
Delivery of Oligonucleotides: Efficiency with Lipid Conjugation and Clinical Outcome



Abstract
:1. Introduction
1.1. Background of Oligonucleotide
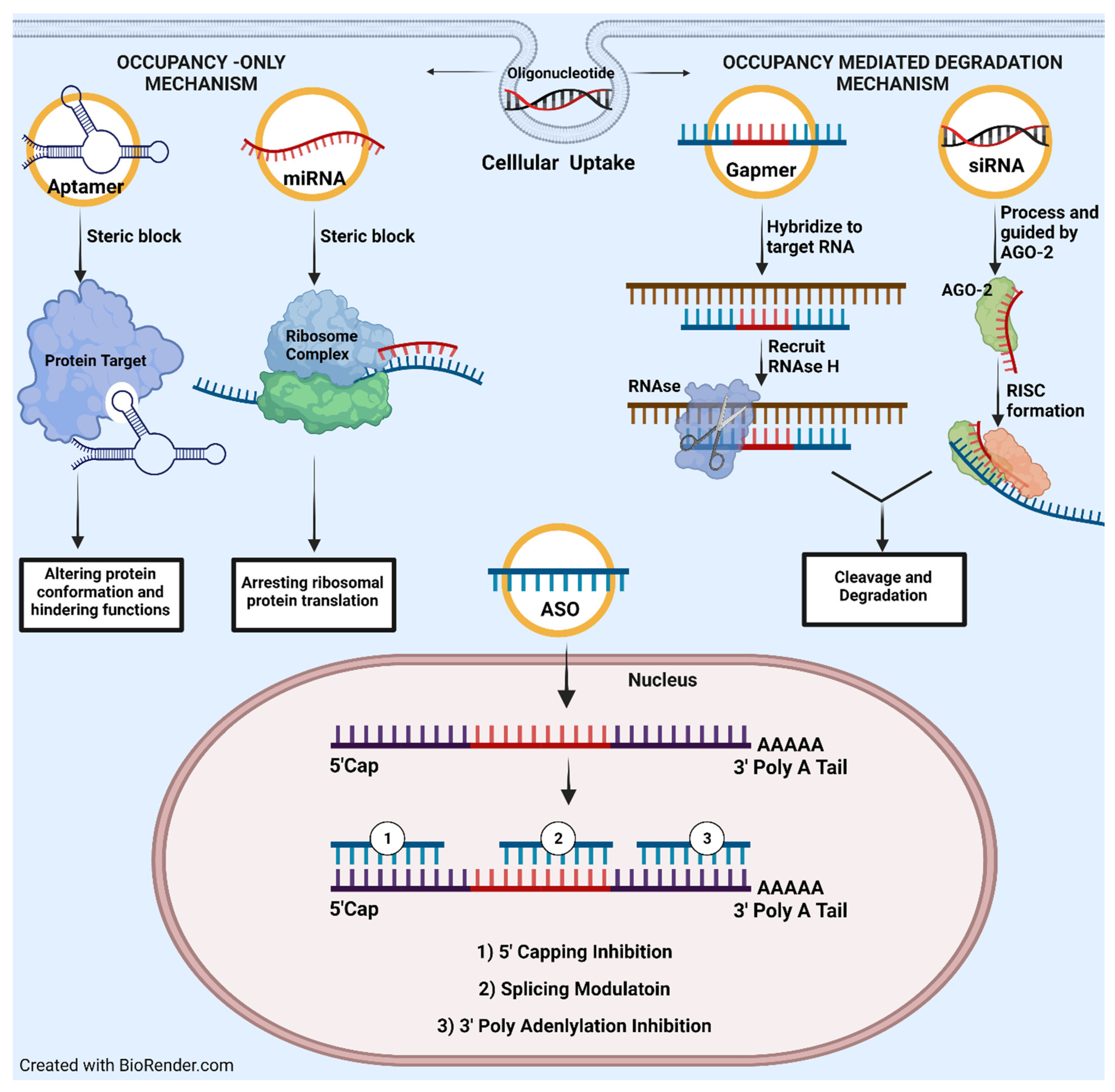
1.2. Oligonucleotide-Based Drug Mechanism of Action
1.3. Biological Barriers That Challenged Druggability/Pharmacokinetic Profile of Oligonucleotide In Vivo
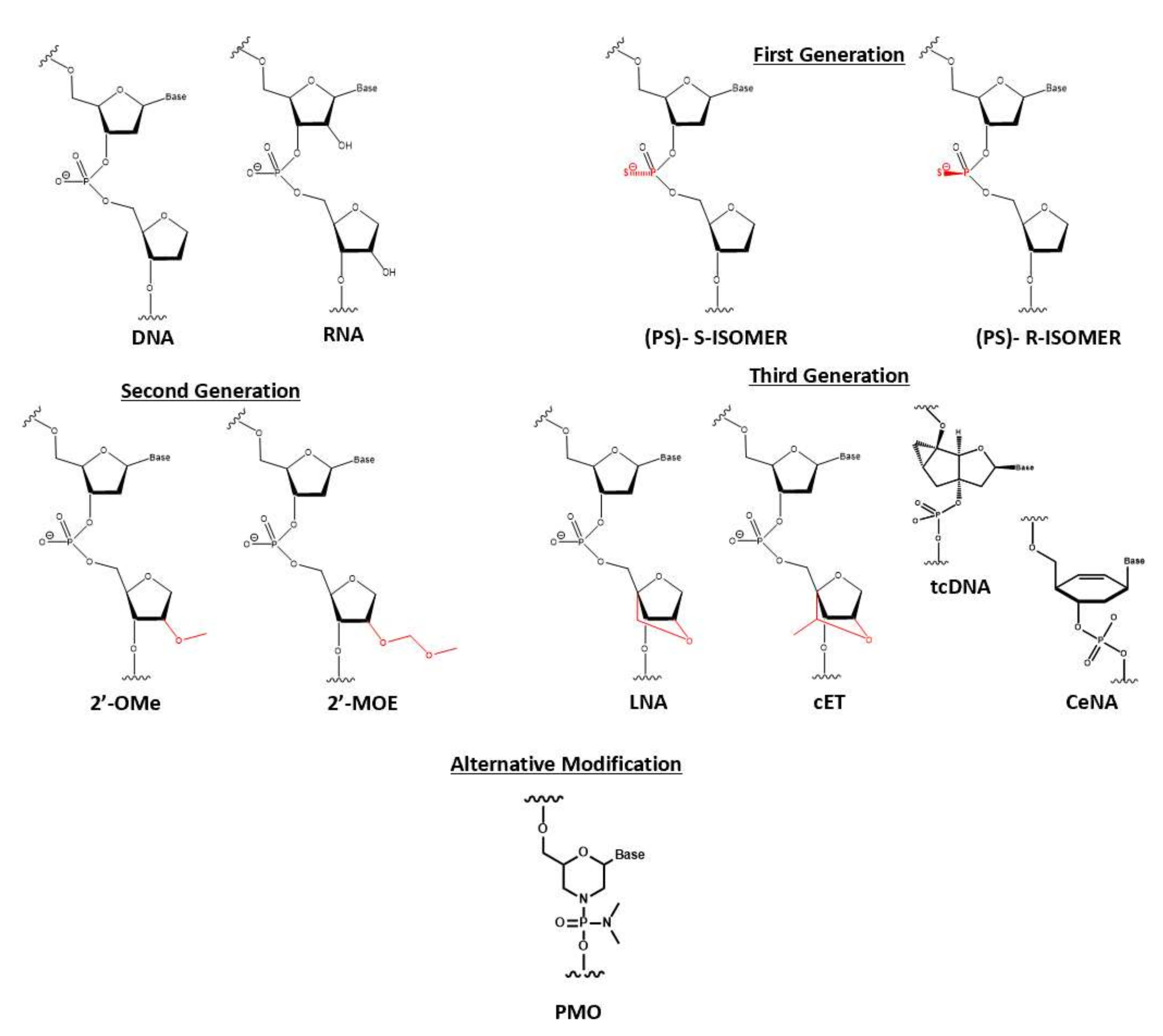
1.4. Early Attempt of Oligonucleotide Chemical Modification
1.5. Lipid-Conjugated Oligonucleotides: Method of Delivery and Example of Conjugation
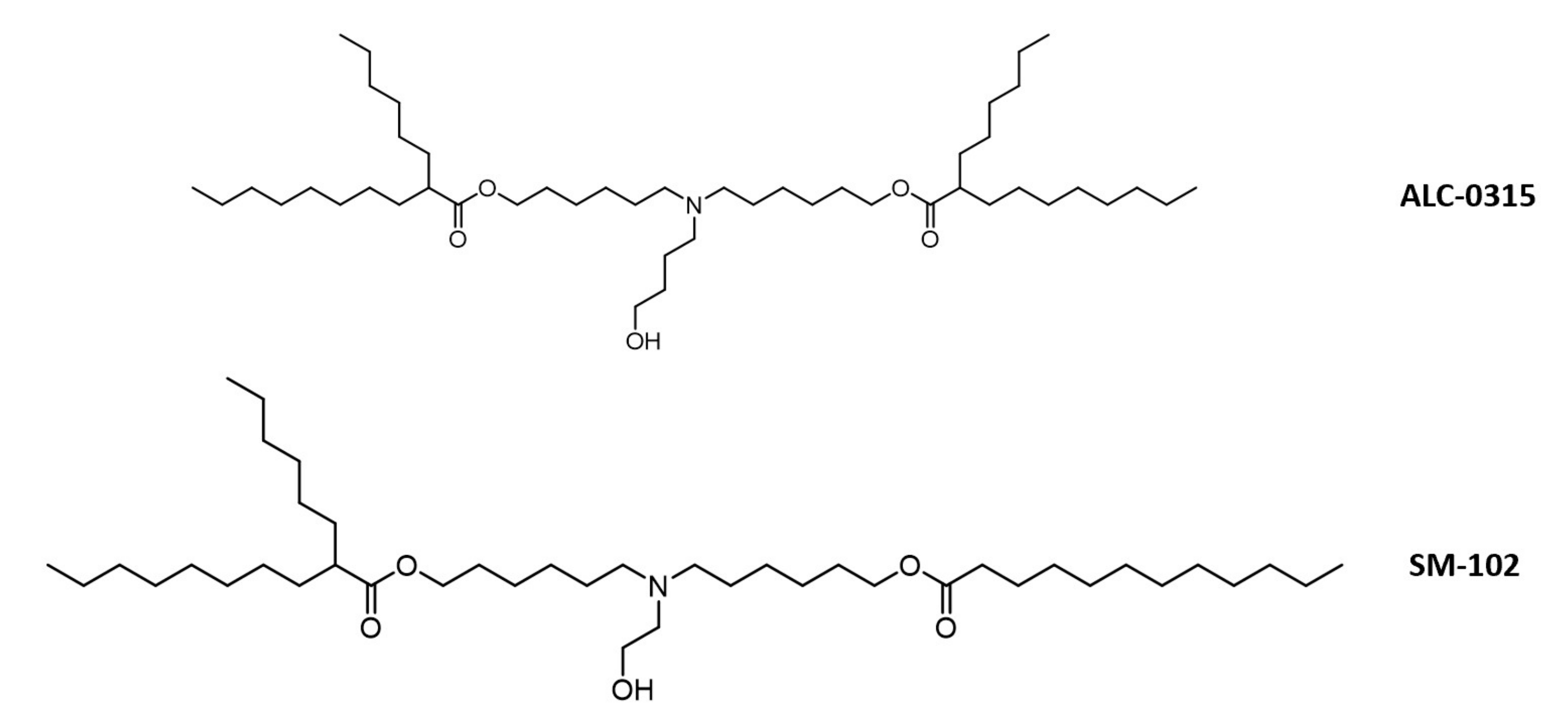
1.5.1. Method of Enhanced Delivery and Lipid-Conjugated Structure
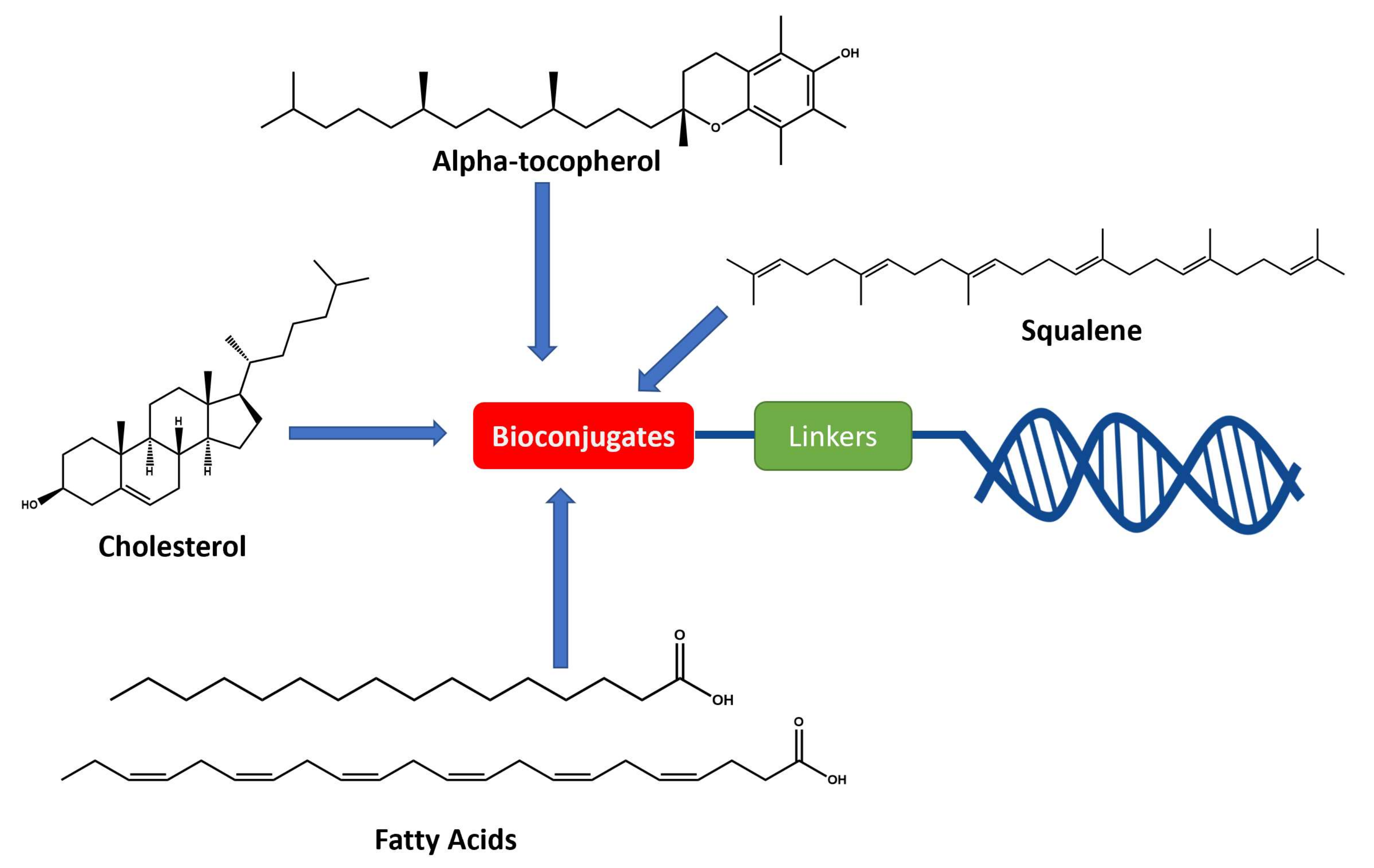
1.5.2. Example of Conjugations
A. Cholesterol Conjugates
B. Fatty Acid Conjugates
C. Vitamin E (α-tocopherol)
D. Squalene
2. Conclusions and Future Outcome
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Crooke, S.T. Molecular Mechanisms of Antisense Oligonucleotides. Nucleic Acid Ther. 2017, 27, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Feng, K.; Li, L.; Yang, L.; Pan, X.; Yazd, H.S.; Cui, C.; Li, J.; Moroz, L.; Sun, Y.; et al. Lipid–Oligonucleotide Conjugates for Bioapplications. Natl. Sci. Rev. 2020, 7, 1933–1953. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of Cationic Lipids and Cationic Polymers in Gene Delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Monnery, B.D.; Wright, M.; Cavill, R.; Hoogenboom, R.; Shaunak, S.; Steinke, J.H.G.; Thanou, M. Cytotoxicity of Polycations: Relationship of Molecular Weight and the Hydrolytic Theory of the Mechanism of Toxicity. Int. J. Pharm. 2017, 521, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Vickers, T.; Baker, B.F.; Cook, P.D.; Zounes, M.; Buckheit, R.W.; Germany, J.; Ecker, D.J. Inhibition of HIV-LTR Gene Expression by Oligonucleotides Targeted to the TAR Element. Nucl. Acids Res. 1991, 19, 3359–3368. [Google Scholar] [CrossRef] [Green Version]
- Donis-Keller, H. Site Specific Enzymatic Cleavage of RNA. Nucl. Acids Res. 1979, 7, 179–192. [Google Scholar] [CrossRef] [Green Version]
- Casey, B.P.; Glazer, P.M. Gene Targeting via Triple-Helix Formation. In Progress in Nucleic Acid Research and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2001; Volume 67, pp. 163–192. ISBN 978-0-12-540067-1. [Google Scholar]
- Rand, T.A.; Petersen, S.; Du, F.; Wang, X. Argonaute2 Cleaves the Anti-Guide Strand of SiRNA during RISC Activation. Cell 2005, 123, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Bumcrot, D.; Manoharan, M.; Koteliansky, V.; Sah, D.W.Y. RNAi Therapeutics: A Potential New Class of Pharmaceutical Drugs. Nat. Chem. Biol. 2006, 2, 711–719. [Google Scholar] [CrossRef]
- De Smet, M.D.; Meenken, C.; van den Horn, G.J. Fomivirsen—a Phosphorothioate Oligonucleotide for the Treatment of CMV Retinitis. Ocul. Immunol. Inflamm. 1999, 7, 189–198. [Google Scholar] [CrossRef]
- Xiong, H.; Veedu, R.N.; Diermeier, S.D. Recent Advances in Oligonucleotide Therapeutics in Oncology. IJMS 2021, 22, 3295. [Google Scholar] [CrossRef]
- Agrawal, S.; Temsamani, J.; Galbraith, W.; Tang, J. Pharmacokinetics of Antisense Oligonucleotides. Clin. Pharmacokinet. 1995, 28, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.; Bauman, J.; Kang, H.; Ming, X. Biological Barriers to Therapy with Antisense and SiRNA Oligonucleotides. Mol. Pharm. 2009, 6, 686–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jason, T.L.H.; Koropatnick, J.; Berg, R.W. Toxicology of Antisense Therapeutics. Toxicol. Appl. Pharmacol. 2004, 201, 66–83. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X. Cellular Uptake and Trafficking of Antisense Oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Geary, R.S. Antisense Oligonucleotide Pharmacokinetics and Metabolism. Expert Opin. Drug Metab. Toxicol. 2009, 5, 381–391. [Google Scholar] [CrossRef]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, Biodistribution and Cell Uptake of Antisense Oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Wójcik, M.; Cieślak, M.; Stec, W.J.; Goding, J.W.; Koziołkiewicz, M. Nucleotide Pyrophosphatase/Phosphodiesterase 1 Is Responsible for Degradation of Antisense Phosphorothioate Oligonucleotides. Oligonucleotides 2007, 17, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Khvorova, A.; Watts, J.K. The Chemical Evolution of Oligonucleotide Therapies of Clinical Utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef]
