
Catching Them Early: Framework Parameters and Progress for Prenatal and Childhood Application of Advanced Therapies

 ,
,  ,
,  , ,
, ,  , and
, and 
Abstract
:1. General Introduction
2. Current Status and Challenges of Advanced Therapy Medicinal Products (ATMPs) and Rationale for Early Interventions
2.1. Defining ATMPs
2.2. Current Key Areas of ATMP Application
2.3. Exemplary ATMP Successes
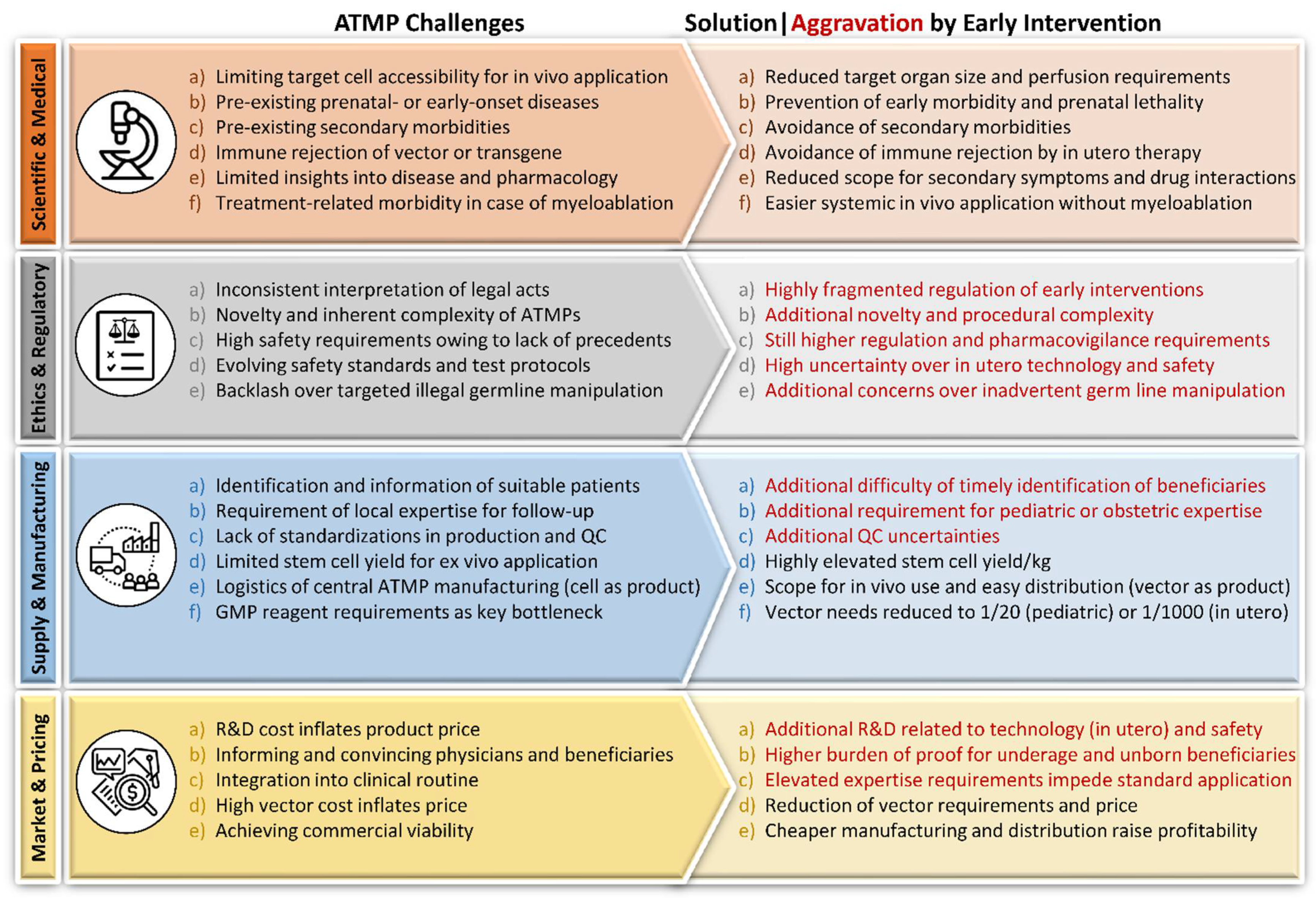
2.4. Challenges of ATMP Application
2.4.1. Scientific and Medical
2.4.2. Ethics and Regulatory
2.4.3. Supply
2.4.4. Manufacturing
2.4.5. Market and Pricing
2.5. The Rationale for Early Intervention
3. Conceptual and Regulatory Framework for Early Interventions
3.1. Pediatric
3.2. In Utero
4. Preclinical Studies of Early Interventions in Animal Models Using ATMPs
4.1. Pediatric
4.2. In Utero
5. Clinical Studies of Early Interventions
5.1. Pediatric
5.1.1. CAR Cells
5.1.2. HSPCs
5.1.3. MSCs

{kind=link}
{kind=link}
{kind=link}
| Cell Type | Target Disease | Drug 1 | Drug Short Description | NCT ID | IU/P/A | n | Ref. |
|---|---|---|---|---|---|---|---|
| CAR-T cell | Relapsed/Refractory HL | CD30.CAR-T | CD30-directed genetically modified autologous T cells | NCT04268706 | P/A | 14 (recruiting) | [227,228] |
| HSPC | TDBT | CTX001 | Autologous CRISPR-Cas9 modified ex vivo CD34+ cells | NCT03655678 | P/A | 15 | [229,230] |
| HSPC | SCD | CTX001 | Autologous CRISPR-Cas9 modified ex vivo CD34+ cells | NCT03745287 | P/A | 7 | [229,230] |
| HSPC | TDBT | OTL-300 | Autologous CD34+ cells transduced ex vivo with a lentiviral vector (GLOBE) encoding the HBB gene. | NCT02453477 NCT03275051 | P/A P/A | 9 9 | [231] [194,232] |
| HSPC | LAD-1 | RPL-201 | Autologous CD34+ cells transduced ex vivo with a lentiviral vector (Chim-CD18-WPRE)encoding the ITGB2 gene | NCT03812263 | P/A | 7 | [233,234,235] |
| HSPC | MPS-IH | OTL-203 | Autologous CD34+ cells transduced ex vivo with a lentiviral vector (IDUA LV) encoding the IDUA gene. | NCT03488394 | P | 8 | [236,237] |
| HSPC | XSCID | MB-107 | Autologous CD34+ cells transduced ex vivo with a lentiviral vector (CL20-i4-EF1α-hγc-OPT) encoding the IL2RG gene. | NCT03315078 NCT01512888 | P/A P | 5 (recruiting) 8 (recruiting) | [238,239] [240,241] |
| HSPC | SCD | ECT-001-CB | UM171-expanded cord blood | NCT04594031 | P/A | Recruiting | [242] |
| HSPC | High-Risk Myeloid Malignancies | ECT-001-CB | UM171-expanded cord blood | NCT04990323 | P/A | Recruiting | [243] |
| HSPC | FA | RP-L102 | Autologous CD34+ cells transduced ex vivo with a lentiviral vector (PGK-FANCA-WPRE) encoding the FANCA gene. | NCT03814408 NCT04248439 NCT04069533 NCT04437771 | P P/A P P/A | 25 (recruiting) 5 (recruiting) 9 | [244,245] [246] [247] [248] |
| T cell | Serious viral infections in allogeneic HSCT recipients | Posoleucel (ALVR-105) | Allogeneic multi-virus specific T lymphocytes | NCT04693637 NCT04390113 | P/A P/A | 12 (recruiting) Recruiting | [249,250] [251] |
| MSC | BPD | Pneumostem® | Intratracheal delivery of umbilical cord MSCs, 1–2 × 107 cells/kg BW | NCT01297205 NCT01632475 NCT01828957 NCT01897987 NCT02023788 NCT02381366 | P P P P P P | 9 9 33 (T) + 33 (C) 62 8 12 | [217] [218] [252,253] [254] [219] [220] |
| MSC | MMC | PMSC-ECM | Placental delivery of PMSC-ECM | NCT04652908 (CuRe) | IU | 35 (T) + 20 (C) (recruiting) | [255] |
| MSC | OI | Boost cells | Intravenous injection of first-trimester-derived allogeneic expanded fetal liver MSCs | NCT03706482 (BOOSTB4) | IU/P | 15 (T) + 15 (C) (recruiting) | [256] |
5.2. In Utero
6. Tools for Success of Early Interventions
6.1. Sources for Cell-Based Therapies
6.2. Traditional Viral and Non-Viral Vectors
6.3. Nanomedicine
7. Non-Technical Considerations for the Routine Application of Early ATMP Interventions
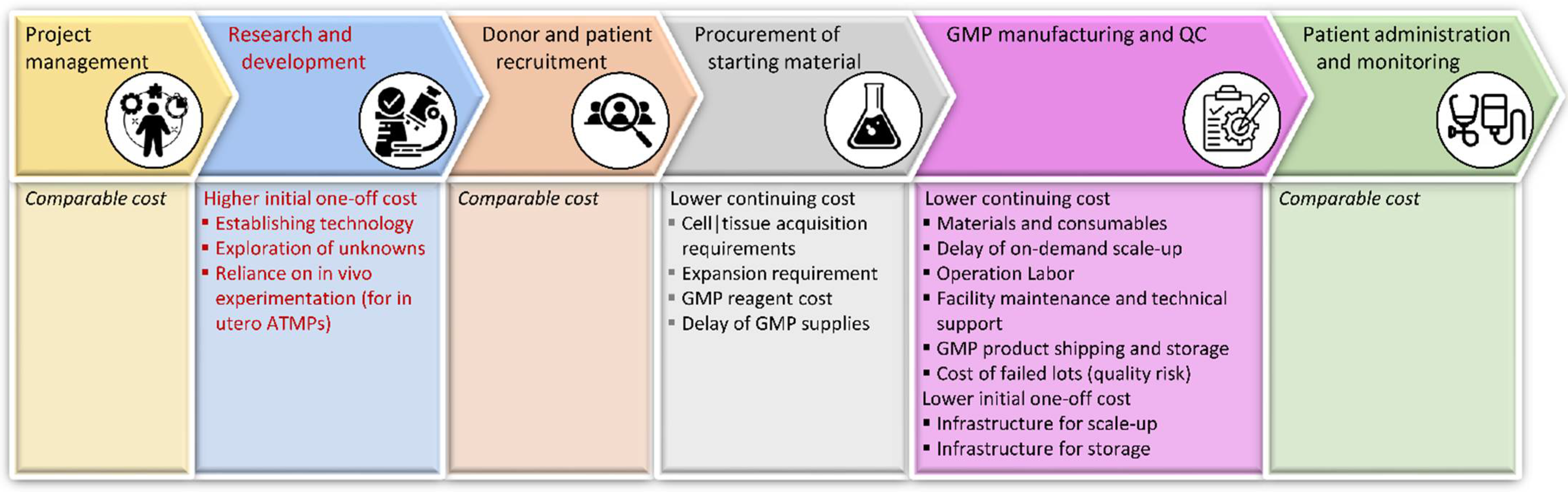
7.1. Financial Considerations
7.2. Ethical and Regulatory Considerations
7.2.1. Pediatric
7.2.2. In Utero
8. Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- European Medicines Agency. Advanced Therapy Medicinal Products: Overview|European Medicines Agency. Available online: https://www.ema.europa.eu/en/human-regulatory/overview/advanced-therapy-medicinal-products-overview (accessed on 11 October 2021).
- European Medicines Agency. Advanced Therapy Classification|European Medicines Agency. Available online: https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/advanced-therapies/advanced-therapy-classification (accessed on 11 October 2021).
- European Medicines Agency. Guidelines Relevant for Advanced Therapy Medicinal Products|European Medicines Agency. Available online: https://www.ema.europa.eu/en/human-regulatory/research-development/advanced-therapies/guidelines-relevant-advanced-therapy-medicinal-products (accessed on 11 October 2021).
- European Medicines Agency. Marketing-Authorisation Procedures for Advanced-Therapy Medicinal Products|European Medicines Agency. Available online: https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/advanced-therapies/marketing-authorisation-procedures-advanced-therapy-medicinal-products (accessed on 11 October 2021).
- European Medicines Agency. Niraparib|Medicines|European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines?search_api_views_fulltext=cancer+niraparib (accessed on 11 October 2021).
- EC Medicines for Children|EC Public Health. Available online: https://ec.europa.eu/health/human-use/paediatric-medicines/ (accessed on 11 October 2021).
- Halioua-Haubold, C.L.; Peyer, J.G.; Smith, J.A.; Arshad, Z.; Scholz, M.; Brindley, D.A.; Maclaren, R.E. Regulatory considerations for gene therapy products in the US, EU, and Japan. Yale J. Biol. Med. 2017, 90, 683–693. [Google Scholar] [PubMed]
- Iglesias-Lopez, C.; Obach, M.; Vallano, A.; Agustí, A. Comparison of regulatory pathways for the approval of advanced therapies in the European Union and the United States. Cytotherapy 2021, 23, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Eder, C.; Wild, C. Technology forecast: Advanced therapies in late clinical research, EMA. approval or clinical application via hospital exemption. J. Mark. Access Health Policy 2019, 7, 1600939. [Google Scholar] [CrossRef] [PubMed]
- Gatline, A. Investor’s Business Daily® – Technology: Can CRISPR and These 3 Small Biotechs Cure 10,000 Diseases? Available online: https://www.investors.com/news/technology/crispr-gene-editing-biotech-companies/ (accessed on 17 May 2018).
- López-Paniagua, M.; de la Mata, A.; Galindo, S.; Blázquez, F.; Calonge, M.; Nieto-Miguel, T. Advanced Therapy Medicinal Products for the Eye: Definitions and Regulatory Framework. Pharmaceutics 2021, 13, 347. [Google Scholar] [CrossRef] [PubMed]
- Ronco, V.; Dilecce, M.; Lanati, E.; Canonico, P.L.; Jommi, C. Price and reimbursement of advanced therapeutic medicinal products in Europe: Are assessment and appraisal diverging from expert recommendations? J. Pharm. Policy Pract. 2021, 14, 30. [Google Scholar] [CrossRef]
- Mebarki, M.; Abadie, C.; Larghero, J.; Cras, A. Human umbilical cord-derived mesenchymal stem/stromal cells: A promising candidate for the development of advanced therapy medicinal products. Stem Cell Res. Ther. 2021, 12, 152. [Google Scholar] [CrossRef]
- Ciccocioppo, R.; Comoli, P.; Astori, G.; del Bufalo, F.; Prapa, M.; Dominici, M.; Locatelli, F. Developing cell therapies as drug products. Br. J. Pharmacol. 2021, 178, 262–279. [Google Scholar] [CrossRef]
- Elverum, K.; Whitman, M. Delivering cellular and gene therapies to patients: Solutions for realizing the potential of the next generation of medicine. Gene Ther. 2020, 27, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Koniali, L.; Lederer, C.W.; Kleanthous, M. Therapy Development by Genome Editing of Hematopoietic Stem Cells. Cells 2021, 10, 1492. [Google Scholar] [CrossRef]
- Whomsley, R.; Palmi Reig, V.; Hidalgo-Simon, A. Environmental risk assessment of advanced therapies containing genetically modified organisms in the EU. Br. J. Clin. Pharmacol. 2021, 87, 2450–2458. [Google Scholar] [CrossRef]
- Lechanteur, C.; Briquet, A.; Bettonville, V.; Baudoux, E.; Beguin, Y. Msc manufacturing for academic clinical trials: From a clinical-grade to a full gmp-compliant process. Cells 2021, 10, 1320. [Google Scholar] [CrossRef] [PubMed]
- Beattie, S. Call for more effective regulation of clinical trials with advanced therapy medicinal products consisting of or containing genetically modified organisms in the European Union. Hum. Gene Ther. 2021, 32, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Scientific Recommendations on Classification of Advanced Therapy Medicinal Products|EMA/140033/2021. Available online: https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/advanced-therapies/advanced-therapy-classification/scientific-recommendations-classification-advanced-therapy-medicinal-products (accessed on 10 December 2021).
