
New Peptide Based Fluconazole Conjugates with Expanded Molecular Targets

 , , , , and
, , , , and
Abstract
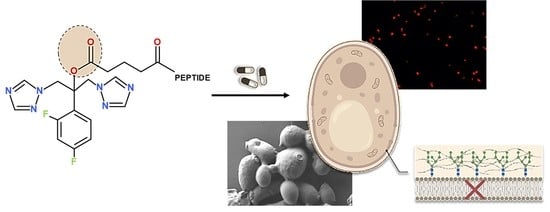
:
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Anti-Candidal In Vitro Activity
2.3. Time Kill Assay
2.4. Membrane Permeabilization Assay
2.5. SEM Analysis of the Compounds’ Effects on C. albicans Cells
2.6. Cytotoxicity
3. Conclusions
4. Materials and Methods
4.1. Solid-Phase Peptide Synthesis (SPPS)
4.2. Synthesis of FLCpOH
4.3. Synthesis of Fluconazole-Based Conjugates
4.4. Antifungal Agents
4.5. Organisms and Culture Conditions
4.6. Determination of Minimum Inhibitory Concentration (MIC) and Minimum Fungicidal Concentration (MFC)
4.7. Time-Kill Assay
4.8. Membrane Permeabilization Assay Using Propidium Iodide Staining
4.9. Scanning Electron Microscopy (SEM)
4.10. Cytotoxicity Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bongomin, F.; Gago, S.; Oladele, R.O.; Denning, D.W. Global and Multi-National Prevalence of Fungal Diseases-Estimate Precision. J. Fungi 2017, 18, 57. [Google Scholar] [CrossRef]
- Pfaller, M.A.; Diekema, D.J. Epidemiology of invasive candidiasis: A persistent public health problem. Clin. Microbiol. Rev. 2007, 20, 133–163. [Google Scholar] [CrossRef] [Green Version]
- Sanglard, D.; Coste, A.; Ferrari, S. Antifungal drug resistance mechanisms in fungal pathogens from the perspective of transcriptional gene regulation. FEMS Yeast Res. 2009, 9, 1029–1050. [Google Scholar] [CrossRef] [Green Version]
- Wall, G.; Lopez-Ribot, J.L. Current Antimycotics, New Prospects, and Future Approaches to Antifungal Therapy. Antibiotics 2020, 9, 445. [Google Scholar] [CrossRef]
- Lepesheva, G.I.; Hargrove, T.Y.; Kleshchenko, Y.; Nes, W.D.; Villalta, F.; Waterman, M.R. CYP51: A major drug target in the cytochrome P450superfamily. Lipids 2008, 43, 1117–1125. [Google Scholar] [CrossRef] [Green Version]
- Lepesheva, G.I.; Waterman, M.R. Structural basis for conservation in the CYP51 family. Biochim. Biophys. Acta 2011, 1814, 88–93. [Google Scholar] [CrossRef] [Green Version]
- Sagatova, A.A.; Keniya, M.V.; Wilson, R.K.; Monk, B.C.; Tyndall, J.D.A. 2015. Structural insights into binding of the antifungal drug fluconazole to Saccharomyces cerevisiae lanosterol 14-demethylase. Antimicrob. Agents Chemother. 2015, 59, 4982–4989. [Google Scholar] [CrossRef] [Green Version]
- Maertens, J.A. History of the development of azole derivatives. Clin. Microbiol. Infect. 2004, 10 (Suppl. S1), 1–10. [Google Scholar] [CrossRef] [Green Version]
- Shafiei, M.; Peyton, L.; Hashemzadeh, M.; Foroumadi, A. History of the development of antifungal azoles: A review on structures, SAR, and mechanism of action. Bioorgan. Chem. 2020, 104, 104240. [Google Scholar] [CrossRef]
- Nett, J.E.; Andes, D.R. Antifungal Agents: Spectrum of Activity, Pharmacology, and Clinical Indications. Infect. Dis. Clin. N. Am. 2016, 30, 51–83. [Google Scholar] [CrossRef]