- Singh, S.K.; Koshkin, A.A.; Wengel, J.; Nielsen, P. LNA (Locked Nucleic Acids): Synthesis and High-Affinity Nucleic Acid Recognition. Chem. Commun. 1998, 455–456. [Google Scholar] [CrossRef]
- Agrawal, S.; Crooke, S.T. Antisense Research and Application; Springer: Berlin, Germany; New York, NY, USA, 1998; pp. 51–101. ISBN 978-3-642-58785-6. [Google Scholar]
- Lima, W.F.; Crooke, S.T. Binding Affinity and Specificity of Escherichia Coli RNase H1: Impact on the Kinetics of Catalysis of Antisense Oligonucleotide−RNA Hybrids. Biochemistry 1997, 36, 390–398. [Google Scholar] [CrossRef]
- Goyenvalle, A.; Leumann, C.; Garcia, L. Therapeutic Potential of Tricyclo-DNA Antisense Oligonucleotides. J. Neuromuscular Dis. 2016, 3, 157–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbeure, B. RNase H Mediated Cleavage of RNA by Cyclohexene Nucleic Acid (CeNA). Nucleic Acids Res. 2001, 29, 4941–4947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nan, Y.; Zhang, Y.-J. Antisense Phosphorodiamidate Morpholino Oligomers as Novel Antiviral Compounds. Front. Microbiol. 2018, 9, 750. [Google Scholar] [CrossRef] [PubMed]
- Frazier, K.S. Antisense Oligonucleotide Therapies: The Promise and the Challenges from a Toxicologic Pathologist’s Perspective. Toxicol. Pathol. 2015, 43, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Zanardi, T.A.; Kim, T.-W.; Shen, L.; Serota, D.; Papagiannis, C.; Park, S.-Y.; Kim, Y.; Henry, S.P. Chronic Toxicity Assessment of 2′- O -Methoxyethyl Antisense Oligonucleotides in Mice. Nucleic Acid Ther. 2018, 28, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Bozzer, S.; Bo, M.D.; Toffoli, G.; Macor, P.; Capolla, S. Nanoparticles-Based Oligonucleotides Delivery in Cancer: Role of Zebrafish as Animal Model. Pharmaceutics 2021, 13, 1106. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, C.; Wang, C.; Jankovic, K.E.; Dong, Y. Lipids and Lipid Derivatives for RNA Delivery. Chem. Rev. 2021, 121, 12181–12277. [Google Scholar] [CrossRef] [PubMed]
- Schoenmaker, L.; Witzigmann, D.; Kulkarni, J.A.; Verbeke, R.; Kersten, G.; Jiskoot, W.; Crommelin, D.J.A. MRNA-Lipid Nanoparticle COVID-19 Vaccines: Structure and Stability. Int. J. Pharm. 2021, 601, 120586. [Google Scholar] [CrossRef]
- Moss, K.H.; Popova, P.; Hadrup, S.R.; Astakhova, K.; Taskova, M. Lipid Nanoparticles for Delivery of Therapeutic RNA Oligonucleotides. Mol. Pharm. 2019, 16, 2265–2277. [Google Scholar] [CrossRef]
- Li, Z.; Rana, T.M. Therapeutic Targeting of MicroRNAs: Current Status and Future Challenges. Nat. Rev. Drug Discov. 2014, 13, 622–638. [Google Scholar] [CrossRef]
- Osborn, M.F.; Coles, A.H.; Biscans, A.; Haraszti, R.A.; Roux, L.; Davis, S.; Ly, S.; Echeverria, D.; Hassler, M.R.; Godinho, B.M.D.C.; et al. Hydrophobicity Drives the Systemic Distribution of Lipid-Conjugated SiRNAs via Lipid Transport Pathways. Nucleic Acids Res. 2019, 47, 1070–1081. [Google Scholar] [CrossRef] [Green Version]
- Hassler, M.R.; Turanov, A.A.; Alterman, J.F.; Haraszti, R.A.; Coles, A.H.; Osborn, M.F.; Echeverria, D.; Nikan, M.; Salomon, W.E.; Roux, L.; et al. Comparison of Partially and Fully Chemically-Modified SiRNA in Conjugate-Mediated Delivery in Vivo. Nucleic Acids Res. 2018, 46, 2185–2196. [Google Scholar] [CrossRef] [PubMed]
- Raouane, M.; Desmaële, D.; Urbinati, G.; Massaad-Massade, L.; Couvreur, P. Lipid Conjugated Oligonucleotides: A Useful Strategy for Delivery. Bioconjugate Chem. 2012, 23, 1091–1104. [Google Scholar] [CrossRef] [PubMed]