- Attico, E.; Sceberras, V.; Pellegrini, G. Approaches for Effective Clinical Application of Stem Cell Transplantation. Curr. Transplant. Rep. 2018, 5, 244–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, C.; Young, C.; Thomas, J.; Trusheim, M. Estimating the Clinical Pipeline of Cell and Gene Therapies and Their Potential Economic Impact on the US Healthcare System. Value Health 2019, 22, 621–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, V.; Seoane-Vazquez, E.; Fawaz, S.; Brown, L.; Rodriguez-Monguio, R. The Landscape of Cellular and Gene Therapy Products: Authorization, Discontinuations, and Cost. Hum. Gene Ther. Clin. Dev. 2019, 30, 102–113. [Google Scholar] [CrossRef]
- Adami, A.; Maher, J. An overview of CAR T-cell clinical trial activity to 2021. Immunother. Adv. 2021, 1, ltab004. [Google Scholar] [CrossRef]
- Marofi, F.; Saleh, M.M.; Rahman, H.S.; Suksatan, W.; Al-Gazally, M.E.; Abdelbasset, W.K.; Thangavelu, L.; Yumashev, A.V.; Hassanzadeh, A.; Yazdanifar, M.; et al. CAR-engineered NK cells; a promising therapeutic option for treatment of hematological malignancies. Stem Cell Res. Ther. 2021, 12, 374. [Google Scholar] [CrossRef]
- Li, Y.-R.; Dunn, Z.S.; Zhou, Y.; Lee, D.; Yang, L. Development of Stem Cell-Derived Immune Cells for Off-the-Shelf Cancer Immunotherapies. Cells 2021, 10, 3497. [Google Scholar] [CrossRef]
- Aiuti, A.; Cattaneo, F.; Galimberti, S.; Benninghoff, U.; Cassani, B.; Callegaro, L.; Scaramuzza, S.; Andolfi, G.; Mirolo, M.; Brigida, I.; et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med. 2009, 360, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Bank, A.; Dorazio, R.; Leboulch, P. A phase I/II clinical trial of beta-globin gene therapy for beta-thalassemia. Ann. N. Y. Acad. Sci. 2005, 1054, 308–316. [Google Scholar] [CrossRef]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Ribeil, J.-A.; Hacein-Bey-Abina, S.; Payen, E.; Magnani, A.; Semeraro, M.; Magrin, E.; Caccavelli, L.; Neven, B.; Bourget, P.; El Nemer, W.; et al. Gene Therapy in a Patient with Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.A.; Walters, M.C.; Kwiatkowski, J.; Rasko, J.E.J.; Ribeil, J.-A.A.; Hongeng, S.; Magrin, E.; Schiller, G.J.; Payen, E.; Semeraro, M.; et al. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2018, 378, 1479–1493. [Google Scholar] [CrossRef] [PubMed]
- CRISPRTX. CRISPR Therapeutics Provides Business Update and Reports Fourth Quarter and Full Year 2020 Financial Results. Available online: http://ir.crisprtx.com/news-releases/news-release-details/crispr-therapeutics-provides-business-update-and-reports-4 (accessed on 1 June 2021).
- Gruhn, B.; Brodt, G.; Ernst, J. Extended Treatment with Mesenchymal Stromal Cells-Frankfurt am Main in a Pediatric Patient with Steroid-refractory Acute Gastrointestinal Graft-Versus-Host Disease: Case Report and Review of the Literature. J. Pediatr. Hematol. Oncol. 2021, 43, e419–e425. [Google Scholar] [CrossRef] [PubMed]
- Buscail, E.; Le Cosquer, G.; Gross, F.; Lebrin, M.; Bugarel, L.; Deraison, C.; Vergnolle, N.; Bournet, B.; Gilletta, C.; Buscail, L. Adipose-derived stem cells in the treatment of perianal fistulas in Crohn’s disease: Rationale, clinical results and perspectives. Int. J. Mol. Sci. 2021, 22, 9967. [Google Scholar] [CrossRef]
- Cuende, N.; Rasko, J.E.J.; Koh, M.B.C.; Dominici, M.; Ikonomou, L. Cell, tissue and gene products with marketing authorization in 2018 worldwide. Cytotherapy 2018, 20, 1401–1413. [Google Scholar] [CrossRef]
- Globerson Levin, A.; Rivière, I.; Eshhar, Z.; Sadelain, M. CAR T cells: Building on the CD19 paradigm. Eur. J. Immunol. 2021, 51, 2151–2163. [Google Scholar] [CrossRef]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Lichtenstein, D.A.; Schischlik, F.; Shao, L.; Steinberg, S.M.; Yates, B.; Wang, H.W.; Wang, Y.; Inglefield, J.; Dulau-Florea, A.; Ceppi, F.; et al. Characterization of HLH-like manifestations as a CRS variant in patients receiving CD22 CAR T cells. Blood 2021, 138, 2469–2484. [Google Scholar] [CrossRef]
- Liu, R.; Cheng, Q.; Kang, L.; Wang, E.; Li, Y.; Zhang, J.; Xiao, H.; Zhang, Y.; Chu, L.; Chen, X.; et al. CD19 or CD20 CAR T-cell Therapy Demonstrates Durable Antitumor Efficacy in Patients with CNS Lymphoma. Hum. Gene Ther. 2022, 33, 318–329. [Google Scholar] [CrossRef]
- Shah, N.; Chari, A.; Scott, E.; Mezzi, K.; Usmani, S.Z. B-cell maturation antigen (BCMA) in multiple myeloma: Rationale for targeting and current therapeutic approaches. Leukemia 2020, 34, 985–1005. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.B.; Lai, X.; Li, R.L.; Ge, C.L.; Zeng, B.Z.; Li, Z.; Fu, Q.F.; Zhao, L.F.; Dong, S.W.; Yang, J.Y.; et al. CD19 and CD30 CAR T-Cell Immunotherapy for High-Risk Classical Hodgkin’s Lymphoma. Front. Oncol. 2021, 10, 607362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, M.; Xiao, X.; Lv, H.; Jiang, Y.; Li, X.; Yuan, T.; Zhao, M. A combination of humanized anti-BCMA and murine anti-CD38 CAR-T cell therapy in patients with relapsed or refractory multiple myeloma. Leuk. Lymphoma 2022, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, R.; Chowdhury, C.R.; Arega, S.; Sen, P.; Ganguly, P.; Ganguly, N. CAR T cell therapy: A new era for cancer treatment (Review). Oncol. Rep. 2019, 42, 2183–2195. [Google Scholar] [CrossRef]
- Tang, Y.; Yin, H.; Zhao, X.; Jin, D.; Liang, Y.; Xiong, T.; Li, L.; Tang, W.; Zhang, J.; Liu, M.; et al. High efficacy and safety of CD38 and BCMA bispecific CAR-T in relapsed or refractory multiple myeloma. J. Exp. Clin. Cancer Res. 2022, 41, 1–15. [Google Scholar] [CrossRef]
- Shah, N.N.; Johnson, B.D.; Schneider, D.; Zhu, F.; Szabo, A.; Keever-Taylor, C.A.; Krueger, W.; Worden, A.A.; Kadan, M.J.; Yim, S.; et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: A phase 1 dose escalation and expansion trial. Nat. Med. 2020, 26, 1569–1575. [Google Scholar] [CrossRef]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef]
- Tucci, F.; Scaramuzza, S.; Aiuti, A.; Mortellaro, A. Update on Clinical Ex Vivo Hematopoietic Stem Cell Gene Therapy for Inherited Monogenic Diseases. Mol. Ther. 2021, 29, 489–504. [Google Scholar] [CrossRef]
- Simaria, A.S.; Hassan, S.; Varadaraju, H.; Rowley, J.; Warren, K.; Vanek, P.; Farid, S.S. Allogeneic cell therapy bioprocess economics and optimization: Single-use cell expansion technologies. Biotechnol. Bioeng. 2014, 111, 69–83. [Google Scholar] [CrossRef] [Green Version]
- Papasavva, P.; Kleanthous, M.; Lederer, C.W. Rare Opportunities: CRISPR/Cas-Based Therapy Development for Rare Genetic Diseases. Mol. Diagn. Ther. 2019, 23, 201–222. [Google Scholar] [CrossRef] [Green Version]
- Papanikolaou, E.; Bosio, A. The Promise and the Hope of Gene Therapy. Front. Genome Ed. 2021, 3, 618346. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Zynteglo. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/zynteglo (accessed on 21 March 2022).
- European Medicines Agency. Skysona. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/skysona (accessed on 21 March 2022).
- European Medicines Agency. Strimvelis. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/strimvelis (accessed on 21 March 2022).
- European Medicines Agency. Libmeldy. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/libmeldy (accessed on 21 March 2022).
- European Medicines Agency. Luxturna. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/luxturna (accessed on 21 March 2022).
- European Medicines Agency. Zolgensma. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/zolgensma (accessed on 21 March 2022).
- European Medicines Agency. Resamirigene Bilparvovec. Available online: https://www.ema.europa.eu/en/medicines/human/paediatric-investigation-plans/emea-002571-pip01-19 (accessed on 21 March 2022).
- Lazarus, H.M.; Haynesworth, S.E.; Gerson, S.L.; Rosenthal, N.S.; Caplan, A.I. Ex vivo expansion and subsequent infusion of human bone marrow-derived stromal progenitor cells (mesenchymal progenitor cells): Implications for therapeutic use. Bone Marrow Transplant. 1995, 16, 557–564. [Google Scholar] [PubMed]
- Jacobsohn, D.A.; Vogelsang, G.B. Acute graft versus host disease. Orphanet J. Rare Dis. 2007, 2, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mac Sweeney, R.; McAuley, D.F. Mesenchymal stem cell therapy in acute lung injury: Is it time for a clinical trial? Thorax 2012, 67, 475–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, S.; Pittenger, M.F. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 2005, 105, 1815–1822. [Google Scholar] [CrossRef] [Green Version]
- Maitra, B.; Szekely, E.; Gjini, K.; Laughlin, M.J.; Dennis, J.; Haynesworth, S.E.; Koç, O.N. Human mesenchymal stem cells support unrelated donor hematopoietic stem cells and suppress T-cell activation. Bone Marrow Transplant. 2004, 33, 597–604. [Google Scholar] [CrossRef] [Green Version]
- Introna, M.; Lucchini, G.; Dander, E.; Galimberti, S.; Rovelli, A.; Balduzzi, A.; Longoni, D.; Pavan, F.; Masciocchi, F.; Algarotti, A.; et al. Treatment of graft versus host disease with mesenchymal stromal cells: A phase I study on 40 adult and pediatric patients. Biol. Blood Marrow Transplant. 2014, 20, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Le Blanc, K.; Frassoni, F.; Ball, L.; Locatelli, F.; Roelofs, H.; Lewis, I.; Lanino, E.; Sundberg, B.; Bernardo, M.E.; Remberger, M.; et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: A phase II study. Lancet 2008, 371, 1579–1586. [Google Scholar] [CrossRef]
- McIntyre, L.A.; Moher, D.; Fergusson, D.A.; Sullivan, K.J.; Mei, S.H.J.; Lalu, M.; Marshall, J.; McLeod, M.; Griffin, G.; Grimshaw, J.; et al. Efficacy of mesenchymal stromal cell therapy for acute lung injury in preclinical animal models: A systematic review. PLoS ONE 2016, 11, e0147170. [Google Scholar] [CrossRef] [Green Version]
- Rojas, M.; Xu, J.; Woods, C.R.; Mora, A.L.; Spears, W.; Roman, J.; Brigham, K.L. Bone marrow-derived mesenchymal stem cells in repair of the injured lung. Am. J. Respir. Cell Mol. Biol. 2005, 33, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Sagar, R.; David, A.; Gotherstrom, C. BOOSTB4 (Boost Brittle Bones before Birth) trial protocol. Prenat. Diagn. 2020, 40, 46. [Google Scholar]
- Otsuru, S.; Desbourdes, L.; Guess, A.J.; Hofmann, T.J.; Relation, T.; Kaito, T.; Dominici, M.; Iwamoto, M.; Horwitz, E.M. Extracellular vesicles released from mesenchymal stromal cells stimulate bone growth in osteogenesis imperfecta. Cytotherapy 2018, 20, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Bai, Y.; Dai, J.; Deng, M.; Zhao, C.; Tian, Z.; Zeng, F.; Liang, W.; Liu, L.; Dong, S. Engineered scaffolds based on mesenchymal stem cells/preosteoclasts extracellular matrix promote bone regeneration. J. Tissue Eng. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kon, E.; Muraglia, A.; Corsi, A.; Bianco, P.; Marcacci, M.; Martin, I.; Boyde, A.; Ruspantini, I.; Chistolini, P.; Rocca, M.; et al. Autologous bone marrow stromal cells loaded onto porous hydroxyapatite ceramic accelerate bone repair in critical-size defects of sheep long bones. J. Biomed. Mater. Res. 2000, 49, 328–337. [Google Scholar] [CrossRef]
- Van Gaalen, S.M.; Dhert, W.J.A.; Van Den Muysenberg, A.; Oner, F.C.; Van Blitterswijk, C.; Verbout, A.J.; De Bruijn, J.D. Bone Tissue Engineering for Spine Fusion: An Experimental Study on Ectopic and Orthotopic Implants in Rats. Tissue Eng. 2004, 10, 231–239. [Google Scholar] [CrossRef]
- Vicinanza, C.; Lombardi, E.; Da Ros, F.; Marangon, M.; Durante, C.; Mazzucato, M.; Agostini, F. Modified mesenchymal stem cells in cancer therapy: A smart weapon requiring upgrades for wider clinical applications. World J. Stem Cells 2022, 14, 54–75. [Google Scholar] [CrossRef]
- Harrell, C.R.; Volarevic, A.; Djonov, V.G.; Jovicic, N.; Volarevic, V. Mesenchymal stem cell: A friend or foe in anti-tumor immunity. Int. J. Mol. Sci. 2021, 22, 12429. [Google Scholar] [CrossRef]
- Premer, C.; Blum, A.; Bellio, M.A.; Schulman, I.H.; Hurwitz, B.E.; Parker, M.; Dermarkarian, C.R.; DiFede, D.L.; Balkan, W.; Khan, A.; et al. Allogeneic Mesenchymal Stem Cells Restore Endothelial Function in Heart Failure by Stimulating Endothelial Progenitor Cells. EBioMedicine 2015, 2, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; He, J.; Zheng, S.; Zhang, K.; Ouyang, Y.; Zhang, Y.; Li, C.; Wu, D. Human umbilical cord mesenchymal stem cells ameliorate acute liver failure by inhibiting apoptosis, inflammation and pyroptosis. Ann. Transl. Med. 2021, 9, 1615. [Google Scholar] [CrossRef]
- Liu, Q.; Lv, C.; Jiang, Y.; Luo, K.; Gao, Y.; Liu, J.; Zhang, X.; Mohammad Omar, J.; Jin, S. From hair to liver: Emerging application of hair follicle mesenchymal stem cell transplantation reverses liver cirrhosis by blocking the TGF-β/Smad signaling pathway to inhibit pathological HSC activation. PeerJ. 2022, 10, e12872. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, L. Mesenchymal stem cells and extracellular vesicles in therapy against kidney diseases. Stem Cell Res. Ther. 2021, 12, 219. [Google Scholar] [CrossRef] [PubMed]
- Mastrolia, I.; Foppiani, E.M.; Murgia, A.; Candini, O.; Samarelli, A.V.; Grisendi, G.; Veronesi, E.; Horwitz, E.M.; Dominici, M. Challenges in Clinical Development of Mesenchymal Stromal/Stem Cells: Concise Review. Stem Cells Transl. Med. 2019, 8, 1135–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherkow, J.S. Controlling CRISPR Through Law: Legal Regimes as Precautionary Principles. Cris. J. 2019, 2, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Daley, G.Q.; Lovell-Badge, R.; Steffann, J. After the Storm—A Responsible Path for Genome Editing. N. Engl. J. Med. 2019, 380, 897–899. [Google Scholar] [CrossRef] [Green Version]
- Cockroft, A.; Wilson, A. Comparability: What we can learn from the review of advanced therapy medicinal products. Regen. Med. 2021, 16, 655–667. [Google Scholar] [CrossRef]
- Coppens, D.G.M.; de Wilde, S.; Guchelaar, H.J.; De Bruin, M.L.; Leufkens, H.G.M.; Meij, P.; Hoekman, J. A decade of marketing approval of gene and cell-based therapies in the United States, European Union and Japan: An evaluation of regulatory decision-making. Cytotherapy 2018, 20, 769–778. [Google Scholar] [CrossRef]
- Gozzo, L.; Romano, G.L.; Romano, F.; Brancati, S.; Longo, L.; Vitale, D.C.; Drago, F. Health Technology Assessment of Advanced Therapy Medicinal Products: Comparison Among 3 European Countries. Front. Pharmacol. 2021, 12, 755052. [Google Scholar] [CrossRef]
- Ten Ham, R.M.T.; Hoekman, J.; Hövels, A.M.; Broekmans, A.W.; Leufkens, H.G.M.; Klungel, O.H. Challenges in Advanced Therapy Medicinal Product Development: A Survey among Companies in Europe. Mol. Ther. Methods Clin. Dev. 2018, 11, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Adair, J.; Sevilla, J.; Heredia, C.; Becker, P.; Kiem, H.-P.; Bueren, J. Lessons Learned from Two Decades of Clinical Trial Experience in Gene Therapy for Fanconi Anemia. Curr. Gene Ther. 2017, 16, 338–348. [Google Scholar] [CrossRef]
- Jossen, V.; Muoio, F.; Panella, S.; Harder, Y.; Tallone, T.; Eibl, R. An approach towards a gmp compliant in-vitro expansion of human adipose stem cells for autologous therapies. Bioengineering 2020, 7, 77. [Google Scholar] [CrossRef]
- Agostini, F.; Vicinanza, C.; Biolo, G.; Spessotto, P.; Da Ros, F.; Lombardi, E.; Durante, C.; Mazzucato, M. Nucleofection of Adipose Mesenchymal Stem/Stromal Cells: Improved Transfection Efficiency for GMP Grade Applications. Cells 2021, 10, 3412. [Google Scholar] [CrossRef]
- Ayati, N.; Saiyarsarai, P.; Nikfar, S. Short and long term impacts of COVID-19 on the pharmaceutical sector. DARU J. Pharm. Sci. 2020, 28, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Plieth, J. The $100,000 Problem Gene Therapy Companies Would Rather Not Mention|Evaluate. Available online: https://www.evaluate.com/vantage/articles/analysis/vantage-points/100000-problem-gene-therapy-companies-would-rather-not (accessed on 31 December 2021).
