
Sparsomycin Exhibits Potent Antiplasmodial Activity In Vitro and In Vivo


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Compounds
2.2. Parasites
2.3. In Vitro Culture of P. falciparum
2.4. In Vitro Inhibition Assay of P. falciparum
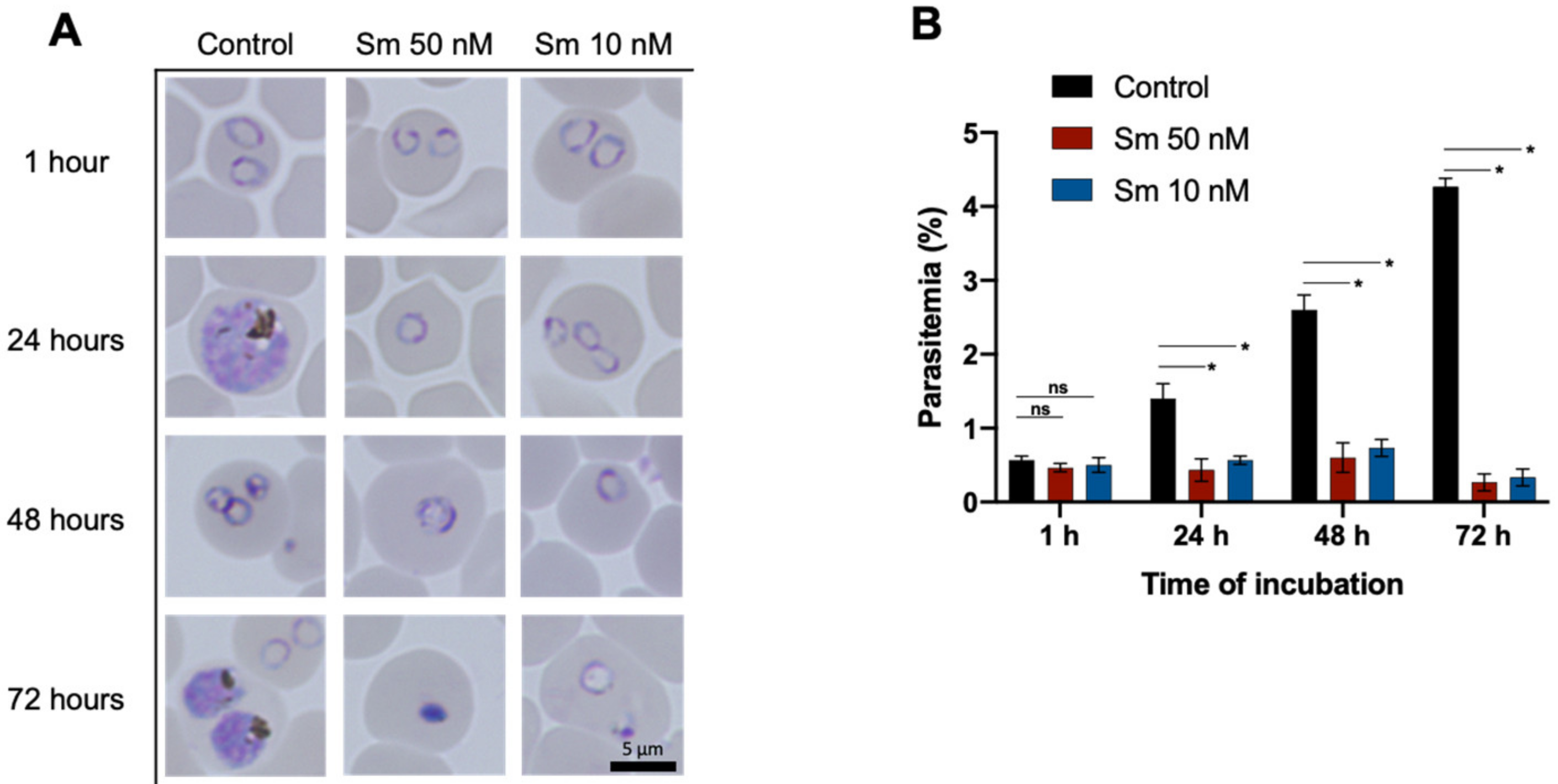
2.5. Microscopic Analysis of Parasitemia and Morphology of P. falciparum 3D7 Treated with Sm In Vitro
2.6. In Vitro Cytotoxicity in Human Cells
2.7. In Vitro Sm Hemolysis Rate in Human Erythrocytes
2.8. Mice and In Vivo Infections
2.9. Statistical Analysis
3. Results
3.1. Effects of Sm on P. falciparum In Vitro
3.2. Effects of Sm on parasitemia and the Morphology of P. falciparum 3D7 In Vitro
3.3. Effect of Sm on P. yoelii 17XNL-Infected Mice
3.4. Effect of Sm on P. berghei ANKA-Infected Mice
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, G.-H.; Gamez, S.; Raban, R.R.; Marshall, J.M.; Alphey, L.; Li, M.; Rasgon, J.L.; Akbari, O.S. Combating Mosquito-Borne Diseases Using Genetic Control Technologies. Nat. Commun. 2021, 12, 4388. [Google Scholar] [CrossRef]
- World Health Organization. World Malaria Report 2021; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- World Health Organization. Malaria Eradication: Benefits, Future Scenarios and Feasibility. A Report of the Strategic Advisory Group on Malaria Eradication; World Health Organization: Geneva, Switzerland, 2019; ISBN 9789240003675. [Google Scholar]
- Talisuna, A.O.; Bloland, P.; D’Alessandro, U. History, Dynamics, and Public Health Importance of Malaria Parasite Resistance. Clin. Microbiol. Rev. 2004, 17, 235–254. [Google Scholar] [CrossRef] [Green Version]
- Ross, L.S.; Fidock, D.A. Elucidating Mechanisms of Drug-Resistant Plasmodium falciparum. Cell Host Microbe 2019, 26, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Argoudelis, A.D.; Herr, R.R. Sparsomycin, a New Antitumor Antibiotic. II. Isolation and Characterization. Antimicrob. Agents Chemother. 1962, 780–786. [Google Scholar]
- Ottenheijm, H.C.J.; Van Den Broek, L.A.G.M.; Ballesta, J.P.G.; Zylicz, Z. 6 Chemical and biological aspects of sparsomycin, an antibiotic, from streptomyces. In Progress in Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 1986; Volume 23, pp. 219–268. [Google Scholar]
- Porse, B.T.; Kirillov, S.V.; Awayez, M.J.; Ottenheijm, H.C.J.; Garrett, R.A. Direct Crosslinking of the Antitumor Antibiotic Sparsomycin, and Its Derivatives, to A2602 in the Peptidyl Transferase Center of 23S-like RRNA within Ribosome-TRNA Complexes. Proc. Natl. Acad. Sci. USA 1999, 96, 9003–9008. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.L.; Moore, P.B.; Steitz, T.A. Structures of Five Antibiotics Bound at the Peptidyl Transferase Center of the Large Ribosomal Subunit. J. Mol. Biol. 2003, 330, 1061–1075. [Google Scholar] [CrossRef]