- Letsinger, R.L.; Zhang, G.R.; Sun, D.K.; Ikeuchi, T.; Sarin, P.S. Cholesteryl-Conjugated Oligonucleotides: Synthesis, Properties, and Activity as Inhibitors of Replication of Human Immunodeficiency Virus in Cell Culture. Proc. Natl. Acad. Sci. USA 1989, 86, 6553–6556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikan, M.; Osborn, M.F.; Coles, A.H.; Godinho, B.M.; Hall, L.M.; Haraszti, R.A.; Hassler, M.R.; Echeverria, D.; Aronin, N.; Khvorova, A. Docosahexaenoic Acid Conjugation Enhances Distribution and Safety of SiRNA upon Local Administration in Mouse Brain. Mol. Ther. Nucleic Acids 2016, 5, e344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stetsenko, D.A.; Gait, M.J. A Convenient Solid-Phase Method for Synthesis of 3‘-Conjugates of Oligonucleotides. Bioconjugate Chem. 2001, 12, 576–586. [Google Scholar] [CrossRef]
- Ueno, Y.; Inoue, T.; Yoshida, M.; Yoshikawa, K.; Shibata, A.; Kitamura, Y.; Kitade, Y. Synthesis of Nuclease-Resistant SiRNAs Possessing Benzene-Phosphate Backbones in Their 3′-Overhang Regions. Bioorganic Med. Chem. Lett. 2008, 18, 5194–5196. [Google Scholar] [CrossRef]
- Guzaev, A.; Lönnberg, H. Solid Support Synthesis of Ester Linked Hydrophobic Conjugates of Oligonucleotides. Tetrahedron 1999, 55, 9101–9116. [Google Scholar] [CrossRef]
- Durand, A.; Brown, T. Synthesis And Properties Of Oligonucleotides Containing A Cholesterol Thymidine Monomer. Nucleosides Nucleotides Nucleic Acids 2007, 26, 785–794. [Google Scholar] [CrossRef]
- Massaad-Massade, L.; Boutary, S.; Caillaud, M.; Gracia, C.; Parola, B.; Gnaouiya, S.B.; Stella, B.; Arpicco, S.; Buchy, E.; Desmaële, D.; et al. New Formulation for the Delivery of Oligonucleotides Using “Clickable” SiRNA-Polyisoprenoid-Conjugated Nanoparticles: Application to Cancers Harboring Fusion Oncogenes. Bioconjugate Chem. 2018, 29, 1961–1972. [Google Scholar] [CrossRef]
- Raouane, M.; Desmaele, D.; Gilbert-Sirieix, M.; Gueutin, C.; Zouhiri, F.; Bourgaux, C.; Lepeltier, E.; Gref, R.; Ben Salah, R.; Clayman, G.; et al. Synthesis, Characterization, and in Vivo Delivery of SiRNA-Squalene Nanoparticles Targeting Fusion Oncogene in Papillary Thyroid Carcinoma. J. Med. Chem. 2011, 54, 4067–4076. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, M.; Tivel, K.L.; Andrade, L.K.; Mohan, V.; Condon, T.P.; Bennett, C.F.; Cook, P.D. Oligonucleotide Conjugates: Alteration of the Pharmacokinetic Properties of Antisense Agents. Null 1995, 14, 969–973. [Google Scholar] [CrossRef]
- Bijsterbosch, M.K. Modulation of Plasma Protein Binding and in Vivo Liver Cell Uptake of Phosphorothioate Oligodeoxynucleotides by Cholesterol Conjugation. Nucleic Acids Res. 2000, 28, 2717–2725. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, C.; Hadwiger, P.; John, M.; Vornlocher, H.-P.; Unverzagt, C. Steroid and Lipid Conjugates of SiRNAs to Enhance Cellular Uptake and Gene Silencing in Liver Cells. Bioorganic Med. Chem. Lett. 2004, 14, 4975–4977. [Google Scholar] [CrossRef] [PubMed]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Geick, A.; Hadwiger, P.; Harborth, J.; et al. Therapeutic Silencing of an Endogenous Gene by Systemic Administration of Modified SiRNAs. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Wolfrum, C.; Shi, S.; Jayaprakash, K.N.; Jayaraman, M.; Wang, G.; Pandey, R.K.; Rajeev, K.G.; Nakayama, T.; Charrise, K.; Ndungo, E.M.; et al. Mechanisms and Optimization of in Vivo Delivery of Lipophilic SiRNAs. Nat. Biotechnol. 2007, 25, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Godinho, B.M.D.C.; Gilbert, J.W.; Haraszti, R.A.; Coles, A.H.; Biscans, A.; Roux, L.; Nikan, M.; Echeverria, D.; Hassler, M.; Khvorova, A. Pharmacokinetic Profiling of Conjugated Therapeutic Oligonucleotides: A High-Throughput Method Based Upon Serial Blood Microsampling Coupled to Peptide Nucleic Acid Hybridization Assay. Nucleic Acid Ther. 2017, 27, 323–334. [Google Scholar] [CrossRef]
- Pei, D.; Buyanova, M. Overcoming Endosomal Entrapment in Drug Delivery. Bioconjugate Chem. 2019, 30, 273–283. [Google Scholar] [CrossRef]
