
Swine as the Animal Model for Testing New Formulations of Anti-Inflammatory Drugs: Carprofen Pharmacokinetics and Bioavailability of the Intramuscular Route

 , ,
, ,  , ,
, ,  ,
, 
Abstract
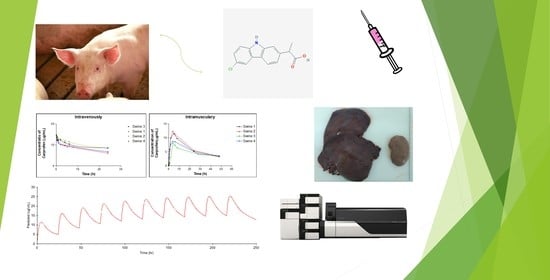
:
1. Introduction
2. Materials and Methods
2.1. Animals and Chemicals
2.2. Experimental Design
2.3. Liquid Chromatography-Mass Spectrometry Analysis
2.3.1. Standard Curve
2.3.2. LC-MS/MS Method
2.3.3. Analytical Validation Assays
2.3.4. Sample Preparation
2.4. Recovery from Liver and Kidney
2.5. Statistical Analysis
2.6. Pharmacokinetic Analysis
2.6.1. Non-Compartmental Pharmacokinetic Analysis
2.6.2. Pharmacokinetic Modeling Using the Population Approach
2.6.3. Model Development
2.6.4. Model Evaluation
2.6.5. Simulations and Model Applicability
3. Results
3.1. Liquid Chromatography-Mass Spectrometry
3.2. Recovery from Liver and Kidney
3.3. Carprofen Plasma/Tissue Concentrations
3.4. Carprofen Pharmacokinetic Analysis
3.4.1. Non-Compartmental Analysis
3.4.2. Population Pharmacokinetic Analysis
3.4.3. Simulations and Model Applicability
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yü, T.F.; Perel, J. Pharmacokinetic and Clinical Studies of Carprofen in Gout. J. Clin. Pharmacol. 1980, 20, 347–351. [Google Scholar] [CrossRef]
- Griswold, D.E.; Adams, J.L. Constitutive Cyclooxygenase (COX-1) and Inducible Cyclooxygenase (COX-2): Rationale for Selective Inhibition and Progress to Date. Med. Res. Rev. 1996, 16, 181–206. [Google Scholar] [CrossRef]
- Kerr, A.C.; Muller, F.; Ferguson, J.; Dawe, R.S.R.S. Occupational carprofen photoallergic contact dermatitis. Br. J. Dermatol. 2008, 159, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.L.; Botting, R.M.; Hla, T. Cyclooxygenase isozymes: The biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 2004, 56, 387–437. [Google Scholar] [CrossRef] [Green Version]
- Sandersoln, R.O.; Beata, C.; Flipo, R.M.; Genevois, J.P.; Macias, C.; Tacke, S.; Innes, J.F. Systematic Review of the Management of Canine Osteoarthitis. Vet. Rec. 2009, 164, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Elitok, B.; Elitok, O.M. Clinical Efficacy of Carprofen as an Adjunct to the Antibacterial Treatment of Bovine Respiratory Disease. J. Vet. Pharmacol. Ther. 2004, 27, 317–320. [Google Scholar] [CrossRef]
- Lockwood, P.W.; Johnson, J.C.; Katz, T.L. Clinical Efficacy of Flunixin: Carprofen and Ketoprofen as Adjuncts to the an-Tibacterial Treatment of Bovine Respiratory Disease. Vet. Rec. 2003, 152, 392–394. [Google Scholar] [CrossRef] [PubMed]
- Vangroenweghe, F.; Duchateau, L.; Boutet, P.; Lekeux, P.; Rainard, P.; Paape, M.J.; Burvenich, C. Effect of Carprofen treatment following experimentally induced Escherichia coli mastitis in primiparous cows. J. Dairy Sci. 2005, 88, 2361–2376. [Google Scholar] [CrossRef] [Green Version]
- Karol, A.M. Nonsteroidal Anti-Inflammatory Analgesics in Pain Management in Dogs and Cats. Can. Vet. J. 1996, 37, 539–543. [Google Scholar]
- Sidler, M.; Fouché, N.; Meth, I.; Hahn, F.; Rechenberg, B.; Kronen, P.W. Transcutaneous Treatment with Vetdrop Sustains the Adjacent Cartilage in a Microfracturing Joint Defect Model in Sheep. BMC Vet. Res. 2014, 10, 268. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, H.M.; Nolte, I.; Kramer, S. Comparison of Analgesic Efficacy of Preoperative or Postoperative Carprofen with or without Preincisional Mepivacaine Epidural Anesthesia in Canine Pelvic or Femoral Fracture Repair. Vet. Surg. 2007, 36, 623–632. [Google Scholar] [CrossRef]