- Gonçalves, E. Advanced therapy medicinal products: Value judgement and ethical evaluation in health technology assessment. Eur. J. Health Econ. 2020, 21, 311–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BioPharma Dive. Available online: https://www.biopharmadive.com/news/bluebird-withdraw-zynteglo-germany-price/598689/ (accessed on 18 May 2021).
- Rackaityte, E.; Halkias, J. Mechanisms of Fetal T Cell Tolerance and Immune Regulation. Front. Immunol. 2020, 11, 588. [Google Scholar] [CrossRef]
- Bose, S.K.; Menon, P.; Peranteau, W.H. In Utero Gene Therapy: Progress and Challenges. Trends Mol. Med. 2021, 27, 728–730. [Google Scholar] [CrossRef]
- European Medicines Agency. Paediatric Regulation|European Medicines Agency. Available online: https://www.ema.europa.eu/en/human-regulatory/overview/paediatric-medicines/paediatric-regulation (accessed on 11 October 2021).
- EUR-Lex-32006R1901-EN-EUR-Lex. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32006R1901 (accessed on 24 February 2022).
- EUR-Lex-32006R1902-EN-EUR-Lex. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32006R1902 (accessed on 24 February 2022).
- European Commission. State of Paediatric Medicines in the EU – 10 Years of the EU Paediatric Regulation; COM(2017)626; European Commission: Brussel, Belgium, 2017. [Google Scholar]
- European Medicines Agency. 10-Year Report to the European Commission – General Report on the Paediatric Regulation; EMA/231225/2015; European Medicines Agency: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Park, P.J.; Colletti, E.; Ozturk, F.; Wood, J.A.; Tellez, J.; Almeida-Porada, G.; Porada, C.D. Factors determining the risk of inadvertent retroviral transduction of male germ cells after in utero gene transfer in sheep. Hum. Gene Ther. 2009, 20, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Porada, G.; Atala, A.; Porada, C.D. In utero stem cell transplantation and gene therapy: Rationale, history, and recent advances toward clinical application. Mol. Ther.-Methods Clin. Dev. 2016, 3, 16020. [Google Scholar] [CrossRef] [Green Version]
- Staud, F.; Karahoda, R. Trophoblast: The central unit of fetal growth, protection and programming. Int. J. Biochem. Cell Biol. 2018, 105, 35–40. [Google Scholar] [CrossRef]
- Sharma, A.; Sah, N.; Kannan, S.; Kannan, R.M. Targeted drug delivery for maternal and perinatal health: Challenges and opportunities. Adv. Drug Deliv. Rev. 2021, 177, 113950. [Google Scholar] [CrossRef]
- Schrepfer, S.; Deuse, T.; Reichenspurner, H.; Fischbein, M.P.; Robbins, R.C.; Pelletier, M.P. Stem Cell Transplantation: The Lung Barrier. Transplant. Proc. 2007, 39, 573–576. [Google Scholar] [CrossRef]
- Nijagal, A.; Le, T.; Wegorzewska, M.; MacKenzie, T.C. A mouse model of in Utero transplantation. J. Vis. Exp. 2010, e2303. [Google Scholar] [CrossRef] [Green Version]
- Mattar, C.N.Z.; Gil-Farina, I.; Rosales, C.; Johana, N.; Tan, Y.Y.W.; McIntosh, J.; Kaeppel, C.; Waddington, S.N.; Biswas, A.; Choolani, M.; et al. In Utero Transfer of Adeno-Associated Viral Vectors Produces Long-Term Factor IX Levels in a Cynomolgus Macaque Model. Mol. Ther. 2017, 25, 1843–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palanki, R.; Peranteau, W.H.; Mitchell, M.J. Delivery technologies for in utero gene therapy. Adv. Drug Deliv. Rev. 2021, 169, 51–62. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. EMEA/273974/2005-Note for Guidance on the Quality, Preclinical and Clinical Aspects of Gene Transfer Medicinal Products. Available online: https://www.ema.europa.eu/ (accessed on 8 November 2018).
- European Commission. Guidelines on Good Clinical Practice specific to Advanced Therapy Medicinal Products; C(2019)7140; European Commission: Brussel, Belgium, 2019. [Google Scholar]
- Jaeggi, E.T.; Carvalho, J.S.; De Groot, E.; Api, O.; Clur, S.A.B.; Rammeloo, L.; McCrindle, B.W.; Ryan, G.; Manlhiot, C.; Blom, N.A. Comparison of transplacental treatment of fetal supraventricular tachyarrhythmias with digoxin, flecainide, and sotalol: Results of a nonrandomized multicenter study. Circulation 2011, 124, 1747–1754. [Google Scholar] [CrossRef] [Green Version]
- Cerveny, L.; Murthi, P.; Staud, F. HIV in pregnancy: Mother-to-child transmission, pharmacotherapy, and toxicity. Biochim. Biophys. Acta-Mol. Basis Dis. 2021, 1867, 166206. [Google Scholar] [CrossRef]
- Korth-Bradley, J.M. The Path to Perfect Pediatric Posology—Drug Development in Pediatrics. J. Clin. Pharmacol. 2018, 58, S48–S57. [Google Scholar] [CrossRef] [Green Version]
- Manual, R.; Ray, L. Breeding Strategies for Maintaining Colonies of Laboratory Mice: A Jackson Laboratory Resource Manual; The Jackson Laboratory: Bar Harbor, ME, USA, 2007; Volume 83, p. 29. [Google Scholar]
- Ayuso, M.; Buyssens, L.; Stroe, M.; Valenzuela, A.; Allegaert, K.; Smits, A.; Annaert, P.; Mulder, A.; Carpentier, S.; Van Ginneken, C.; et al. The neonatal and juvenile pig in pediatric drug discovery and development. Pharmaceutics 2021, 13, 44. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Kiem, H.P. Large animal models of hematopoietic stem cell gene therapy. Gene Ther. 2010, 17, 939–948. [Google Scholar] [CrossRef] [Green Version]
- Story, B.D.; Miller, M.E.; Bradbury, A.M.; Million, E.D.; Duan, D.; Taghian, T.; Faissler, D.; Fernau, D.; Beecy, S.J.; Gray-Edwards, H.L. Canine Models of Inherited Musculoskeletal and Neurodegenerative Diseases. Front. Vet. Sci. 2020, 7, 80. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Niu, Y.; Ji, W. Genome editing in nonhuman primates: Approach to generating human disease models. J. Intern. Med. 2016, 280, 246–251. [Google Scholar] [CrossRef]
- Sasaki, E.; Sakakibara, Y.; Kumita, W.; Ito, R.; Nozu, R.; Inoue, T.; Katano, I.; Okahara, N.; Okahara, J.; Shimizu, Y.; et al. Generation of a Nonhuman Primate Model of Severe Combined Immunodeficiency Using Highly Efficient Genome Editing. Cell Stem Cell 2016, 19, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Gray-Edwards, H.L.; Randle, A.N.; Maitland, S.A.; Benatti, H.R.; Hubbard, S.M.; Canning, P.F.; Vogel, M.B.; Brunson, B.L.; Hwang, M.; Ellis, L.E.; et al. Adeno-Associated Virus Gene Therapy in a Sheep Model of Tay-Sachs Disease. Hum. Gene Ther. 2018, 29, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Kleine Holthaus, S.M.; Aristorena, M.; Maswood, R.; Semenyuk, O.; Hoke, J.; Hare, A.; Smith, A.J.; Mole, S.E.; Ali, R.R. Gene Therapy Targeting the Inner Retina Rescues the Retinal Phenotype in a Mouse Model of CLN3 Batten Disease. Hum. Gene Ther. 2020, 31, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Dufour, V.L.; Cideciyan, A.V.; Ye, G.J.; Swider, M.; Newmark, J.A.; Timmers, A.M.; Robinson, P.M.; Knop, D.R.; Chulay, J.D.; et al. Dose Range Finding Studies with Two RPGR Transgenes in a Canine Model of X-Linked Retinitis Pigmentosa Treated with Subretinal Gene Therapy. Hum. Gene Ther. 2020, 31, 743–755. [Google Scholar] [CrossRef]
- Hastings, M.L.; Brigande, J.V. Fetal gene therapy and pharmacotherapy to treat congenital hearing loss and vestibular dysfunction. Hear. Res. 2020, 394, 107931. [Google Scholar] [CrossRef]
- Fåne, A.; Myhre, M.R.; Inderberg, E.M.; Wälchli, S. In vivo experimental mouse model to test CD19CAR T cells generated with different methods. Methods Cell Biol. 2022, 167, 149–161. [Google Scholar] [CrossRef]
- Wang, Y.; Buck, A.; Grimaud, M.; Culhane, A.C.; Kodangattil, S.; Razimbaud, C.; Bonal, D.M.; De Nguyen, Q.; Zhu, Z.; Wei, K.; et al. Anti-CAIX BBζ CAR4/8 T cells exhibit superior efficacy in a ccRCC mouse model. Mol. Ther.-Oncolytics 2022, 24, 385–399. [Google Scholar] [CrossRef]
- Daher, M.; Basar, R.; Gokdemir, E.; Baran, N.; Uprety, N.; Nunez Cortes, A.K.; Mendt, M.; Kerbauy, L.N.; Banerjee, P.P.; Shanley, M.; et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood 2021, 137, 624–636. [Google Scholar] [CrossRef]
- Makkouk, A.; Yang, X.C.; Barca, T.; Lucas, A.; Turkoz, M.; Wong, J.T.S.; Nishimoto, K.P.; Brodey, M.M.; Tabrizizad, M.; Gundurao, S.R.Y.; et al. Off-the-shelf Vδ 1 gamma delta T cells engineered with glypican-3 (GPC-3)-specific chimeric antigen receptor (CAR) and soluble IL-15 display robust antitumor efficacy against hepatocellular carcinoma. J. Immunother. Cancer 2021, 9, e003441. [Google Scholar] [CrossRef]
- Goyama, S.; Wunderlich, M.; Mulloy, J.C. Xenograft models for normal and malignant stem cells. Blood 2015, 125, 2630–2640. [Google Scholar] [CrossRef] [Green Version]
- Radtke, S.; Humbert, O.; Kiem, H.P. Mouse models in hematopoietic stem cell gene therapy and genome editing. Biochem. Pharmacol. 2020, 174, 113692. [Google Scholar] [CrossRef] [PubMed]
- Mortellaro, A.; Hernandez, R.J.; Guerrini, M.M.; Carlucci, F.; Tabucchi, A.; Ponzoni, M.; Sanvito, F.; Doglioni, C.; Di Serio, C.; Biasco, L.; et al. Ex vivo gene therapy with lentiviral vectors rescues adenosine deaminase (ADA)-deficient mice and corrects their immune and metabolic defects. Blood 2006, 108, 2979–2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, H.W.J.; Molina, J.G.; Dimina, D.; Zhong, H.; Jacobson, M.; Chan, L.-N.L.; Chan, T.-S.; Lee, J.J.; Blackburn, M.R. A 3 Adenosine Receptor Signaling Contributes to Airway Inflammation and Mucus Production in Adenosine Deaminase-Deficient Mice. J. Immunol. 2004, 173, 1380–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackburn, M.R.; Datta, S.K.; Kellems, R.E. Adenosine deaminase-deficient mice generated using a two-stage genetic engineering strategy exhibit a combined immunodeficiency. J. Biol. Chem. 1998, 273, 5093–5100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walia, J.S.; Altaleb, N.; Bello, A.; Kruck, C.; LaFave, M.C.; Varshney, G.K.; Burgess, S.M.; Chowdhury, B.; Hurlbut, D.; Hemming, R.; et al. Long-term correction of Sandhoff disease following intravenous delivery of rAAV9 to mouse neonates. Mol. Ther. 2015, 23, 414–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massaro, G.; Geard, A.F.; Liu, W.; Coombe-tennant, O.; Waddington, S.N.; Baruteau, J.; Gissen, P.; Rahim, A.A. Gene therapy for lysosomal storage disorders: Ongoing studies and clinical development. Biomolecules 2021, 11, 611. [Google Scholar] [CrossRef] [PubMed]
- Scaramuzza, S.; Biasco, L.; Ripamonti, A.; Castiello, M.C.; Loperfido, M.; Draghici, E.; Hernandez, R.J.; Benedicenti, F.; Radrizzani, M.; Salomoni, M.; et al. Preclinical Safety and Efficacy of human CD34 + Cells transduced with lentiviral vector for the treatment of wiskott-aldrich syndrome. Mol. Ther. 2013, 21, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Huo, Y.; McConnell, S.C.; Liu, S.; Zhang, T.; Yang, R.; Ren, J.; Ryan, T.M. Humanized mouse models of Cooley’s anemia: Correct fetal-to-adult hemoglobin switching, disease onset, and disease pathology. Ann. N. Y. Acad. Sci. 2010, 1202, 45–51. [Google Scholar] [CrossRef]
- Huo, Y.; McConnell, S.C.; Liu, S.R.; Yang, R.; Zhang, T.T.; Sun, C.W.; Wu, L.C.; Ryan, T.M. Humanized Mouse Model of Cooley’s Anemia. J. Biol. Chem. 2009, 284, 4889–4896. [Google Scholar] [CrossRef] [Green Version]
- Ciavatta, D.J.; Ryan, T.M.; Farmer, S.C.; Townes, T.M. Mouse model of human beta zero thalassemia: Targeted deletion of the mouse beta maj-and beta min-globin genes in embryonic stem cells. Proc. Natl. Acad. Sci. USA 1995, 92, 9259–9263. [Google Scholar] [CrossRef] [Green Version]
- Shangaris, P.; Loukogeorgakis, S.P.; Subramaniam, S.; Flouri, C.; Jackson, L.H.; Wang, W.; Blundell, M.P.; Liu, S.; Eaton, S.; Bakhamis, N.; et al. In Utero Gene Therapy (IUGT) Using GLOBE Lentiviral Vector Phenotypically Corrects the Heterozygous Humanised Mouse Model and Its Progress Can Be Monitored Using MRI Techniques. Sci. Rep. 2019, 9, 11592. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; McConnell, S.C.; Ryan, T.M. Preclinical transfusion-dependent humanized mouse model of beta thalassemia major. Blood 2009, 113, 4763–4770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casal, M.; Haskins, M. Large animal models and gene therapy. Eur. J. Hum. Genet. 2006, 14, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Chinnadurai, R.; Ng, S.; Velu, V.; Galipeau, J. Challenges in animal modelling of mesenchymal stromal cell therapy for inflammatory bowel disease. World J. Gastroenterol. 2015, 21, 4779–4787. [Google Scholar] [CrossRef]
- Chinnadurai, R.; Garcia, M.A.; Sakurai, Y.; Lam, W.A.; Kirk, A.D.; Galipeau, J.; Copland, I.B. Actin cytoskeletal disruption following cryopreservation alters the biodistribution of human mesenchymal stromal cells in vivo. Stem Cell Rep. 2014, 3, 60–72. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.K.; Lim, S.H.; Chung, I.S.; Park, Y.; Park, M.J.; Kim, J.Y.; Kim, Y.G.; Hong, J.T.; Kim, Y.; Han, S.-B. Preclinical Efficacy and Mechanisms of Mesenchymal Stem Cells in Animal Models of Autoimmune Diseases. Immune Netw. 2014, 14, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Zhu, K.; Guo, Y.; Wang, E.; Huang, J. Evaluation of animal models of Crohn’s disease with anal fistula (Review). Exp. Ther. Med. 2021, 22, 974. [Google Scholar] [CrossRef]
- Harman, R.M.; Marx, C.; Van de Walle, G.R. Translational Animal Models Provide Insight Into Mesenchymal Stromal Cell (MSC) Secretome Therapy. Front. Cell Dev. Biol. 2021, 9, 654885. [Google Scholar] [CrossRef]
