faraday Discuss. 2016, 187, 273–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batista De Carvalho, A.L.M.; Medeiros, P.S.C.; Costa, F.M.; Ribeiro, V.P.; Sousa, J.B.; Diniz, C.; Marques, M.P.M. Anti-invasive and anti-proliferative synergism between docetaxel and a polynuclear pd-spermine agent. PLoS ONE 2016, 11, e0167218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tummala, R.; Diegelman, P.; Fiuza, S.M.; Batista de Carvalho, L.A.; Marques, M.P.; Kramer, D.L.; Clark, K.; Vujcic, S.; Porter, C.W.; Pendyala, L. Characterization of Pt-, Pd-spermine complexes for their effect on polyamine pathway and cisplatin re-sistance in A2780 ovarian carcinoma cells. Oncol. Rep. 2010, 24, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro-Ranninger, C.; Zamora, F.; Masaguer, J.; Perez, J.; Gonzalez, V.M.; Alonso, C. Palladium(II) compounds of putrescine and spermine. Synthesis, characterization, and DNA-binding and antitumor properties. J. Inorg. Biochem. 1993, 52, 37–49. [Google Scholar] [CrossRef]
- Vojtek, M.; Marques, M.P.M.; Ferreira, I.M.P.L.V.O.; Mota-Filipe, H.; Diniz, C. Anticancer activity of palladium-based complexes against triple negative breast cancer. Drug Discov. Today 2019, 24, 1044–1058. [Google Scholar] [CrossRef]
- Martins, A.S.; Batista de Carvalho, A.L.M.; Lamego, I.; Marques, M.P.M.; Gil, A.M. Cytotoxicity of platinum and palladium chelates against osteosarcoma. Chem. Sel. 2020, 5, 5993–6000. [Google Scholar] [CrossRef]
- Soares, A.; Fiuza, S.; Goncalves, M.; de Carvalho, L.A.E.B.; Marques, M.P.; Urbano, A. Effect of the metal center on the antitumor activity of the analogous dinuclear spermine chelates (PdCl2)2(spermine) and (PtCl2)2(spermine). Lett. Drug Des. Discov. 2007, 4, 460–463. [Google Scholar] [CrossRef] [Green Version]
- Carneiro, T.J.; Araújo, R.; Vojtek, M.; Gonçalves-Monteiro, S.; Diniz, C.; Batista de Carvalho, A.L.M.; Marques, M.P.M.; Gil, A.M. Novel Insights into Mice Multi-Organ Metabolism upon Exposure to a Potential Anticancer Pd(II)-Agent. Metabolites 2021, 11, 114. [Google Scholar] [CrossRef]
- Sarpong-Kumankomah, S.; Gailer, J. Application of a Novel Metallomics Tool to Probe the Fate of Metal-Based Anticancer Drugs in Blood Plasma: Potential, Challenges and Prospects. Curr. Top. Med. Chem. 2021, 2, 48–58. [Google Scholar] [CrossRef]
