
Application of Machine Learning Classification to Improve the Performance of Vancomycin Therapeutic Drug Monitoring


Abstract
:1. Introduction
2. Materials and Methods
2.1. Classifier Development
2.1.1. PK Models for the Classifier
2.1.2. Virtual Patients for the Classifier
2.1.3. Features and Labels
2.1.4. Classification Model
2.2. Validation of TDM Performance
2.2.1. PK Models and Virtual Patients for Validation
2.2.2. PK Parameter Estimation
2.2.3. ML Application
2.2.4. Performance Evaluation
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Filippone, E.J.; Kraft, W.K.; Farber, J.L. The nephrotoxicity of vancomycin. Clin. Pharmacol. Ther. 2017, 102, 459–469. [Google Scholar] [CrossRef]
- Matzke, G.; Zhanel, G.; Guay, D. Clinical pharmacokinetics of vancomycin. Clin. Pharmacokinet. 1986, 11, 257–282. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A. Therapeutic Drug Monitoring: Newer Drugs and Biomarkers; Academic Press: Cambridge, MA, USA, 2012. [Google Scholar]
- Rybak, M.J.; Le, J.; Lodise, T.P.; Levine, D.P.; Bradley, J.S.; Liu, C.; Mueller, B.A.; Pai, M.P.; Wong-Beringer, A.; Rotschafer, J.C. Therapeutic monitoring of vancomycin for serious methicillin-resistant Staphylococcus aureus infections: A revised consensus guideline and review by the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, the Pediatric Infectious Diseases Society, and the Society of Infectious Diseases Pharmacists. Clin. Infect. Dis. 2020, 71, 1361–1364. [Google Scholar] [PubMed]
- Drennan, P.; Doogue, M.; van Hal, S.J.; Chin, P. Bayesian therapeutic drug monitoring software: Past, present and future. Int. J. Pharmacokinet. 2018, 3, 109–114. [Google Scholar] [CrossRef]
- Broeker, A.; Nardecchia, M.; Klinker, K.; Derendorf, H.; Day, R.; Marriott, D.; Carland, J.; Stocker, S.; Wicha, S. Towards precision dosing of vancomycin: A systematic evaluation of pharmacometric models for Bayesian forecasting. Clin. Microbiol. Infect. 2019, 25, 1286.e1–1286.e7. [Google Scholar] [CrossRef]
