
Oil-Based Delivery Control Release System Targeted to the Later Part of the Gastrointestinal Tract—A Mechanistic Study


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation and Characterization of Pancreatin Extract
2.3. In Vitro Method
2.4. Oil Formulations Investigated
2.4.1. Bulk Retarders
2.4.2. Ethylcellulose
2.4.3. Interfacial Retarders

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interfacial Retarders | Molecular Mass (g/mol) | CMC * (wt ppm) |
|---|---|---|
| Monooleate | 356 | 1.4 |
| Polysorbate 80 | 1310 | 1.6 |
| Poloxamer 407 | 12,600 | 35 |
| C12E5 | 406 | 28 |
| Cetearyl glucoside | 404 | 0.40 |
2.5. Droplet Size
2.6. Interfacial Tension
2.7. Rheological Analysis
2.8. Texture Analyzer
3. Results
3.1. Hypothesis 1: Change in the Consistency of the Oil Phase Influences the Lipolysis
3.2. Hypothesis 2: Thermodynamic Activity of Oil Phase Influences the Lipolysis
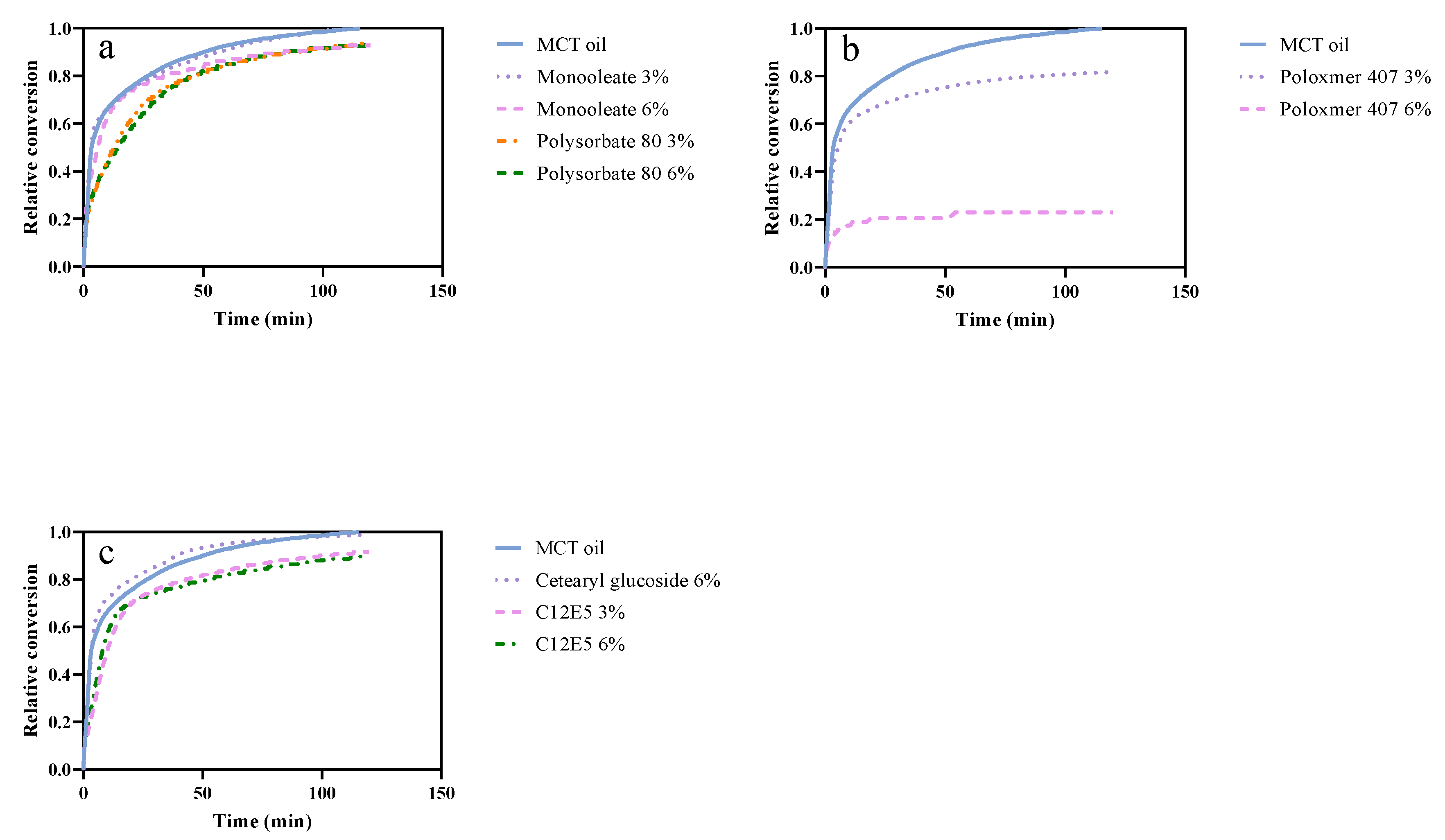
3.3. Hypothesis 3: The Composition of the Interface Influences the Lipolysis
4. Discussion
4.1. Hypothesis 1: Change in the Consistency of the Oil Phase Influences the Lipolyze
4.2. Hypothesis 2: Thermodynamic Activity of Oil Phase Influences the Lipolysis
4.3. Hypothesis 3: The Composition of the Interface Influences the Lipolysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pouton, C.W. Self-emulsifying drug delivery systems: Assessment of the efficiency of emulsification. Int. J. Pharm. 1985, 27, 335–348. [Google Scholar] [CrossRef]
- Jannin, V.; Musakhanian, J.; Marchaud, D. Approaches for the development of solid and semi-solid lipid-based formulations. Adv. Drug Deliv. Rev. 2008, 60, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Serajuddin, A.T.M. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Gershanik, T.; Benita, S. Self-dispersing lipid formulations for improving oral absorption of lipophilic drugs. Eur. J. Pharm. Biopharm. 2000, 50, 179–188. [Google Scholar] [CrossRef]
- Gursoy, R.N.; Benita, S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed. Pharmacother. 2004, 58, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Pouton, C.W. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef]
- Sharma, S.; Sinha, V.R. Current pharmaceutical strategies for efficient site specific delivery in inflamed distal intestinal mucosa. J. Control. Release 2018, 272, 97–106. [Google Scholar] [CrossRef]
- Krishnaiah, Y.S.R.; Satyanarayana, V.; Dinesh Kumar, B.; Karthikeyan, R.S.; Bhaskar, P. In vivo pharmacokinetics in human volunteers: Oral administered guar gum-based colon-targeted 5-fluorouracil tablets. Eur. J. Pharm. Sci. 2003, 19, 355–362. [Google Scholar] [CrossRef]
- Murakami, T.; Takano, M. Intestinal efflux transporters and drug absorption. Expert Opin. Drug Metab. Toxicol. 2008, 4, 923–939. [Google Scholar] [CrossRef]
- McConnell, E.L.; Liu, F.; Basit, A.W. Colonic treatments and targets: Issues and opportunities. J. Drug Target. 2009, 17, 335–363. [Google Scholar] [CrossRef]