- Furst, D.E.; Caldwell, J.R.; Klugman, M.P.; Enthoven, D.; Rittweger, K.; Scheer, R.; Sarkissian, E.; Dromgoole, S. Serum Concentration and Dose-Response Relationships for Carprofen in Rheumatoid Arthritis. Clin. Pharmacol. Ther. 1988, 44, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Jalava, S. A Double Blind Crossover Study of Carprofen and Indomethacin in Patients with Rheumatoid Arthritis. Scand. J. Rheumatol. 1983, 12, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Knusel, O. Double-Blind Comparative Clinical Trial of Carprofen versus Diclofenac-Na in Patients with Coxarthrosis. Eur. J. Rheumatol. Inflamm. 1982, 5, 536–542. [Google Scholar]
- Gimeno, A.; Mestres-Truyol, J.; Ojeda-Montes, M.J.; Macip, G.; Saldivar-Espinoza, B.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallvé, S. Prediction of Novel Inhibitors of the Main Protease (M-pro) of SARS-CoV-2 through Consensus Docking and Drug Reposition. Int. J. Mol. Sci. 2020, 21, 3793. [Google Scholar] [CrossRef]
- Fujiyama, A.; Watanabe, H.; Toyoda, A.; Taylor, T.D.; Itoh, T.; Tsai, S.F.; Sakaki, Y. Construction and Analysis of a Human-Chimpanzee Comparative Clone Map. Science 2002, 295, 131–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Zhang, Q.; Deng, M.; Miao, J.; Guo, Y.; Gao, W.; Cui, Q. An analysis of human microRNA and disease associations. PLoS ONE 2008, 3, e3420. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, S.; Tricklebank, P.A.L.; Frean, S.; Lees, P. Pharmacokinetics of Carprofen Enantiomers in Equine Plasma and Synovial Fluid–A Comparison with Ketoprofen. J. Vet. Pharmacol. Ther. 1992, 22, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Lees, P.; Aliabadi, F.S.; Landoni, M.F. Pharmacodynamics and Enantioselective Pharmacokinetics of Racemic Carprofen in the Horse. J. Vet. Pharmacol. Ther. 2003, 25, 433–448. [Google Scholar] [CrossRef]
- Mealey, K.L.; Matthews, N.S.; Peck, K.E.; Burchfield, M.L.; Bennett, B.S.; Taylor, T.S. Carprofen after Administration of Racemic Carprofen in Donkeys and Horses. Am. J. Vet. Res. 2004, 65, 1479–1482. [Google Scholar] [CrossRef]
- Brentnall, C.; Cheng, Z.; McKellar, Q.A.; Lees, P. Influence of Oxytetracycline on Carprofen Pharmacodynamics and Pharmacokinetics in Calves. J. Vet. Pharmacol. Ther. 2013, 36, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Brentnall, C.; Cheng, Z.; McKellar, Q.A.; Lees, P. Pharmacokinetic–Pharmacodynamic Integration and Modelling of Oxy-Tetracycline Administered Alone and in Combination with Carprofen in Calves. Res. Vet. Sci. 2013, 94, 687–694. [Google Scholar] [CrossRef]
- Cheng, Z.; Nolan, A.; Monteiro, A.; McKellar, Q. Enantioselective Pharmacokinetics and Cyclo-Oxygenase Inhibition of Carprofen and Carprofen Enantiomers in Sheep. J. Vet. Pharmacol. 2003, 26, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, M.G.; Taylor, I.T.; Craigmill, A.L.; Tell, L.A. Enantioselective Pharmacokinetics of Racemic Carprofen in New Zealand White Rabbits. J. Vet. Pharmacol. Ther. 2008, 31, 423–430. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; Wang, L.; Xia, H. Ocular Pharmacokinetics and Availability of Topically Applied Baicalein in Rabbits. Curr. Eye Res. 2009, 34, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Fourie, T.; Cromarty, D.; Duncan, N.; Wolter, K.N. The Safety and Pharmacokinetics of Carprofen, Flunixin and Phenylbutazone in the Cape Vulture (Gyps Coprotheres) Following Oral Exposure. PLoS ONE 2015, 10, e0141419. [Google Scholar]