- Guo, T.; van Hest, R.M.; Roggeveen, L.F.; Fleuren, L.M.; Thoral, P.J.; Bosman, R.J.; van der Voort, P.H.; Girbes, A.R.; Mathot, R.A.; Elbers, P.W. External evaluation of population pharmacokinetic models of vancomycin in large cohorts of intensive care unit patients. Antimicrob. Agents Chemother. 2019, 63, e02543-18. [Google Scholar] [CrossRef] [Green Version]
- Rodvold, K.A. 60 plus years later and we are still trying to learn how to dose vancomycin. Clin. Infect. Dis. 2020, 70, 1546–1549. [Google Scholar] [CrossRef]
- Uster, D.W.; Stocker, S.L.; Carland, J.E.; Brett, J.; Marriott, D.J.; Day, R.O.; Wicha, S.G. A model averaging/selection approach improves the predictive performance of model-informed precision dosing: Vancomycin as a case study. Clin. Pharmacol. Ther. 2021, 109, 175–183. [Google Scholar] [CrossRef]
- Aoki, Y.; Röshammar, D.; Hamrén, B.; Hooker, A.C. Model selection and averaging of nonlinear mixed-effect models for robust phase III dose selection. J. Pharmacokinet. Pharmacodyn. 2017, 44, 581–597. [Google Scholar] [CrossRef] [Green Version]
- Buckland, S.T.; Burnham, K.P.; Augustin, N.H. Model selection: An integral part of inference. Biometrics 1997, 53, 603–618. [Google Scholar] [CrossRef]
- Badillo, S.; Banfai, B.; Birzele, F.; Davydov, I.I.; Hutchinson, L.; Kam-Thong, T.; Siebourg-Polster, J.; Steiert, B.; Zhang, J.D. An introduction to machine learning. Clin. Pharmacol. Ther. 2020, 107, 871–885. [Google Scholar] [CrossRef] [Green Version]
- Ngiam, K.Y.; Khor, W. Big data and machine learning algorithms for health-care delivery. Lancet Oncol. 2019, 20, e262–e273. [Google Scholar] [CrossRef]
- Huang, X.; Yu, Z.; Bu, S.; Lin, Z.; Hao, X.; He, W.; Yu, P.; Wang, Z.; Gao, F.; Zhang, J. An ensemble model for prediction of vancomycin trough concentrations in pediatric patients. DrugDes. Dev. Ther. 2021, 15, 1549. [Google Scholar] [CrossRef]
- Hughes, J.H.; Keizer, R.J. A hybrid machine learning/pharmacokinetic approach outperforms maximum a posteriori Bayesian estimation by selectively flattening model priors. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 1150–1160. [Google Scholar] [CrossRef]
- Woillard, J.B.; Labriffe, M.; Debord, J.; Marquet, P. Tacrolimus exposure prediction using machine learning. Clin. Pharmacol. Ther. 2021, 110, 361–369. [Google Scholar] [CrossRef]