- Wahlgren, M.; Axenstrand, M.; Håkansson, Å.; Marefati, A.; Lomstein Pedersen, B. In Vitro Methods to Study Colon Release: State of the Art and An Outlook on New Strategies for Better In-Vitro Biorelevant Release Media. Pharmaceutics 2019, 11, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, T.; Paterson, R.; Moore, V.; Carlsson, A.; Abrahamsson, B.; Basit, A.W. The gastrointestinal microbiota as a site for the biotransformation of drugs. Int. J. Pharm. 2008, 363, 1–25. [Google Scholar] [CrossRef]
- Fukui, E.; Miyamura, N.; Kobayashi, M. An in vitro investigation of the suitability of press-coated tablets with hydroxypropylmethylcellulose acetate succinate (HPMCAS) and hydrophobic additives in the outer shell for colon targeting. J. Control. Release 2001, 70, 97–107. [Google Scholar] [CrossRef]
- Gazzaniga, A.; Iamartino, P.; Maffione, G.; Sangalli, M.E. Oral delayed-release system for colonic specific delivery. Int. J. Pharm. 1994, 108, 77–83. [Google Scholar] [CrossRef]
- Ibekwe, V.C.; Khela, M.K.; Evans, D.F.; Basit, A.W. A new concept in colonic drug targeting: A combined pH-responsive and bacterially-triggered drug delivery technology. Aliment. Pharmacol. Ther. 2008, 28, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Nugent, S.G.; Kumar, D.; Rampton, D.S.; Evans, D.F. Intestinal luminal pH in inflammatory bowel disease: Possible determinants and implications for therapy with aminosalicylates and other drugs. Gut 2001, 48, 571. [Google Scholar] [CrossRef] [PubMed]
- Zechner, R.; Zimmermann, R.; Eichmann, T.O.; Kohlwein, S.D.; Haemmerle, G.; Lass, A.; Madeo, F. Fat Signals—Lipases and Lipolysis in Lipid Metabolism and Signaling. Cell Metab. 2012, 15, 279–291. [Google Scholar] [CrossRef] [Green Version]
- Champe, P.C.; Harvey, R.A.; Ferrier, D.R. Biochemistry; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2005. [Google Scholar]
- Patton, J.S.; Carey, M.C. Inhibition of human pancreatic lipase-colipase activity by mixed bile salt-phospholipid micelles. Am. J. Physiol. 1981, 241, G328–G336. [Google Scholar] [CrossRef]
- Kawase, T.; Hashimoto, T.; Fujii, T.; Minagawa, M. Studies on the Effects of Surfactants on Lipase Activity. J. Jpn. Oil Chem. Soc. 1985, 34, 530–538. [Google Scholar] [CrossRef]
- Skagerlind, P.; Jansson, M.; Bergenståhl, B.; Hult, K. Binding of Rhizomucor miehei lipase to emulsion interfaces and its interference with surfactants. Colloids Surf. B Biointerfaces 1995, 4, 129–135. [Google Scholar] [CrossRef]
- Delorme, V.; Dhouib, R.; Canaan, S.; Fotiadu, F.; Carrière, F.; Cavalier, J.-F. Effects of Surfactants on Lipase Structure, Activity, and Inhibition. Pharm. Res. 2011, 28, 1831–1842. [Google Scholar] [CrossRef] [PubMed]
- Heck, A.M.; Yanovski, J.A.; Calis, K.A. Orlistat, a New Lipase Inhibitor for the Management of Obesity. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2000, 20, 270–279. [Google Scholar] [CrossRef]
- Torcello-Gómez, A.; Wulff-Pérez, M.; Gálvez-Ruiz, M.J.; Martín-Rodríguez, A.; Cabrerizo-Vílchez, M.; Maldonado-Valderrama, J. Block copolymers at interfaces: Interactions with physiological media. Adv. Colloid Interface Sci. 2014, 206, 414–427. [Google Scholar] [CrossRef]