- Zheng, Y.; Tai, W. Insight into the SiRNA Transmembrane Delivery—From Cholesterol Conjugating to Tagging. WIREs Nanomed. Nanobiotechnol. 2020, 12, e1606. [Google Scholar] [CrossRef]
- Du Rietz, H.; Hedlund, H.; Wilhelmson, S.; Nordenfelt, P.; Wittrup, A. Imaging Small Molecule-Induced Endosomal Escape of SiRNA. Nat. Commun. 2020, 11, 1809. [Google Scholar] [CrossRef]
- Biscans, A.; Coles, A.; Haraszti, R.; Echeverria, D.; Hassler, M.; Osborn, M.; Khvorova, A. Diverse Lipid Conjugates for Functional Extra-Hepatic SiRNA Delivery in vivo. Nucleic Acids Res. 2019, 47, 1082–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindholm, M.W. PCSK9 LNA Antisense Oligonucleotides Induce Sustained Reduction of LDL Cholesterol in Nonhuman Primates. Cell Ther. 2012, 20, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, C.S.J.; Wiklander, O.P.B.; Larsson, L.; Moreno, P.M.D.; Parini, P.; Lundin, K.E.; Smith, C.I.E. RNA Therapeutics Inactivate PCSK9 by Inducing a Unique Intracellular Retention Form. J. Mol. Cell. Cardiol. 2015, 8, 186–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poelgeest, E.P.; Hodges, M.R.; Moerland, M.; Tessier, Y.; Levin, A.A.; Persson, R.; Lindholm, M.W.; Erichsen, K.D.; Ørum, H.; Cohen, A.F.; et al. Antisense-Mediated Reduction of Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9): A First-in-Human Randomized, Placebo-Controlled Trial. Br. J. Clin. Pharmacol. 2015, 80, 1350–1361. [Google Scholar] [CrossRef] [Green Version]
- Crooke, R.M. An Apolipoprotein B Antisense Oligonucleotide Lowers LDL Cholesterol in Hyperlipidemic Mice without Causing Hepatic Steatosis. J. Lipid Res. 2005, 13, 872–884. [Google Scholar] [CrossRef] [Green Version]
- Mullick, A.E.; Fu, W.; Graham, M.J.; Lee, R.G.; Witchell, D.; Bell, T.A.; Whipple, C.P.; Crooke, R.M. Antisense Oligonucleotide Reduction of ApoB-Ameliorated Atherosclerosis in LDL Receptor-Deficient Mice. J. Lipid Res. 2011, 52, 885–896. [Google Scholar] [CrossRef] [Green Version]
- Agarwala, A.; Jones, P.; Nambi, V. The Role of Antisense Oligonucleotide Therapy in Patients with Familial Hypercholesterolemia: Risks, Benefits, and Management Recommendations. Curr. Atheroscler. Rep. 2015, 8, 467. [Google Scholar] [CrossRef]
- Santos, R.D.; Raal, F.J.; Catapano, A.L.; Witztum, J.L.; Steinhagen-Thiessen, E.; Tsimikas, S. Mipomersen, an Antisense Oligonucleotide to Apolipoprotein B-100, Reduces Lipoprotein(a) in Various Populations With Hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2015, 11, 689–699. [Google Scholar] [CrossRef] [Green Version]
- Wada, S.; Yasuhara, H.; Wada, F.; Sawamura, M.; Waki, R.; Yamamoto, T.; Harada-Shiba, M.; Obika, S. Evaluation of the Effects of Chemically Different Linkers on Hepatic Accumulations, Cell Tropism and Gene Silencing Ability of Cholesterol-Conjugated Antisense Oligonucleotides. J. Control. Release 2016, 226, 57–65. [Google Scholar] [CrossRef]
- Nakajima, M.; Kasuya, T.; Yokota, S.; Onishi, R.; Ikehara, T.; Kugimiya, A.; Watanabe, A. Gene Silencing Activity and Hepatic Accumulation of Antisense Oligonucleotides Bearing Cholesterol-Conjugated Thiono Triester at the Gap Region. Nucleic Acid Ther. 2017, 27, 232–237. [Google Scholar] [CrossRef]
- Watanabe, A.; Nakajima, M.; Kasuya, T.; Onishi, R.; Kitade, N.; Mayumi, K.; Ikehara, T.; Kugimiya, A. Comparative Characterization of Hepatic Distribution and MRNA Reduction of Antisense Oligonucleotides Conjugated with Triantennary N-Acetyl Galactosamine and Lipophilic Ligands Targeting Apolipoprotein B. J. Pharmacol. Exp. Ther. 2016, 357, 320–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.M.; Xia, Y.; Dai, W.; Han, H.Y.; Dong, Y.X.; Cai, J.; Zeng, X.; Luo, F.Y.; Yang, T.; Li, Y.Z.; et al. Cholesterol-Conjugated Let-7amimics: Antitumor Efficacy on Hepatocellular Carcinoma in Vitro and in a Preclinical Orthotopic Xenograft Model of Systemic Therapy. BMC Cancer 2014, 14, 889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anthony, V.; Skach, W.R. Molecular Mechanism of P-Glycoprotein Assembly into Cellular Membranes. Curr Protein Pept. Sci. 2002, 3, 485–501. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wu, D.; Bui, T.; Ho, R.J.Y. A Novel Human Multidrug Resistance Gene MDR1 Variant G571A (G191R) Modulates Cancer Drug Resistance and Efflux Transport. J. Pharmacol. Exp. Ther. 2008, 327, 474–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova, N.S.; Chernikov, I.V.; Meschaninova, M.I.; Dovydenko, I.S.; Venyaminova, A.G.; Zenkova, M.A.; Vlassov, V.V.; Chernolovskaya, E.L. Carrier-Free Cellular Uptake and the Gene-Silencing Activity of the Lipophilic SiRNAs Is Strongly Affected by the Length of the Linker between SiRNA and Lipophilic Group. Nucleic Acids Res. 2012, 40, 2330–2344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernikov, I.V.; Gladkikh, D.V.; Karelina, U.A.; Meschaninova, M.I.; Ven’yaminova, A.G.; Vlassov, V.V.; Chernolovskaya, E.L. Trimeric Small Interfering RNAs and Their Cholesterol-Containing Conjugates Exhibit Improved Accumulation in Tumors, but Dramatically Reduced Silencing Activity. Molecules 2020, 25, 1877. [Google Scholar] [CrossRef] [PubMed]
- DiFiglia, M.; Sena-Esteves, M.; Chase, K.; Sapp, E.; Pfister, E.; Sass, M.; Yoder, J.; Reeves, P.; Pandey, R.K.; Rajeev, K.G.; et al. Therapeutic Silencing of Mutant Huntingtin with SiRNA Attenuates Striatal and Cortical Neuropathology and Behavioral Deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 17204–17209. [Google Scholar] [CrossRef] [Green Version]
- Alterman, J.F.; Hall, L.M.; Coles, A.H.; Hassler, M.R.; Didiot, M.-C.; Chase, K.; Abraham, J.; Sottosanti, E.; Johnson, E.; Sapp, E.; et al. Hydrophobically Modified SiRNAs Silence Huntingtin MRNA in Primary Neurons and Mouse Brain. Mol. Ther.—Nucleic Acids 2015, 4, e266. [Google Scholar] [CrossRef]
- Yuan, H.; Lanting, L.; Xu, Z.-G.; Li, S.-L.; Swiderski, P.; Putta, S.; Jonnalagadda, M.; Kato, M.; Natarajan, R. Effects of Cholesterol-Tagged Small Interfering RNAs Targeting 12/15-Lipoxygenase on Parameters of Diabetic Nephropathy in a Mouse Model of Type 1 Diabetes. Am. J. Physiol.—Ren. Physiol. 2008, 295, F605–F617. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Navarro, F.; Lal, A.; Basar, E.; Pandey, R.K.; Manoharan, M.; Feng, Y.; Lee, S.J.; Lieberman, J.; Palliser, D. Durable Protection from Herpes Simplex Virus-2 Transmission Following Intravaginal Application of SiRNAs Targeting Both a Viral and Host Gene. Cell Host Microbe 2009, 5, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Rozema, D.B.; Lewis, D.L.; Wakefield, D.H.; Wong, S.C.; Klein, J.J.; Roesch, P.L.; Bertin, S.L.; Reppen, T.W.; Chu, Q.; Blokhin, A.V.; et al. Dynamic PolyConjugates for Targeted in Vivo Delivery of SiRNA to Hepatocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 12982–12987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, A.M.; Stolk, J.; Bals, R.; Lickliter, J.D.; Hamilton, J.; Christianson, D.R.; Given, B.D.; Burdon, J.G.; Loomba, R.; Stoller, J.K.; et al. Hepatic-Targeted RNA Interference Provides Robust and Persistent Knockdown of Alpha-1 Antitrypsin Levels in ZZ Patients. J. Hepatol. 2018, 69, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Chang, C.; Kim, J.H.; Oh, C.T.; Lee, H.N.; Lee, C.; Oh, D.; Lee, C.; Kim, B.; Hong, S.W.; et al. Development of Cell-Penetrating Asymmetric Interfering RNA Targeting Connective Tissue Growth Factor. J. Investig. Dermatol. 2016, 136, 2305–2313. [Google Scholar] [CrossRef] [PubMed]
- Choe, J.Y.; Son, D.S.; Kim, Y.; Lee, J.; Shin, H.; Kim, W.J.; Kang, Y.G.; Dua, P.; Hong, S.W.; Park, J.H.; et al. L-Type Calcium Channel Blocker Enhances Cellular Delivery and Gene Silencing Potency of Cell-Penetrating Asymmetric SiRNAs. Mol. Pharm. 2020, 17, 777–786. [Google Scholar] [CrossRef]
- Prakash, T.P.; Mullick, A.E.; Lee, R.G.; Yu, J.; Yeh, S.T.; Low, A.; Chappell, A.E.; Østergaard, M.E.; Murray, S.; Gaus, H.J.; et al. Fatty Acid Conjugation Enhances Potency of Antisense Oligonucleotides in Muscle. Nucleic Acids Res. 2019, 47, 6029–6044. [Google Scholar] [CrossRef]