- Aljutayli, A.; Marsot, A.; Nekka, F. An update on population pharmacokinetic analyses of vancomycin, part I: In adults. Clin. Pharmacokinet. 2020, 59, 671–698. [Google Scholar] [CrossRef]
- Marsot, A.; Boulamery, A.; Bruguerolle, B.; Simon, N. Vancomycin. Clin. Pharmacokinet. 2012, 51, 1–13. [Google Scholar] [CrossRef]
- Lim, H.S.; Chong, Y.; Noh, Y.H.; Jung, J.A.; Kim, Y. Exploration of optimal dosing regimens of vancomycin in patients infected with methicillin-resistant Staphylococcus aureus by modeling and simulation. J. Clin. Pharm. Ther. 2014, 39, 196–203. [Google Scholar] [CrossRef]
- Llopis-Salvia, P.; Jimenez-Torres, N. Population pharmacokinetic parameters of vancomycin in critically ill patients. J. Clin. Pharm. Ther. 2006, 31, 447–454. [Google Scholar] [CrossRef]
- Moore, J.; Healy, J.; Thoma, B.; Peahota, M.; Ahamadi, M.; Schmidt, L.; Cavarocchi, N.; Kraft, W. A population pharmacokinetic model for vancomycin in adult patients receiving extracorporeal membrane oxygenation therapy. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 495–502. [Google Scholar] [CrossRef]
- Mulla, H.; Pooboni, S. Population pharmacokinetics of vancomycin in patients receiving extracorporeal membrane oxygenation. Br. J. Clin. Pharmacol. 2005, 60, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Okada, A.; Kariya, M.; Irie, K.; Okada, Y.; Hiramoto, N.; Hashimoto, H.; Kajioka, R.; Maruyama, C.; Kasai, H.; Hamori, M. Population pharmacokinetics of vancomycin in patients undergoing allogeneic hematopoietic stem-cell transplantation. J. Clin. Pharmacol. 2018, 58, 1140–1149. [Google Scholar] [CrossRef]
- Purwonugroho, T.A.; Chulavatnatol, S.; Preechagoon, Y.; Chindavijak, B.; Malathum, K.; Bunuparadah, P. Population pharmacokinetics of vancomycin in Thai patients. Sci. World J. 2012, 2012, 762649. [Google Scholar] [CrossRef]