- Crevoisier, C. Pharmacokinetic Properties of Carprofen in Humans. Eur. J. Rheumatol. Inflamm. 1982, 5, 492–502. [Google Scholar]
- Lipscomb, V.J.; AliAbadi, F.S.; Lees, P.; Pead, M.J.; Muir, P. Clinical Efficacy and Pharmacokinetics of Carprofen in the Treatment of Dogs with Osteoarthritis. Vet. Rec. 2002, 150, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Messenger, K.M.; Wofford, J.A.; Papich, M.G. Carprofen Pharmacokinetics in Plasma and in Control and Inflamed Canine Tissue Fluid Using in Vivo Ultrafiltration. J. Vet. Pharmacol. Ther. 2016, 39, 32–39. [Google Scholar] [CrossRef]
- Parton, K.; Balmer, T.V.; Boyle, J.; Whittem, T.; MacHon, R. The Pharmacokinetics and Effects of Intravenously Adminis-Trated Carprofen and Salicylate on Gastrointestinal Mucosa and Selected Biochemical Measurements in Healthy Cats. J. Vet. Pharmacol. Ther. 2000, 23, 73–79. [Google Scholar] [CrossRef]
- Taylor, P.M.; Delatour, P.; Landoni, F.M.; Deal, C.; Pickett, C.; Aliabadi, F.S.; Foot, R.; Lees, P. Phamacodynamics and Enantioselective Pharmacokinetics of Carprofen in the Cat. Res. Vet. Sci. 1996, 60, 144–151. [Google Scholar] [CrossRef]
- Bošnjak, M.; Kržan, M.; Lampreht, T.U.; Dolenc, J.; Čemažar, M.; Seliškar, A. Pharmacokinetics of Carprofen in Anaesthetized Pigs: A Preliminary Study. Vet. Anaesth. Analg. 2021, 48, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Christianson, C.C.; Johnson, C.J.; Needham, S.R. The Advantages of Microflow LC-MS/MS Compared with Conventional HPLC-MS/MS for the Analysis of Methotrexate from Human Plasma. Bioanalysis 2013, 5, 1387–1396. [Google Scholar] [CrossRef]
- Li, R.; Ran, R.; Li, Q.; Huang, Y.; Gu, Y.; Si, D. Development of an LC–MS/MS Method for the Quantitation of Deoxy-Glycychloxazol in Rat Plasma and Its Application in Pharmacokinetic Study. J. Pharm. Anal. 2016, 6, 184–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jedziniak, P.; Szprengier-Juszkiewicz, T.; Pietruk, K.; Sledzi’snka, E.; Zmudzki, J. Determination of Non-Steroidal an-Ti-Nflammatori Drugs and Theri Metabolitse in Milk by Liquid Chromatrography-Tandem Mass Spectrometry. Anal. Bioanal. Chem. 2012, 403, 2955–2963. [Google Scholar] [CrossRef] [PubMed]
- Iwakawa, S.; Suganuma, T.; Lee, S.-F.; Spahn, H.; Benet, L.Z.; Lin, E.T. Direct Determination of Diastereomeric Carprofen Glucuronides in Human Plasma and Urine and Preliminary Measurements of Stereoselective Metabolic and Renal Elimination after Oral Administration of Carprofen in Man. Drug Metab. Dispos. 1989, 17, 414–419. [Google Scholar]
- FDA. Bioanalytical Method Validation. Guidance for Industry. 2018. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 10 January 2020).
- FDA. Analytical Procedures and Methods Validation for Drugs and Biologics. Guidance for Industry. 2015. Available online: https://www.fda.gov/media/87801/download (accessed on 10 January 2020).
- EMA ICH Q2(R1): Note for Guidance on Validation of Analytical Procedures: Text and Methodology. 1995. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-2-r1-validation-analytical-procedures-text-methodology-step-5_en.pdf (accessed on 10 January 2020).