- Berkow, E.L.; Lockhart, S.R. Fluconazole resistance in Candida species: A current perspective. Infect. Drug Resist. 2017, 10, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Lockhart, S.R.; Etienne, K.A.; Vallabhaneni, S.; Farooqi, J.; Chowdhary, A.; Govender, N.P.; Colombo, A.L.; Calvo, B.; Cuomo, C.A.; Desjardins, C.A.; et al. Simultaneous Emergence of Multidrug-Resistant Candida auris on 3 Continents Confirmed by Whole-Genome Sequencing and Epidemiological Analyses. Clin. Infect. Dis. 2017, 64, 134–140. [Google Scholar] [CrossRef] [Green Version]
- Gomathi, A.; Babu, S.N.; Thilagam, T.G. Fluconazole-induced hepatotoxicity in a tertiary care hospital among patients with dermatophytosis. Natl. J. Physiol. Pharm. Pharmacol. 2021, 11, 109–112. [Google Scholar]
- Song, J.C.; Deresinski, S. Hepatotoxicity of antifungal agents. Curr. Opin. Investig. Drugs 2005, 6, 170–177. [Google Scholar]
- Hornik, C.D.; Bondi, D.S.; Greene, N.M.; Cober, M.P.; John, B. Review of Fluconazole Treatment and Prophylaxis for Invasive Candidiasis in Neonates. J. Pediatr. Pharmacol. Ther. 2021, 26, 115–122. [Google Scholar] [CrossRef]
- Story, K.; Sobel, R. Fluconazole Prophylaxis in Prevention of Symptomatic Candida Vaginitis. Curr. Infect. Dis. Rep. 2020, 22, 2. [Google Scholar] [CrossRef]
- Vitale, R.G. Role of Antifungal Combinations in Difficult to Treat Candida Infections. J. Fungi 2021, 7, 731. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, X.; Gao, L.; Wang, L.; Song, F.; Zhang, L.; Wan, Y. The synergistic antifungal activity of resveratrol with azoles against Candida albicans. Lett. Appl. Microbiol. 2021, 72, 688–697. [Google Scholar] [CrossRef]
- Xu, J.; Liu, R.; Sun, F.; An, L.; Shang, Z.; Kong, L.; Yang, M. Eucalyptal D Enhances the Antifungal Effect of Fluconazole on Fluconazole-Resistant Candida albicans by Competitively Inhibiting Efflux Pump. Front. Cell. Infect. Microbiol. 2019, 9, 211. [Google Scholar] [CrossRef]
- Sun, L.M.; Liao, K.; Liang, S.; Yu, P.H.; Wang, D.Y. Synergistic activity of magnolol with azoles and its possible antifungal mechanism against Candida albicans. J. Appl. Microbiol. 2015, 118, 826–838. [Google Scholar] [CrossRef]
- Gucwa, K.; Kusznierewicz, B.; Milewski, S.; Van Dijck, P.; Szweda, P. Antifungal Activity and Synergism with Azoles of Polish Propolis. Pathogens 2018, 7, 56. [Google Scholar] [CrossRef] [Green Version]
- Ptaszyńska, N.; Gucwa, K.; Olkiewicz, K.; Heldt, M.; Serocki, M.; Stupak, A.; Martynow, D.; Dębowski, D.; Gitlin-Domagalska, A.; Lica, J.; et al. Conjugates of Ciprofloxacin and Levofloxacin with Cell-Penetrating Peptide Exhibit Antifungal Activity and Mammalian Cytotoxicity. Int. J. Mol. Sci. 2020, 21, 34696. [Google Scholar] [CrossRef]
- Ptaszyńska, N.; Olkiewicz, K.; Okońska, J.; Gucwa, K.; Łęgowska, A.; Gitlin-Domagalska, A.; Dębowski, D.; Lica, J.; Heldt, M.; Milewski, S.; et al. Peptide Conjugates of Lactoferricin Analogues and Antimicrobials—Design, Chemical Synthesis, And Evaluation of Antimicrobial Activity and Mammalian Cytotoxicity. Peptides 2019, 117, 170079. [Google Scholar] [CrossRef]
- Ptaszyńska, N.; Gucwa, K.; Łęgowska, A.; Dębowski, D.; Gitlin-Domagalska, A.; Lica, J.; Heldt, M.; Martynow, D.; Olszewski, M.; Milewski, S.; et al. Antimicrobial activity of chimera peptides composed of human neutrophil peptide 1 (HNP-1) truncated analogues and bovine lactoferrampin. Bioconjug. Chem. 2018, 29, 3060–3071. [Google Scholar] [CrossRef]