- Chappell, A.E.; Gaus, H.J.; Berdeja, A.; Gupta, R.; Jo, M.; Prakash, T.P.; Oestergaard, M.; Swayze, E.E.; Seth, P.P. Mechanisms of Palmitic Acid-Conjugated Antisense Oligonucleotide Distribution in Mice. Nucleic Acids Res. 2020, 48, 4382–4395. [Google Scholar] [CrossRef]
- Biscans, A.; Coles, A.; Echeverria, D.; Khvorova, A. The Valency of Fatty Acid Conjugates Impacts SiRNA Pharmacokinetics, Distribution, and Efficacy in Vivo. J. Control. Release 2019, 302, 116–125. [Google Scholar] [CrossRef]
- Elkina, Y.; von Haehling, S.; Anker, S.D.; Springer, J. The Role of Myostatin in Muscle Wasting: An Overview. J. Cachexia Sarcopenia Muscle 2011, 2, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Pirruccello-Straub, M.; Jackson, J.; Wawersik, S.; Webster, M.T.; Salta, L.; Long, K.; McConaughy, W.; Capili, A.; Boston, C.; Carven, G.J.; et al. Blocking Extracellular Activation of Myostatin as a Strategy for Treating Muscle Wasting. Sci. Rep. 2018, 8, 2292. [Google Scholar] [CrossRef]
- Kobayashi, M.; Kasamatsu, S.; Shinozaki, S.; Yasuhara, S.; Kaneki, M. Myostatin Deficiency Not Only Prevents Muscle Wasting but Also Improves Survival in Septic Mice. Am. J. Physiol. -Endocrinol. Metab. 2021, 320, E150–E159. [Google Scholar] [CrossRef]
- St. Andre, M.; Johnson, M.; Bansal, P.N.; Wellen, J.; Robertson, A.; Opsahl, A.; Burch, P.M.; Bialek, P.; Morris, C.; Owens, J. A Mouse Anti-Myostatin Antibody Increases Muscle Mass and Improves Muscle Strength and Contractility in the Mdx Mouse Model of Duchenne Muscular Dystrophy and Its Humanized Equivalent, Domagrozumab (PF-06252616), Increases Muscle Volume in Cynomolgus Monkeys. Skelet. Muscle 2017, 7, 25. [Google Scholar] [CrossRef]
- Wagner, K.R. The Elusive Promise of Myostatin Inhibition for Muscular Dystrophy. Curr. Opin. Neurol. 2020, 33, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Mariot, V.; Le Guiner, C.; Barthélémy, I.; Montus, M.; Blot, S.; Torelli, S.; Morgan, J.; Muntoni, F.; Voit, T.; Dumonceaux, J. Myostatin Is a Quantifiable Biomarker for Monitoring Pharmaco-Gene Therapy in Duchenne Muscular Dystrophy. Mol. Ther.—Methods Clin. Dev. 2020, 18, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Biscans, A.; Caiazzi, J.; McHugh, N.; Hariharan, V.; Muhuri, M.; Khvorova, A. Docosanoic Acid Conjugation to SiRNA Enables Functional and Safe Delivery to Skeletal and Cardiac Muscles. Mol. Ther. 2021, 29, 1382–1394. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.; Maruyama, R.; Yokota, T. Eteplirsen in the Treatment of Duchenne Muscular Dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurster, C.D.; Ludolph, A.C. Nusinersen for Spinal Muscular Atrophy. Ther. Adv. Neurol. Disord. 2018, 11, 1756285618754459. [Google Scholar] [CrossRef] [Green Version]
- Scoto, M.; Finkel, R.; Mercuri, E.; Muntoni, F. Genetic Therapies for Inherited Neuromuscular Disorders. Lancet Child Adolesc. Health 2018, 2, 600–609. [Google Scholar] [CrossRef]
- Carver, M.P.; Charleston, J.S.; Shanks, C.; Zhang, J.; Mense, M.; Sharma, A.K.; Kaur, H.; Sazani, P. Toxicological Characterization of Exon Skipping Phosphorodiamidate Morpholino Oligomers (PMOs) in Non-Human Primates. J. Neuromuscul. Dis. 2016, 30, 381–393. [Google Scholar] [CrossRef]
- Marian, C.O.; Cho, S.K.; McEllin, B.M.; Maher, E.A.; Hatanpaa, K.J.; Madden, C.J.; Mickey, B.E.; Wright, W.E.; Shay, J.W.; Bachoo, R.M. The Telomerase Antagonist, Imetelstat, Efficiently Targets Glioblastoma Tumor-Initiating Cells Leading to Decreased Proliferation and Tumor Growth. Clin. Cancer Res. 2010, 16, 154–163. [Google Scholar] [CrossRef] [Green Version]
- Shammas, M.A.; Koley, H.; Bertheau, R.C.; Neri, P.; Fulciniti, M.; Tassone, P.; Blotta, S.; Protopopov, A.; Mitsiades, C.; Batchu, R.B.; et al. Telomerase Inhibitor GRN163L Inhibits Myeloma Cell Growth in Vitro and in Vivo. Leukemia 2008, 22, 1410–1418. [Google Scholar] [CrossRef]