- Sanchez, J.; Dominguez, A.; Lane, J.; Anderson, P.; Capparelli, E.; Cornejo-Bravo, J. Population pharmacokinetics of vancomycin in adult and geriatric patients: Comparison of eleven approaches. Int. J. Clin. Pharmacol. Ther. 2010, 48, 525–533. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kuzuya, T.; Baba, H.; Yamada, K.; Nabeshima, T. Population pharmacokinetic analysis of vancomycin in patients with gram-positive infections and the influence of infectious disease type. J. Clin. Pharm. Ther. 2009, 34, 473–483. [Google Scholar] [CrossRef]
- Yasuhara, M.; Iga, T.; Zenda, H.; Okumura, K.; Oguma, T.; Yano, Y.; Hori, R. Population pharmacokinetics of vancomycin in Japanese adult patients. Ther. Drug Monit. 1998, 20, 139–148. [Google Scholar] [CrossRef]
- Vancomycin HCl Injection; [Package Insert]; HK Inno.N Co.: Seoul, Korea, 2020.
- Baron, K.T.; Hindmarsh, A.; Petzold, L.; Gillespie, B.; Margossian, C.; Pastoor, D. mrgsolve: Simulate from ODE-Based Population PK/PD and Systems Pharmacology Models. 2019. Available online: https://github.com/metrumresearchgroup/mrgsolve (accessed on 27 March 2022).
- Therneau, T.; Atkinson, B. rpart: Recursive Partitioning and Regression Trees. R Package Version 4.1–15. 2019. Available online: https://github.com/bethatkinson/rpart (accessed on 27 March 2022).
- Wright, M.N.; Wager, S.; Probst, P.; Wright, M.M.N. Package ‘ranger’. Version 0.11. 2019, Volume 2. Available online: https://github.com/imbs-hl/ranger (accessed on 27 March 2022).
- Chen, T.; He, T.; Benesty, M.; Khotilovich, V. Package ‘xgboost’. R Version. 2019, Volume 90. Available online: https://github.com/dmlc/xgboost (accessed on 27 March 2022).
- Bischl, B.; Lang, M.; Kotthoff, L.; Schiffner, J.; Richter, J.; Studerus, E.; Casalicchio, G.; Jones, Z.M. mlr: Machine learning in R. J. Mach. Learn. Res. 2016, 17, 5938–5942. [Google Scholar]
- Bae, S.H.; Yim, D.-S.; Lee, H.; Park, A.-R.; Kwon, J.-E.; Sumiko, H.; Han, S. Application of pharmacometrics in pharmacotherapy: Open-source software for vancomycin therapeutic drug management. Pharmaceutics 2019, 11, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolton, M.; Xu, H.; Cheong, E.; Maitz, P.; Kennedy, P.; Gottlieb, T.; Buono, E.; McLachlan, A.J. Vancomycin pharmacokinetics in patients with severe burn injuries. Burns 2010, 36, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Goti, V.; Chaturvedula, A.; Fossler, M.J.; Mok, S.; Jacob, J.T. Hospitalized patients with and without hemodialysis have markedly different vancomycin pharmacokinetics: A population pharmacokinetic model-based analysis. Ther. Drug Monit. 