- Oboh, G.; Akomolafe, T.L.; Adefegha, S.A.; Adetuyi, A.O. Attenuation of cyclophosphamide-induced neurotoxicity in rat by yellow dye extract from root of Brimstone tree (Morinda lucida). Exp. Toxicol. Pathol. 2012, 64, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Karavelioglu, E.; Gonul, Y.; Aksit, H.; Boyaci, M.G.; Karademir, M.; Simsek, N.; Guven, M.; Atalay, T.; Rakip, U. Cabazitaxel causes a dose-dependent central nervous system toxicity in rats. J. Neurol. Sci. 2016, 360, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Pondugula, S.R.; Majrashi, M.; Almaghrabi, M.; Ramesh, S.; Abbott, K.L.; Govindarajulu, M.; Gill, K.; Fahoury, E.; Narayanan, N.; Desai, D.; et al. Oroxylum Indicum ameliorates chemotherapy induced cognitive impairment. PLoS ONE 2021, 16, e0252522. [Google Scholar] [CrossRef] [PubMed]
- Vairano, M.; Graziani, G.; Tentori, L.; Tringali, G.; Navarra, P.; Dello Russo, C. Primary cultures of microglial cells for testing toxicity of anticancer drugs. Toxicol. Lett. 2004, 148, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare Mannelli, L.; Tenci, B.; Zanardelli, M.; Failli, P.; Ghelardini, C. α7 Nicotinic Receptor Promotes the Neuroprotective Functions of Astrocytes against Oxaliplatin Neurotoxicity. Neural Plast. 2015, 2015, 396908. [Google Scholar] [CrossRef] [Green Version]
- Veloso, C.F.; Machado, A.K.; Cadoná, F.C.; Azzolin, V.F.; Cruz, I.B.M.; Silveira, A.F. Neuroprotective Effects of Guarana (Paullinia cupana Mart.) against Vincristine in Vitro Exposure. J. Prev. Alzheimers Dis. 2018, 5, 65–70. [Google Scholar] [CrossRef]
- Kim, J.; Chen, C.H.; Yang, J.; Mochly-Rosen, D. Aldehyde dehydrogenase 2*2 knock-in mice show increased reactive oxygen species production in response to cisplatin treatment. J. Biomed. Sci. 2017, 24, 33. [Google Scholar] [CrossRef] [Green Version]
- Onk, D.; Mammadov, R.; Suleyman, B.; Cimen, F.K.; Cankaya, M.; Gul, V.; Altuner, D.; Senol, O.; Kadioglu, Y.; Malkoc, I.; et al. The effect of thiamine and its metabolites on peripheral neuropathic pain induced by cisplatin in rats. Exp. Anim. 2018, 67, 259–269. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zheng, M.; Sah, S.K.; Mishra, A.; Singh, Y. Neuroprotective influence of sitagliptin against cisplatin-induced neurotoxicity, biochemical and behavioral alterations in Wistar rats. Mol. Cell. Biochem. 2019, 455, 91–97. [Google Scholar] [CrossRef]
- Hardej, D.; Trombetta, L.D. The effects of ebselen on cisplatin and diethyldithiocarbamate (DDC) cytotoxicity in rat hippocampal astrocytes. Toxicol. Lett. 2002, 131, 215–226. [Google Scholar] [CrossRef]