- Shammas, M.A.; Qazi, A.; Batchu, R.B.; Bertheau, R.C.; Wong, J.Y.Y.; Rao, M.Y.; Prasad, M.; Chanda, D.; Ponnazhagan, S.; Anderson, K.C.; et al. Telomere Maintenance in Laser Capture Microdissection-Purified Barrett’s Adenocarcinoma Cells and Effect of Telomerase Inhibition in Vivo. Clin. Cancer Res. 2008, 14, 4971–4980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gellert, G.C.; Dikmen, Z.G.; Wright, W.E.; Gryaznov, S.; Shay, J.W. Effects of a Novel Telomerase Inhibitor, GRN163L, in Human Breast Cancer. Breast Cancer Res. Treat. 2006, 96, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Dikmen, Z.G.; Gellert, G.C.; Jackson, S.; Gryaznov, S.; Tressler, R.; Dogan, P.; Wright, W.E.; Shay, J.W. In Vivo Inhibition of Lung Cancer by GRN163L: A Novel Human Telomerase Inhibitor. Cancer Res. 2005, 65, 7866–7873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djojosubroto, M.W.; Chin, A.C.; Go, N.; Schaetzlein, S.; Manns, M.P.; Gryaznov, S.; Harley, C.B.; Rudolph, K.L. Telomerase Antagonists GRN163 and GRN163L Inhibit Tumor Growth and Increase Chemosensitivity of Human Hepatoma. Hepatology 2005, 42, 1127–1136. [Google Scholar] [CrossRef]
- Herbert, B.-S.; Gellert, G.C.; Hochreiter, A.; Pongracz, K.; Wright, W.E.; Zielinska, D.; Chin, A.C.; Harley, C.B.; Shay, J.W.; Gryaznov, S.M. Lipid Modification of GRN163, an N3′ → P5′ Thio-Phosphoramidate Oligonucleotide, Enhances the Potency of Telomerase Inhibition. Oncogene 2005, 24, 5262–5268. [Google Scholar] [CrossRef] [Green Version]
- Benizri, S.; Gissot, A.; Martin, A.; Vialet, B.; Grinstaff, M.W.; Barthélémy, P. Bioconjugated Oligonucleotides: Recent Developments and Therapeutic Applications. Bioconjugate Chem. 2019, 30, 366–383. [Google Scholar] [CrossRef]
- Burchett, K.M.; Yan, Y.; Ouellette, M.M. Telomerase Inhibitor Imetelstat (GRN163L) Limits the Lifespan of Human Pancreatic Cancer Cells. PLoS ONE 2014, 9, e85155. [Google Scholar] [CrossRef] [Green Version]
- Kauss, T.; Arpin, C.; Bientz, L.; Vinh Nguyen, P.; Vialet, B.; Benizri, S.; Barthélémy, P. Lipid Oligonucleotides as a New Strategy for Tackling the Antibiotic Resistance. Sci. Rep. 2020, 10, 1054. [Google Scholar] [CrossRef]
- Nishina, T.; Numata, J.; Nishina, K.; Yoshida-Tanaka, K.; Nitta, K.; Piao, W.; Iwata, R.; Ito, S.; Kuwahara, H.; Wada, T.; et al. Chimeric Antisense Oligonucleotide Conjugated to α-Tocopherol. Mol. Ther.—Nucleic Acids 2015, 4, e220. [Google Scholar] [CrossRef]
- Orengo, J.P.; Chambon, P.; Metzger, D.; Mosier, D.R.; Snipes, G.J.; Cooper, T.A. Expanded CTG Repeats within the DMPK 3’ UTR Causes Severe Skeletal Muscle Wasting in an Inducible Mouse Model for Myotonic Dystrophy. Proc. Natl. Acad. Sci. USA 2008, 105, 2646–2651. [Google Scholar] [CrossRef] [Green Version]
- Østergaard, M.E.; Jackson, M.; Low, A.; Chappell, A.E.; Lee, R.G.; Peralta, R.Q.; Yu, J.; Kinberger, G.A.; Dan, A.; Carty, R.; et al. Conjugation of Hydrophobic Moieties Enhances Potency of Antisense Oligonucleotides in the Muscle of Rodents and Non-Human Primates. Nucleic Acids Res. 2019, 47, 6045–6058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, C.A.; Castanotto, D. FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther. 2017, 25, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Type | Modification | Mechanism | Indication/Target | FDA Approval |
|---|---|---|---|---|---|
| Fomivirsen | ASO | 21 nt PS DNA | RNase H1 | Cytomegalovirus retinitis/CMV UL123 | Aug 1998 |
| Pegaptanib | Aptamer | 27 nt 2ʹ-F/2ʹ-OME PEGylated | Blocking binding | Neovascular age-related macular degeneration/VEGF-165 | Dec 2004 |
| Mipomersen | ASO Gapmer | 20 nt PS 2ʹ-MOE | RNase H1 | Homozygous familial Hypercholesterolemia/APOB | Jan 2013 |
| Defibrotide | ssDNA and dsDNA | Mixture of PO | Non single sequence dependent based mechanism | hepatic veno-occlusive disease | Mar 2016 |
| Nusinersen | Steric block ASO | 18 nt PS 2′-MOE | Splicing, intron 7 | Spinal muscular atrophy/SMN2 exon 7 | Dec 2016 |
| Eteplirsen | Steric block ASO | 30 nt PMO | Splicing, exon 51 | Duchenne muscular dystrophy/DMD exon 51 | Sep 2016 |