- Ptaszyńska, N.; Gucwa, K.; Olkiewicz, K.; Łȩgowska, A.; Okońska, J.; Ruczyński, J.; Gitlin-Domagalska, A.; Dȩbowski, D.; Milewski, S.; Rolka, K. Antibiotic-Based Conjugates Containing Antimicrobial HLopt2 Peptide: Design, Synthesis, Antimicrobial and Cytotoxic Activities. ACS Chem. Biol. 2019, 14, 2233–2242. [Google Scholar] [CrossRef]
- Pooga, M.; Hällbrink, M.; Zorko, M.; Langel, U. Cell penetration by transportan. FASEB J. 1998, 12, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Gou, Y.; Zhao, Q.; Li, S.; Zhang, W.; Song, J.; Mou, L.; Li, J.; Wang, K.; Zhang, B.; et al. Antimicrobial activities and action mechanism studies of transportan 10 and its analogues against multidrug-resistant bacteria. J. Pept. Sci. 2015, 21, 599–607. [Google Scholar] [CrossRef]
- Szweda, P.; Gucwa, K.; Romanowska, E.; Dzierzanowska-Fangrat, K.; Naumiuk, Ł.; Brillowska-Dabrowska, A.; Wojciechowska-Koszko, I.; Milewski, S. Mechanisms of azole resistance among clinical isolates of Candida glabrata in Poland. J. Med. Microbiol. 2015, 64, 610–619. [Google Scholar] [CrossRef]
- Song, J.; Kai, M.; Zhang, W.; Zhang, J.; Liu, L.; Zhang, B.; Liu, X.; Wang, R. Cellular uptake of transportan 10 and its analogs in live cells: Selectivity and structure–activity relationship studies. Peptides 2011, 32, 1934–1941. [Google Scholar] [CrossRef]
- Wong, S.S.W.; Kao, R.Y.T.; Yuen, K.Y.; Wang, Y.; Yang, D.; Samaranayake, L.P.; Seneviratne, C.J. In Vitro and In Vivo Activity of a Novel Antifungal Small Molecule against Candida Infections. PLoS ONE 2014, 9, e85836. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Tangadanchu, V.K.R.; Bheemanaboina, R.R.Y.; Cheng, Y.; Zhou, C. Novel carbazole-triazole conjugates as DNA-targeting membrane active potentia-tors against clinical isolated fungi. Eur. J. Med. Chem. 2018, 155, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Elias, R.; Benhamou, R.I.; Jaber, Q.Z.; Dorot, O.; Zada, S.L.; Oved, K.; Pichinuk, E.; Fridman, M. Antifungal activity, mode of action variability, and subcellular dis-tribution of coumarin-based antifungal azoles. Eur. J. Med. Chem. 2019, 179, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Aneja, B.; Irfan, M.; Kapil, C.; Jairajpuri, M.A.; Maguire, R.; Kavanagh, K.; Rizvi, M.M.; Manzoor, N.; Azam, A.; Abid, M. Effect of novel triazole-amino acid hybrids on growth and virulence of Candida species: In vitro and in vivo studies. Org. Biomol. Chem. 2016, 14, 10599–10619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagniez, F.; Lebouvier, N.; Na, Y.M.; Ourliac-Garnier, I.; Picot, C.; Le Borgne, M.; Le Pape, P. Biological exploration of a novel 1,2,4-triazole-indole hybrid molecule as antifungal agent. J. Enzym. Inhib. Med. Chem. 2020, 35, 398–403. [Google Scholar] [CrossRef]
- Thamban Chandrika, N.; Shrestha, S.K.; Ngo, H.X.; Tsodikov, O.V.; Howard, K.C.; Garneau-Tsodikova, S. Alkylated Piperazines and Piperazine-Azole Hybrids as Antifungal Agents. J. Med. Chem. 2018, 61, 158–173. [Google Scholar] [CrossRef]
- Han, G.; Liu, N.; Li, C.; Tu, J.; Li, Z.; Sheng, C. Discovery of Novel Fungal Lanosterol 14α-Demethylase (CYP51)/Histone Deacetylase Dual Inhibitors to Treat Azole-Resistant Candidiasis. J. Med. Chem. 2020, 63, 5341–5359. [Google Scholar] [CrossRef]