- De Castro, F.; Benedetti, M.; Del Coco, L.; Fanizzi, F.P. NMR-based metabolomics in metal-based drug research. Molecules 2019, 24, 2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.; Hasan, M.R. Cancer Metabolism and Drug Resistance. Metabolites 2015, 5, 571–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 2016, 15, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Pristner, M.; Warth, B. Drug-Exposome Interactions: The Next Frontier in Precision Medicine. Trends Pharmacol. Sci. 2020, 41, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- St-Coeur, P.D.; Poitras, J.J.; Cuperlovic-Culf, M.; Touaibia, M.; Morin, P., Jr. Investigating a signature of temozolomide resistance in GBM cell lines using metabolomics. J. Neurooncol. 2015, 125, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Mirbahai, L.; Wilson, M.; Shaw, C.S.; McConville, C.; Malcomson, R.D.; Griffin, J.L.; Kauppinen, R.A.; Peet, A.C. 1H magnetic resonance spectroscopy metabolites as biomarkers for cell cycle arrest and cell death in rat glioma cells. Int. J. Biochem. Cell Biol. 2011, 43, 990–1001. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Wilson, M.; Mirbahai, L.; McConville, C.; Arvanitis, T.N.; Griffin, J.L.; Kauppinen, R.A.; Peet, A.C. In vitro metabonomic study detects increases in UDP-GlcNAc and UDP-GalNAc, as early phase markers of cisplatin treatment response in brain tumor cells. J. Proteome Res. 2011, 10, 3493–3500. [Google Scholar] [CrossRef]
- Li, M.; Ren, T.; Lin, M.; Wang, Z.; Zhang, J. Integrated proteomic and metabolomic profiling the global response of rat glioma model by temozolomide treatment. J. Proteom. 2020, 211, 103578. [Google Scholar] [CrossRef]
- Bandu, R.; Kim, H.J.; Mok, H.J.; Kim, K.P. Liquid Chromatography Electrospray Ionization Tandem Mass Spectrometric (LC/ESI-MS/MS) Study for the Identification and Characterization of In Vivo Metabolites of Cisplatin in Rat Kidney Cancer Tissues: Hydrogen/Deuterium (H/D) Exchange Study. RSC Adv. 2015, 5, 89951–89958. [Google Scholar] [CrossRef]
- Brierley, D.I.; Harman, J.R.; Giallourou, N.; Leishman, E.; Roashan, A.E.; Mellows, B.A.D.; Bradshaw, H.B.; Swann, J.R.; Patel, K.; Whalley, B.J.; et al. Chemotherapy-induced cachexia dysregulates hypothalamic and systemic lipoamines and is attenuated by cannabigerol. J. Cachexia Sarcopenia Muscle 2019, 10, 844–859. [Google Scholar] [CrossRef]