2018, 40, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Medellín-Garibay, S.E.; Ortiz-Martín, B.; Rueda-Naharro, A.; García, B.; Romano-Moreno, S.; Barcia, E. Pharmacokinetics of vancomycin and dosing recommendations for trauma patients. J. Antimicrob. Chemother. 2016, 71, 471–479. [Google Scholar] [CrossRef] [Green Version]
- Le Louedec, F.; Puisset, F.; Thomas, F.; Chatelut, É.; White-Koning, M. Easy and reliable maximum a posteriori Bayesian estimation of pharmacokinetic parameters with the open-source R package mapbayr. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 1208–1220. [Google Scholar] [CrossRef]
- Kuhn, M.; Wing, J.; Weston, S.; Williams, A.; Keefer, C.; Engelhardt, A.; Cooper, T.; Mayer, Z.; Kenkel, B.; Team, R.C. Package ‘caret’. R J. 2020, 223, 7. [Google Scholar]
- Kuhn, M.; Johnson, K. Feature Engineering and Selection: A Practical Approach for Predictive Models; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- D’Argenio, D.Z. Optimal sampling times for pharmacokinetic experiments. J. Pharmacokinet. Biopharm. 1981, 9, 739–756. [Google Scholar] [CrossRef]
- Hastie, T.; Tibshirani, R.; Friedman, J. Model Assessment and Selection. In The Elements of Statistical Learning; Springer: Berlin/Heidelberg, Germany, 2009; pp. 219–259. [Google Scholar]
- Chen, T.; Guestrin, C. Xgboost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
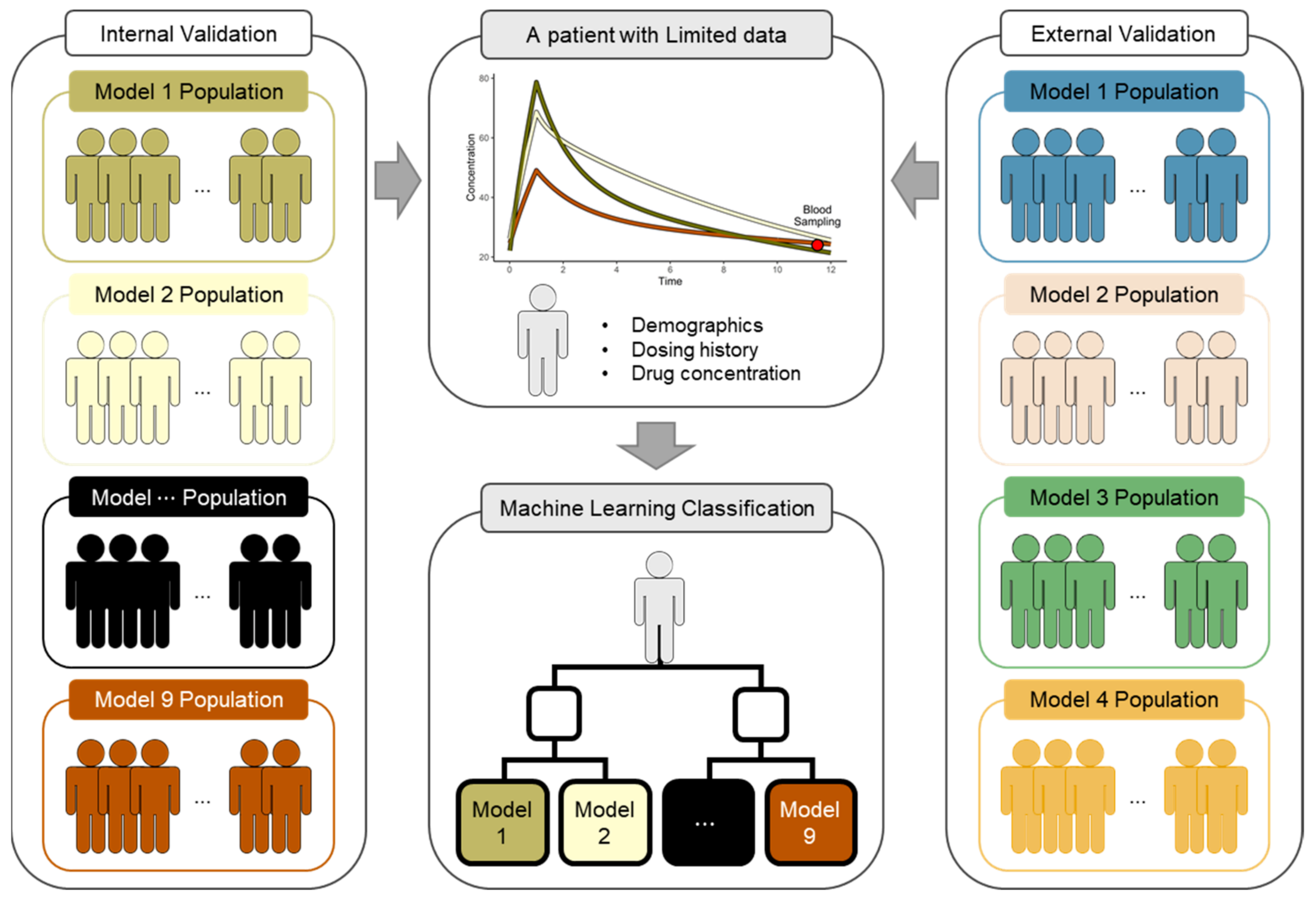



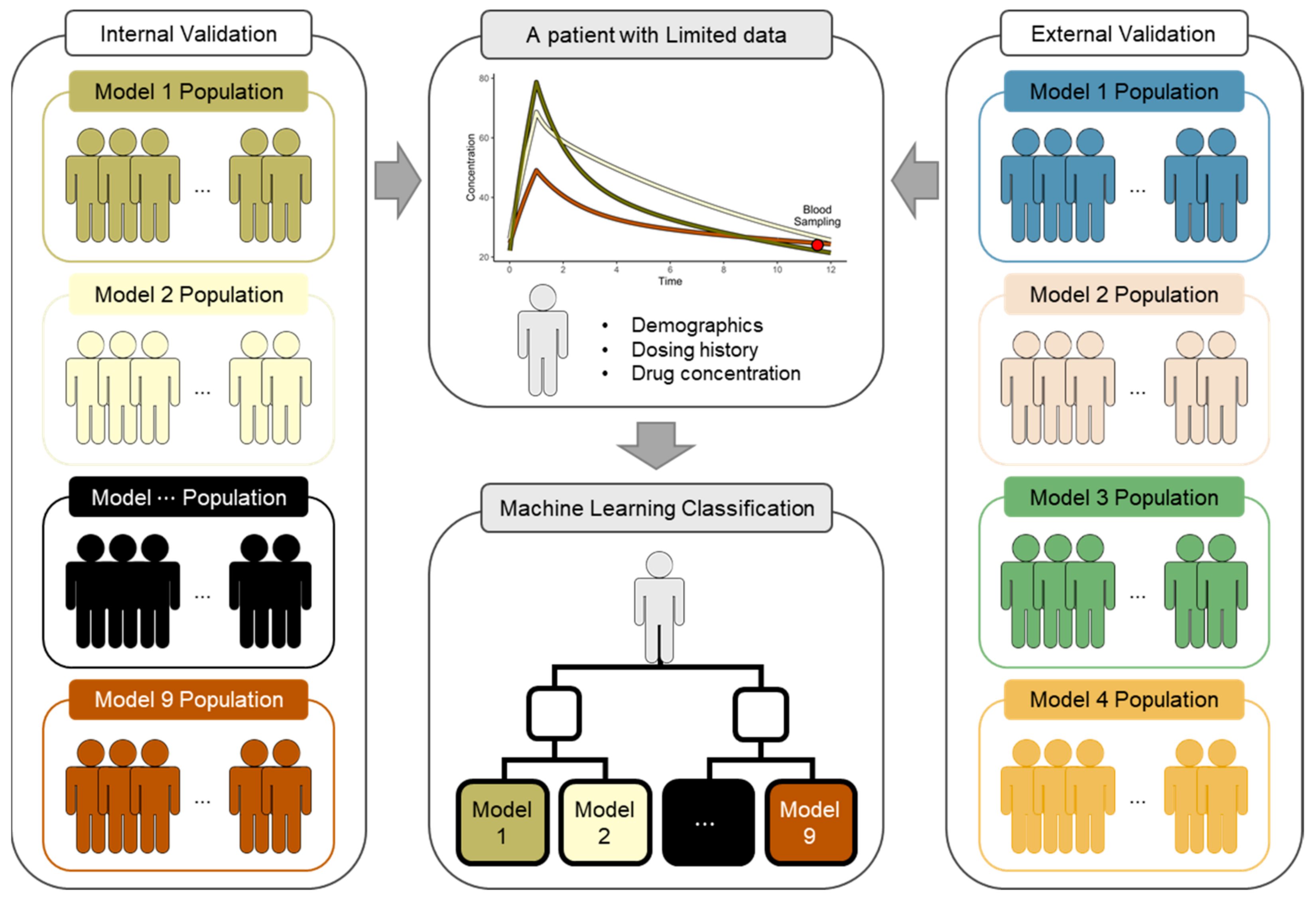{kind=link}
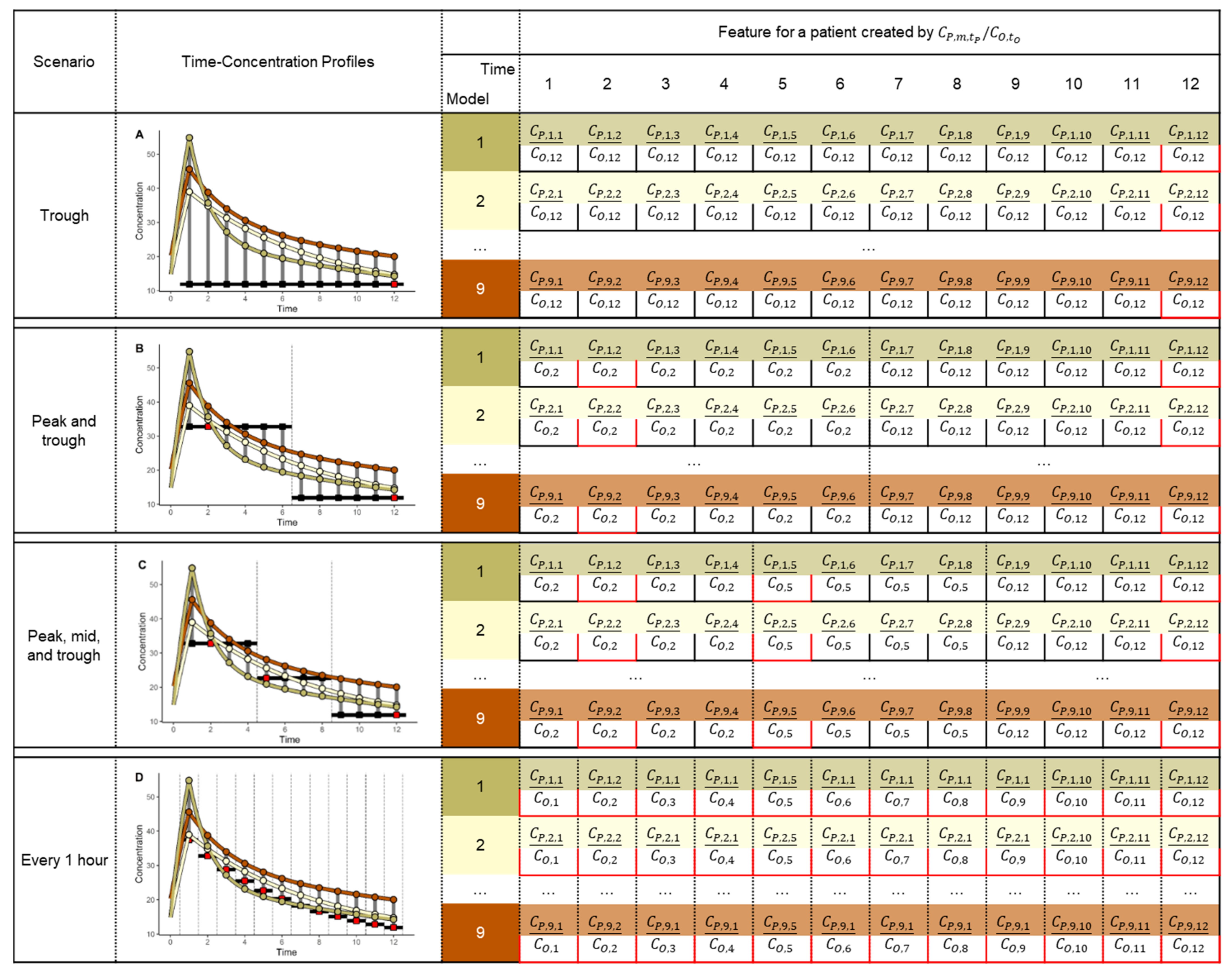{kind=link}
{kind=link}
{kind=link}
| Scenarios | Trough (%) | Peak and Trough (%) | Peak, Mid, and Trough (%) | One-HourInterval (%) |
|---|---|---|---|---|
| Decision Tree | ||||
| Single Dose | 21.0 | 22.2 | 30.5 | 31.1 |
| Steady State | 16.8 | 20.7 | 22.9 | 27.0 |
| Random Forest | ||||
| Single Dose | 23.4 | 30.7 | 42.6 | 68.6 |