- Fang, X.F.; Li, D.; Tangadanchu, V.K.R.; Gopala, L.; Gao, W.W.; Zhou, C.H. Novel potentially antifungal hybrids of 5-flucytosine and fluconazole: Design, synthesis and bioactive evaluation. Bioorgan. Med. Chem. Lett. 2017, 22, 4964–4969. [Google Scholar] [CrossRef]
- Franz, R.; Ruhnke, M.; Morschhäuser, J. Molecular aspects of fluconazole resistance development in Candida albicans. Mycoses 1999, 42, 453–458. [Google Scholar] [CrossRef]
- Rex, J.H. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts; Approved Standard, 3rd ed.; CLSI Document M27-A3; Clinical & Laboratory Standards Institute: Wayne, PA, USA, 2008. [Google Scholar]
- Klepser, M.E.; Ernst, E.J.; Lewis, R.E.; Ernst, M.E.; Pfaller, M.A. Influence of test conditions on antifungal time-kill curve results: Proposal for standardized methods. Antimicrob. Agents Chemother. 1998, 42, 1207–1212. [Google Scholar] [CrossRef] [Green Version]
- Drab, M. Phage Aggregation-Dispersion by Ions: Striving beyond Antibacterial Therapy. Trends Biotechnol. 2018, 36, 875–881. [Google Scholar] [CrossRef]
- Drab, M.; Krajniak, J.; Grzelakowski, K. The New Methodology and Chemical Contrast Observation by Use of the Energy-Selective Back-Scattered Electron Detector. Microsc. Microanal. 2016, 22, 1369–1373. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Calculated Monoisotopic Molecular Weight | Measured Accurate Mass [M + H]+ [m/z] | tR (min) | Yield % |
|---|---|---|---|---|
| FLCpOH | 420.2 | 421.1 | 16.5 | 38% |
| LFcinB(2-11)-NH2 | 1494.8 | 1495.8 | 14.3 | 95% |
| LFacinB[Nle1,11]-NH2 | 2016.2 | 2017.6 | 17.4 | 95% |
| TP10-7-NH2 | 2278.5 | 2278.5 | 22.4 | 90% |
| TP10-NH2 | 2180.4 | 2181.4 | 27.0 | 80% |
| HLopt2-NH2 | 1606.9 | 1607.9 | 12.4 | 95% |
| FLCpOH-LFacinB(2-11)-NH2 | 1896.8 | 1897.9 | 19.5 | 56% |
| FLCpOH-LFacinB[Nle1,11]-NH2 | 2418.4 | 2419.8 | 20.8 | 53% |
| FLCpOH-TP10-7-NH2 | 2679.5 | 2680.8 | 27.8 | 45% |
| FLCpOH-TP10-NH2 | 2582.6 | 2583.3 | 29.5. | 50% |
| FLCpOH-HLopt2-NH2 | 2008.9 | 2010.0 | 17.5 | 49% |
| Yeast Strains | MIC * and MFC ** [µM] | Compounds | |||||||
|---|---|---|---|---|---|---|---|---|---|
| FLCpOH | FLCpOH-TP10-NH2 | FLCpOH-TP10-7-NH2 | FLCpOH-LFacinB[Nle1,11]-NH2 | FLCpOH-LFacinB(2-11)-NH2 | FLCpOH-HLopt2-NH2 | Fluconazole (FLC) | Amphotericin B (AmpB) | ||
| Candida glabrata DSM 6128 | MIC50 | >250 | 62 | 190 | 250 | >250 | >250 | 62 | <0.1 |
| MIC90 | >250 | 125 | 250 | >250 | >250 | >250 | 250 | <0.1 | |
| MFC | - | - | - | - | - | - | - | <0.1 | |
| Candida krusei DSM 11226 | MIC50 | >250 | 62 | 31 | 31 | 31 | >250 | 70 | <0.1 |
| MIC90 | >250 | 125 | 62 | 62 | 62 | >250 | 125 | <0.1 | |
| MFC | - | 250 | 62 | - | 125 | - | - | <0.1 | |
| Candida albicans SC 5314 | MIC50 | >250 | 8 | 8 | 23 | 62 | 62 | <8 | <0.1 |
| MIC90 | >250 | 15 | 15 | 31 | 125 | 125 | <8 | <0.1 | |
| MFC | - | 31 | 15 | 250 | 250 | - | - | <0.1 | |
| Candida albicans ATCC 10231 | MIC50 | >250 | 15 | 15 | 45 | 125 | 125 | <8 | <0.1 |
| MIC90 | >250 | 31 | 31 | 62 | 250 | 250 | <8 | <0.1 | |
| MFC | - | 62 | 62 | - | - | - | - | <0.1 | |
| Candida albicans B3 | MIC50 | >250 | 31 | 31 | 45 | 125 | >250 | - | <0.1 |