- Lamego, I.; Marques, M.P.M.; Duarte, I.F.; Martins, A.S.; Oliveira, H.; Gil, A.M. Impact of the Pd2Spermine chelate on osteosarcoma metabolism: An NMR metabolomics study. J. Proteome Res. 2017, 16, 1773–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, A.S.; Batista de Carvalho, A.L.M.; Marques, M.P.M.; Gil, A.M. Response of Osteosarcoma Cell Metabolism to Platinum and Palladium Chelates as Potential New Drugs. Molecules 2021, 26, 4805. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, T.J.; Araújo, R.; Vojtek, M.; Gonçalves-Monteiro, S.; Batista de Carvalho, A.L.M.; Marques, M.P.M.; Diniz, C.; Gil, A.M. Impact of the Pd2Spm (Spermine) Complex on the Metabolism of Triple-Negative Breast Cancer Tumors of a Xenograft Mouse Model. Int. J. Mol. Sci. 2021, 22, 10775. [Google Scholar] [CrossRef] [PubMed]
- Codina, G.; Caubet, A.; López, C.; Moreno, V.; Molins, E. Palladium(II) and platinum(II) polyamine complexes: X-ray crystal structures of (SP-4-2) chloro{N-[(3-amino-κN)propyl]propane-1,3-diamine-κN,κN′}palladium(1+) tetrachloropalladate (2–) (2:1) and (R,S)-tetrachloro[μ-(spermine)]dipalladium(II) (={μ {N,N′-Bis[(3-amino-κN)propyl]butane-1,4-diamine-κN:κN′}}tetrachlorodipalladium). Helv. Chim. Acta 1999, 82, 1025–1037. [Google Scholar] [CrossRef]
- Marques, M.P.M.; Batista de Carvalho, A.L.M.; Mamede, A.P.; Santos, I.P.; Garcia Sakai, V.; Dopplapudi, A.; Cinque, G.; Wolna, M.; Gardner, P.; Batista de Carvalho, L.A.E. Chemotherapeutic targets in osteosarcoma—Insights from synchrotron-microFTIR and quasi-elastic neutron scattering. J. Phys. Chem. B 2019, 123, 6968–6979. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef]
- Beckonert, O.; Keun, H.C.; Ebbels, T.M.D.; Bundy, J.G.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat. Protoc. 2007, 2, 2692–2703. [Google Scholar] [CrossRef]
- Lin, C.Y.; Wu, H.; Tjeerdema, R.S.; Viant, M.R. Evaluation of metabolite extraction strategies from tissue samples using NMR metabolomics. Metabolomics 2007, 3, 55–67. [Google Scholar] [CrossRef]
- Le Belle, J.E.; Harris, N.G.; Williams, S.R.; Bhakoo, K.K. A comparison of cell and tissue extraction techniques using high-resolution 1H-NMR spectroscopy. NMR Biomed. 2002, 15, 37–44. [Google Scholar] [CrossRef]
- Wu, H.; Southam, A.D.; Hines, A.; Viant, M.R. High-throughput tissue extraction protocol for NMR- and MS-based metabolomics. Anal. Biochem. 2008, 372, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0-The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- As Berben, L.; Sereika, S.M.; Engberg, S. Effect size estimation: Methods and examples. Int. J. Nurs. Stud. 2012, 49, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Liu, X.; Zhu, W.; Guan, S.; Feng, R.; Zhang, H.; Liu, Q.; Sun, P.; Lin, D.; Zhang, N.; Shen, J. Metabolomic analysis of anti-hypoxia and anti-anxiety effects of Fu Fang Jin Jing Oral Liquid. PLoS ONE 2013, 8, e78281. [Google Scholar] [CrossRef] [Green Version]