- Nelli, M.R.; Heitmeier, K.N.; Looper, R.E. Dissecting the Nucleoside Antibiotics as Universal Translation Inhibitors. Acc. Chem. Res. 2021, 54, 2798–2811. [Google Scholar] [CrossRef]
- Burgers, L.D.; Fürst, R. Natural Products as Drugs and Tools for Influencing Core Processes of Eukaryotic MRNA Translation. Pharmacol. Res. 2021, 170, 105535. [Google Scholar] [CrossRef] [PubMed]
- Mcfarlane, J.R.; Yanoff, M.; Scheie, H.G. Toxic Retinopathy Following Sparsomycin Therapy. Arch. Ophthalmol. 1966, 76, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Schmeing, T.M.; Huang, K.S.; Kitchen, D.E.; Strobel, S.A.; Steitz, T.A. Structural Insights into the Roles of Water and the 2′ Hydroxyl of the P Site TRNA in the Peptidyl Transferase Reaction. Mol. Cell 2005, 20, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Bashan, A.; Agmon, I.; Zarivach, R.; Schluenzen, F.; Harms, J.; Berisio, R.; Bartels, H.; Franceschi, F.; Auerbach, T.; Hansen, H.A.S.; et al. Structural Basis of the Ribosomal Machinery for Peptide Bond Formation, Translocation, and Nascent Chain Progression. Mol. Cell 2003, 11, 91–102. [Google Scholar] [CrossRef]
- Prugnolle, F.; Durand, P.; Ollomo, B.; Duval, L.; Ariey, F.; Arnathau, C.; Gonzalez, J.P.; Leroy, E.; Renaud, F. A Fresh Look at the Origin of Plasmodium falciparum, the Most Malignant Malaria Agent. PLoS Pathog. 2011, 7, e1001283. [Google Scholar] [CrossRef]
- Ding, X.C.; Ubben, D.; Wells, T.N. A Framework for Assessing the Risk of Resistance for Anti-Malarials in Development. Malar. J. 2012, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, R.; Awasthi, V.; Thakur, R.S.; Pande, V.; Chattopadhyay, D.; Das, J. CD4+ICOS+Foxp3+: A Sub-Population of Regulatory T Cells Contribute to Malaria Pathogenesis. Malar. J. 2022, 21, 1–10. [Google Scholar] [CrossRef]
- Leesombun, A.; Iijima, M.; Pagmadulam, B.; Orkhon, B.; Doi, H.; Issiki, K.; Sawa, R.; Nihei, C.-i.; Nishikawa, Y. Metacytofilin Has Potent Anti-Malarial Activity. Parasitol. Int. 2021, 81, 102267. [Google Scholar] [CrossRef]
- Smilkstein, M.; Sriwilaijaroen, N.; Kelly, J.X.; Wilairat, P.; Riscoe, M. Simple and Inexpensive Fluorescence-Based Technique for High-Throughput Antimalarial Drug Screening. Antimicrob. Agents Chemother. 2004, 48, 1803–1806. [Google Scholar] [CrossRef] [Green Version]
- Le Manach, C.; Scheurer, C.; Sax, S.; Schleiferböck, S.; Cabrera, D.G.; Younis, Y.; Paquet, T.; Street, L.; Smith, P.; Ding, X.C.; et al. Fast in Vitro Methods to Determine the Speed of Action and the Stage-Specificity of Anti-Malarials in Plasmodium falciparum. Malar. J. 2013, 12, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Evans, B.C.; Nelson, C.E.; Yu, S.S.; Beavers, K.R.; Kim, A.J.; Li, H.; Nelson, H.M.; Giorgio, T.D.; Duvall, C.L. Ex Vivo Red Blood Cell Hemolysis Assay for the Evaluation of PH-Responsive Endosomolytic Agents for Cytosolic Delivery of Biomacromolecular Drugs. J. Vis. Exp. 2013, 73, e50166. [Google Scholar] [CrossRef] [Green Version]
