
Design and Optimization of Pioglitazone Hydrochloride Self-Nanoemulsifying Drug Delivery System (SNEDDS) Incorporated into an Orally Disintegrating Tablet


 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Design of PGZ Liquid Self-Nanoemulsifying Drug Delivery Systems (L-SNEDDS)
2.2.1. PGZ Equilibrium Solubility Study
2.2.2. Construction of Pseudo-Ternary Phase Diagrams
2.2.3. Preparation of PGZ L-SNEDDS
2.2.4. Assessment of PGZ L-SNEDDS
Visual Inspection, Self-Emulsification Time and Robustness
Droplet Size (PS) and Polydispersity Index (PDI)
Percentage of Content
L-SNEDDS In Vitro Release
L-SNEDDS Equilibrium Solubility
Thermodynamic Stability
2.3. Preparation and Characterization of Solidified Self-Nanoemulsifying Formulations (S-SNEDDS)
2.3.1. Determination of Optimum L-SNEDDS: Adsorbent Ratio
2.3.2. Differential Scanning Calorimetry (DSC)
2.3.3. Fourier Transform Infrared Spectroscopy (FT-IR)
2.3.4. X-ray Diffraction (XRD)
2.3.5. Scanning Electron Microscopy (SEM)
2.4. Preparation and Characterization of Orally Disintegrating Tablets (ODT)
2.4.1. Hardness, Friability and Disintegration Testing
2.4.2. Content Uniformity and Weight Variation
2.4.3. In Vitro Release Study
2.4.4. Pharmacokinetic Study in Healthy Rats
2.4.5. Pharmacodynamic Study in Diabetic Rats
2.4.6. Accelerated Stability Studies
2.5. Statistical Analysis
3. Results
3.1. PGZ Solubility Study
3.2. Construction of Pseudo-Ternary Phase Diagrams
3.3. Assessment of PGZ L-SNEDDS
3.3.1. Visual Inspection, Self-Emulsification Time and Robustness
3.3.2. Droplet Size (PS) and Polydispersity Index (PDI)
3.3.3. Percentage of Content
3.3.4. L-SNEDDS Equilibrium Solubility
3.3.5. Thermodynamic Stability
3.4. Optimization and Characterization of Solidified Self-Nanoemulsifying Formulations (S-SNEDDS)
3.4.1. Micrometric Properties for Determination of Optimum L-SNEDDS: Adsorbent Ratio
3.4.2. Differential Scanning Calorimetry (DSC)
3.4.3. Fourier Transform Infrared Spectroscopy (FT-IR)
3.4.4. X-ray Powder Diffraction (XRPD)
3.4.5. Scanning Electron Microscopy (SEM)
3.5. Characterization of Oral Disintegrating SNEDDS Tablets (T-SNEDDS)
3.5.1. Tablet Quality Control Assessment
3.5.2. In Vitro Release Studies
In Vitro Release of Liquid SNEDDS (L-SNEDDS)
In Vitro Release of Solidified Formulation (S-SNEDDS)
In Vitro Release of Orally Disintegrating Tablets (T-SNEDDS)
3.5.3. In Vivo Studies
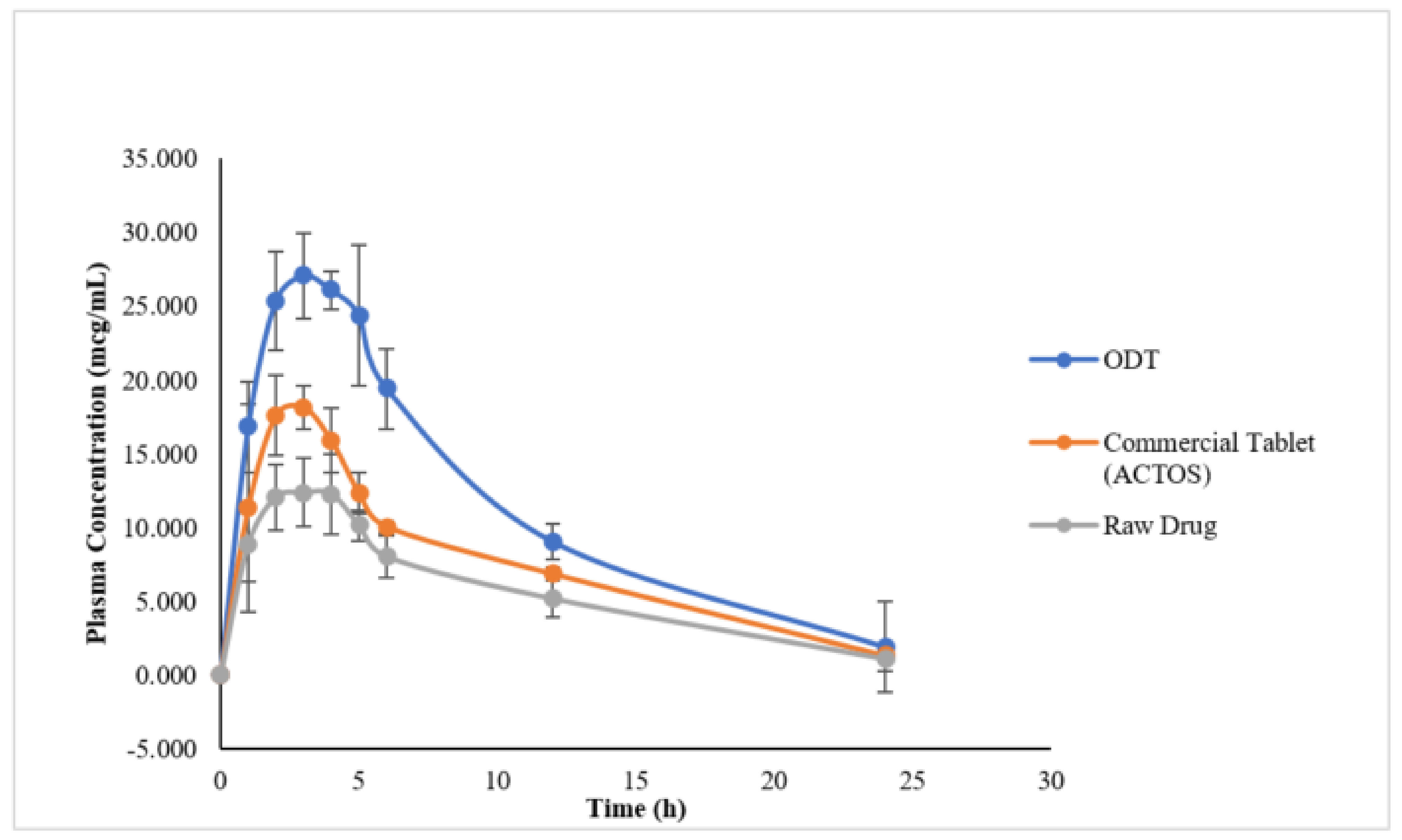
Pharmacokinetic Study in Healthy Rats
Pharmacodynamic Study in Diabetic Rats