- Diémé, B.; Lefèvre, A.; Nadal-Desbarats, L.; Galineau, L.; Madji Hounoum, B.; Montigny, F.; Blasco, H.; Andres, C.R.; Emond, P.; Mavel, S. Workflow methodology for rat brain metabolome exploration using NMR, LC-MS and GC-MS analytical platforms. J. Pharm. Biomed. Anal. 2017, 142, 270–278. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, X.; Du, G.; Li, Z.; Qin, X. Brain metabonomics study of the antidepressant-like effect of Xiaoyaosan on the CUMS-depression rats by 1H NMR analysis. J. Ethnopharmacol. 2019, 235, 141–154. [Google Scholar] [CrossRef]
- Gonzalez-Riano, C.; Garcia, A.; Barbas, C. Metabolomics studies in brain tissue: A review. J. Pharm. Biomed. Anal. 2016, 130, 141–168. [Google Scholar] [CrossRef]
- Baslow, M.H. N-acetylaspartate in the vertebrate brain: Metabolism and function. Neurochem. Res. 2003, 28, 941–953. [Google Scholar] [CrossRef]
- Moffett, J.R.; Ross, B.; Arun, P.; Madhavarao, C.N.; Namboodiri, A.M. N-Acetylaspartate in the CNS: From neurodiagnostics to neurobiology. Prog. Neurobiol. 2007, 81, 89–131. [Google Scholar] [CrossRef] [Green Version]
- Sperringer, J.E.; Addington, A.; Hutson, S.M. Branched-Chain Amino Acids and Brain Metabolism. Neurochem. Res. 2017, 42, 1697–1709. [Google Scholar] [CrossRef]
- Itapa, P.L.; Pesi, R. Nucleoside recycling in the brain and the nucleosidome: A complex metabolic and molecular cross-talk between the extracellular nucleotide cascade system and the intracellular nucleoside salvage. Metabolomics 2011, 12, 22. [Google Scholar] [CrossRef]
- Garcia-Gil, M.; Camici, M.; Allegrini, S.; Pesi, R.; Petrotto, E.; Tozzi, M.G. Emerging Role of Purine Metabolizing Enzymes in Brain Function and Tumors. Int. J. Mol. Sci. 2018, 19, 3598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, F.; Hirrlinger, J. The NAD+ /NADH redox state in astrocytes: Independent control of the NAD+ and NADH content. J. Neurosci. Res. 2011, 89, 1956–1964. [Google Scholar] [CrossRef] [PubMed]
- Tayebati, S.K.; Amenta, F. Choline-containing phospholipids: Relevance to brain functional pathways. Clin. Chem. Lab. Med. 2013, 51, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Tracey, T.J.; Steyn, F.J.; Wolvetang, E.J.; Ngo, S.T. Neuronal Lipid Metabolism: Multiple Pathways Driving Functional Outcomes in Health and Disease. Front. Mol. Neurosci. 2018, 11, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietzke, M.; Meiser, J.; Vazquez, A. Formate metabolism in health and disease. Mol. Metab. 2020, 33, 23–37. [Google Scholar] [CrossRef]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef]