- Hou, H.; Zhang, L.; Duan, L.; Liu, Y.; Han, Z.; Li, Z.; Cao, X. Spatio-Temporal Metabolokinetics and Efficacy of Human Placenta-Derived Mesenchymal Stem/Stromal Cells on Mice with Refractory Crohn’s-like Enterocutaneous Fistula. Stem Cell Rev. Rep. 2020, 16, 1292–1304. [Google Scholar] [CrossRef]
- Li, Q.; Lian, Y.; Deng, Y.; Chen, J.; Wu, T.; Lai, X.; Zheng, B.; Qiu, C.; Peng, Y.; Li, W.; et al. mRNA-engineered mesenchymal stromal cells expressing CXCR2 enhances cell migration and improves recovery in IBD. Mol. Ther.-Nucleic Acids 2021, 26, 222–236. [Google Scholar] [CrossRef]
- Hansen, M.; Stahl, L.; Heider, A.; Hilger, N.; Sack, U.; Kirschner, A.; Cross, M.; Fricke, S. Reduction of Graft-versus-Host-Disease in NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ. (NSG) Mice by Cotransplantation of Syngeneic Human Umbilical Cord-Derived Mesenchymal Stromal Cells: M. Transplant. Cell. Ther. 2021, 27, 658.e1–658.e10. [Google Scholar] [CrossRef] [PubMed]
- Augustine, S.; Cheng, W.; Avey, M.T.; Chan, M.L.; Lingappa, S.M.C.; Hutton, B.; Thébaud, B. Are all stem cells equal? Systematic review, evidence map, and meta-analyses of preclinical stem cell-based therapies for bronchopulmonary dysplasia. Stem Cells Transl. Med. 2020, 9, 158–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ee, M.T.; Thébaud, B. The Therapeutic Potential of Stem Cells for Bronchopulmonary Dysplasia: “It’s About Time” or “Not so Fast”? Curr. Pediatr. Rev. 2018, 14, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.; Baveja, R.; Liang, O.D.; Fernandez-Gonzalez, A.; Lee, C.; Mitsialis, S.A.; Kourembanas, S. Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease. Am. J. Respir. Crit. Care Med. 2009, 180, 1122–1130. [Google Scholar] [CrossRef] [Green Version]
- Tropea, K.A.; Leder, E.; Aslam, M.; Lau, A.N.; Raiser, D.M.; Lee, J.H.; Balasubramaniam, V.; Fredenburgh, L.E.; Mitsialis, S.A.; Kourembanas, S.; et al. Bronchioalveolar stem cells increase after mesenchymal stromal cell treatment in a mouse model of bronchopulmonary dysplasia. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2012, 302, 829–837. [Google Scholar] [CrossRef]
- Hansmann, G.; Fernandez-Gonzalez, A.; Aslam, M.; Vitali, S.H.; Martin, T.; Alex Mitsialis, S.; Kourembanas, S. Mesenchymal stem cell-mediated reversal of bronchopulmonary dysplasia and associated pulmonary hypertension. Pulm. Circ. 2012, 2, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Fang, J.; Su, H.; Yang, M.; Lai, W.; Mai, Y.; Wu, Y. Bone marrow mesenchymal stem cells attenuate lung inflammation of hyperoxic newborn rats. Pediatr. Transplant. 2012, 16, 589–598. [Google Scholar] [CrossRef]
- Khemani, R.G.; Smith, L.; Lopez-Fernandez, Y.M.; Kwok, J.; Morzov, R.; Klein, M.J.; Yehya, N.; Willson, D.; Kneyber, M.C.J.; Lillie, J.; et al. Paediatric acute respiratory distress syndrome incidence and epidemiology (PARDIE): An international, observational study. Lancet Respir. Med. 2019, 7, 115–128. [Google Scholar] [CrossRef]
- Behnke, J.; Kremer, S.; Shahzad, T.; Chao, C.M.; Böttcher-Friebertshäuser, E.; Morty, R.E.; Bellusci, S.; Ehrhardt, H. MSC based therapies—new perspectives for the injured lung. J. Clin. Med. 2020, 9, 682. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Luo, L.; Tian, R.; Yu, C. A review and update for registered clinical studies of stem cells for non-tumorous and non-hematological diseases. Regen. Ther. 2021, 18, 355–362. [Google Scholar] [CrossRef]
- Sondhi, D.; Peterson, D.A.; Edelstein, A.M.; del Fierro, K.; Hackett, N.R.; Crystal, R.G. Survival advantage of neonatal CNS gene transfer for late infantile neuronal ceroid lipofuscinosis. Exp. Neurol. 2008, 213, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.S.; Li, H.; Cao, C.; Sikoglu, E.M.; Denninger, A.R.; Su, Q.; Eaton, S.; Liso Navarro, A.A.; Xie, J.; Szucs, S.; et al. A single intravenous rAAV injection as late as P20 achieves efficacious and sustained CNS gene therapy in Canavan mice. Mol. Ther. 2013, 21, 2136–2147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foust, K.D.; Wang, X.; McGovern, V.L.; Braun, L.; Bevan, A.K.; Haidet, A.M.; Le, T.T.; Morales, P.R.; Rich, M.M.; Burghes, A.H.M.; et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 2010, 28, 271–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrera-Salazar, M.A.; Roskelley, E.M.; Bu, J.; Hodges, B.L.; Yew, N.; Dodge, J.C.; Shihabuddin, L.S.; Sohar, I.; Sleat, D.E.; Scheule, R.K.; et al. Timing of therapeutic intervention determines functional and survival outcomes in a mouse model of late infantile batten disease. Mol. Ther. 2007, 15, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Cataldi, M.P.; Ware, T.A.; Zaraspe, K.; Meadows, A.S.; Murrey, D.A.; McCarty, D.M. Functional correction of neurological and somatic disorders at later stages of disease in MPS IIIA mice by systemic scAAV9-hSGSH gene delivery. Mol. Ther.-Methods Clin. Dev. 2016, 3, 16036. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.; Rafuse, M.; Selvakumar, P.P.; Tan, W. Effects of recipient age, heparin release and allogeneic bone marrow-derived stromal cells on vascular graft remodeling. Acta Biomater. 2021, 125, 172–182. [Google Scholar] [CrossRef]
- Themis, M.; Waddington, S.N.; Schmidt, M.; von Kalle, C.; Wang, Y.; Al-Allaf, F.; Gregory, L.G.; Nivsarkar, M.; Themis, M.; Holder, M.V.; et al. Oncogenesis following delivery of a nonprimate lentiviral gene therapy vector to fetal and neonatal mice. Mol. Ther. 2005, 12, 763–771. [Google Scholar] [CrossRef]
- Nowrouzi, A.; Cheung, W.T.; Li, T.; Zhang, X.; Arens, A.; Paruzynski, A.; Waddington, S.N.; Osejindu, E.; Reja, S.; von Kalle, C.; et al. The fetal mouse is a sensitive genotoxicity model that exposes lentiviral-associated mutagenesis resulting in liver oncogenesis. Mol. Ther. 2013, 21, 324–337. [Google Scholar] [CrossRef] [Green Version]
- Riley, R.S.; Kashyap, M.V.; Billingsley, M.M.; White, B.; Alameh, M.G.; Bose, S.K.; Zoltick, P.W.; Li, H.; Zhang, R.; Cheng, A.Y.; et al. Ionizable lipid nanoparticles for in utero mRNA delivery. Sci. Adv. 2021, 7, eaba1028. [Google Scholar] [CrossRef]
- Borrell, V.; Yoshimura, Y.; Callaway, E.M. Targeted gene delivery to telencephalic inhibitory neurons by directional in utero electroporation. J. Neurosci. Methods 2005, 143, 151–158. [Google Scholar] [CrossRef]
- Joyeux, L.; Danzer, E.; Limberis, M.P.; Zoltick, P.W.; Radu, A.; Flake, A.W.; Davey, M.G. In utero lung gene transfer using adeno-associated viral and lentiviral vectors in mice. Hum. Gene Ther. Methods 2014, 25, 197–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabatino, D.E.; MacKenzie, T.C.; Peranteau, W.; Edmonson, S.; Campagnoli, C.; Liu, Y.L.; Flake, A.W.; High, K.A. Persistent expression of hF.IX after tolerance induction by in utero or neonatal administration of AAV-1-F.IX in hemophilia B mice. Mol. Ther. 2007, 15, 1677–1685. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.K.; White, B.M.; Kashyap, M.V.; Dave, A.; De Bie, F.R.; Li, H.; Singh, K.; Menon, P.; Wang, T.; Teerdhala, S.; et al. In utero adenine base editing corrects multi-organ pathology in a lethal lysosomal storage disease. Nat. Commun. 2021, 12, 4291. [Google Scholar] [CrossRef] [PubMed]
- Massaro, G.; Mattar, C.N.Z.; Wong, A.M.S.; Sirka, E.; Buckley, S.M.K.; Herbert, B.R.; Karlsson, S.; Perocheau, D.P.; Burke, D.; Heales, S.; et al. Fetal gene therapy for neurodegenerative disease of infants. Nat. Med. 2018, 24, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Rossidis, A.C.; Stratigis, J.D.; Chadwick, A.C.; Hartman, H.A.; Ahn, N.J.; Li, H.; Singh, K.; Coons, B.E.; Li, L.; Lv, W.; et al. In utero CRISPR-mediated therapeutic editing of metabolic genes. Nat. Med. 2018, 24, 1513–1518. [Google Scholar] [CrossRef]
- Chan, J.K.Y.Y.; Gil-Farina, I.; Johana, N.; Rosales, C.; Tan, Y.W.; Ceiler, J.; Mcintosh, J.; Ogden, B.; Waddington, S.N.; Schmidt, M.; et al. Therapeutic expression of human clotting factors IX and × following adeno-associated viral vector-mediated intrauterine gene transfer in early-gestation fetal macaques. FASEB J. 2019, 33, 3954–3967. [Google Scholar] [CrossRef] [Green Version]
- Alapati, D.; Zacharias, W.J.; Hartman, H.A.; Rossidis, A.C.; Stratigis, J.D.; Ahn, N.J.; Coons, B.; Zhou, S.; Li, H.; Singh, K.; et al. In utero gene editing for monogenic lung disease. Sci. Transl. Med. 2019, 11, eaav8375. [Google Scholar] [CrossRef] [Green Version]
- Dighe, N.M.; Tan, K.W.; Tan, L.G.; Shaw, S.S.W.; Buckley, S.M.K.; Sandikin, D.; Johana, N.; Tan, Y.W.; Biswas, A.; Choolani, M.; et al. A comparison of intrauterine hemopoietic cell transplantation and lentiviral gene transfer for the correction of severe β-thalassemia in a HbbTh3/+ murine model. Exp. Hematol. 2018, 62, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Ricciardi, A.S.; Bahal, R.; Farrelly, J.S.; Quijano, E.; Bianchi, A.H.; Luks, V.L.; Putman, R.; López-Giráldez, F.; Coşkun, S.; Song, E.; et al. In utero nanoparticle delivery for site-specific genome editing. Nat. Commun. 2018, 9, 2481. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Gao, K.; Wang, C.; Pivetti, C.; Lankford, L.; Farmer, D.; Wang, A. In Utero Transplantation of Placenta-Derived Mesenchymal Stromal Cells for Potential Fetal Treatment of Hemophilia A. Cell Transplant. 2018, 27, 130–139. [Google Scholar] [CrossRef]
- Hayashi, S.; Abdulmalik, O.; Peranteau, W.H.; Ashizuka, S.; Campagnoli, C.; Chen, Q.; Horiuchi, K.; Asakura, T.; Flake, A.W. Mixed chimerism following in utero hematopoietic stem cell transplantation in murine models of hemoglobinopathy. Exp. Hematol. 2003, 31, 176–184. [Google Scholar] [CrossRef]
- Meza, N.W.; Alonso-Ferrero, M.E.; Navarro, S.; Quintana-Bustamante, O.; Valeri, A.; Garcia-Gomez, M.; Bueren, J.A.; Bautista, J.M.; Segovia, J.C. Rescue of pyruvate kinase deficiency in mice by gene therapy using the human isoenzyme. Mol. Ther. 2009, 17, 2000–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loukogeorgakis, S.P.; Shangaris, P.; Bertin, E.; Franzin, C.; Piccoli, M.; Pozzobon, M.; Subramaniam, S.; Tedeschi, A.; Kim, A.G.; Li, H.; et al. In Utero Transplantation of Expanded Autologous Amniotic Fluid Stem Cells Results in Long-Term Hematopoietic Engraftment. Stem Cells 2019, 37, 1176–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shangaris, P.; Loukogeorgakis, S.P.; Blundell, M.P.; Petra, E.; Shaw, S.W.; Ramachandra, D.L.; Maghsoudlou, P.; Urbani, L.; Thrasher, A.J.; De Coppi, P.; et al. Long-Term Hematopoietic Engraftment of Congenic Amniotic Fluid Stem Cells After in Utero Intraperitoneal Transplantation to Immune Competent Mice. Stem Cells Dev. 2018, 27, 515–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, M.; Muramatsu, H.; Nakano, M.; Ito, H.; Inoie, M.; Tomizuka, Y.; Inoue, M.; Yoshimoto, S. Experience of using cultured epithelial autografts for the extensive burn wounds in eight patients. Ann. Plast. Surg. 2014, 73, 25–29. [Google Scholar] [CrossRef]
- Hoburg, A.; Löer, I.; Körsmeier, K.; Siebold, R.; Niemeyer, P.; Fickert, S.; Ruhnau, K. Matrix-Associated Autologous Chondrocyte Implantation Is an Effective Treatment at Midterm Follow-up in Adolescents and Young Adults. Orthop. J. Sport. Med. 2019, 7, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, C. Gintuit cell therapy approval signals shift at US regulator. Nat. Biotechnol. 2012, 30, 479. [Google Scholar] [CrossRef]
- Takaya, K.; Kato, T.; Ishii, T.; Sakai, S.; Okabe, K.; Aramaki-Hattori, N.; Asou, T.; Kishi, K. Clinical Analysis of Cultured Epidermal Autograft (JACE) Transplantation for Giant Congenital Melanocytic Nevus. Plast. Reconstr. Surg.-Glob. Open 2021, 9, e3380. [Google Scholar] [CrossRef]
- Eudy, M.; Eudy, C.L.; Roy, S. Apligraf as an Alternative to Skin Grafting in the Pediatric Population. Cureus 2021, 13, e16226. [Google Scholar] [CrossRef]
- Mavilio, F.; Pellegrini, G.; Ferrari, S.; Di Nunzio, F.; Di Iorio, E.; Recchia, A.; Maruggi, G.; Ferrari, G.; Provasi, E.; Bonini, C.; et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat. Med. 2006, 12, 1397–1402. [Google Scholar] [CrossRef]
- Hirsch, T.; Rothoeft, T.; Teig, N.; Bauer, J.W.; Pellegrini, G.; De Rosa, L.; Scaglione, D.; Reichelt, J.; Klausegger, A.; Kneisz, D.; et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature 2017, 551, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Di, W.L.; Lwin, S.M.; Petrova, A.; Bernadis, C.; Syed, F.; Farzaneh, F.; Moulding, D.; Martinez, A.E.; Sebire, N.J.; Rampling, D.; et al. Generation and Clinical Application of Gene-Modified Autologous Epidermal Sheets in Netherton Syndrome: Lessons Learned from a Phase 1 Trial. Hum. Gene Ther. 2019, 30, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Siprashvili, Z.; Nguyen, N.T.; Gorell, E.S.; Loutit, K.; Khuu, P.; Furukawa, L.K.; Lorenz, H.P.; Leung, T.H.; Keene, D.R.; Rieger, K.E.; et al. Safety and Wound Outcomes Following Genetically Corrected Autologous Epidermal Grafts in Patients with Recessive Dystrophic Epidermolysis Bullosa. JAMA 2016, 316, 1808–1817. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. PRIME: Priority Medicines|European Medicines Agency. Available online: https://www.ema.europa.eu/en/human-regulatory/research-development/prime-priority-medicines (accessed on 30 December 2021).