3.5.4. Stability Studies
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scientific Discussion, European Medicines Agency. Available online: www.emeuropeasia.org (accessed on 1 December 2020).
- Charman, S.A.; Charman, W.N.; Rogge, M.C.; Wilson, T.D.; Dutko, F.J.; Pouton, C.W. Self-emulsifying drug delivery systems: Formulation and biopharmaceutic evaluation of an investigational lipophilic compound. Pharm. Res. 1992, 9, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.J.H.; Pouton, C.W.; Cuine, J.F.; Charman, W.N. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv. Drug Deliv. Rev. 2008, 60, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Lu, Y.; Qi, J.; Nie, S.; Hu, F.; Pan, W.; Wu, W. Solid self-nanoemulsifying cyclosporin A pellets prepared by fluid-bed coating: Preparation, characterization and in vitro redispersibility. Int. J. Nanomed. 2011, 6, 795. [Google Scholar]
- Gumaste, S.G.; Dalrymple, D.M.; Serajuddin, A.T. Development of solid SEDDS, V: Compaction and drug release properties of tablets prepared by adsorbing lipid-based formulations onto Neusilin® US2. Pharm. Res. 2013, 30, 3186–3199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahba, A.A.-W.; Ahmed, A.R.; Alanazi, F.K.; Mohsin, K.; Abdel-Rahman, S.I. Multi-layer self-nanoemulsifying pellets: An innovative drug delivery system for the poorly water-soluble drug cinnarizine. AAPS Pharmscitech 2018, 19, 2087–2102. [Google Scholar] [CrossRef]
- Khanfar, M.; Al-Nimry, S. Stabilization and amorphization of lovastatin using different types of silica. AAPS PharmSciTech 2017, 18, 2358–2367. [Google Scholar] [CrossRef]
- Abd-Elhakeem, E.; Teaima, M.H.; Abdelbary, G.A.; El Mahrouk, G.M. Bioavailability enhanced clopidogrel-loaded solid SNEDDS: Development and in-vitro/in-vivo characterization. J. Drug Deliv. Sci. Technol. 2019, 49, 603–614. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Tan, A.; Santos, L.; Ngo, D.; Edwards, G.A.; Porter, C.J.; Prestidge, C.A.; Boyd, B.J. Silica–lipid hybrid (SLH) formulations enhance the oral bioavailability and efficacy of celecoxib: An in vivo evaluation. J. Control. Release 2013, 167, 85–91. [Google Scholar] [CrossRef]
- Midha, K.; Nagpal, M.; Aggarwal, G.; Singh, T.G. Development of dispersible self-microemulsifying tablet of atorvastatin. Pharm. Methods 2015, 6, 9–25. [Google Scholar] [CrossRef] [Green Version]
- Asthana, A.; Aggarwal, S.; Asthana, G. Oral dispersible tablets: Novel technology and development. Int. J. Pharm. Sci. Rev. Res. 2013, 20, 193–199. [Google Scholar]