| Milasen | ASO | 22 nt PS 2′-MOE | Splicing | Batten disease/CLN7 | Jan 2018 |
| Patisiran | siRNA LNP formulation | 19 + 2 nt 2′-OME | Ago2 | Hereditary transthyretin amyloidosis, polyneuropathy-TTR | Aug 2018 |
| Inotersen | Gapmer ASO | 20 nt PS 2ʹ-MOE | RNase H1 | hereditary transthyretin amyloidosis, polyneuropathy/TTR | Oct 2018 |
| Givosiran | Dicer substrate siRNA | 21/23 nt- GalNAc conjugate | Ago2 | Acute hepatic porphyria ALAS1 | Nov 2019 |
| Golodirsen | Steric block ASO | 25 nt PMO | Splicing, exon 53 | Duchenne muscular dystrophy/DMD exon 53 | Dec 2019 |
| Viltolarsen | ASO | 21 nt-PMO | Splicing | Duchenne muscular dystrophy/DMD exon 53 | Aug 2020 |
| Casimersen | ASO | 22-PMO | Splicing | Duchenne muscular dystrophy/DMD Exon 45 | Feb 2021 |
| Inclisiran | siRNA | 21/23 nt- GalNAc conjugate | Ago2 | Hypercholesterolaemia/PCSKK9 | Dec 2021 |
| Parameter (Units) | Chol-hsiRNA |
|---|---|
| k(min−1) | 0.0013 |
| t1/2α(min) | 515.8 |
| t1/2β (min) | 33.2 |
| Cmax (µg/mL) | 753.4 |
| AUC0–48 h (µg/mL·min) | 54,532.5 |
| AUC0-inf (µg/mL·min) | 54,807.5 |
| MRT0-inf (min) | 156.9 |
| Vz (mL) | 6.8 |
| Cl (mL/min) | 0.0091 |
| Sequences | Spacer | Conjugates (X) | Source |
|---|---|---|---|
| Passenger 5’-CUUACCGACUGGAAGA-3’-X Guide 3’-CCGGACGGGAGCGCCGAAUGGCUGACCUUCU-5 | N/A | Cholesterol | Hwang et al. |
| X-5’-TAGGGTTAGACAA-3’ | 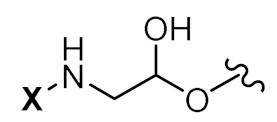 | Palmitic acid (16C) | Herbert et al. |
| X-5’-TCAACAATAAATACCGAGG-3’ | 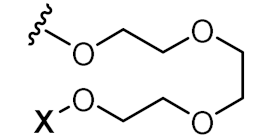 | α-tocopherol | Østergaard et al. |
| Sense X-5’-GGAGGAACUCUCCUGAUGAAU-3’ Anti-sense 5’-UCAUCAGGAGAGUUCCUGCCG-3’ | 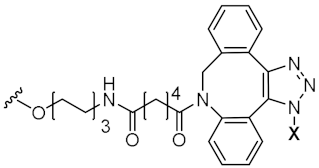 | Squalene | Massaad-Massade et al. |
| Sequence | Conjugates (X = 5’-end) | Albumin Ki (µM) | LDL Ki (µM) | HDL Ki (µM) |
|---|---|---|---|---|
| GCATTCTAATAGCAGC | None | 24.00 | N/A | N/A |
| X- GCATTCTAATAGCAGC | C8 (Octanoyl) | 2.20 | 11.80 | 5.80 |
| X- GCATTCTAATAGCAGC | C10 (Decanoyl) | 4.99 | 3.20 | 1.70 |
| X- GCATTCTAATAGCAGC | C12 (Dodecanoyl) | 3.22 | 1.30 | 0.75 |
| X- GCATTCTAATAGCAGC | C14 (Myristoyl) | 1.97 | 0.36 | 0.79 |
| X- GCATTCTAATAGCAGC | C16 (Palmitoyl) | 0.92 | 0.13 | 0.79 |
| X- GCATTCTAATAGCAGC | C18 (Stearoyl) | 0.85 | 0.17 | 0.66 |
| X- GCATTCTAATAGCAGC | C20 (Eicosanoyl) | 0.91 | 0.22 | 1.26 |
| X- GCATTCTAATAGCAGC | C22 (Docosanoyl) | 0.97 | 0.31 | 1.30 |
| Parameter (Units) | Monomeric Myr | Dimeric Myr | Trimeric Myr |
|---|---|---|---|
| kabs (min−1) | 0.0562 | 0.0213 | 0.0341 |
| t1/2 abs (min) | 12.3 | 32.5 | 20.3 |
| kβ (min−1) | 0.0218 | 0.0050 | 0.0015 |
| t1/2 β (min) | 54.1 | 139.0 | 465.3 |
| Tmax (min) | 60 | 120 | 120 |
| Cmax (µg/mL) | 21.4 | 6.1 | 0.9 |
| AUC (µg/mL*min) | 3768.1 | 3511.1 | 984.6 |
| MRT (min) | 644.7 | 1534.5 | 2009.6 |
| Parameter | Toc-17-mer ASO |
|---|---|
| AUC (∞) (ug/mL·min) | 379 ± 14 |
| CLtot (mL/min/g) | 0.0079 ± 0.0005 |
| MRT (min) | 32 ± 1 |
| Vdss (mL/g) | 0.252 ± 0.023 |
| Kα (min−1) | 0.0571 ± 0.0041 |
| Kβ (min−1) | 0.00272 ± 0.00137 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, P.; Weldemichael, T.; Liu, Z.; Li, H.-y. Delivery of Oligonucleotides: Efficiency with Lipid Conjugation and Clinical Outcome. Pharmaceutics 2022, 14, 342. https://doi.org/10.3390/pharmaceutics14020342
Tran P, Weldemichael T, Liu Z, Li H-y. Delivery of Oligonucleotides: Efficiency with Lipid Conjugation and Clinical Outcome. Pharmaceutics. 2022; 14(2):342. https://doi.org/10.3390/pharmaceutics14020342
Chicago/Turabian StyleTran, Phuc, Tsigereda Weldemichael, Zhichao Liu, and Hong-yu Li. 2022. "Delivery of Oligonucleotides: Efficiency with Lipid Conjugation and Clinical Outcome" Pharmaceutics 14, no. 2: 342. https://doi.org/10.3390/pharmaceutics14020342
APA StyleTran, P., Weldemichael, T., Liu, Z., & Li, H.-y. (2022). Delivery of Oligonucleotides: Efficiency with Lipid Conjugation and Clinical Outcome. Pharmaceutics, 14(2), 342. https://doi.org/10.3390/pharmaceutics14020342