- Zylicz, Z.; Wagener, D.J.T.; van Rennes, H.; van der Kleijn, E.; Lelieveld, P.; van den Broek, L.A.G.M.; Ottenheijm, H.C.J. In Vivo Antitumor Activity of Sparsomycin and Its Analogues in Eight Murine Tumor Models. Invest. New Drugs 1988, 6, 285–292. [Google Scholar] [CrossRef]
- Chen, M.; Theander, T.G.; Christensen, S.B.; Hviid, L.; Zhai, L.; Kharazmi, A. Licochalcone A, a New Antimalarial Agent, Inhibits in Vitro Growth of the Human Malaria Parasite Plasmodium falciparum and Protects Mice from P. yoelii Infection. Antimicrob. Agents Chemother. 1994, 38, 1470–1475. [Google Scholar] [CrossRef] [Green Version]
- Peters, W.; Portus, J.H.; Robinson, B.L. The Chemotherapy of Rodent Malaria, XXII: The Value of Drug-Resistant Strains of P. berghei in Screening for Blood Schizontocidal Activity. Ann. Trop. Med. Parasitol. 1975, 69, 155–171. [Google Scholar] [CrossRef]
- Fredrick, K.; Noller, H.F. Catalysis of Ribosomal Translocation by Sparsomycin. Science 2003, 300, 1159–1162. [Google Scholar] [CrossRef]
- Prokhorova, I.V.; Akulich, K.A.; Makeeva, D.S.; Osterman, I.A.; Skvortsov, D.A.; Sergiev, P.V.; Dontsova, O.A.; Yusupova, G.; Yusupov, M.M.; Dmitriev, S.E. Amicoumacin A Induces Cancer Cell Death by Targeting the Eukaryotic Ribosome. Sci. Rep. 2016, 6, 27720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilles, A.; Frechin, L.; Natchiar, K.; Biondani, G.; von Loeffelholz, O.; Holvec, S.; Malaval, J.L.; Winum, J.Y.; Klaholz, B.P.; Peyron, J.F. Targeting the Human 80S Ribosome in Cancer: From Structure to Function and Drug Design for Innovative Adjuvant Therapeutic Strategies. Cells 2020, 9, 629. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.X.; Chandramohanadas, R.; Shyong-Wei Tan, K. High-Content Screening of the Medicines for Malaria Venture Pathogen Box for Plasmodium falciparum Digestive Vacuole-Disrupting Molecules Reveals Valuable Starting Points for Drug Discovery. Antimicrob. Agents Chemother. 2018, 62, e02031-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuno, K.; Burrows, J.N.; Duncan, K.; Van Huijsduijnen, R.H.; Kaneko, T.; Kita, K.; Mowbray, C.E.; Schmatz, D.; Warner, P.; Slingsby, B.T. Hit and Lead Criteria in Drug Discovery for Infectious Diseases of the Developing World. Nat. Rev. Drug Discov. 2015, 14, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Goyal, T.; Schmotzer, C.L. Validation of Hemolysis Index Thresholds Optimizes Detection of Clinically Significant Hemolysis. Am. J. Clin. Pathol. 2015, 143, 579–583. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, C.M.; Garcia, V.E.; Ahyong, V.; DeRisi, J.L. The Plasmodium Falciparum Cytoplasmic Translation Apparatus: A Promising Therapeutic Target Not yet Exploited by Clinically Approved Anti-Malarials. Malar. J. 2018, 17, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, K.E.; Habib, S.; Frugier, M.; Hoen, R.; Khan, S.; Pham, J.S.; de Pouplana, L.R.; Royo, M.; Santos, M.A.S.; Sharma, A.; et al. Protein Translation in Plasmodium Parasites. Trends Parasitol. 2011, 27, 467–476. [Google Scholar] [CrossRef]