- Higuchi, T. A phase solubility technique. Adv. Anal. Chem. Instrum. 1965, 4, 117–211. [Google Scholar]
- El-Laithy, H.M.; Basalious, E.B.; El-Hoseiny, B.M.; Adel, M.M. Novel self-nanoemulsifying self-nanosuspension (SNESNS) for enhancing oral bioavailability of diacerein: Simultaneous portal blood absorption and lymphatic delivery. Int. J. Pharm. 2015, 490, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Sjöholm, E.; Sandler, N. Additive manufacturing of personalized orodispersible warfarin films. Int. J. Pharm. 2019, 564, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, Y.; Feng, N.; Xu, J. Preparation and evaluation of self-microemulsifying drug delivery system of oridonin. Int. J. Pharm. 2008, 355, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Ammar, H.; Ghorab, M.; Mostafa, D.M.; Ghoneim, A.M. Self-nanoemulsifying drug delivary system for sertraline hydrochloride: Design, preparation and characterization. Future 2013, 1, 1. [Google Scholar]
- Yin, Y.M.; Cui, F.D.; Mu, C.F.; Choi, M.K.; Kim, J.S.; Chung, S.J.; Shim, C.K.; Kim, D.D. Docetaxel microemulsion for enhanced oral bioavailability: Preparation and in vitro and in vivo evaluation. J. Control. Release 2009, 140, 86–94. [Google Scholar] [CrossRef]
- Mohd, A.B.; Sanka, K.; Bandi, S.; Diwan, P.V.; Shastri, N. Solid self-nanoemulsifying drug delivery system (S-SNEDDS) for oral delivery of glimepiride: Development and antidiabetic activity in albino rabbits. Drug Deliv. 2015, 22, 499–508. [Google Scholar] [CrossRef]
- U.S. Food and Drug Database. Available online: www.accessdata.fda.gov (accessed on 1 February 2009).
- Shafiq, S.; Shakeel, F.; Talegaonkar, S.; Ahmad, F.J.; Khar, R.K.; Ali, M. Development and bioavailability assessment of ramipril nanoemulsion formulation. Eur. J. Pharm. Biopharm. 2007, 66, 227–243. [Google Scholar] [CrossRef]
- Pathade, P.; Imran, M.; Bairagi, V.; Ahire, Y. Development and validation of stability indicating UV spectrophotometric method for the estimation of sitagliptin phosphate in bulk and tablet dosage form. J. Pharm. Res. 2011, 4, 871–873. [Google Scholar]
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef]
- The ICH Guidelines for Stability Testing of Active Pharmaceutical Ingredients and Finished Pharmaceutical Products (Annex10). Available online: https://database.ich.org/sites/default/files/Q1F_Stability_Guideline_WHO_2018.pdf (accessed on 1 March 2017).