- Li, Y.; McClements, D.J. Inhibition of lipase-catalyzed hydrolysis of emulsified triglyceride oils by low-molecular weight surfactants under simulated gastrointestinal conditions. Eur. J. Pharm. Biopharm. 2011, 79, 423–431. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, Z.; Zhang, H.; Decker, E.A.; McClements, D.J. Influence of lipid type on gastrointestinal fate of oil-in-water emulsions: In vitro digestion study. Food Res. Int. 2015, 75, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClements, D.J.; Decker, E.A.; Park, Y. Controlling Lipid Bioavailability through Physicochemical and Structural Approaches. Crit. Rev. Food Sci. Nutr. 2008, 49, 48–67. [Google Scholar] [CrossRef] [PubMed]
- Dressman, J.B.; Krämer, J. Pharmaceutical Dissolution Testing; Taylor & Francis Boca Raton: Boca Raton, FL, USA, 2005. [Google Scholar]
- Fatouros, D.G.; Mullertz, A. In vitro lipid digestion models in design of drug delivery systems for enhancing oral bioavailability. Expert Opin. Drug Metab. Toxicol. 2008, 4, 65–76. [Google Scholar] [CrossRef]
- Carino, S.R.; Sperry, D.C.; Hawley, M. Relative bioavailability estimation of carbamazepine crystal forms using an artificial stomach-duodenum model. J. Pharm. Sci. 2006, 95, 116–125. [Google Scholar] [CrossRef]
- Brouwers, J.; Anneveld, B.; Goudappel, G.-J.; Duchateau, G.; Annaert, P.; Augustijns, P.; Zeijdner, E. Food-dependent disintegration of immediate release fosamprenavir tablets: In vitro evaluation using magnetic resonance imaging and a dynamic gastrointestinal system. Eur. J. Pharm. Biopharm. 2011, 77, 313–319. [Google Scholar] [CrossRef]
- Verwei, M.; Arkbåge, K.; Havenaar, R.; van den Berg, H.; Witthöft, C.; Schaafsma, G. Folic acid and 5-methyltetrahydrofolate in fortified milk are bioaccessible as determined in a dynamic in vitro gastrointestinal model. J. Nutr. 2003, 133, 2377–2383. [Google Scholar] [CrossRef] [Green Version]
- Christensen, J.Ø.; Schultz, K.; Mollgaard, B.; Kristensen, H.G.; Mullertz, A. Solubilisation of poorly water-soluble drugs during in vitro lipolysis of medium- and long-chain triacylglycerols. Eur. J. Pharm. Sci. 2004, 23, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Zangenberg, N.H.; Müllertz, A.; Kristensen, H.G.; Hovgaard, L. A dynamic in vitro lipolysis model: I. Controlling the rate of lipolysis by continuous addition of calcium. Eur. J. Pharm. Sci. 2001, 14, 115–122. [Google Scholar] [CrossRef]
- Borgström, B.; Erlanson, C. Pancreatic Lipase and Co-Lipase. Eur. J. Biochem. 1973, 37, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Patton, J.S.; Vetter, R.D.; Hamosh, M.; Borgstrom, B.; Lindstrom, M.; Carey, M.C. The Light Microscopy of Triglyceride Digestion. Food Struct. 1985, 4, 5. [Google Scholar]
- Liao, T.H.; Hamosh, P.; Hamosh, M. Gastric lipolysis in the developing rat ontogeny of the lipases active in the stomach. Biochim. Biophys. Acta Lipids Lipid Metab. 1983, 754, 1–9. [Google Scholar] [CrossRef]