| Steady State | 19.1 | 27.0 | 33.3 | 54.4 |
| XGBoost | ||||
| Single Dose | 24.6 | 31.8 | 42.7 | 71.6 |
| Steady State | 20.8 | 27.8 | 33.7 | 56.6 |
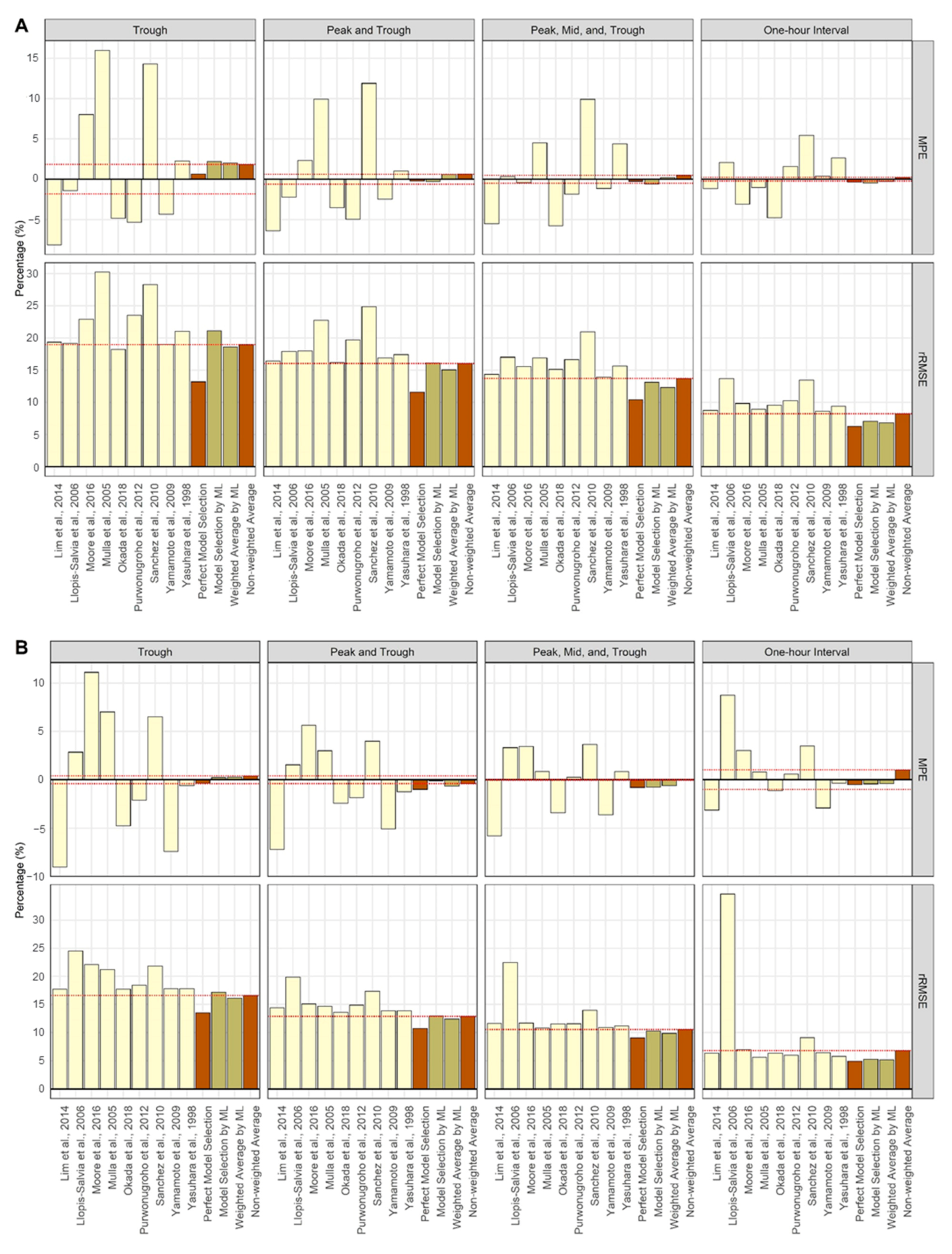
| Measures | ||||||||
|---|---|---|---|---|---|---|---|---|
| Scenarios | Trough | Peak and Trough | Peak, Mid, and Trough | One-Hour Interval | Trough | Peak and Trough | Peak, Mid, and Trough | One-Hour Interval |
| Single Dose Model | ||||||||
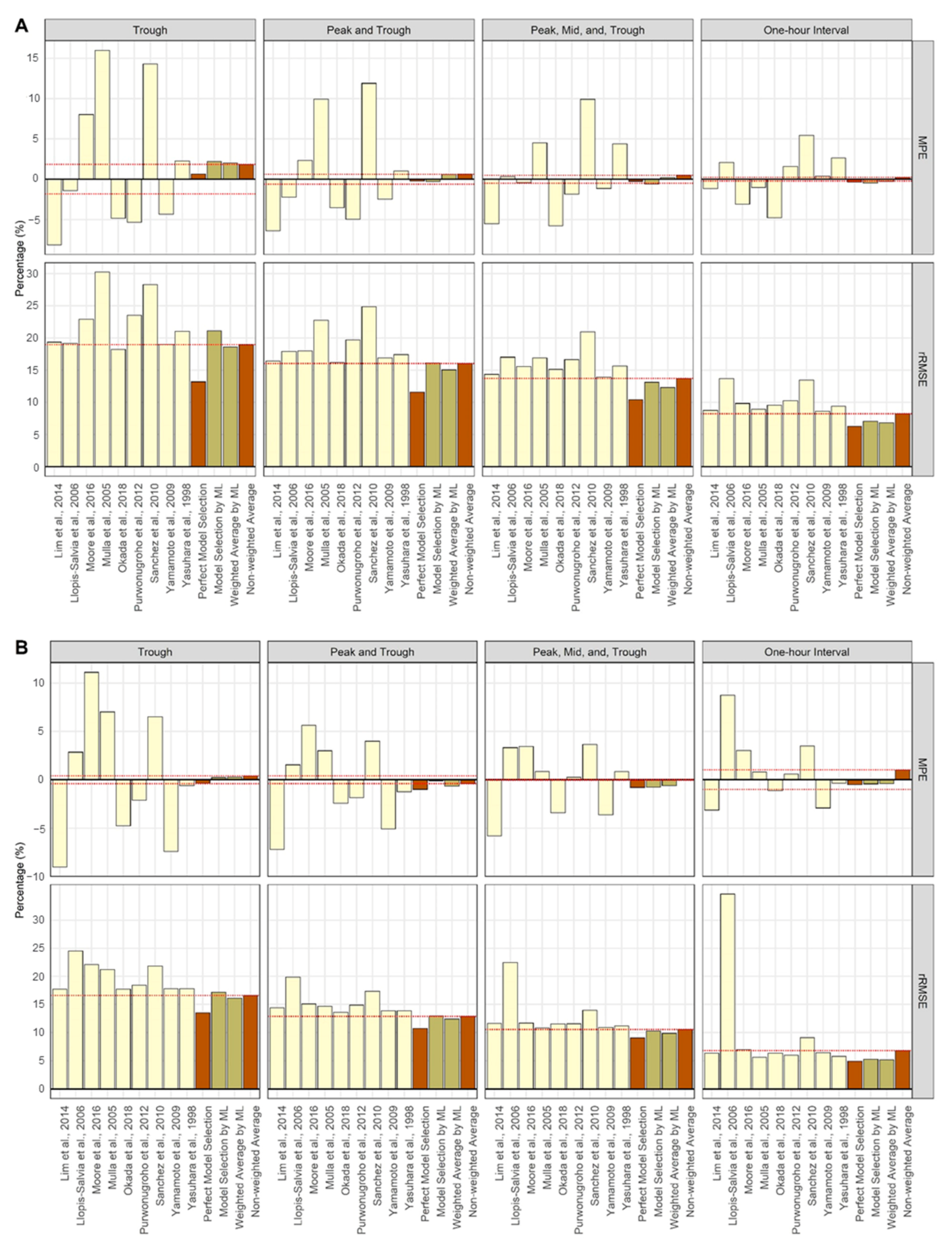
| Lim et al., 2014 [19] | −8.16 | −6.40 | −5.50 | −1.15 | 19.36 | 16.40 | 14.36 | 8.75 |
| Llopis-Salvia et al., 2006 [20] | −1.39 | −2.24 | 0.32 | 2.10 | 19.18 | 17.92 | 17.03 | 13.70 |
| Moore et al., 2016 [21] | 8.02 | 2.31 | −0.44 | −3.11 | 22.93 | 17.98 | 15.58 | 9.83 |
| Mulla et al., 2005 [22] | 15.97 | 9.91 | 4.49 | −1.02 | 30.25 | 22.75 | 16.93 | 8.97 |
| Okada et al., 2018 [23] | −4.86 | −3.53 | −5.76 | −4.79 | 18.25 | 16.23 | 15.14 | 9.56 |
| Purwonugroho et al., 2012 [24] | −5.33 | −4.96 | −1.86 | 1.59 | 23.52 | 19.72 | 16.63 | 10.25 |
| Sánchez et al., 2010 [25] | 14.31 | 11.88 | 9.90 | 5.42 | 28.29 | 24.89 | 20.94 | 13.46 |
| Yamamoto et al., 2009 [26] | −4.32 | −2.45 | −1.13 | 0.38 | 19.03 | 16.89 | 13.94 | 8.64 |
| Yasuhara et al., 1998 [27] | 2.27 | 1.01 | 4.40 | 2.64 | 21.05 | 17.42 | 15.64 | 9.39 |
| Perfect Model Selection | 0.65 | −0.23 | −0.26 | −0.35 | 13.19 | 11.62 | 10.44 | 6.25 |
| Model Selection by ML | 2.22 | −0.31 | −0.60 | −0.46 | 21.10 | 16.15 | 13.12 | 7.09 |
| Weighted Average by ML | 2.00 | 0.59 | 0.19 | −0.27 | 18.60 | 15.07 | 12.32 | 6.84 |
| Non-weighted average | 1.83 | 0.61 | 0.49 | 0.23 | 18.97 | 16.04 | 13.71 | 8.22 |
| Steady-State Model | ||||||||
| Lim et al., 2014 [19] | −9.02 | −7.21 | −5.78 | −3.13 | 17.70 | 14.40 | 11.60 | 6.33 |
| Llopis-Salvia et al., 2006 [20] | 2.85 | 1.55 | 3.32 | 8.73 | 24.55 | 19.89 | 22.46 | 34.66 |
| Moore et al., 2016 [21] | 11.12 | 5.65 | 3.45 | 3.02 | 22.06 | 15.08 | 11.68 | 7.00 |
| Mulla et al., 2005 [22] | 7.02 | 3.00 | 0.87 | 0.82 | 21.23 | 14.63 | 10.80 | 5.63 |
| Okada et al., 2018 [23] | −4.74 | −2.40 | −3.39 | −1.14 | 17.73 | 13.57 | 11.55 | 6.35 |