- Inacio, P. Amicus Discontinues Gene Therapy Program for CLN6 Batten Disease. Available online: https://battendiseasenews.com/2022/01/21/amicus-discontinues-cln6-gene-therapy-program/ (accessed on 28 February 2022).
- Pearson, A.D.J.; Rossig, C.; Lesa, G.; Diede, S.J.; Weiner, S.; Anderson, J.; Gray, J.; Geoerger, B.; Minard-Colin, V.; Marshall, L.V.; et al. ACCELERATE and European Medicines Agency. Paediatric Strategy Forum for medicinal product development of checkpoint inhibitors for use in combination therapy in paediatric patients. Eur. J. Cancer 2020, 127, 52–66. [Google Scholar] [CrossRef] [Green Version]
- Buckland, K.F.; Bobby Gaspar, H. Gene and cell therapy for children--new medicines, new challenges? Adv. Drug Deliv. Rev. 2014, 73, 162–169. [Google Scholar] [CrossRef] [Green Version]
- Marktel, S.; Scaramuzza, S.; Cicalese, M.P.; Giglio, F.; Galimberti, S.; Lidonnici, M.R.; Calbi, V.; Assanelli, A.; Bernardo, M.E.; Rossi, C.; et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent ß-thalassemia. Nat. Med. 2019, 25, 234–241. [Google Scholar] [CrossRef]
- DeWeerdt, S. Prenatal gene therapy offers the earliest possible cure. Nature 2018, 564, S6–S8. [Google Scholar] [CrossRef]
- Escolar, M.L.; Poe, M.D.; Provenzale, J.M.; Richards, K.C.; Allison, J.; Wood, S.; Wenger, D.A.; Pietryga, D.; Wall, D.; Champagne, M.; et al. Transplantation of Umbilical-Cord Blood in Babies with Infantile Krabbe’s Disease. N. Engl. J. Med. 2005, 352, 2069–2081. [Google Scholar] [CrossRef] [Green Version]
- Gray, S.J. Timing of Gene Therapy Interventions: The Earlier, the Better. Mol. Ther. 2016, 24, 1017–1018. [Google Scholar] [CrossRef] [Green Version]
- Garrison, L.P.; Jackson, T.; Paul, D.; Kenston, M. Value-based pricing for emerging gene therapies: The economic case for a higher cost-effectiveness threshold. J. Manag. Care Spec. Pharm. 2019, 25, 793–799. [Google Scholar] [CrossRef]
- Conti, R.; Gruber, J.; Ollendorf, D.; Neumann, P. Valuing Rare Pediatric Drugs: An Economics Perspective. SSRN Electron. J. 2021. NBER Working Paper No. w27978. [Google Scholar] [CrossRef]
- Bolous, N.S.; Chen, Y.; Wang, H.; Davidoff, A.M.; Devidas, M.; Jacobs, T.W.; Meagher, M.M.; Nathwani, A.C.; Neufeld, E.J.; Piras, B.A.; et al. The cost-effectiveness of gene therapy for severe hemophilia B: A microsimulation study from the United States perspective. Blood 2021, 138, 1677–1690. [Google Scholar] [CrossRef] [PubMed]
- Aiuti, A.; Roncarolo, M.G.; Naldini, L. Gene therapy for ADA-SCID, the first marketing approval of an ex vivo gene therapy in Europe: Paving the road for the next generation of advanced therapy medicinal products. EMBO Mol. Med. 2017, 9, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, F.; Calbi, V.; Natali Sora, M.G.; Sessa, M.; Baldoli, C.; Rancoita, P.M.V.; Ciotti, F.; Sarzana, M.; Fraschini, M.; Zambon, A.A.; et al. Lentiviral haematopoietic stem-cell gene therapy for early-onset metachromatic leukodystrophy: Long-term results from a non-randomised, open-label, phase 1/2 trial and expanded access. Lancet 2022, 399, 372–383. [Google Scholar] [CrossRef]
- Matteini, F.; Mulaw, M.A.; Florian, M.C. Aging of the Hematopoietic Stem Cell Niche: New Tools to Answer an Old Question. Front. Immunol. 2021, 12, 738204. [Google Scholar] [CrossRef]
- Garcia, O.; Carraro, G.; Navarro, S.; Bertoncello, I.; McQualter, J.; Driscoll, B.; Jesudason, E.; Warburton, D. Cell-based therapies for lung disease. Br. Med. Bull. 2012, 101, 147–161. [Google Scholar] [CrossRef] [Green Version]
- Alofisel|European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/alofisel (accessed on 30 December 2021).
- Kuçi, Z.; Bönig, H.; Kreyenberg, H.; Bunos, M.; Jauch, A.; Janssen, J.W.G.; Škifić, M.; Michel, K.; Eising, B.; Lucchini, G.; et al. Mesenchymal stromal cells from pooled mononuclear cells of multiple bone marrow donors as rescue therapy in pediatric severe steroid-refractory graft-versus-host disease: A multicenter survey. Haematologica 2016, 101, 985–994. [Google Scholar] [CrossRef]
- Kurtzberg, J.; Prockop, S.; Teira, P.; Bittencourt, H.; Lewis, V.; Chan, K.W.; Horn, B.; Yu, L.; Talano, J.A.; Nemecek, E.; et al. Allogeneic human mesenchymal stem cell therapy (Remestemcel-L, Prochymal) as a rescue agent for severe refractory acute graft-versus-host disease in pediatric patients. Biol. Blood Marrow Transplant. 2014, 20, 229–235. [Google Scholar] [CrossRef] [Green Version]
- Lucchini, G.; Introna, M.; Dander, E.; Rovelli, A.; Balduzzi, A.; Bonanomi, S.; Salvadè, A.; Capelli, C.; Belotti, D.; Gaipa, G.; et al. Platelet-lysate-expanded mesenchymal stromal cells as a salvage therapy for severe resistant graft-versus-host disease in a pediatric population. Biol. Blood Marrow Transplant. 2010, 16, 1293–1301. [Google Scholar] [CrossRef] [Green Version]
- Prasad, V.K.; Lucas, K.G.; Kleiner, G.I.; Talano, J.A.M.; Jacobsohn, D.; Broadwater, G.; Monroy, R.; Kurtzberg, J. Efficacy and Safety of Ex Vivo Cultured Adult Human Mesenchymal Stem Cells (ProchymalTM) in Pediatric Patients with Severe Refractory Acute Graft-Versus-Host Disease in a Compassionate Use Study. Biol. Blood Marrow Transplant. 2011, 17, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Zilberberg, J.; Friedman, T.M.; Korngold, R.; Szabolcs, P.; Visani, G.; Locatelli, F.; Kleiner, G.; Nishida, T.; Onizuka, M.; Inamoto, Y.; et al. Treatment Of Steroid-Refractory Acute GVHD with Mesenchymal Stem Cells Improves Outcomes In Pediatric Patients; Results Of The Pediatric Subset In A Phase III Randomized, Placebo-Controlled Study. Biol. Blood Marrow Transplant. 2010, 16, S298. [Google Scholar] [CrossRef] [Green Version]
- MacMillan, M.L.; Blazar, B.R.; DeFor, T.E.; Wagner, J.E. Transplantation of ex-vivo culture-expanded parental haploidentical mesenchymal stem cells to promote engraftment in pediatric recipients of unrelated donor umbilical cord blood: Results of a phase I-II clinical trial. Bone Marrow Transplant. 2009, 43, 447–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Lee, M.W.; Yoo, K.H.; Kim, D.S.; Son, M.H.; Sung, K.W.; Cheuh, H.; Choi, S.J.; Oh, W.; Yang, Y.S.; et al. Co-transplantation of third-party umbilical cord blood-derived MSCs promotes engraftment in children undergoing unrelated umbilical cord blood transplantation. Bone Marrow Transplant. 2013, 48, 1040–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardo, M.E.; Ball, L.M.; Cometa, A.M.; Roelofs, H.; Zecca, M.; Avanzini, M.A.; Bertaina, A.; Vinti, L.; Lankester, A.; MacCario, R.; et al. Co-infusion of ex vivo-expanded, parental MSCs prevents life-threatening acute GVHD, but does not reduce the risk of graft failure in pediatric patients undergoing allogeneic umbilical cord blood transplantation. Bone Marrow Transplant. 2011, 46, 200–207. [Google Scholar] [CrossRef] [Green Version]
- Voynow, J.A. “New” bronchopulmonary dysplasia and chronic lung disease. Paediatr. Respir. Rev. 2017, 24, 17–18. [Google Scholar] [CrossRef]
- Möbius, M.A.; Thébaud, B. Cell Therapy for Bronchopulmonary Dysplasia: Promises and Perils. Paediatr. Respir. Rev. 2016, 20, 33–41. [Google Scholar] [CrossRef]
- Fujinaga, H.; Baker, C.D.; Ryan, S.L.; Markham, N.E.; Seedorf, G.J.; Balasubramaniam, V.; Abman, S.H. Hyperoxia disrupts vascular endothelial growth factor-nitric oxide signaling and decreases growth of endothelial colony-forming cells from preterm infants. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2009, 297, 1160–1169. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.S.; Ahn, S.Y.; Yoo, H.S.; Sung, S.I.; Choi, S.J.; Oh, W.I.; Park, W.S. Mesenchymal Stem Cells for Bronchopulmonary Dysplasia: Phase 1 Dose-Escalation Clinical Trial. J. Pediatr. 2014, 164, 966–972.e6. [Google Scholar] [CrossRef]
- Ahn, S.Y.; Chang, Y.S.; Kim, J.H.; Sung, S.I.; Park, W.S. Two-Year Follow-Up Outcomes of Premature Infants Enrolled in the Phase I Trial of Mesenchymal Stem Cells Transplantation for Bronchopulmonary Dysplasia. J. Pediatr. 2017, 185, 49–54.e2. [Google Scholar] [CrossRef] [Green Version]
- Medipost Co Ltd. Long-Term Safety and Efficacy Follow-Up Study of PNEUMOSTEM® in Patients Who Completed PNEUMOSTEM® Phase-I Study. Available online: https://clinicaltrials.gov/ct2/show/NCT02023788 (accessed on 8 December 2021).
- Powell, S.B.; Silvestri, J.M. Safety of Intratracheal Administration of Human Umbilical Cord Blood Derived Mesenchymal Stromal Cells in Extremely Low Birth Weight Preterm Infants. J. Pediatr. 2019, 210, 209–213.e2. [Google Scholar] [CrossRef]
- Jouvet, P.; Thomas, N.J.; Willson, D.F.; Erickson, S.; Khemani, R.; Smith, L.; Zimmerman, J.; Dahmer, M.; Flori, H.; Quasney, M.; et al. Pediatric Acute Respiratory Distress Syndrome: Consensus Recommendations from the Pediatric Acute Lung Injury Consensus Conference. Pediatr. Crit. Care Med. 2015, 16, 428–439. [Google Scholar] [CrossRef] [Green Version]
- Heidemann, S.M.; Nair, A.; Bulut, Y.; Sapru, A. Pathophysiology and Management of Acute Respiratory Distress Syndrome in Children. Pediatr. Clin. N. Am. 2017, 64, 1017–1037. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.T.; Chambers, R.C.; Liu, K.D. Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2017, 377, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.G.; Liu, K.D.; Zhuo, H.; Caballero, L.; McMillan, M.; Fang, X.; Cosgrove, K.; Vojnik, R.; Calfee, C.S.; Lee, J.W.; et al. Mesenchymal stem (stromal) cells for treatment of ARDS: A phase 1 clinical trial. Lancet Respir. Med. 2015, 3, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Weiss, D.J. Cell-based therapies for acute respiratory distress syndrome. Lancet Respir. Med. 2019, 7, 105–106. [Google Scholar] [CrossRef]
- Matthay, M.A.; Calfee, C.S.; Zhuo, H.; Thompson, B.T.; Wilson, J.G.; Levitt, J.E.; Rogers, A.J.; Gotts, J.E.; Wiener-Kronish, J.P.; Bajwa, E.K.; et al. Treatment with allogeneic mesenchymal stromal cells for moderate to severe acute respiratory distress syndrome (START study): A randomised phase 2a safety trial. Lancet Respir. Med. 2019, 7, 154–162. [Google Scholar] [CrossRef]
- Ahmed, S.; Flinn, I.W.; Mei, M.; Riedell, P.A.; Armand, P.; Grover, N.S.; Engert, A.; Lapteva, N.; Nadler, P.I.; Myo, A.; et al. Safety and Efficacy Profile of Autologous CD30.CAR-T-Cell Therapy in Patients with Relapsed or Refractory Classical Hodgkin Lymphoma (CHARIOT Trial). Blood 2021, 138, 3847–3850. [Google Scholar] [CrossRef]
- Tessa Therapeutics. Phase 2 Study Evaluating Autologous CD30.CAR-T Cells in Patients with Relapsed/Refractory Hodgkin Lymphoma (CHARIOT). Available online: https://clinicaltrials.gov/ct2/show/NCT04268706 (accessed on 21 March 2022).
- Nichols, H.; Eides, R. Vertex and CRISPR Therapeutics Present New Data in 22 Patients with Greater than 3 Months Follow-Up Post-Treatment with Investigational CRISPR/Cas9 Gene-Editing Therapy, CTX001TM. Available online: https://investors.vrtx.com/news-releases/news-release-details/vertex-and-crispr-therapeutics-present-new-data-22-patients (accessed on 21 March 2022).
- CRISPRTX. A Safety and Efficacy Study Evaluating CTX001 in Subjects with Severe Sickle Cell Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT03745287 (accessed on 21 March 2022).
- Aiuiti, A and IRCCS San Raffaele. Gene Therapy for Transfusion Dependent Beta-thalassemia (TIGET-BTHAL). Available online: https://clinicaltrials.gov/ct2/show/NCT02453477 (accessed on 27 February 2022).