- Wang, L.; Dong, J.; Chen, J.; Eastoe, J.; Li, X. Design and optimization of a new self-nanoemulsifying drug delivery system. J. Colloid Interface Sci. 2009, 330, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Madan, J.R.; Sudarshan, B.; Kadam, V.S.; Kama, D. Formulation and development of self-microemulsifying drug delivery system of pioglitazone. Asian J. Pharm. 2014, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Mohsin, K.; Long, M.A.; Pouton, C.W. Design of lipid-based formulations for oral administration of poorly water-soluble drugs: Precipitation of drug after dispersion of formulations in aqueous solution. J. Pharm. Sci. 2009, 98, 3582–3595. [Google Scholar] [CrossRef] [PubMed]
- Shahba, A.; Ahmed, A.R.; Mohsin, K.; Abdel-Rahman, S.I.; Alanazi, F.K. Solidification of cinnarizine self-nanoemulsifying drug delivery systems by fluid bed coating: Optimization of the process and formulation variables. Pharm. Int. J. Pharm. Sci. 2017, 72, 143–151. [Google Scholar]
- Al-Khattawi, A.; Mohammed, A.R. Compressed orally disintegrating tablets: Excipients evolution and formulation strategies. Expert Opin. Drug Deliv. 2013, 10, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Manivannan, R. Oral disintegrating tablets: A future compaction. Drug Invent. Today 2009, 1, 61–65. [Google Scholar]
- Dobetti, L. Fast-melting tablets: Developments and technologies. Pharm. Technol. 2001, 12, 44. [Google Scholar]
- Guidance for Industry: Orally Disintegrating Tablets. FDA CDER; FDA/Center for Drug Evaluation and Research: Silver Spring, MD, USA, 2008. Available online: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070578.pdf (accessed on 1 October 2012).
- Deshmukh, A.; Kulkarni, S. Solid self-microemulsifying drug delivery system of ritonavir. Drug Dev. Ind. Pharm. 2014, 40, 477–487. [Google Scholar] [CrossRef]
- Basalious, E.B.; Shawky, N.; Badr-Eldin, S.M. SNEDDS containing bioenhancers for improvement of dissolution and oral absorption of lacidipine. I: Development and optimization. Int. J. Pharm. 2010, 391, 203–211. [Google Scholar] [CrossRef]
- Kommuru, T.; Gurley, B.; Khan, M.; Reddy, I. Self-emulsifying drug delivery systems (SEDDS) of coenzyme Q10: Formulation development and bioavailability assessment. Int. J. Pharm. 2001, 212, 233–246. [Google Scholar] [CrossRef]
- Elnaggar, Y.S.; El-Massik, M.A.; Abdallah, O.Y. Self-nanoemulsifying drug delivery systems of tamoxifen citrate: Design and optimization. Int. J. Pharm. 2009, 380, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Rane, S.S.; Anderson, B.D. What determines drug solubility in lipid vehicles: Is it predictable? Adv. Drug Deliv. Rev. 2008, 60, 638–656. [Google Scholar] [CrossRef]
- Smail, S.S.; Ghareeb, M.M.; Omer, H.K.; Al-Kinani, A.A.; Alany, R.G. Studies on surfactants, cosurfactants, and oils for prospective use in formulation of ketorolac tromethamine ophthalmic nanoemulsions. Pharmaceutics 2021, 13, 467. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.R.; Aghav, S.; Sukre, G.; Kumar, M. Determination of required HLB of Capryol 90. J. Dispers. Sci. Technol. 2014, 35, 161–167. [Google Scholar] [CrossRef]