| Purwonugroho et al., 2012 [24] | −2.11 | −1.84 | 0.26 | 0.60 | 18.45 | 14.89 | 11.57 | 6.00 |
| Sánchez et al., 2010 [25] | 6.52 | 4.00 | 3.64 | 3.48 | 21.83 | 17.32 | 13.98 | 9.08 |
| Yamamoto et al., 2009 [26] | −7.40 | −5.06 | −3.62 | −2.91 | 17.78 | 13.87 | 10.92 | 6.43 |
| Yasuhara et al., 1998 [27] | −0.59 | −1.25 | 0.84 | −0.36 | 17.81 | 13.86 | 11.19 | 5.78 |
| Perfect Model Selection | −0.35 | −0.99 | −0.79 | −0.48 | 13.51 | 10.76 | 9.07 | 4.94 |
| Model Selection by ML | 0.25 | −0.11 | −0.77 | −0.42 | 17.17 | 12.95 | 10.28 | 5.28 |
| Weighted Average by ML | 0.27 | −0.64 | −0.61 | −0.40 | 16.11 | 12.40 | 9.87 | 5.18 |
| Non-weighted Average | 0.41 | −0.39 | −0.05 | 1.01 | 16.59 | 12.87 | 10.56 | 6.80 |
| Measures | ||||||||
|---|---|---|---|---|---|---|---|---|
| Scenarios | Trough | Peak and Trough | Peak, Mid and Trough | One-hour Interval | Trough | Peak and Trough | Peak, Mid and Trough | One-hour Interval |
| Single Dose Model | ||||||||
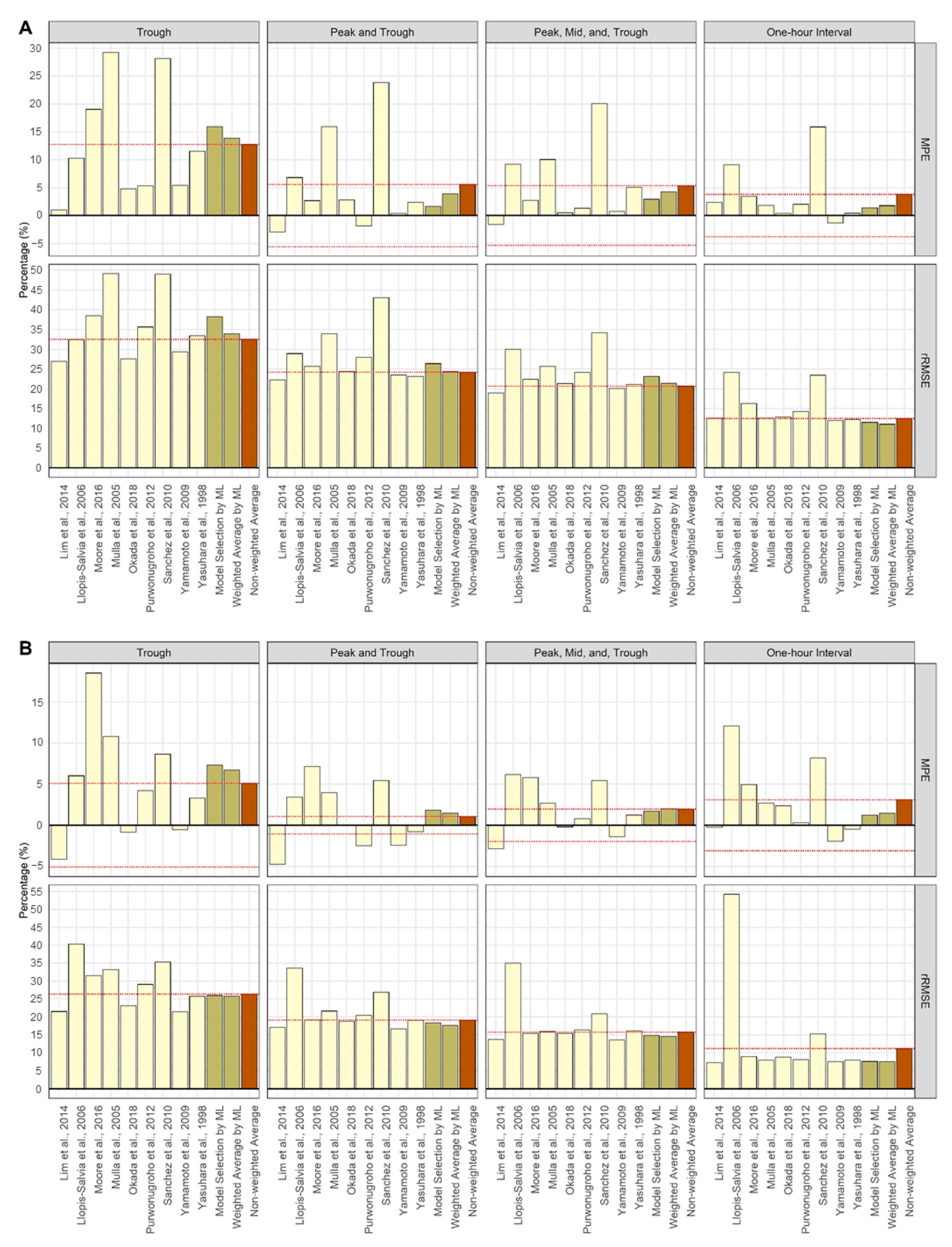
| Lim et al., 2014 [19] | 1.03 | −2.92 | −1.56 | 2.35 | 26.98 | 22.23 | 18.97 | 12.62 |
| Llopis-Salvia et al., 2006 [20] | 10.26 | 6.81 | 9.22 | 9.15 | 32.41 | 28.91 | 29.97 | 24.11 |
| Moore et al., 2016 [21] | 19.04 | 2.69 | 2.73 | 3.41 | 38.48 | 25.67 | 22.43 | 16.30 |
| Mulla et al., 2005 [22] | 29.24 | 15.97 | 10.05 | 1.81 | 49.13 | 33.94 | 25.69 | 12.62 |
| Okada et al., 2018 [23] | 4.77 | 2.78 | 0.52 | 0.40 | 27.58 | 24.37 | 21.31 | 12.88 |
| Purwonugroho et al., 2012 [24] | 5.31 | −1.82 | 1.32 | 2.05 | 35.64 | 27.92 | 24.13 | 14.28 |
| Sánchez et al., 2010 [25] | 28.13 | 23.84 | 20.07 | 15.89 | 48.98 | 43.01 | 34.17 | 23.44 |
| Yamamoto et al., 2009 [26] | 5.42 | 0.42 | 0.77 | −1.33 | 29.37 | 23.46 | 20.07 | 11.96 |
| Yasuhara et al., 1998 [27] | 11.54 | 2.40 | 5.07 | 0.43 | 33.43 | 23.16 | 21.08 | 12.22 |
| Model Selection by ML | 15.91 | 1.65 | 2.95 | 1.37 | 38.21 | 26.37 | 23.11 | 11.53 |
| Weighted Average by ML | 13.90 | 3.89 | 4.27 | 1.78 | 33.91 | 24.34 | 21.41 | 11.04 |
| Non-weighted average | 12.75 | 5.58 | 5.36 | 3.79 | 32.49 | 24.20 | 20.67 | 12.49 |
| Steady-State Model | ||||||||