- Orchard Therapeutics. Long-Term Follow-Up of Subjects Treated with OTL-300 for Transfusion-Dependent Beta-Thalassemia Study (TIGET-BTHAL). Available online: https://clinicaltrials.gov/ct2/show/NCT03275051 (accessed on 21 March 2022).
- Kohn, D.B.; Booth, C.; Sevilla, J.; Rao, G.R.; Almarza, E.; Terrazas, D.; Nicoletti, E.; Fernandes, A.; Kuo, C.; de Oliveira, S.; et al. A Phase 1/2 Study of Lentiviral-Mediated Ex-Vivo Gene Therapy for Pediatric Patients with Severe Leukocyte Adhesion Deficiency-I (LAD-I): Interim Results. Blood 2021, 138, 2932. [Google Scholar] [CrossRef]
- Kohn, D.B.; Rao, G.R.; Almarza, E.; Terrazas, D.; Nicoletti, E.; Fernandes, A.; Kuo, C.; De Oliveira, S.N.; Moore, T.B.; Law, K.M.; et al. A Phase 1/2 Study of Lentiviral-Mediated Ex-Vivo Gene Therapy for Pediatric Patients with Severe Leukocyte Adhesion Deficiency-I (LAD-I): Results from Phase 1. Blood 2020, 136, 15. [Google Scholar] [CrossRef]
- Rocket Pharmaceuticals Inc. A Clinical Trial to Evaluate the Safety and Efficacy of RP-L201 in Subjects with Leukocyte Adhesion Deficiency-I. Available online: https://clinicaltrials.gov/ct2/show/NCT03812263 (accessed on 21 March 2022).
- Aiuiti, A and IRCCS San Raffaele. Gene Therapy with Modified Autologous Hematopoietic Stem Cells for the Treatment of Patients with Mucopolysaccharidosis Type I, Hurler Variant (TigetT10_MPSIH). Available online: https://clinicaltrials.gov/ct2/show/NCT03488394 (accessed on 21 March 2022).
- Gentner, B.; Tucci, F.; Galimberti, S.; Fumagalli, F.; De Pellegrin, M.; Silvani, P.; Camesasca, C.; Pontesilli, S.; Darin, S.; Ciotti, F.; et al. Hematopoietic Stem-and Progenitor-Cell Gene Therapy for Hurler Syndrome. N. Engl. J. Med. 2021, 385, 1929–1940. [Google Scholar] [CrossRef]
- National Institutes of Health Clinical Center (CC) and National Institute of Allergy and Infectious Diseases (NIAID). Lentiviral Gene Transfer for Treatment of Children Older than 2 Years of Age with X-Linked Severe Combined Immunodeficiency (LVXSCID-OC). Available online: https://clinicaltrials.gov/ct2/show/NCT01306019 (accessed on 21 March 2022).
- De Ravin, S.S.; Anaya O’Brien, S.; Kwatemaa, N.; Theobald, N.; Liu, S.; Lee, J.; Kardava, L.; Liu, T.; Goldman, F.; Moir, S.; et al. Enhanced Transduction Lentivector Gene Therapy for Treatment of Older Patients with X-Linked Severe Combined Immunodeficiency. Blood 2019, 134 (Suppl. 1), 608. [Google Scholar] [CrossRef]
- Mamcarz, E.; Zhou, S.; Lockey, T.; Abdelsamed, H.; Cross, S.J.; Kang, G.; Ma, Z.; Condori, J.; Dowdy, J.; Triplett, B.; et al. Lentiviral gene therapy combined with low-dose busulfan in infants with SCID-X1. N. Engl. J. Med. 2019, 380, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- St. Jude Children′s Research Hospital. Gene Transfer for X-Linked Severe Combined Immunodeficiency in Newly Diagnosed Infants (LVXSCID-ND). Available online: https://clinicaltrials.gov/ct2/show/NCT01512888 (accessed on 21 March 2022).
- ExCellThera Inc. US Phase I Study of ECT-001-CB in Patients with Sickle-Cell Disease. Available online: https://clinicaltrials.gov/ct2/show/results/NCT04594031 (accessed on 21 March 2022).
- ExCellThera Inc. US Study of ECT-001-CB in Pediatric and Young Adult Patients with High-Risk Myeloid Malignancies. Available online: https://clinicaltrials.gov/ct2/show/NCT04990323 (accessed on 21 March 2022).
- Rocket Pharmaceuticals Inc. A Clinical Trial to Evaluate the Safety of RP-L102 in Pediatric Subjects with Fanconi Anemia Subtype A. Available online: https://clinicaltrials.gov/ct2/show/NCT03814408 (accessed on 21 March 2022).
- Czechowicz, A.; Roncarolo, M.G.; Beard, B.C.; Law, K.; Nicoletti, E.; Río, P.; Bueren, J.A.; Schwartz, J.D.; Soni, S. Changing the Natural History of Fanconi Anemia Complementation Group-A with Gene Therapy: Early Results of U.S. Phase I Study of Lentiviral-Mediated Ex-VivoFANCA Gene Insertion in Human Stem and Progenitor Cells. Blood 2019, 134, 3350. [Google Scholar] [CrossRef]
- Rocket Pharmaceuticals Inc. Gene Therapy for Fanconi Anemia, Complementation Group A. Available online: https://clinicaltrials.gov/ct2/show/NCT04248439 (accessed on 21 March 2022).
- Rocket Pharmaceuticals Inc. Lentiviral-Mediated Gene Therapy for Pediatric Patients with Fanconi Anemia Subtype A. Available online: https://clinicaltrials.gov/ct2/show/NCT04069533 (accessed on 21 March 2022).
- bluebird bio. Longterm Follow-Up of Subjects with Hemoglobinopathies Treated with Ex Vivo Gene Therapy. Available online: https://clinicaltrials.gov/ct2/show/NCT02633943 (accessed on 21 March 2022).
- AlloVir. Study of Viralym-M (ALVR105) for Multi-Virus Prevention in Patients Post-Allogeneic Hematopoietic Cell Transplant. Available online: https://clinicaltrials.gov/ct2/show/NCT04693637 (accessed on 21 March 2022).
- Dadwal, S.S.; Shuster, M.; Myers, G.D.; Boundy, K.; Warren, M.; Stoner, E.; Truong, T.; Hill, J.A. Posoleucel (ALVR105), an Off-the-Shelf, Multivirus-Specific T-Cell Therapy, for the Prevention of Viral Infections Post-HCT: Results from an Open-Label Cohort of a Phase 2 Trial. Blood 2021, 138, 1760. [Google Scholar] [CrossRef]
- AlloVir. Study to Evaluate Viralym-M (ALVR105) for the Treatment of Virus-Associated Hemorrhagic Cystitis (HC). Available online: https://clinicaltrials.gov/ct2/show/NCT04390113 (accessed on 21 March 2022).
- Elbashir, S.M.; Martinez, J.; Patkaniowska, A.; Lendeckel, W.; Tuschl, T. Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J. 2001, 20, 6877–6888. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.Y.; Chang, Y.S.; Lee, M.H.; Sung, S.I.; Lee, B.S.; Kim, K.S.; Kim, A.R.; Park, W.S. Stem cells for bronchopulmonary dysplasia in preterm infants: A randomized controlled phase II trial. Stem Cells Transl. Med. 2021, 10, 1129–1137. [Google Scholar] [CrossRef]
- Medipost Co Ltd. Follow-Up Safety and Efficacy Evaluation on Subjects Who Completed PNEUMOSTEM® Phase-II Clinical Trial. Available online: https://clinicaltrials.gov/ct2/show/NCT01897987 (accessed on 8 December 2021).
- Lee Farmer, D.; University of California, Davis. Cellular Therapy for In Utero Repair of Myelomeningocele—The CuRe Trial. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04652908 (accessed on 11 December 2021).
- Götherström, C and Karolinska Institutet. Boost Brittle Bones before Birth (BOOSTB4). Available online: https://www.clinicaltrials.gov/ct2/show/NCT03706482 (accessed on 11 December 2021).
- Schneider, H.; Faschingbauer, F.; Schuepbach-Mallepell, S.; Körber, I.; Wohlfart, S.; Dick, A.; Wahlbuhl, M.; Kowalczyk-Quintas, C.; Vigolo, M.; Kirby, N.; et al. Prenatal Correction of X-Linked Hypohidrotic Ectodermal Dysplasia. N. Engl. J. Med. 2018, 378, 1604–1610. [Google Scholar] [CrossRef]
- Kreger, E.M.; Singer, S.T.; Witt, R.G.; Sweeters, N.; Lianoglou, B.; Lal, A.; Mackenzie, T.C.; Vichinsky, E. Favorable outcomes after in utero transfusion in fetuses with alpha thalassemia major: A case series and review of the literature. Prenat. Diagn. 2016, 36, 1242–1249. [Google Scholar] [CrossRef]
- Mackenzie, T.; University of California, San Francisco. In Utero Hematopoietic Stem Cell Transplantation for Alpha-Thalassemia Major (ATM). Available online: https://www.clinicaltrials.gov/ct2/show/NCT02986698 (accessed on 11 December 2021).
- MacKenzie, T.C.; Frascoli, M.; Sper, R.; Lianoglou, B.R.; Gonzalez Velez, J.; Dvorak, C.C.; Kharbanda, S.; Vichinsky, E. In Utero Stem Cell Transplantation in Patients with Alpha Thalassemia Major: Interim Results of a Phase 1 Clinical Trial. Blood 2020, 136 (Suppl. 1), 1. [Google Scholar] [CrossRef]
- Dimitri, P.; Pignataro, V.; Lupo, M.; Bonifazi, D.; Henke, M.; Musazzi, U.M.; Ernst, F.; Minghetti, P.; Redaelli, D.F.; Antimisiaris, S.G.; et al. Medical device development for children and young people—reviewing the challenges and opportunities. Pharmaceutics 2021, 13, 2178. [Google Scholar] [CrossRef]
- Fesnak, A.; O’Doherty, U. Clinical development and manufacture of chimeric antigen receptor T cells and the role of leukapheresis. Eur. Oncol. Haematol. 2017, 13, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Cattoglio, C.; Facchini, G.; Sartori, D.; Antonelli, A.; Miccio, A.; Cassani, B.; Schmidt, M.; Von Kalle, C.; Howe, S.; Thrasher, A.J.; et al. Hot spots of retroviral integration in human CD34+ hematopoietic cells. Blood 2007, 110, 1770–1778. [Google Scholar] [CrossRef] [PubMed]
- Uchida, N.; Sutton, R.E.; Friera, A.M.; He, D.; Reitsma, M.J.; Chang, W.C.; Veres, G.; Scollay, R.; Weissman, I.L. HIV, but not murine leukemia virus, vectors mediate high efficiency gene transfer into freshly isolated G0/G1 human hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 1998, 95, 11939–11944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Zhao, X.; Li, Z.; Hu, Y.; Wang, H. From CAR-T Cells to CAR-NK Cells: A Developing Immunotherapy Method for Hematological Malignancies. Front. Oncol. 2021, 11, 720501. [Google Scholar] [CrossRef] [PubMed]
- Tonn, T.; Becker, S.; Esser, R.; Schwabe, D.; Seifried, E. Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J. Hematother. Stem Cell Res. 2001, 10, 535–544. [Google Scholar] [CrossRef]
- Bjordahl, R.; Gaidarova, S.; Goodridge, J.P.; Mahmood, S.; Bonello, G.; Robinson, M.; Ruller, C.; Pribadi, M.; Lee, T.; Abujarour, R.; et al. FT576: A Novel Multiplexed Engineered Off-the-Shelf Natural Killer Cell Immunotherapy for the Dual-Targeting of CD38 and Bcma for the Treatment of Multiple Myeloma. Blood 2019, 134, 3214. [Google Scholar] [CrossRef]
- Goodridge, J.P.; Mahmood, S.; Zhu, H.; Gaidarova, S.; Blum, R.; Bjordahl, R.; Cichocki, F.; Chu, H.; Bonello, G.; Lee, T.; et al. FT596: Translation of First-of-Kind Multi-Antigen Targeted Off-the-Shelf CAR-NK Cell with Engineered Persistence for the Treatment of B Cell Malignancies. Blood 2019, 134, 301. [Google Scholar] [CrossRef]
- Haspel, R.; Miller, K. Hematopoietic Stem Cells: Source Matters. Curr. Stem Cell Res. Ther. 2008, 3, 229–236. [Google Scholar] [CrossRef]
- Styczynski, J.; Balduzzi, A.; Gil, L.; Labopin, M.; Hamladji, R.M.; Marktel, S.; Yesilipek, M.A.; Fagioli, F.; Ehlert, K.; Matulova, M.; et al. Risk of complications during hematopoietic stem cell collection in pediatric sibling donors: A prospective European Group for Blood and Marrow Transplantation Pediatric Diseases Working Party study. Blood 2012, 119, 2935–2942. [Google Scholar] [CrossRef]
- Drabko, K. Autologous hematopoietic stem cell transplantation (auto-HSCT) in children in Poland: 2021 indications and practice. Acta Haematol. Pol. 2021, 52, 234–236. [Google Scholar] [CrossRef]
- Ohara, Y.; Ohto, H.; Tasaki, T.; Sano, H.; Mochizuki, K.; Akaihata, M.; Kobayashi, S.; Waragai, T.; Ito, M.; Hosoya, M.; et al. Comprehensive technical and patient-care optimization in the management of pediatric apheresis for peripheral blood stem cell harvesting. Transfus. Apher. Sci. 2016, 55, 338–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karakukcu, M.; Unal, E. Stem cell mobilization and collection from pediatric patients and healthy children. Transfus. Apher. Sci. 2015, 53, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Lipton, J.M. Peripheral blood as a stem cell source for hematopoietic cell transplantation in children: Is the effort in vein? Pediatr. Transplant. 2003, 7, 65–70. [Google Scholar] [CrossRef] [PubMed]
- DiPersio, J.F.; Karpova, D.; Rettig, M.P. Mobilized peripheral blood: An updated perspective. F1000Research 2019, 8, 2125. [Google Scholar] [CrossRef] [Green Version]
- Luzzi, J.R.; Borba, C.C.; Miyaji, S.C.; Brito, C.A.; Navarro-Xavier, R.; Dinardo, C.L. Reduced volume of red blood cell priming is safe for pediatric patients undergoing therapeutic plasma exchange. Transfus. Apher. Sci. 2021, 60, 103005. [Google Scholar] [CrossRef]
- Luo, C.; Wang, L.; Wu, G.; Huang, X.; Zhang, Y.; Ma, Y.; Xie, M.; Sun, Y.; Huang, Y.; Huang, Z.; et al. Comparison of the efficacy of hematopoietic stem cell mobilization regimens: A systematic review and network meta-analysis of preclinical studies. Stem Cell Res. Ther. 2021, 12, 310. [Google Scholar] [CrossRef]
- Tajer, P.; Pike-Overzet, K.; Arias, S.; Havenga, M.; Staal, F. Ex Vivo Expansion of Hematopoietic Stem Cells for Therapeutic Purposes: Lessons from Development and the Niche. Cells 2019, 8, 169. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, K.; Urbinati, F.; Romero, Z.; Campo-Fernandez, B.; Kaufman, M.L.; Cooper, A.R.; Masiuk, K.; Hollis, R.P.; Kohn, D.B. Enrichment of human hematopoietic stem/progenitor cells facilitates transduction for stem cell gene therapy. Stem Cells 2015, 33, 1532–1542. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Xu, J. Immune modulation by mesenchymal stem cells. Cell Prolif. 2020, 53, e12712. [Google Scholar] [CrossRef]
- Ullah, I.; Subbarao, R.B.; Rho, G.J. Human mesenchymal stem cells-Current trends and future prospective. Biosci. Rep. 2015, 35, e00191. [Google Scholar] [CrossRef]