- Hussain, T.; Yogavel, M.; Sharma, A. Inhibition of Protein Synthesis and Malaria Parasite Development by Drug Targeting of Methionyl-TRNA Synthetases. Antimicrob. Agents Chemother. 2015, 59, 1856–1867. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, P.J. Azithromycin for Malaria? Am. J. Trop. Med. Hyg. 2016, 95, 2–4. [Google Scholar] [CrossRef] [Green Version]
- Budimulja, A.S.; Syafruddin; Tapchaisri, P.; Wilairat, P.; Marzuki, S. The Sensitivity of Plasmodium Protein Synthesis to Prokaryotic Ribosomal Inhibitors. Mol. Biochem. Parasitol. 1997, 84, 137–141. [Google Scholar] [CrossRef]
- Dahl, E.L.; Shock, J.L.; Shenai, B.R.; Gut, J.; DeRisi, J.L.; Rosenthal, P.J. Tetracyclines Specifically Target the Apicoplast of the Malaria Parasite Plasmodium falciparum. Antimicrob. Agents Chemother. 2006, 50, 3124–3131. [Google Scholar] [CrossRef] [Green Version]
- Zylicz, Z.; Theo Wagener, D.J.; del Moral, P.F.; van Rennes, H.; Wessels, J.M.C.; Winograd, B.; van der Kleijn, E.; Vree, T.B.; van Haelst, U.; van den Broek, L.A.G.M.; et al. Pharmacokinetics and Toxicology of Sparsomycin in Beagle Dogs. Cancer Chemother. Pharmacol. 1987, 20, 115–124. [Google Scholar] [CrossRef]
- Zylicz, Z. Sparsomycin and Its Analouges: A Preclinical Study on Novel Anticancer Drugs; University Hospital Nijmegen: Nijmegen, The Netherlands, 1988. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 P. falciparum (nM) | Resistance Index | CC50 HFF (μM) | SI for P. falciparum | RBC Hemolysis Rate (%) at 100 μM | ||
|---|---|---|---|---|---|---|---|
| 3D7 | K1 | 3D7 | K1 | ||||
| Sparsomycin | 12.07 ± 4.41 a | 25.43 ± 8.15 a | 2.1 | 1.14 ± 0.03 | 94.45 | 44.83 | 1.04 ± 0.23 |
| Artemisinin | 13.18 ± 2.66 a | 19.89 ± 1.51 a | 1.4 | 153.00 ± 30.76 | 10,983.49 | 7692.31 | 1.03 ± 0.46 |
| Chloroquine | 26.20 ± 3.66 b | 740.07 ± 95.67 b | 28.24 | 20.71 ± 6.80 | 790.46 | 27.98 | 0.71 ± 0.35 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ariefta, N.R.; Pagmadulam, B.; Nihei, C.-i.; Nishikawa, Y. Sparsomycin Exhibits Potent Antiplasmodial Activity In Vitro and In Vivo. Pharmaceutics 2022, 14, 544. https://doi.org/10.3390/pharmaceutics14030544
Ariefta NR, Pagmadulam B, Nihei C-i, Nishikawa Y. Sparsomycin Exhibits Potent Antiplasmodial Activity In Vitro and In Vivo. Pharmaceutics. 2022; 14(3):544. https://doi.org/10.3390/pharmaceutics14030544
Chicago/Turabian StyleAriefta, Nanang Rudianto, Baldorj Pagmadulam, Coh-ichi Nihei, and Yoshifumi Nishikawa. 2022. "Sparsomycin Exhibits Potent Antiplasmodial Activity In Vitro and In Vivo" Pharmaceutics 14, no. 3: 544. https://doi.org/10.3390/pharmaceutics14030544