- Legen, I.; Peternel, L.; Novak, S.; Homar, M.; Rozman, P.; Klancar, U. Self-Microemulsifying Drug Delivery System of Abiraterone or Abiraterone. Acetate. Patent WO2014009434, 10 July 2013. [Google Scholar]
- Kang, B.K.; Lee, J.S.; Chon, S.K.; Jeong, S.Y.; Yuk, S.H.; Khang, G.; Lee, H.B.; Cho, S.H. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int. J. Pharm. 2004, 274, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Eid, A.M.M.; Baie, S.H.; Arafat, O.M. The effect of surfactant blends on the production of self-emulsifying system. Int. J. Pharm. Front. Res. 2012, 2, 21–31. [Google Scholar]
- Constantinides, P.P.; Scalart, J.P.; Lancaster, C.; Marcello, J.; Marks, G.; Ellens, H.; Smith, P.L. Formulation and intestinal absorption enhancement evaluation of water-in-oil microemulsions incorporating medium-chain glycerides. Pharm. Res. 1994, 11, 1385–1390. [Google Scholar] [CrossRef]
- Alhasani, K.F.; Kazi, M.; Ibrahim, M.A.; Shahba, A.A.; Alanazi, F.K. Self-nanoemulsifying ramipril tablets: A novel delivery system for the enhancement of drug dissolution and stability. Int. J. Nanomed. 2019, 14, 5435. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Singh, S.K.; Khursheed, R.; Pandey, N.K.; Kumar, B.; Kumar, R.; Kumari, Y.; Kaur, G.; Clarisse, A.; Awasthi, A.; et al. Impact of solidification on micromeritic properties and dissolution rate of self-nanoemulsifying delivery system loaded with docosahexaenoic acid. Drug Dev. Ind. Pharm. 2020, 46, 597–605. [Google Scholar] [CrossRef]
- Verma, S.; Singh, S.K.; Verma, P.R.P. Solidified SNEDDS of loratadine: Formulation using hydrophilic and hydrophobic grades of Aerosil®, pharmacokinetic evaluations and in vivo–in silico predictions using GastroPlus™. RSC Adv. 2016, 6, 3099–3116. [Google Scholar] [CrossRef]
- Hespeler, D.; El Nomeiri, S.; Kaltenbach, J.; Müller, R.H. Nanoporous smartPearls for dermal application–Identification of optimal silica types and a scalable production process as prerequisites for marketed products. Beilstein J. Nanotechnol. 2019, 10, 1666–1678. [Google Scholar] [CrossRef] [PubMed]
- Choudhari, Y.; Reddy, U.; Monsuur, F.; Pauly, T.; Hoefer, H.; McCarthy, W. Comparative evaluation of porous silica-based carriers for lipids and liquid drug formulations. Open Mater. Sci. 2014, 1, 61–74. [Google Scholar] [CrossRef]
- Kharechkina, E.S.; Nikiforova, A.B.; Belosludtsev, K.N.; Rokitskaya, T.I.; Antonenko, Y.N.; Kruglov, A.G. Pioglitazone Is a Mild Carrier-Dependent Uncoupler of Oxidative Phosphorylation and a Modulator of Mitochondrial Permeability Transition. Pharmaceuticals 2021, 14, 1045. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Solubility (mg/mL ± SD) |
|---|---|
| Campul MCM | 32.93 ± 0.826 |
| Oleic acid | 23.05 ± 0.286 |
| Olive oil | 16.4863 ± 0.437 |
| Soybean oil | 17.9653 ± 0.676 |