- Nehlin, J.O.; Jafari, A.; Tencerova, M.; Kassem, M. Aging and lineage allocation changes of bone marrow skeletal (stromal)stem cells. Bone 2019, 123, 265–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, Y.; Shin, T.H.; Kim, H.S. Current strategies to enhance adipose stem cell function: An update. Int. J. Mol. Sci. 2019, 20, 3827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuca-Warnawin, E.; Skalska, U.; Janicka, I.; Musiałowicz, U.; Bonek, K.; Głuszko, P.; Szczęsny, P.; Olesińska, M.; Kontny, E. The Phenotype and Secretory Activity of Adipose-Derived Mesenchymal Stem Cells (ASCs) of Patients with Rheumatic Diseases. Cells 2019, 8, 1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, D.C.; Chang, Y.H.; Shyu, W.C.; Lin, S.Z. Human umbilical cord mesenchymal stem cells: A new era for stem cell therapy. Cell Transplant. 2015, 24, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.A.; Schanz, S.; Windrem, M.S. Stem cell-based strategies for treating pediatric disorders of myelin. Hum Mol. Genet 2008, 17, R76–R83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Tang, L.; Zhou, Y. Subretinal Injection: A Review on the Novel Route of Therapeutic Delivery for Vitreoretinal Diseases. Ophthalmic Res. 2017, 58, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Ramachandra, D.L.; Shaw, S.S.W.; Shangaris, P.; Loukogeorgakis, S.; Guillot, P.V.; De Coppi, P.; David, A.L. In utero therapy for congenital disorders using amniotic fluid stem cells. Front. Pharmacol. 2014, 5, 270. [Google Scholar] [CrossRef] [Green Version]
- Biological, K. Non Viral Vectors in Gene Therapy-An Overview. J. Clin. Diagn. Res. 2015, 9, GE01–GE06. [Google Scholar] [CrossRef]
- Boulaiz, H.; Marchal, J.A.; Prados, J.; Melguizo, C.; Aránega, A. Non-viral and viral vectors for gene therapy. Cell. Mol. Biol. 2005, 51, 3–22. [Google Scholar]
- Richter, M.; Stone, D.; Miao, C.; Humbert, O.; Kiem, H.P.; Papayannopoulou, T.; Lieber, A. In Vivo Hematopoietic Stem Cell Transduction. Hematol. Oncol. Clin. N. Am. 2017, 31, 771–785. [Google Scholar] [CrossRef]
- Murai, N.; Ohtaki, H.; Watanabe, J.; Xu, Z.; Sasaki, S.; Yagura, K.; Shioda, S.; Nagasaka, S.; Honda, K.; Izumizaki, M. Intrapancreatic injection of human bone marrow-derived mesenchymal stem/stromal cells alleviates hyperglycemia and modulates the macrophage state in streptozotocin-induced type 1 diabetic mice. PLoS ONE 2017, 12, e0186637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Georgakopoulou, A.; Mishra, A.; Gil, S.; Hawkins, R.D.; Yannaki, E.; Lieber, A. In vivo HSPC gene therapy with base editors allows for efficient reactivation of fetal γ-globin in β-YAC mice. Blood Adv. 2021, 5, 1122–1135. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, S.; Lange, S.; Werner, J.; Machka, C.; Neumann, K.; Knuebel, G.; Vogel, H.; Lindner, I.; Glass, Ä.; Escobar, H.M.; et al. Engraftment Effects after Intra-Bone Marrow versus Intravenous Allogeneic Stem Cell Transplantation in a Reduced-Intensity Conditioning Dog Leukocyte Antigen-Identical Canine Model. Transplant. Cell. Ther. 2021, 28, 70.e1–70.e5. [Google Scholar] [CrossRef] [PubMed]
- Greber, U.F.; Gomez-Gonzalez, A. Adenovirus-a blueprint for gene delivery. Curr. Opin. Virol. 2021, 48, 49–56. [Google Scholar] [CrossRef]
- Chen, W.; Yao, S.; Wan, J.; Tian, Y.; Huang, L.; Wang, S.; Akter, F.; Wu, Y.; Yao, Y.; Zhang, X. BBB-crossing adeno-associated virus vector: An excellent gene delivery tool for CNS disease treatment. J. Control. Release 2021, 333, 129–138. [Google Scholar] [CrossRef]
- Jacobs, L.; De Smidt, E.; Geukens, N.; Declerck, P.; Hollevoet, K. Electroporation outperforms in vivo-jetPEI for intratumoral DNA-based reporter gene transfer. Sci. Rep. 2020, 10, 19532. [Google Scholar] [CrossRef]
- Kerstan, A.; Niebergall-Roth, E.; Esterlechner, J.; Schröder, H.M.; Gasser, M.; Waaga-Gasser, A.M.; Goebeler, M.; Rak, K.; Schrüfer, P.; Endres, S.; et al. Ex vivo-expanded highly pure ABCB5+ mesenchymal stromal cells as Good Manufacturing Practice-compliant autologous advanced therapy medicinal product for clinical use: Process validation and first in-human data. Cytotherapy 2021, 23, 165–175. [Google Scholar] [CrossRef]
- Weiss, R.; Gerdes, W.; Berthold, R.; Sack, U.; Koehl, U.; Hauschildt, S.; Grahnert, A. Comparison of Three CD3-Specific Separation Methods Leading to Labeled and Label-Free T Cells. Cells 2021, 10, 2824. [Google Scholar] [CrossRef]
- Han, L.; Zhou, J.; Li, L.; Zhou, K.; Zhao, L.; Zhu, X.; Yin, Q.; Li, Y.; You, H.; Zhang, J.; et al. Culturing adequate CAR-T cells from less peripheral blood to treat B-cell malignancies. Cancer Biol. Med. 2021, 18, 1066–1079. [Google Scholar] [CrossRef]
- Patsali, P.; Turchiano, G.; Papasavva, P.; Romito, M.; Loucari, C.C.; Stephanou, C.; Christou, S.; Sitarou, M.; Mussolino, C.; Cornu, T.I.; et al. Correction of IVS I-110(G>A) β-thalassemia by CRISPR/Cas-and TALEN-mediated disruption of aberrant regulatory elements in human hematopoietic stem and progenitor cells. Haematologica 2019, 104, e497–e501. [Google Scholar] [CrossRef] [Green Version]
- Stephanou, C.; Papasavva, P.; Zachariou, M.; Patsali, P.; Epitropou, M.; Ladas, P.; Al-Abdulla, R.; Christou, S.; Antoniou, M.N.; Lederer, C.W.; et al. Suitability of small diagnostic peripheral-blood samples for cell-therapy studies. Cytotherapy 2017, 19, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Robbins, G.M.; Wang, M.; Pomeroy, E.J.; Moriarity, B.S. Nonviral genome engineering of natural killer cells. Stem Cell Res. Ther. 2021, 12, 350. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Choi, J.H.; Kim, S.H.; Park, H.; Lee, D.; Kim, G.J. Efficacy of Gene Modification in Placenta-Derived Mesenchymal Stem Cells Based on Nonviral Electroporation. Int. J. Stem Cells 2021, 14, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Holstein, M.; Mesa-Nuñez, C.; Miskey, C.; Almarza, E.; Poletti, V.; Schmeer, M.; Grueso, E.; Ordóñez Flores, J.C.; Kobelt, D.; Walther, W.; et al. Efficient Non-viral Gene Delivery into Human Hematopoietic Stem Cells by Minicircle Sleeping Beauty Transposon Vectors. Mol. Ther. 2018, 26, 1137–1153. [Google Scholar] [CrossRef] [Green Version]
- Lattanzi, A.; Meneghini, V.; Pavani, G.; Amor, F.; Ramadier, S.; Felix, T.; Antoniani, C.; Masson, C.; Alibeu, O.; Lee, C.; et al. Optimization of CRISPR/Cas9 Delivery to Human Hematopoietic Stem and Progenitor Cells for Therapeutic Genomic Rearrangements. Mol. Ther. 2019, 27, 137–150. [Google Scholar] [CrossRef] [Green Version]
- Russkamp, N.F.; Myburgh, R.; Kiefer, J.D.; Neri, D.; Manz, M.G. Anti-CD117 immunotherapy to eliminate hematopoietic and leukemia stem cells. Exp. Hematol. 2021, 95, 31–45. [Google Scholar] [CrossRef]
- Mangeot, P.E.; Risson, V.; Fusil, F.; Marnef, A.; Laurent, E.; Blin, J.; Mournetas, V.; Massouridès, E.; Sohier, T.J.M.; Corbin, A.; et al. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat. Commun. 2019, 10, 45. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Psatha, N.; Wang, H.; Singh, M.; Samal, H.B.; Zhang, W.; Ehrhardt, A.; Izsvák, Z.; Papayannopoulou, T.; Lieber, A. Integrating HDAd5/35++ Vectors as a New Platform for HSC Gene Therapy of Hemoglobinopathies. Mol. Ther.-Methods Clin. Dev. 2018, 9, 142–152. [Google Scholar] [CrossRef] [Green Version]
- Banskota, S.; Raguram, A.; Suh, S.; Du, S.W.; Davis, J.R.; Choi, E.H.; Wang, X.; Nielsen, S.C.; Newby, G.A.; Randolph, P.B.; et al. Engineered virus-like particles for efficient in vivo delivery of therapeutic proteins. Cell 2022, 185, 250–265. [Google Scholar] [CrossRef]
- Zu, H.; Gao, D. Non-viral Vectors in Gene Therapy: Recent Development, Challenges, and Prospects. AAPS J. 2021, 23, 78. [Google Scholar] [CrossRef]
- Mangeot, P.E.; Guiguettaz, L.; Sohier, T.J.M.; Ricci, E.P. Delivery of the Cas9/sgRNA Ribonucleoprotein Complex in Immortalized and Primary Cells via Virus-like Particles (“Nanoblades”). J. Vis. Exp. 2021, 169, e62245. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Urip, B.A.; Zhang, Z.; Ngan, C.C.; Feng, B. Evolving AAV-delivered therapeutics towards ultimate cures. J. Mol. Med. 2021, 99, 593–617. [Google Scholar] [CrossRef] [PubMed]
- Definition-Nanomaterials-Environment-European Commission. Available online: https://ec.europa.eu/environment/chemicals/nanotech/faq/definition_en.htm (accessed on 13 January 2021).
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- DeLong, R.K.; Reynolds, C.M.; Malcolm, Y.; Schaeffer, A.; Severs, T.; Wanekaya, A. Functionalized gold nanoparticles for the binding, stabilization, and delivery of therapeutic DNA, RNA, and other biological macromolecules. Nanotechnol. Sci. Appl. 2010, 3, 53–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, R.; Alwani, S.; Badea, I. Polymeric nanoparticles in gene therapy: New avenues of design and optimization for delivery applications. Polymers 2019, 11, 745. [Google Scholar] [CrossRef] [Green Version]
- Tetro, N.; Moushaev, S.; Rubinchik-Stern, M.; Eyal, S. The Placental Barrier: The Gate and the Fate in Drug Distribution. Pharm. Res. 2018, 35, 71. [Google Scholar] [CrossRef]
- Muoth, C.; Aengenheister, L.; Kucki, M.; Wick, P.; Buerki-Thurnherr, T. Nanoparticle transport across the placental barrier: Pushing the field forward. Nanomedicine 2016, 11, 941–957. [Google Scholar] [CrossRef]
- Cruz, L.J.; van Dijk, T.; Vepris, O.; Li, T.M.W.Y.; Schomann, T.; Baldazzi, F.; Kurita, R.; Nakamura, Y.; Grosveld, F.; Philipsen, S.; et al. PLGA-Nanoparticles for Intracellular Delivery of the CRISPR-Complex to Elevate Fetal Globin Expression in Erythroid Cells. Biomaterials 2021, 268, 120580. [Google Scholar] [CrossRef]
- King, A.; Ndifon, C.; Lui, S.; Widdows, K.; Kotamraju, V.R.; Agemy, L.; Teesalu, T.; Glazier, J.D.; Cellesi, F.; Tirelli, N.; et al. Tumor-homing peptides as tools for targeted delivery of payloads to the placenta. Sci. Adv. 2016, 2, e1600349. [Google Scholar] [CrossRef] [Green Version]
- Kaitu’u-Lino, T.J.; Pattison, S.; Ye, L.; Tuohey, L.; Sluka, P.; MacDiarmid, J.; Brahmbhatt, H.; Johns, T.; Horne, A.W.; Brown, J.; et al. Targeted nanoparticle delivery of doxorubicin into placental tissues to treat ectopic pregnancies. Endocrinology 2013, 154, 911–919. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Li, H.; Xue, J.; Chen, P.; Zhou, Q.; Zhang, C. Nanoparticle-Mediated Simultaneous Downregulation of Placental Nrf2 and sFlt1 Improves Maternal and Fetal Outcomes in a Preeclampsia Mouse Model. ACS Biomater. Sci. Eng. 2020, 6, 5866–5873. [Google Scholar] [CrossRef] [PubMed]
- EUR-Lex-32007R1394-EN-EUR-Lex. Available online: https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=CELEX%3A32007R1394 (accessed on 13 January 2021).
- Promising mRNA Tech Comes with Regulatory, CMC Headaches|RAPS. Available online: https://www.raps.org/news-and-articles/news-articles/2020/11/euro-convergence-regulatory-and-cmc-considerations (accessed on 13 January 2021).
- Singh, V.P.; McKinney, S.; Gerton, J.L. Persistent DNA Damage and Senescence in the Placenta Impacts Developmental Outcomes of Embryos. Dev. Cell 2020, 54, 333–347.e7. [Google Scholar] [CrossRef]
- Pritchard, N.; Kaitu’u-Lino, T.; Harris, L.; Tong, S.; Hannan, N. Nanoparticles in pregnancy: The next frontier in reproductive therapeutics. Hum. Reprod. Update 2020, 27, 280–304. [Google Scholar] [CrossRef]
- Irvin-Choy, N.S.; Nelson, K.M.; Gleghorn, J.P.; Day, E.S. Design of nanomaterials for applications in maternal/fetal medicine. J. Mater. Chem. B 2020, 8, 6548–6561. [Google Scholar] [CrossRef]
- Tsukamoto, M.; Ochiya, T.; Yoshida, S.; Sugimura, T.; Terada, M. Gene transfer and expression in progeny after intravenous DNA injection into pregnant mice. Nat. Genet. 1995, 9, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Cornford, E.M.; Hyman, S.; Cornford, M.E.; Chytrova, G.; Rhee, J.; Suzuki, T.; Yamagata, T.; Yamakawa, K.; Penichet, M.L.; Pardridge, W.M. Non-invasive gene targeting to the fetal brain after intravenous administration and transplacental transfer of plasmid DNA using PEGylated immunoliposomes. J. Drug Target. 2016, 24, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Ellah, N.A.; Taylor, L.; Troja, W.; Owens, K.; Ayres, N.; Pauletti, G.; Jones, H. Development of non-viral, trophoblast-specific gene delivery for placental therapy. PLoS ONE 2015, 10, e0140879. [Google Scholar] [CrossRef]
- Giubilato, E.; Cazzagon, V.; Amorim, M.J.B.; Blosi, M.; Bouillard, J.; Bouwmeester, H.; Costa, A.L.; Fadeel, B.; Fernandes, T.F.; Fito, C.; et al. Risk Management Framework for Nano-Biomaterials Used in Medical Devices and Advanced Therapy Medicinal Products. Materials 2020, 13, 4532. [Google Scholar] [CrossRef] [PubMed]
- BIOmaterial RIsk MAnagement|BIORIMA Project|H2020|CORDIS|European Commission. Available online: https://cordis.europa.eu/project/id/760928 (accessed on 13 January 2021).