- Certara, L.P. Phoenix-64 Build 8.1.0.3530; Certara: Princeton, NJ, USA; Available online: https://www.certara.com/ (accessed on 29 March 2022).
- Bauer, R.J. Improvements and New Estimation Methods in NONMEM 7 for PK/PD Population Analysis; Bauer, R.J., Ludden, T.M., Eds.; Icon Development Solutions: Ellicot City, MD, USA, 2009. [Google Scholar]
- Jonsson, E.N.; Karlsson, M.O. Xpose: An S-PLUS Based Population Pharmacokinetic/Pharmacodynamic Model Building Aid for NONMEM. Comput. Meth Programs Biomed. 1998, 58, 51–64. [Google Scholar] [CrossRef]
- Lindbom, L.; Pihlgren, P.; Jonsson, N. PsN-Toolkit: A Collection of Computer Intensive Statistical Methods for Non-Linear Mixed Effect Modeling Using NONMEM. Comput. Meth Programs Biomed. 2005, 79, 241–257. [Google Scholar] [CrossRef]
- Yamaoka, T.; Nakagawa, T.; Uno, T. Application of Akaike’s Information Criterion (AIC) in the Evalua-Tion of Linear Pharmacokinetics Equations. J. Pharm. Biopharm 1978, 6, 165–175. [Google Scholar] [CrossRef]
- Anderson, B.J.; Holford, N.H. Mechanistic Basis of Using Body Size and Maturation to Predict Clear-Ance in Humans. Drug Metab Pharm. 2009, 24, 25–36. [Google Scholar] [CrossRef]
- Savic, R.M.; Karlsson, M.O. Importance of Shrinkage in Empirical Bayes Estimates for Diagnostics: Problems and Solutions. AAPS J. 2009, 11, 558–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SIM, W.I. The Visual Predictive Check Superiority to Standard Diagnostic (Rorschach) Plots. J. Clin. Investig. 1963, 42, 1542–1551. [Google Scholar]
- Davies, B.; Morris, T. Physiological Parameters in Laboratory Animals and Humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef] [PubMed]
- Lees, P.; Landoni, M.F.; Giraudel, J.; Toutain, P.L. Pharmacodynamics and Pharmacokinetics of Nonsteroidal Anti-Inflammatory Drugs in Species of Veterinary Interest. J. Vet. Pharmacol. Ther. 2004, 27, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, T.H.; Madden, J.C.; Naidoo, V.; Walker, C.H. Comparative Metabolism as a Key Driver of Wildlife Species Sensitivity to Human and Veterinary Pharmaceuticals. Phil. Trans. R. Sci. B 2014, 369, 20130583. [Google Scholar] [CrossRef] [Green Version]
- Ten Have, G.A.M.; Bostl, M.C.F.; Suyk-Wiertsl, J.C.A.W.; Van den Bogaard, A.E.J.M.; Deutz, N.E.P. Simultaneous Measurement of Metabolic Flux in Portally-Drained Viscera, Liver, Spleen, Kidney and Hindquarter in the Conscious Pig. Lab. Anim. 1996, 30, 347–358. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T1 | T2 | |||||
|---|---|---|---|---|---|---|
| Units (ppb) | 10 | 250 | 750 | 10 | 250 | 750 |
| Mean | 9180.11 | 161,6303.90 | 463,8409.44 | 2223.12 | 398,172.36 | 115,8660.25 |
| SD | 161.62 | 18078.15 | 51578.04 | 41.39 | 1613.87 | 9652.78 |
| RSD | 1.76 | 1.12 | 1.11 | 1.86 | 0.41 | 0.83 |
| T1 | T2 | |||||
|---|---|---|---|---|---|---|
| Sample Name | Mean | SD | RSD | Mean | SD | RSD |
| RD_ppb_1 | 98.20 | 9.37 | 9.55 | 98.58 | 8.01 | 8.13 |
| RD_ppb_10 | 117.75 | 2.87 | 2.44 | 114.75 | 2.87 | 2.50 |
| RD_ppb_100 | 96.38 | 0.92 | 0.96 | 94.15 | 1.28 | 1.36 |