| Caproyl 90 | 44.6897 ± 0.879 |
| Cremophor® RH40 | 9.2712 ± 0.970 |
| Labrafil ®M1944CS | 16.1616 ± 0.812 |
| Labrasol | 13.6003 ± 0.380 |
| Tween ®80 | 35.533 ± 0.826 |
| Cremophore EL | 48.3765 ± 0.602 |
| Propylene Glycol | 249.99 ± 1.08 |
| Transcutol® HP | 109.379 ± 0.687 |
| System No. | Formula No. | Oil % | Surfactant % | Co- Surfactant % |
|---|---|---|---|---|
| (I) Oil: Capryol 90 Surfactant: Cremophore EL Cosurfactant: Transcutol HP | S14 | 20 | 20 | 60 |
| S18 | 40 | 30 | 30 | |
| S19 | 30 | 30 | 40 | |
| S21 | 10 | 30 | 60 | |
| (II) Oil: Capryol 90 Surfactant: Cremophore EL Cosurfactant: Propylene Glycol | S14 | 20 | 20 | 60 |
| S21 | 10 | 30 | 60 | |
| S24 | 30 | 40 | 30 | |
| S30 | 10 | 50 | 40 | |
| (III) Oil: Capmul MCM Surfactant: Cremophore EL Cosurfactant: Propylene Glycol | S14 | 20 | 20 | 60 |
| S23 | 40 | 40 | 20 | |
| S24 | 30 | 40 | 30 | |
| S30 | 10 | 50 | 40 |
| Formulation No. | Emulsification Time (sec ± SD) | % T | Particle Size (nm ± SD) | PDI | Grade | % Drug Content | Solubility (mg/g) |
|---|---|---|---|---|---|---|---|
| S14-i | 34.06 ± 0.51 | 99.27% | 36.85 ± 0.40 | 0.151 | A | 98.12 ± 2.89 | 18.47 ± 0.34 |
| S18-i | 49.67 ± 0.65 | 95.97% | 61.82 ± 0.40 | 0.11 | B | 100.04 ± 1.67 | 21.57 ± 0.45 |
| S19-i | 45.78 ± 0.52 | 98.53% | 48.97 ± 0.20 | 0.07 | A | 94.28 ± 1.67 | 32.68 ± 1.41 |
| S21-i | 29.29 ± 0.49 | 98.63% | 40.21 ±0.85 | 0.56 | A | 92.35 ± 2.89 | 28.96 ± 0.27 |
| S14-ii | 31.24 ± 0.62 | 100.3% | 22.1 ± 1.58 | 0.27 | A | 110.13 ± 0.15 | 177.12 ± 0.59 |
| S21-ii | 32.47 ± 0.32 | 98.93% | 20.72 ± 0.57 | 0.271 | A | 98.92 ± 0.11 | 164.24 ± 0.16 |
| S24-ii | 60.53 ± 1.42 | 83.93% | 52.97 ± 0.41 | 0.218 | B | 99.95 ± 0.43 | 69.76 ± 0.63 |
| S30-ii | 49.83 ±0.30 | 100.1% | 15.58 ± 0.48 | 0.185 | A | 99.18 ± 0.18 | 127.74 ± 0.54 |
| S14-iii | 15.25 ± 0.80 | 96% | 121.97 ± 1.02 | 0.379 | A | 99.03 ± 0.41 | 202.56 ± 0.63 |
| S23-iii | 29.18 ± 0.90 | 90.4% | 349.73 ± 11.37 | 0.421 | B | 98.48 ± 0.36 | 135.82 ± 1.89 |
| S24-iii | 21.35 ± 0.91 | 93.5% | 124.2 ± 13.8 | 0.362 | A | 95.95 ± 1.9 | 186.18 ± 0.49 |
| S30-iii | 19.75 ± 0.61 | 99.9% | 14.12 ± 0.06 | 0.053 | A | 94.52 ± 1.44 | 191.85 ± 0.62 |
| Formulation No. | Observations Based on Thermodynamic Stability Tests | ||
|---|---|---|---|
| H/C | Cent | F/T | |
| S14-i | ✓ | ✓ | ✓ |
| S18-i | ✓ | ✓ | ✕ |
| S19-i | ✕ | ✕ | ✕ |
| S21-i | ✓ | ✓ | ✕ |
| S14-ii | ✓ | ✓ | ✓ |
| S21-ii | ✓ | ✓ | ✓ |
| S24-ii | ✓ | ✓ | ✕ |
| S30-ii | ✓ | ✓ | ✓ |
| S14-iii | ✓ | ✓ | ✓ |
| S23-iii | ✓ | ✕ | ✕ |
| S24-iii | ✓ | ✓ | ✓ |
| S30-iii | ✓ | ✓ | ✓ |
| Formulation No. | L-SNEDDS Composition | Adsorbent Type | (L-SNEDD: Adsorbent) |
|---|---|---|---|
| S-SNEDDS-1 | 15 mg PGZ in 1 g S14-iii (20% Capmul MCM, 20% Cremophor EL and 60% Propylene Glycol) | Neusilin® US2 | 1:0.5 |
| S-SNEDDS-2 | Neusilin® UFL2 | 1:0.5 | |
| S-SNEDDS-3 | Syloid® 244FP | 1:0.5 | |
| S-SNEDDS-4 | Aerosil® PH200 | 1:0.5 |
| Formula No. | Ratio (L-SNEDDS: SYL) | Appearance | ρbulk | ρtap | HR | CI | θ | Flowability |