| MIC90 | >250 | 62 | 62 | 62 | 250 | >250 | 15 | <0.1 | |
| MFC | - | 62 | 62 | - | - | - | - | <0.1 | |
| Candida albicans B4 | MIC50 | >250 | 62 | 45 | 250 | 190 | >250 | - | <0.1 |
| MIC90 | >250 | 125 | 62 | >250 | 250 | >250 | 62 | <0.1 | |
| MFC | - | 125 | 62 | - | - | - | - | <0.1 | |
| Candida albicans Gu4 | MIC50 | >250 | 45 | 23 | 125 | >250 | >250 | - | <0.1 |
| MIC90 | >250 | 62 | 31 | 250 | >250 | >250 | 31 | <0.1 | |
| MFC | - | 62 | 31 | - | - | - | - | <0.1 | |
| Candida albicans Gu5 | MIC50 | >250 | 62 | 31 | >250 | >250 | >250 | - | <0.1 |
| MIC90 | >250 | 125 | 62 | >250 | >250 | >250 | >250 | <0.1 | |
| MFC | - | 62 | 62 | - | - | - | - | <0.1 | |
| Candida albicans 48 | MIC50 | >250 | 45 | 15 | 250 | 250 | 250 | - | <0.1 |
| MIC90 | >250 | 62 | 31 | >250 | >250 | >250 | >250 | <0.1 | |
| MFC | - | 250 | 125 | - | - | - | - | <0.1 | |
| Candida albicans 138 | MIC50 | >250 | 45 | 23 | 23 | 190 | >250 | - | <0.1 |
| MIC90 | >250 | 62 | 31 | 31 | 250 | >250 | >250 | <0.1 | |
| MFC | - | 250 | 125 | - | - | - | - | <0.1 | |
| Candida albicans 190 | MIC50 | >250 | 45 | 23 | 90 | 190 | 250 | - | <0.1 |
| MIC90 | >250 | 62 | 31 | 125 | 250 | >250 | >250 | <0.1 | |
| MFC | - | 250 | 125 | - | - | - | - | <0.1 | |
| Candida albicans 247 | MIC50 | >250 | 45 | 23 | 45 | 190 | >250 | - | <0.1 |
| MIC90 | >250 | 62 | 31 | 62 | 250 | >250 | >250 | <0.1 | |
| MFC | - | 250 | 125 | - | - | - | - | <0.1 | |
| Candida albicans 574 | MIC50 | >250 | 45 | 23 | 45 | 190 | 250 | - | <0.1 |
| MIC90 | >250 | 62 | 31 | 62 | 250 | >250 | >250 | <0.1 | |
| MFC | - | - | 250 | - | - | - | - | <0.1 | |
| Candida albicans 604 | MIC50 | >250 | 45 | 23 | 45 | 190 | >250 | - | <0.1 |
| MIC90 | >250 | 62 | 31 | 90 | 250 | >250 | >250 | <0.1 | |
| MFC | - | 250 | 250 | - | - | - | - | <0.1 | |
| Compound | Hs27 | HUVEC | ||
|---|---|---|---|---|
| IC50 | IC90 | IC50 | IC90 | |
| FLC | >100 | >100 | >100 | >100 |
| FLCpOH | >100 | >100 | >100 | >100 |
| FLCpOH-TP10-NH2 | 12.00 ± 0.93 | 28.81 ± 0.87 | 14.45 ± 1.13 | 34.30 ± 1.00 |
| FLCpOH-TP10-7-NH2 | 13.07 ± 0.35 | 29.78 ± 0.29 | 15.16 ± 1.26 | 34.03 ± 1.90 |
| FLCpOH-LFacinB[Nle1,11]-NH2 | >100 | >100 | 75.58 ± 1.73 | >100 |
| FLCpOH-LFacinB(2-11)-NH2 | >100 | >100 | >100 | >100 |
| FLCpOH-HLopt2-NH2 | >100 | >100 | >100 | >100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brankiewicz, W.; Okońska, J.; Serbakowska, K.; Lica, J.; Drab, M.; Ptaszyńska, N.; Łęgowska, A.; Rolka, K.; Szweda, P. New Peptide Based Fluconazole Conjugates with Expanded Molecular Targets. Pharmaceutics 2022, 14, 693. https://doi.org/10.3390/pharmaceutics14040693
Brankiewicz W, Okońska J, Serbakowska K, Lica J, Drab M, Ptaszyńska N, Łęgowska A, Rolka K, Szweda P. New Peptide Based Fluconazole Conjugates with Expanded Molecular Targets. Pharmaceutics. 2022; 14(4):693. https://doi.org/10.3390/pharmaceutics14040693
Chicago/Turabian StyleBrankiewicz, Wioletta, Joanna Okońska, Katarzyna Serbakowska, Jan Lica, Marek Drab, Natalia Ptaszyńska, Anna Łęgowska, Krzysztof Rolka, and Piotr Szweda. 2022. "New Peptide Based Fluconazole Conjugates with Expanded Molecular Targets" Pharmaceutics 14, no. 4: 693. https://doi.org/10.3390/pharmaceutics14040693