- Janeiro, M.H.; Ramírez, M.J.; Milagro, F.I.; Martínez, J.A.; Solas, M. Implication of Trimethylamine N-Oxide (TMAO) in Disease: Potential Biomarker or New Therapeutic Target. Nutrients 2018, 10, 1398. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Slupsky, C.M. Metabolic Fingerprint of Dimethyl Sulfone (DMSO2) in Microbial−Mammalian Co-metabolism. J. Proteome Res. 2014, 13, 5281–5292. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
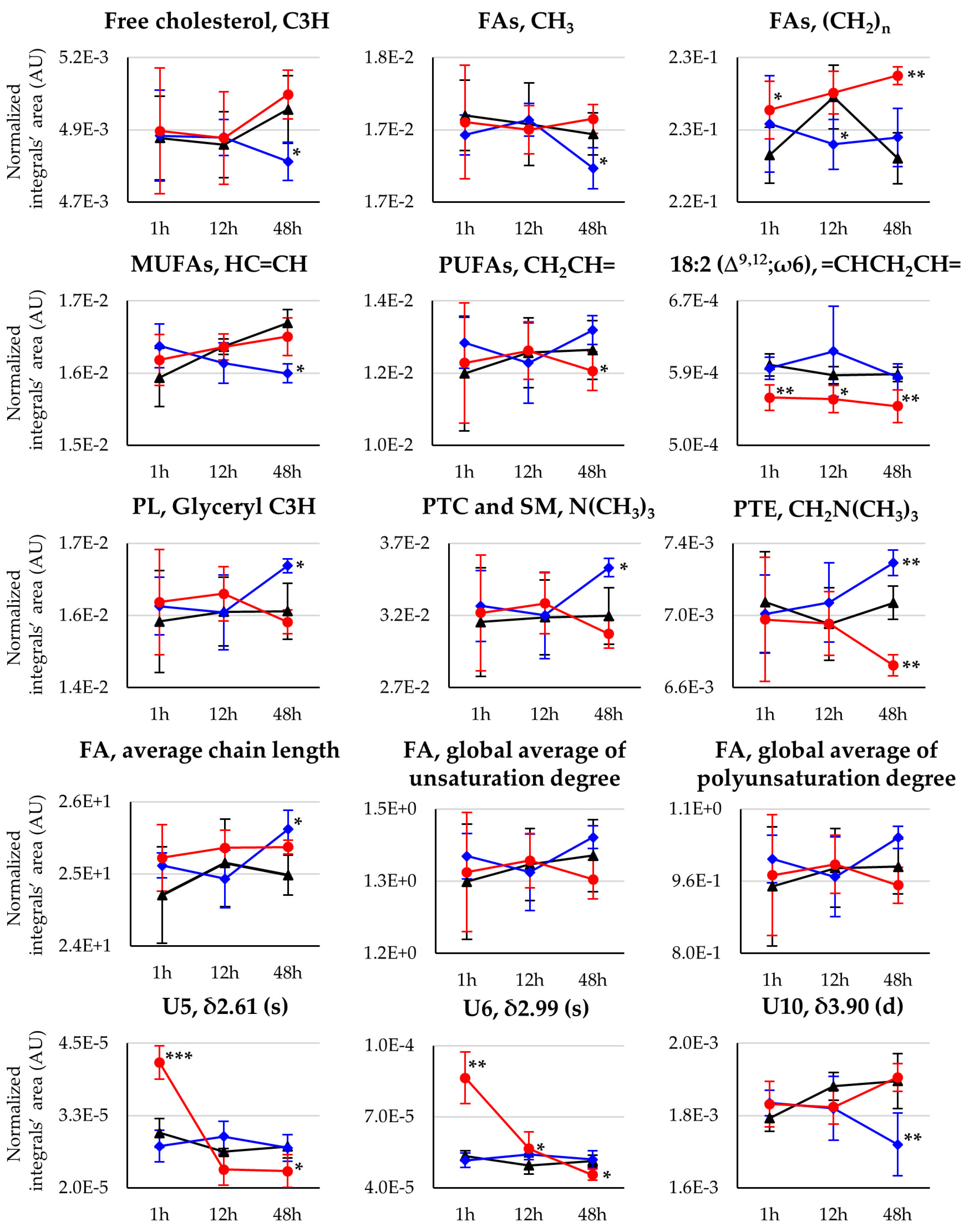{kind=link}
| cDDP vs. Controls | Pd2Spm vs. Controls | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Metabolite Family/Assignment | δH/ppm (Multiplicity) | 1 h | 12 h | 48 h | 1 h | 12 h | 48 h | |||||||
| ES ± Error | p-Value | ES ± Error | p-Value | ES ± Error | p-Value | ES ± Error | p-Value | ES ± Error | p-Value | ES ± Error | P-Value | |||
| Amino acids and derivatives | Ala | 1.48 (d) | — | — | −2.3 ± 1.6 | 1.7 × 10−2 | — | — | — | — | — | — | — | — |
| Asp, Ans | 2.80 (dd) | — | — | −2.0 ± 1.5 | 1.7 × 10−2 | — | — | — | — | — | — | — | — | |
| Gln | 2.45 (m) | — | — | 2.7 ± 1.7 | 2.9 × 10−3 a | — | — | — | — | — | — | — | — | |
| Leu | 0.96 (t) | — | — | −3.4 ± 1.9 | 8.5 × 10−4 a | — | — | — | — | — | — | — | — | |
| NAA | 2.03 (s) | — | — | — | — | 2.2 ± 1.7 | 2.1 × 10−2 | — | — | — | — | — | — | |
| Val | 1.05 (d) | — | — | −2.6 ± 1.7 | 8.8 × 10−3 | — | — | — | — | — | — | — | — | |
| Choline derivatives | Cho | 3.20 (s) | −2.7 ± 1.7 | 2.9 × 10−3 a | — | — | — | — | −2.1 ± 1.6 | 1.3 × 10−2 | — | — | — | — |
| GPC | 3.23 (s) | — | — | — | — | — | — | 2.4 ± 1.6 | 5.8 × 10−3 a | — | — | — | — | |
| Nucleotides and derivatives | Ado | 4.29 (q) | — | — | — | — | −1.6 ± 1.5 | 4.6 × 10−2 | −2.7 ± 1.7 | 1.1 × 10−2 | — | — | — | — |
| ADP | 8.54 (s) | 2.1 ± 1.5 | 1.2 × 10−2 | — | — | 3.2 ± 2.0 | 3.1 × 10−3 a | 3.7 ± 2.0 | 4.8 × 10−4 a | — | — | — | — | |
| AMP | 8.61 (s) | 2.1 ± 1.6 | 1.8 × 10−2 | — | — | — | — | 2.9 ± 1.8 | 6.0 × 10−3 a | — | — | — | — | |
| HX | 8.20 (s) | — | — | — | — | — | — | — | — | — | — | 1.7 ± 1.5 | 3.2 × 10−2 | |
| IMP | 8.58 (s) | — | — | — | — | 2.6 ± 1.8 | 7.2 × 10−3 | — | — | — | — | — | — | |
| Ino | 8.35 (s) | — | — | — | — | — | — | −3.0 ± 1.8 | 3.8 × 10−3 a | −1.5 ± 1.4 | 4.7 × 10−2 | — | — | |
| NAD+ | 8.43 (s) | 1.8 ± 1.5 | 2.3 × 10−2 | — | — | — | — | 2.3 ± 1.6 | 1.1 × 10−2 | — | — | — | — | |