- Dressman, J.B.; Amidon, G.L.; Reppas, C.; Shah, V.P. Dissolution Testing as a Prognostic Tool for Oral Drug Absorption: Immediate Release Dosage Forms. Pharm. Res. 1998, 15, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Fatouros, D.G.; Walrand, I.; Bergenstahl, B.; Müllertz, A. Colloidal Structures in Media Simulating Intestinal Fed State Conditions with and Without Lipolysis Products. Pharm. Res. 2008, 26, 361. [Google Scholar] [CrossRef]
- Benito-Gallo, P.; Franceschetto, A.; Wong, J.C.M.; Marlow, M.; Zann, V.; Scholes, P.; Gershkovich, P. Chain length affects pancreatic lipase activity and the extent and pH–time profile of triglyceride lipolysis. Eur. J. Pharm. Biopharm. 2015, 93, 353–362. [Google Scholar] [CrossRef]
- Williams, H.D.; Sassene, P.; Kleberg, K.; Bakala-N’Goma, J.-C.; Calderone, M.; Jannin, V.; Igonin, A.; Partheil, A.; Marchaud, D.; Jule, E.; et al. Toward the Establishment of Standardized In Vitro Tests for Lipid-Based Formulations, Part 1: Method Parameterization and Comparison of In Vitro Digestion Profiles Across a Range of Representative Formulations. J. Pharm. Sci. 2012, 101, 3360–3380. [Google Scholar] [CrossRef]
- Vithani, K.; Hawley, A.; Jannin, V.; Pouton, C.; Boyd, B.J. Solubilisation behaviour of poorly water-soluble drugs during digestion of solid SMEDDS. Eur. J. Pharm. Biopharm. 2018, 130, 236–246. [Google Scholar] [CrossRef]
- Fernandez, S.; Jannin, V.; Rodier, J.-D.; Ritter, N.; Mahler, B.; Carrière, F. Comparative study on digestive lipase activities on the self emulsifying excipient Labrasol®, medium chain glycerides and PEG esters. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2007, 1771, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Desnuelle, P.; Savary, P. Specificities of lipases. J. Lipid Res. 1963, 4, 369–384. [Google Scholar] [CrossRef]
- O’Sullivan, C.M.; Davidovich-Pinhas, M.; Wright, A.J.; Barbut, S.; Marangoni, A.G. Ethylcellulose oleogels for lipophilic bioactive delivery—Effect of oleogelation on in vitro bioaccessibility and stability of beta-carotene. Food Funct. 2017, 8, 1438–1451. [Google Scholar] [CrossRef] [PubMed]
- Barauskas, J.; Cervin, C.; Jankunec, M.; Špandyreva, M.; Ribokaitė, K.; Tiberg, F.; Johnsson, M. Interactions of lipid-based liquid crystalline nanoparticles with model and cell membranes. Int. J. Pharm. 2010, 391, 284–291. [Google Scholar] [CrossRef]
- Mandal, A.B.; Nair, B.U.; Ramaswamy, D. Determination of the critical micelle concentration of surfactants and the partition coefficient of an electrochemical probe by using cyclic voltammetry. Langmuir 1988, 4, 736–739. [Google Scholar] [CrossRef]
- Shubhra, Q.T.H.; Tóth, J.; Gyenis, J.; Feczkó, T. Poloxamers for Surface Modification of Hydrophobic Drug Carriers and Their Effects on Drug Delivery. Polym. Rev. 2014, 54, 112–138. [Google Scholar] [CrossRef]
- Gentile, L.; Rossi, C.O.; Olsson, U.; Ranieri, G.A. Effect of shear rates on the MLV formation and MLV stability region in the C12E5/D2O system: Rheology and rheo-NMR and rheo-SANS experiments. Langmuir 2011, 27, 2088–2092. [Google Scholar] [CrossRef]