| Lim et al., 2014 [19] | −4.15 | −4.73 | −2.86 | −0.25 | 21.48 | 17.13 | 13.76 | 7.28 |
| Llopis-Salvia et al., 2006 [20] | 6.02 | 3.43 | 6.16 | 12.11 | 40.31 | 33.66 | 35.01 | 54.27 |
| Moore et al., 2016 [21] | 18.54 | 7.16 | 5.82 | 4.97 | 31.51 | 19.18 | 15.40 | 8.94 |
| Mulla et al., 2005 [22] | 10.82 | 3.97 | 2.67 | 2.69 | 33.25 | 21.63 | 15.89 | 7.97 |
| Okada et al., 2018 [23] | −0.85 | −0.05 | −0.20 | 2.37 | 23.11 | 18.78 | 15.38 | 8.83 |
| Purwonugroho et al., 2012 [24] | 4.21 | −2.49 | 0.79 | 0.32 | 29.06 | 20.40 | 16.35 | 8.11 |
| Sánchez et al., 2010 [25] | 8.66 | 5.46 | 5.43 | 8.17 | 35.34 | 26.83 | 20.84 | 15.27 |
| Yamamoto et al., 2009 [26] | −0.56 | −2.45 | −1.41 | −1.94 | 21.37 | 16.68 | 13.64 | 7.59 |
| Yasuhara et al., 1998 [27] | 3.28 | −0.77 | 1.26 | −0.49 | 25.72 | 19.08 | 16.15 | 7.98 |
| Model Selection by ML | 7.32 | 1.84 | 1.73 | 1.21 | 25.97 | 18.34 | 14.83 | 7.62 |
| Weighted Average by ML | 6.74 | 1.48 | 2.02 | 1.46 | 25.80 | 17.68 | 14.56 | 7.61 |
| Non-weighted Average | 5.11 | 1.06 | 1.96 | 3.11 | 26.34 | 19.06 | 15.77 | 11.20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.; Song, M.; Han, J.; Lee, D.; Kim, B.-H. Application of Machine Learning Classification to Improve the Performance of Vancomycin Therapeutic Drug Monitoring. Pharmaceutics 2022, 14, 1023. https://doi.org/10.3390/pharmaceutics14051023
Lee S, Song M, Han J, Lee D, Kim B-H. Application of Machine Learning Classification to Improve the Performance of Vancomycin Therapeutic Drug Monitoring. Pharmaceutics. 2022; 14(5):1023. https://doi.org/10.3390/pharmaceutics14051023
Chicago/Turabian StyleLee, Sooyoung, Moonsik Song, Jongdae Han, Donghwan Lee, and Bo-Hyung Kim. 2022. "Application of Machine Learning Classification to Improve the Performance of Vancomycin Therapeutic Drug Monitoring" Pharmaceutics 14, no. 5: 1023. https://doi.org/10.3390/pharmaceutics14051023
APA StyleLee, S., Song, M., Han, J., Lee, D., & Kim, B.-H. (2022). Application of Machine Learning Classification to Improve the Performance of Vancomycin Therapeutic Drug Monitoring. Pharmaceutics, 14(5), 1023. https://doi.org/10.3390/pharmaceutics14051023