| Organic acids | Formate | 8.46 (s) | — | — | 5.2 ± 2.6 | 5.8 × 10−4 a | 15.7 ± 7.4 | 6.7 × 10−8 a | 6.8 ± 3.2 | 3.3 × 10−5 a | 6.5 ± 3.1 | 1.8 × 10−5 a | 3.9 ± 2.2 | 2.0 × 10−3 a |
| Other compounds | Acetone | 2.24 (s) | 2.7 ± 1.7 | 3.4 × 10−3 a | — | — | — | — | — | — | — | — | — | — |
| DMA | 2.73 (s) | — | — | — | — | — | — | 4.4 ± 2.3 | 2.2 × 10−3 a | — | — | — | — | |
| DMSO2 † | 3.15 (s) | — | — | 2.4 ± 1.6 | 7.9 × 10−3 | — | — | 14.3 ± 6.4 | 7.9 × 10−3 | 3.4 ± 1.9 | 5.8 × 10−3 a | — | — | |
| Unassigned resonances | U1 | 0.80 (t) | — | — | −1.5 ± 1.4 | 4.6 × 10−2 | — | — | −2.2 ± 1.6 | 1.1 × 10−2 | — | — | — | — |
| U2 | 2.98 (d ‡) | — | — | 2.9 ± 1.8 | 2.3 × 10−3 a | — | — | 2.1 ± 1.6 | 1.2 × 10−2 | 2.4 ± 1.6 | 1.1 × 10−2 | — | — | |
| U3 | 3.89 (d) | — | — | −2.4 ± 1.6 | 5.4 × 10−3 a | — | — | — | — | — | — | — | — | |
| cDDP vs. Controls | Pd2Spm vs. Controls | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Metabolite Family/Assignment | δH/ppm (Multiplicity) | 1 h | 12 h | 48 h | 1 h | 12 h | 48 h | |||||||
| ES ± Error | p-Value | ES ± Error | p-Value | ES ± Error | p-Value | ES ± Error | p-Value | ES ± Error | p-Value | ES ± Error | p-Value | |||
| Free cholesterol | C3H | 3.53 (m) | — | — | — | — | −2.0 ± 1.6 | 3.2 × 10−2 | — | — | — | — | — | — |
| Fatty acids | CH3 | 0.89 (br) | — | — | — | — | −1.6 ± 1.5 | 4.8 × 10−2 | — | — | — | — | — | — |
| Saturated, (CH2)n | 1.25 (br) | — | — | −1.7 ± 1.4 | 3.2 × 10−2 | — | — | 1.8 ± 1.5 | 2.1 × 10−2 | — | — | 3.3 ± 2.0 | 2.2 × 10−3 | |
| MUFAs, HC=CH | 5.34 (m) | — | — | — | — | −4.5 ± 2.5 | 1.6 × 10−2 | — | — | — | — | — | — | |
| PUFAs, CH2CH= | 2.05 (m) | — | — | — | — | — | — | — | — | — | — | −1.8 ± 1.6 | 3.0 × 10−2 | |
| 18:2 (Δ9,12; ω6), =CHCH2CH= | 2.77 (t) | — | — | — | — | — | — | −2.8 ± 1.7 | 2.4 × 10−3 | −2.1 ± 1.6 | 1.3 × 10−2 | −2.4 ± 1.7 | 8.8 × 10−3 | |
| Phospholipids | -CH2N(CH3)3 | 3.75 (br) | — | — | — | — | 1.8 ± 1.6 | 3.5 × 10−2 | 1.8 ± 1.5 | 2.7 × 10−2 | 1.7 ± 1.5 | 4.1 × 10−2 | 1.7 ± 1.5 | 4.5 × 10−2 |
| -POCH2 | 4.38 (br) | — | — | — | — | 3.3 ± 2.0 | 1.8 × 10−2 | — | — | — | — | — | — | |
| Glyceryl C3H2 | 3.94 (br) | — | — | — | — | 2.4 ± 1.7 | 4.4 × 10−2 | — | — | — | — | — | — | |
| PTC & SM, N(CH3)3 | 3.29–3.31 | — | — | — | — | 2.5 ± 1.7 | 3.7 × 10−2 | — | — | — | — | — | — | |
| PTE CH2[(NH3)+] | 3.15 (br) | — | — | — | — | 2.7 ± 1.8 | 9.0 × 10−3 | — | — | — | — | −4.6 ± 2.5 | 1.5 × 10−3 | |
| PTE [(NH3)+] | 8.60 (br) | — | — | — | — | — | — | — | — | — | — | −1.9 ± 1.6 | 2.6 × 10−2 | |