- Armand, M.; Borel, P.; Dubois, C.; Senft, M.; Peyrot, J.; Salducci, J.; Lafont, H.; Lairon, D. Characterization of emulsions and lipolysis of dietary lipids in the human stomach. Am. J. Physiol. 1994, 266, G372–G381. [Google Scholar] [CrossRef]
- Reis, P.; Holmberg, K.; Miller, R.; Leser, M.E.; Raab, T.; Watzke, H.J. Lipase reaction at interfaces as self-limiting processes. Comptes Rendus Chim. 2009, 12, 163–170. [Google Scholar] [CrossRef]
- Brockman, H.L. Kinetic behavior of the pancreatic lipase-colipase-lipid system. Biochimie 2000, 82, 987–995. [Google Scholar] [CrossRef]
- Borgström, B. On the interactions between pancreatic lipase and colipase and the substrate, and the importance of bile salts. J. Lipid Res. 1975, 16, 411–417. [Google Scholar] [CrossRef]
- Wannerberger, K.; Wahlgren, M.; Arnebrant, T. Adsorption from lipase-surfactant solutions onto methylated silica surfaces. Colloids Surf. B Biointerfaces 1996, 6, 27–36. [Google Scholar] [CrossRef]
- Verger, R.; Pattus, F.; Pieroni, G.; Riviere, C.; Ferrato, F.; Leonardi, J.; Dargent, B. Regulation by the “interfacial quality” of some biological activities. Colloids Surf. 1984, 10, 163–180. [Google Scholar] [CrossRef]
- Gargouri, Y.; Julien, R.; Bois, A.G.; Verger, R.; Sarda, L. Studies on the detergent inhibition of pancreatic lipase activity. J. Lipid Res. 1983, 24, 1336–1342. [Google Scholar] [CrossRef]
- Christiansen, A.; Backensfeld, T.; Weitschies, W. Effects of non-ionic surfactants on in vitro triglyceride digestion and their susceptibility to digestion by pancreatic enzymes. Eur. J. Pharm. Sci. 2010, 41, 376–382. [Google Scholar] [CrossRef]
- Reis, P.; Holmberg, K.; Watzke, H.; Leser, M.E.; Miller, R. Lipases at interfaces: A review. Adv. Colloid Interface Sci. 2009, 147–148, 237–250. [Google Scholar] [CrossRef]
- Stolnik, S.; Daudali, B.; Arien, A.; Whetstone, J.; Heald, C.R.; Garnett, M.C.; Davis, S.S.; Illum, L. The effect of surface coverage and conformation of poly(ethylene oxide) (PEO) chains of poloxamer 407 on the biological fate of model colloidal drug carriers. Biochim. Biophys. Acta Biomembr. 2001, 1514, 261–279. [Google Scholar] [CrossRef] [Green Version]
- Porter, C.J.H.; Moghimi, S.M.; Illum, L.; Davis, S.S. The polyoxyethylene/polyoxypropylene block co-polymer Poloxamer-407 selectively redirects intravenously injected microspheres to sinusoidal endothelial cells of rabbit bone marrow. FEBS Lett. 1992, 305, 62–66. [Google Scholar] [CrossRef] [Green Version]
- de Gennes, P.G. Polymers at an interface; a simplified view. Adv. Colloid Interface Sci. 1987, 27, 189–209. [Google Scholar] [CrossRef]
- Santander-Ortega, M.J.; Jódar-Reyes, A.B.; Csaba, N.; Bastos-González, D.; Ortega-Vinuesa, J.L. Colloidal stability of Pluronic F68-coated PLGA nanoparticles: A variety of stabilisation mechanisms. J. Colloid Interface Sci. 2006, 302, 522–529. [Google Scholar] [CrossRef]