- Caplan, H.; Olson, S.D.; Kumar, A.; George, M.; Prabhakara, K.S.; Wenzel, P.; Bedi, S.; Toledano-Furman, N.E.; Triolo, F.; Kamhieh-Milz, J.; et al. Mesenchymal Stromal Cell Therapeutic Delivery: Translational Challenges to Clinical Application. Front. Immunol. 2019, 10, 1645. [Google Scholar] [CrossRef]
- Bonig, H.; Kuçi, Z.; Kuçi, S.; Bakhtiar, S.; Basu, O.; Bug, G.; Dennis, M.; Greil, J.; Barta, A.; Kállay, K.M.; et al. Children and Adults with Refractory Acute Graft-versus-Host Disease Respond to Treatment with the Mesenchymal Stromal Cell Preparation “MSC-FFM”-Outcome Report of 92 Patients. Cells 2019, 8, 1577. [Google Scholar] [CrossRef] [Green Version]
- Namba, F. Mesenchymal stem cells for the prevention of bronchopulmonary dysplasia. Pediatr. Int. 2019, 61, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Delhove, J.; Osenk, I.; Prichard, I.; Donnelley, M. Public Acceptability of Gene Therapy and Gene Editing for Human Use: A Systematic Review. Hum. Gene Ther. 2020, 31, 20–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seoane-Vazquez, E.; Shukla, V.; Rodriguez-Monguio, R. Innovation and competition in advanced therapy medicinal products. EMBO Mol. Med. 2019, 11, e9992. [Google Scholar] [CrossRef] [PubMed]
- Lipsitz, Y.Y.; Milligan, W.D.; Fitzpatrick, I.; Stalmeijer, E.; Farid, S.S.; Tan, K.Y.; Smith, D.; Perry, R.; Carmen, J.; Chen, A.; et al. A roadmap for cost-of-goods planning to guide economic production of cell therapy products. Cytotherapy 2017, 19, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Walpole, S.C.; Prieto-Merino, D.; Edwards, P.; Cleland, J.; Stevens, G.; Roberts, I. The weight of nations: An estimation of adult human biomass. BMC Public Health 2012, 12, 439. [Google Scholar] [CrossRef] [Green Version]
- Janssen, P.A.; Thiessen, P.; Klein, M.C.; Whitfield, M.F.; Macnab, Y.C.; Cullis-Kuhl, S.C. Standards for the measurement of birth weight, length and head circumference at term in neonates of European, Chinese and South Asian ancestry. Open Med. 2007, 1, e74–e88. [Google Scholar]
- Shaw, S.W.S.S.; Blundell, M.P.; Pipino, C.; Shangaris, P.; Maghsoudlou, P.; Ramachandra, D.L.; Georgiades, F.; Boyd, M.; Thrasher, A.J.; Porada, C.D.; et al. Sheep CD34+ amniotic fluid cells have hematopoietic potential and engraft after autologous in utero transplantation. Stem Cells 2015, 33, 122–132. [Google Scholar] [CrossRef]
- Aurich, B.; Jacqz-Aigrain, E. Drug safety in translational paediatric research: Practical points to consider for paediatric safety profiling and protocol development: A scoping review. Pharmaceutics 2021, 13, 695. [Google Scholar] [CrossRef]
- Ceci, A.; Felisi, M.; Baiardi, P.; Bonifazi, F.; Catapano, M.; Giaquinto, C.; Nicolosi, A.; Sturkenboom, M.; Neubert, A.; Wong, I. Medicines for children licensed by the European Medicines Agency. (EMEA): The balance after 10 years. Eur. J. Clin. Pharmacol. 2006, 62, 947–952. [Google Scholar] [CrossRef]
- Giannuzzi, V.; Conte, R.; Landi, A.; Ottomano, S.A.; Bonifazi, D.; Baiardi, P.; Bonifazi, F.; Ceci, A. Orphan medicinal products in Europe and United States to cover needs of patients with rare diseases: An increased common effort is to be foreseen. Orphanet J. Rare Dis. 2017, 12, 64. [Google Scholar] [CrossRef] [Green Version]
- TEDDY – European Network of Excellence for Paediatric Clinical Research. European Paediatric Medicines Database (EPMD). Available online: https://www.teddynetwork.net/european-paediatric-medicines-database-epmd/ (accessed on 26 October 2021).
- Pierce, G.F. Uncertainty in an era of transformative therapy for haemophilia: Addressing the unknowns. Haemophilia 2021, 27, 103–113. [Google Scholar] [CrossRef]
- Brooks, S.P.; Bubela, T. Application of protection motivation theory to clinical trial enrolment for pediatric chronic conditions. BMC Pediatr. 2020, 20, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushman, F.D. Retroviral Insertional Mutagenesis in Humans: Evidence for Four Genetic Mechanisms Promoting Expansion of Cell Clones. Mol. Ther. 2020, 28, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Gene therapy needs a long-term approach. Nat. Med. 2021, 27, 563. [CrossRef] [PubMed]
- Blattner, G.; Cavazza, A.; Thrasher, A.J.; Turchiano, G. Gene Editing and Genotoxicity: Targeting the Off-Targets. Front. Genome Ed. 2020, 2, 613252. [Google Scholar] [CrossRef]
- Almeida-Porada, G.; Waddington, S.N.; Chan, J.K.Y.; Peranteau, W.H.; MacKenzie, T.; Porada, C.D. In Utero Gene Therapy Consensus Statement from the IFeTIS. Mol. Ther. 2019, 27, 705–707. [Google Scholar] [CrossRef] [Green Version]
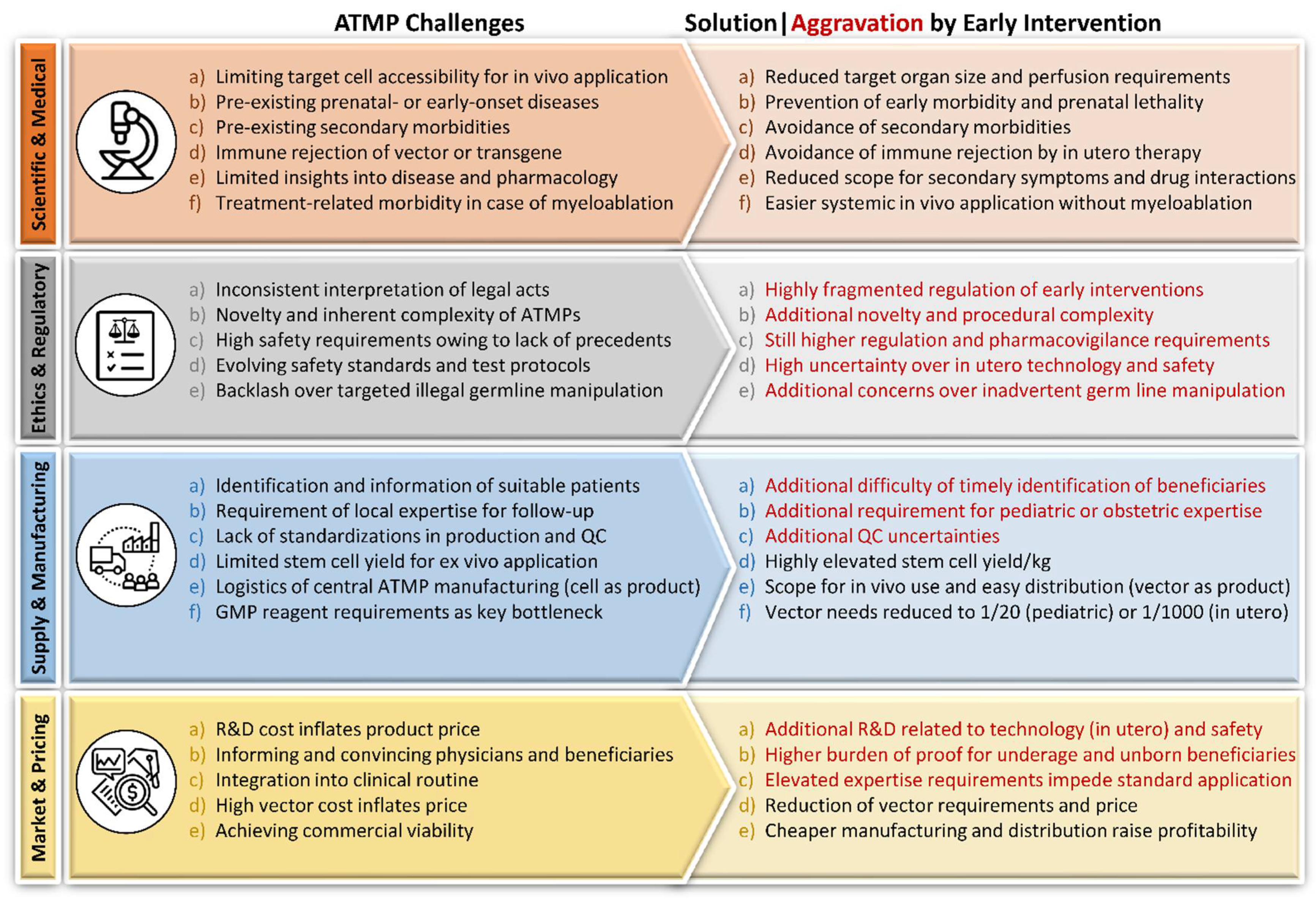
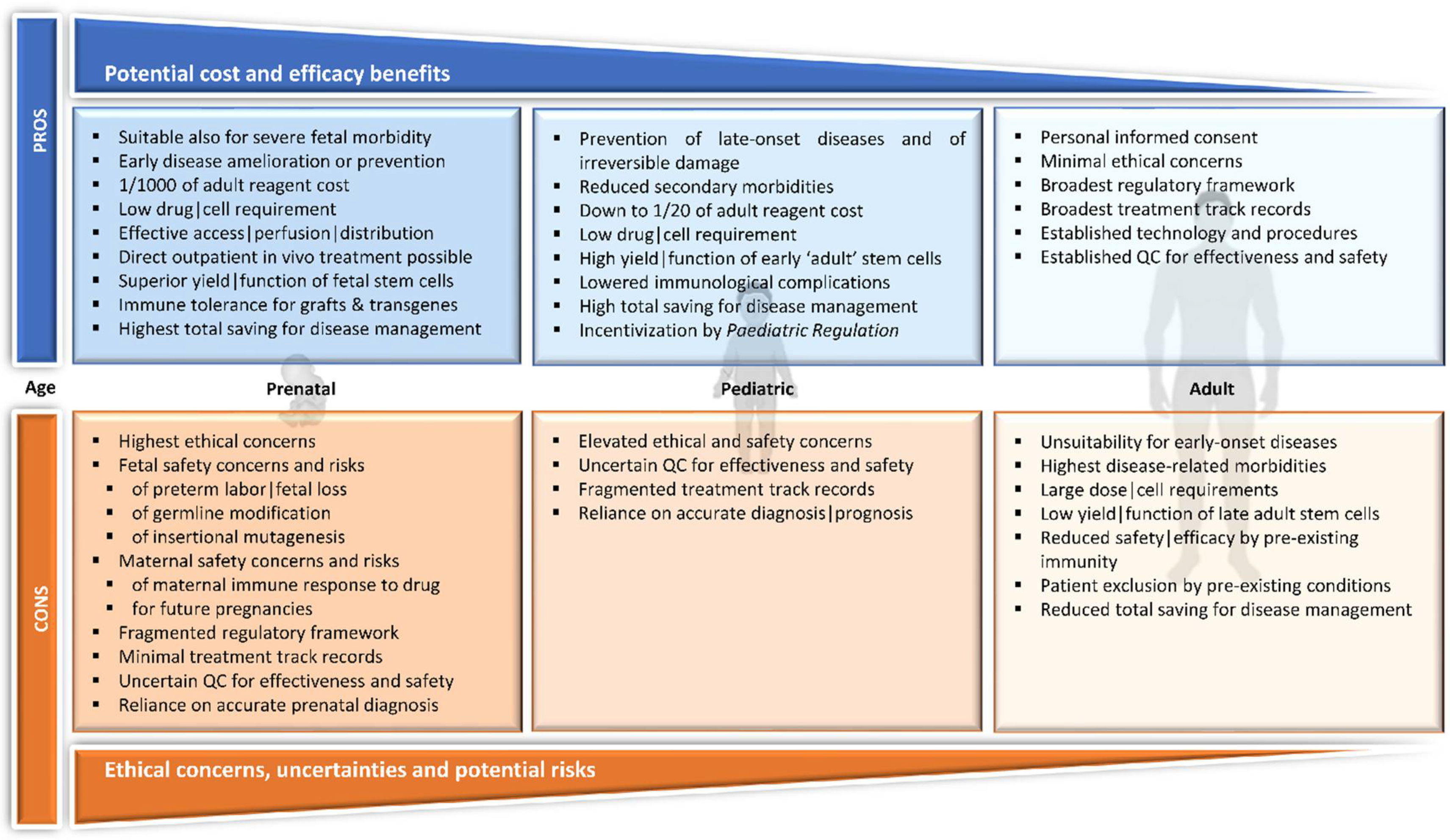
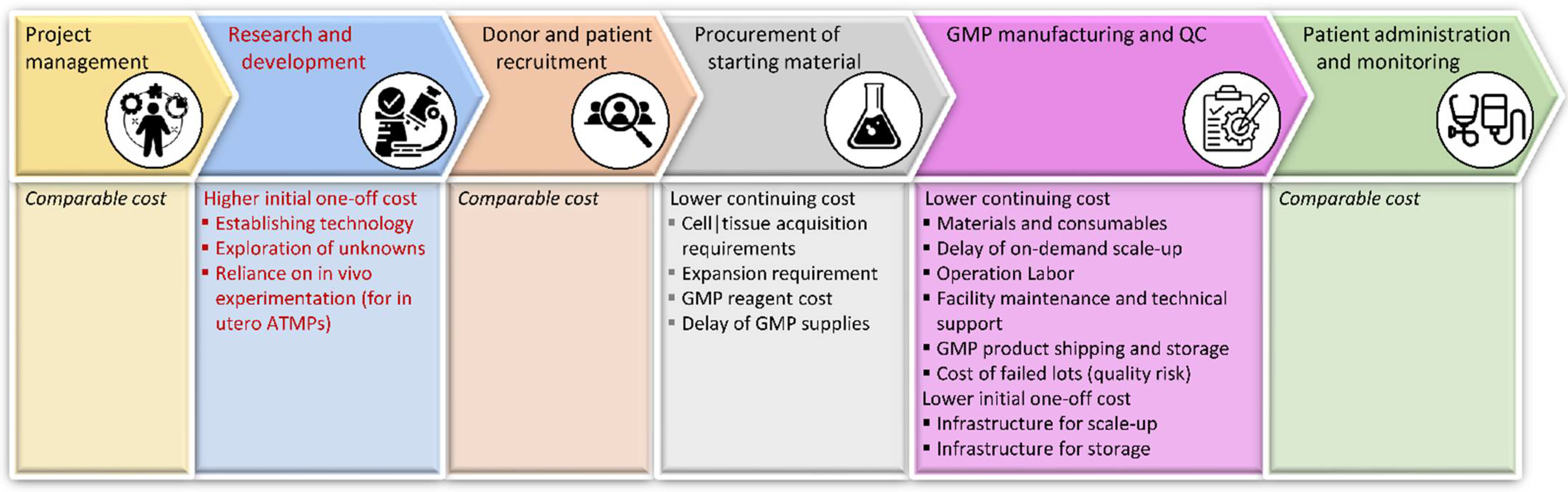
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lederer, C.W.; Koniali, L.; Buerki-Thurnherr, T.; Papasavva, P.L.; La Grutta, S.; Licari, A.; Staud, F.; Bonifazi, D.; Kleanthous, M. Catching Them Early: Framework Parameters and Progress for Prenatal and Childhood Application of Advanced Therapies. Pharmaceutics 2022, 14, 793. https://doi.org/10.3390/pharmaceutics14040793
Lederer CW, Koniali L, Buerki-Thurnherr T, Papasavva PL, La Grutta S, Licari A, Staud F, Bonifazi D, Kleanthous M. Catching Them Early: Framework Parameters and Progress for Prenatal and Childhood Application of Advanced Therapies. Pharmaceutics. 2022; 14(4):793. https://doi.org/10.3390/pharmaceutics14040793
Chicago/Turabian StyleLederer, Carsten W., Lola Koniali, Tina Buerki-Thurnherr, Panayiota L. Papasavva, Stefania La Grutta, Amelia Licari, Frantisek Staud, Donato Bonifazi, and Marina Kleanthous. 2022. "Catching Them Early: Framework Parameters and Progress for Prenatal and Childhood Application of Advanced Therapies" Pharmaceutics 14, no. 4: 793. https://doi.org/10.3390/pharmaceutics14040793