| Unassigned resonances | U1 | 0.54 (d) | −1.6 ± 1.4 | 3.7 × 10−2 | — | — | — | — | −2.1 ± 1.5 | 1.3 × 10−2 | — | — | −2.9 ± 1.9 | 1.4 × 10−2 |
| U2 | 0.60 (d) | −1.8 ± 1.5 | 3.2 × 10−2 | — | — | — | — | — | — | −1.9 ± 1.5 | 1.6 × 10−2 | −3.7 ± 2.1 | 8.9 × 10−3 | |
| U3 | 0.64 (s ‡) | — | — | — | — | — | — | — | — | −1.8 ± 1.5 | 2.1 × 10−2 | — | — | |
| U4 | 2.18 (d) | −1.7 ± 1.4 | 3.6 × 10−2 | — | — | — | — | — | — | — | — | — | — | |
| U5 | 2.61 (s) | — | — | — | — | — | — | 4.5 ± 2.3 | 1.1 × 10−4 a | — | — | −1.7 ± 1.5 | 3.2 × 10−2 | |
| U6 | 2.99 (s) | — | — | 1.6 ± 1.4 | 4.6 × 10−2 | — | — | 4.2 ± 2.2 | 2.0 × 10−3 | — | — | −2.5 ± 1.8 | 3.2 × 10−2 | |
| U7 | 3.49 (s) | 2.0 ± 1.5 | 1.4 × 10−2 | — | — | −2.7 ± 1.8 | 1.5 × 10−2 | — | — | — | — | −2.1 ± 1.6 | 4.6 × 10−2 | |
| U8 | 3.64 (s) | — | — | — | — | — | — | — | — | — | — | −2.6 ± 1.8 | 6.0 × 10−3 | |
| U9 | 3.84 (d) | — | — | — | — | — | — | — | — | −1.7 ± 1.4 | 3.1 × 10−2 | — | — | |
| U10 | 3.90 (br) | — | — | — | — | −4.8 ± 2.6 | 1.2 × 10−3 | — | — | — | — | — | — | |
| U11 | 5.29 (t) | — | — | — | — | — | — | — | — | −2.4 ± 1.6 | 5.9 × 10−3 | — | — | |
| U12 | 8.34 (br) | — | — | — | — | −2.4 ± 1.7 | 8.4 × 10−3 | — | — | — | — | 1.9 ± 1.6 | 2.4 × 10−2 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carneiro, T.J.; Vojtek, M.; Gonçalves-Monteiro, S.; Neves, J.R.; Carvalho, A.L.M.B.d.; Marques, M.P.M.; Diniz, C.; Gil, A.M. Metabolic Impact of Anticancer Drugs Pd2Spermine and Cisplatin on the Brain of Healthy Mice. Pharmaceutics 2022, 14, 259. https://doi.org/10.3390/pharmaceutics14020259
Carneiro TJ, Vojtek M, Gonçalves-Monteiro S, Neves JR, Carvalho ALMBd, Marques MPM, Diniz C, Gil AM. Metabolic Impact of Anticancer Drugs Pd2Spermine and Cisplatin on the Brain of Healthy Mice. Pharmaceutics. 2022; 14(2):259. https://doi.org/10.3390/pharmaceutics14020259
Chicago/Turabian StyleCarneiro, Tatiana J., Martin Vojtek, Salomé Gonçalves-Monteiro, João R. Neves, Ana L. M. Batista de Carvalho, Maria Paula M. Marques, Carmen Diniz, and Ana M. Gil. 2022. "Metabolic Impact of Anticancer Drugs Pd2Spermine and Cisplatin on the Brain of Healthy Mice" Pharmaceutics 14, no. 2: 259. https://doi.org/10.3390/pharmaceutics14020259
APA StyleCarneiro, T. J., Vojtek, M., Gonçalves-Monteiro, S., Neves, J. R., Carvalho, A. L. M. B. d., Marques, M. P. M., Diniz, C., & Gil, A. M. (2022). Metabolic Impact of Anticancer Drugs Pd2Spermine and Cisplatin on the Brain of Healthy Mice. Pharmaceutics, 14(2), 259. https://doi.org/10.3390/pharmaceutics14020259