| RD_ppb_250 | 103.25 | 1.50 | 1.45 | 102.50 | 1.73 | 1.69 |
| RD_ppb_500 | 95.28 | 1.47 | 1.54 | 95.43 | 2.07 | 2.17 |
| RD_ppb_750 | 94.33 | 2.96 | 3.14 | 96.08 | 4.36 | 4.54 |
| RD_ppb_1000 | 94.70 | 1.66 | 1.75 | 98.80 | 2.31 | 2.33 |
| Tissue | Concentration (µg/g) 1 |
|---|---|
| Plasma | 3.26 (3.21–3.60) |
| Liver | 0.19 (0.001–0.37) |
| Kidney | 0.29 (0.22–0.37) |
| Parameter | Intravenous Administration | Intramuscular Administration |
|---|---|---|
| λz (h−1) | 0.019 ± 0.005 | 0.024 ± 0.005 |
| t1/2λz (h) | 36.34 ± 7.88 | 29.73 ± 5.56 |
| AUC (µg/mL)·h | 620.07 ± 168.10 | 403.57 ± 16.33 |
| AUCextrap (%) | 62.35 ± 7.07 | 33.58 ± 7.22 |
| CL (mL/h/kg) | 6.86 ± 2.01 | - |
| Vi (L/kg) | 0.265 ± 0.015 | - |
| Vss (L/kg) | 0.342 ± 0.027 | - |
| Vdarea (L/kg) | 0.342 ± 0.027 | - |
| Cmax (µg/mL) | 15.11 ± 0.86 | 10.14 ± 2.11 |
| Tmax (h) | - | 4.0 (4.0–5.0) |
| F (%) | - | 69.34 ± 21.00 |
| Parameter | Units | Final Model Parameter Estimate (RSE%) | |
|---|---|---|---|
| Disposition parameters | CL | L/h | 0.255 (20.75) |
| VC | L | 7.11 (25.60) | |
| CLD | L/h | 127.00 (21.50) | |
| VP | L | 2.51 (92.43) | |
| Release/Absorption parameters | F | % | 90.80 (11.89) |
| Ka | h−1 | 0.471 (49.26) | |
| Inter-individual variability | IIVCL | % | 33.17 (110.00) |
| IIVVc | % | 33.47 (220.00) | |
| IIVKa | % | 95.86 (54.00) | |
| Residual variability | Proportional | % | 24.23 (21.47) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Segura, L.; Boix-Montañes, A.; Mallandrich, M.; Parra-Coca, A.; Soriano-Ruiz, J.L.; Calpena, A.C.; Gimeno, Á.; Bellido, D.; Colom, H. Swine as the Animal Model for Testing New Formulations of Anti-Inflammatory Drugs: Carprofen Pharmacokinetics and Bioavailability of the Intramuscular Route. Pharmaceutics 2022, 14, 1045. https://doi.org/10.3390/pharmaceutics14051045
Gómez-Segura L, Boix-Montañes A, Mallandrich M, Parra-Coca A, Soriano-Ruiz JL, Calpena AC, Gimeno Á, Bellido D, Colom H. Swine as the Animal Model for Testing New Formulations of Anti-Inflammatory Drugs: Carprofen Pharmacokinetics and Bioavailability of the Intramuscular Route. Pharmaceutics. 2022; 14(5):1045. https://doi.org/10.3390/pharmaceutics14051045
Chicago/Turabian StyleGómez-Segura, Lidia, Antoni Boix-Montañes, Mireia Mallandrich, Alexander Parra-Coca, José L. Soriano-Ruiz, Ana Cristina Calpena, Álvaro Gimeno, David Bellido, and Helena Colom. 2022. "Swine as the Animal Model for Testing New Formulations of Anti-Inflammatory Drugs: Carprofen Pharmacokinetics and Bioavailability of the Intramuscular Route" Pharmaceutics 14, no. 5: 1045. https://doi.org/10.3390/pharmaceutics14051045
APA StyleGómez-Segura, L., Boix-Montañes, A., Mallandrich, M., Parra-Coca, A., Soriano-Ruiz, J. L., Calpena, A. C., Gimeno, Á., Bellido, D., & Colom, H. (2022). Swine as the Animal Model for Testing New Formulations of Anti-Inflammatory Drugs: Carprofen Pharmacokinetics and Bioavailability of the Intramuscular Route. Pharmaceutics, 14(5), 1045. https://doi.org/10.3390/pharmaceutics14051045