|---|---|---|---|---|---|---|---|---|
| R1 | 1:0.25 | Caking and Wet | 0.13 | 0.18 | 1.38 | 27 | 50.2 | Poor |
| R2 | 1:0.5 | Fine and Dry | 0.17 | 0.20 | 1.18 | 15 | 35.3 | Good |
| R3 | 1:1 | Fine and Dry | 0.12 | 0.14 | 1.16 | 14.28 | 36.7 | Good |
| R4 | 1:2 | Dusty | 0.085 | 0.09 | 1.05 | 5.56 | 27.7 | Excellent |
| Formulation | % Composition | Weight (mg) | |
|---|---|---|---|
| L-SNEDDS | Capmul MCM 20%, Cremophor EL 20%, PG 60% | 100 mg | |
| S-SNEDSS | L-SNEDDS: SYL (1:0.5) | 50 mg | |
| ODT excipients | Mannitol, Acdisol, lactose, Prosolv, Mg stearate | ~350 mg | |
| Ingredients | ODT-1 | ODT-2 | ODT-3 |
| Drug incorporated in S-SNEDDS | 110 | 110 | 110 |
| Lactose | - | 335 | - |
| Mannitol | - | - | 335 |
| Acdisol | - | 50 | 50 |
| Prosolv | 385 | - | - |
| Mg stearate | 5 | 5 | 5 |
| Total Weight (mg) | 500 | 500 | 500 |
| Parameter | n * | Results |
|---|---|---|
| Diameter (mm) | 10 | 13.41± 0.01 |
| Thickness (mm) | 10 | 3.08 ± 0.01 |
| Weight Variation (%) | 10 | 0.042% |
| Content Uniformity % | 10 | 99.92% ± 1.2 |
| Friability % | 13 | 0.68% |
| Hardness (kp) | 6 | 5.73 ± 0.63 |
| Disintegration Time (sec) | 6 | 28.36 ± 0.95 |
| Pharmacokinetic Parameter | ODT | ACTOS | Raw PGZ |
|---|---|---|---|
| Cmax (µg/mL) | 27.77 ± 2.55 | 17.40 ± 3.38 | 12.91 ± 2.24 |
| Tmax (h) | 3.4 ± 1.14 | 3.0 ± 1.00 | 3.0 ± 0.89 |
| AUC0−∞ (µgh/mL) | 313.05 | 185.32 | 151.36 |
| AUMC0−∞ (µg h2/mL) | 3461.02 | 1738.71 | 1652.14 |
| Kel (1/hr) | 0.18 ± 0.082 | 0.117 ± 0.013 | 0.126 ± 0.05 |
| t1/2 (h) | 5.57 ± 4.94 | 5.95 ± 0.719 | 6.85 ± 4.35 |
| MRT (h) | 7.19 ± 0.96 | 7.87 ± 0.59 | 7.725 ± 0.84 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teaima, M.; Hababeh, S.; Khanfar, M.; Alanazi, F.; Alshora, D.; El-Nabarawi, M. Design and Optimization of Pioglitazone Hydrochloride Self-Nanoemulsifying Drug Delivery System (SNEDDS) Incorporated into an Orally Disintegrating Tablet. Pharmaceutics 2022, 14, 425. https://doi.org/10.3390/pharmaceutics14020425
Teaima M, Hababeh S, Khanfar M, Alanazi F, Alshora D, El-Nabarawi M. Design and Optimization of Pioglitazone Hydrochloride Self-Nanoemulsifying Drug Delivery System (SNEDDS) Incorporated into an Orally Disintegrating Tablet. Pharmaceutics. 2022; 14(2):425. https://doi.org/10.3390/pharmaceutics14020425
Chicago/Turabian StyleTeaima, Mahmoud, Sandra Hababeh, Mai Khanfar, Fares Alanazi, Doaa Alshora, and Mohammed El-Nabarawi. 2022. "Design and Optimization of Pioglitazone Hydrochloride Self-Nanoemulsifying Drug Delivery System (SNEDDS) Incorporated into an Orally Disintegrating Tablet" Pharmaceutics 14, no. 2: 425. https://doi.org/10.3390/pharmaceutics14020425
APA StyleTeaima, M., Hababeh, S., Khanfar, M., Alanazi, F., Alshora, D., & El-Nabarawi, M. (2022). Design and Optimization of Pioglitazone Hydrochloride Self-Nanoemulsifying Drug Delivery System (SNEDDS) Incorporated into an Orally Disintegrating Tablet. Pharmaceutics, 14(2), 425. https://doi.org/10.3390/pharmaceutics14020425