- Wulff-Pérez, M.; de Vicente, J.; Martín-Rodríguez, A.; Gálvez-Ruiz, M.J. Controlling lipolysis through steric surfactants: New insights on the controlled degradation of submicron emulsions after oral and intravenous administration. Int. J. Pharm. 2012, 423, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Johnston, T.P.; Goldberg, I.J. Inhibition of pancreatic lipase by poloxamer 407 may provide an adjunct treatment strategy for weight loss. J. Pharm. Pharmacol. 2006, 58, 1099–1105. [Google Scholar] [CrossRef] [PubMed]


| Name | Reference Concentration * | Concentration after Modification |
|---|---|---|
| Bile extract | 1.5–6.6 mmol/L | 8 mmol/L |
| Phosphatidylcholine (soybean origin) | 0 | 2 mmol/L |
| NaCl | 150 mmol/L | 150 mmol/L |
| Ca2+ | 4–12 mmol/L | 15 mmol/L |
| Trizma-maleate | 2 mmol/L | 2 mmol/L |
| Pancreatin | 270–1340 USP Unit | 675 TBU |
| Total volume | 300 mL | 100 mL |
| Retarders | MCT Oil | Dimethicon 25 cSt, 16% | Dimethicon 25 cSt, 32% | Dimethicon 750 cSt, 16% | Dimethicon 750 cSt, 32% | PGPR, 16% | PGPR, 32% | Ethylcellulose 300 cP, 10% |
|---|---|---|---|---|---|---|---|---|
| Viscosity (mPa·s−1) | 16 | 13 | 14 | 26 | 49–34 | 36 | 80 | Too high to measure |
| Viscosity ratios to MCT oil | 1 | 0.81 | 0.87 | 1.6 | 3.0–2.1 * | 2.25 | 5 | |
| Conversion (2 h) () ** | 100% | 98% | 91% | 92% | 89% | 98% | 98% | 38% |
| t50% (min) | 3.4 | 3.4 | 3.8 | 3.4 | 13.8 | 1.8 | 2.0 | >120 min |
| Retarders | Hexadecane 16% | Hexadecane 32% | Dimethicon 25 cSt, 32% |
|---|---|---|---|
| Thermodynamic activity | 0.73 | 0.58 | N.A. |
| Viscosity ratio to MCT oil | 0.69 | 0.48 | 0.87 |
| Conversion (2 h) () | 83% | 83% | 92% |
| t50% (min) | 2.7 | 4.4 | 3.8 |
| Retarders | Monooleate 3% | Monooleate 6% | Polysorbate 80, 3% | Polysorbate 80, 6% | Poloxamer 407, 3% | Poloxamer 407, 6% | C12E5 3% | C12E5 6% | Cetearyl Glucoside 6% |
|---|---|---|---|---|---|---|---|---|---|
| Interfacial tension after stability (mN·m−1) | N.A. | N.A. | 4 | 4 | 6 | 6 | <2 * | <2 * | N.A. |
| Conversion (2 h) () | 100% | 92% | 93% | 93% | 81% | 23% | 91% | 90% | 98% |
| t50% (min) | 3.1 | 6.0 | 12.6 | 14.5 | 5.4 | >120 min | 10 | 8.1 | 3.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Wahlgren, M.; Bergenståhl, B. Oil-Based Delivery Control Release System Targeted to the Later Part of the Gastrointestinal Tract—A Mechanistic Study. Pharmaceutics 2022, 14, 896. https://doi.org/10.3390/pharmaceutics14050896
Zhang L, Wahlgren M, Bergenståhl B. Oil-Based Delivery Control Release System Targeted to the Later Part of the Gastrointestinal Tract—A Mechanistic Study. Pharmaceutics. 2022; 14(5):896. https://doi.org/10.3390/pharmaceutics14050896
Chicago/Turabian StyleZhang, Lingping, Marie Wahlgren, and Björn Bergenståhl. 2022. "Oil-Based Delivery Control Release System Targeted to the Later Part of the Gastrointestinal Tract—A Mechanistic Study" Pharmaceutics 14, no. 5: 896. https://doi.org/10.3390/pharmaceutics14050896
APA StyleZhang, L., Wahlgren, M., & Bergenståhl, B. (2022). Oil-Based Delivery Control Release System Targeted to the Later Part of the Gastrointestinal Tract—A Mechanistic Study. Pharmaceutics, 14(5), 896. https://doi.org/10.3390/pharmaceutics14050896