
Gene Therapy for Mitochondrial Diseases: Current Status and Future Perspective


 , , ,
, , ,  and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. General Considerations and Technical Aspects
2.1. Non-Viral Approaches
2.2. Viral Approaches
2.2.1. Lentiviral Vectors
2.2.2. Adenoviral Vectors
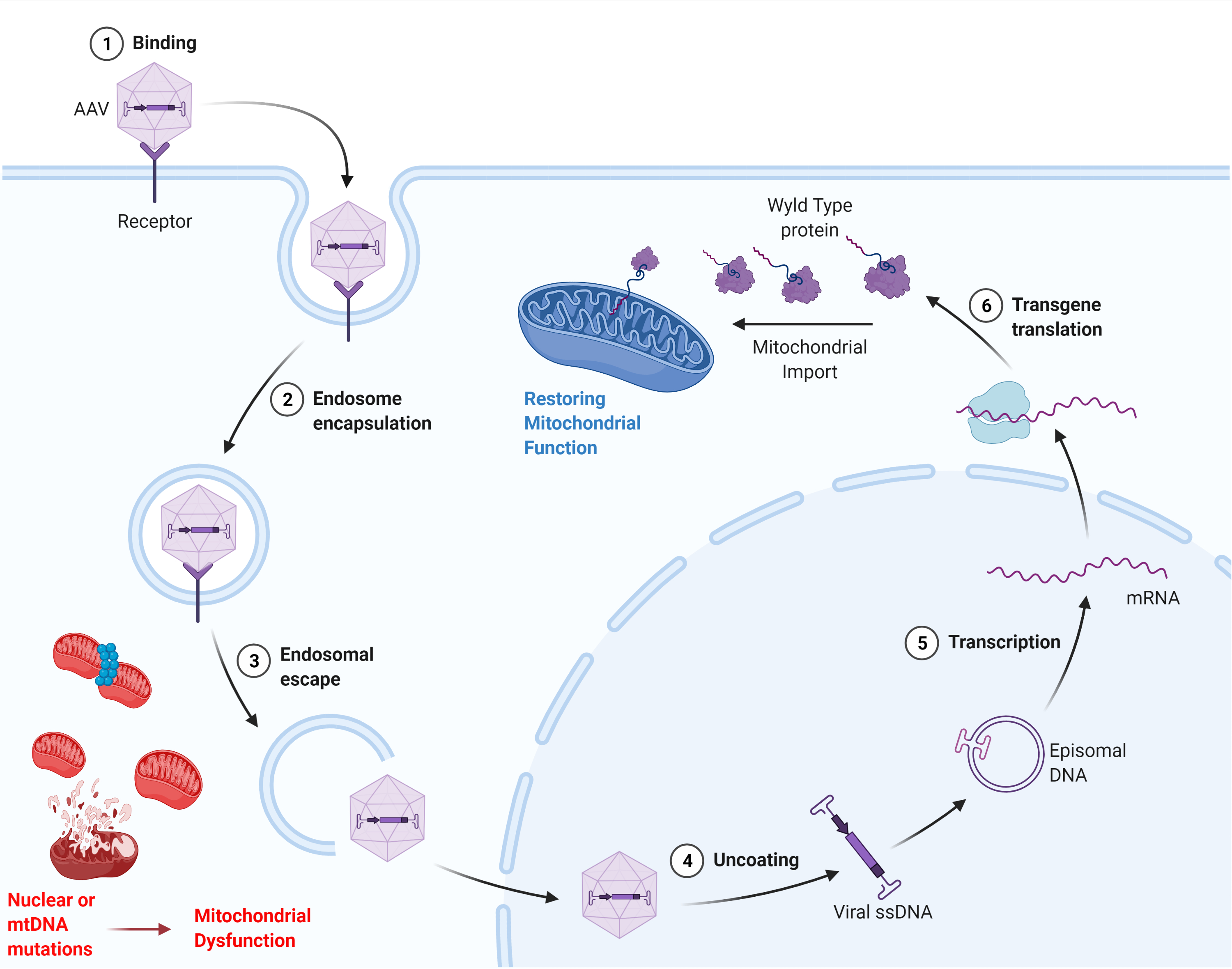
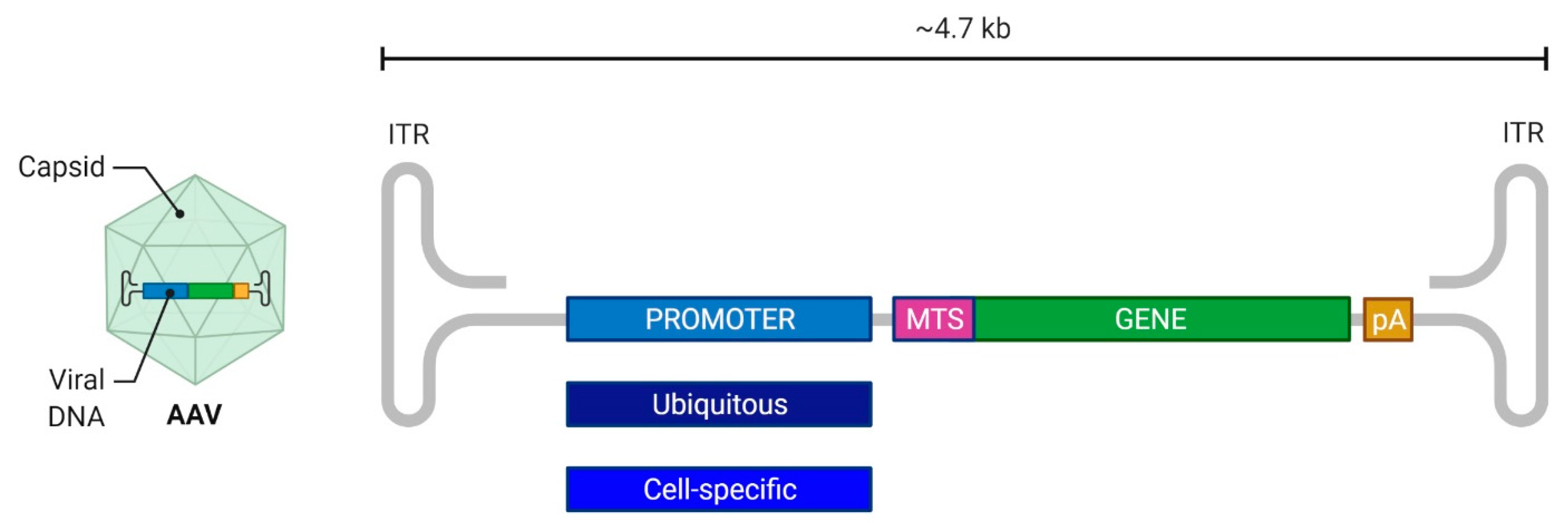
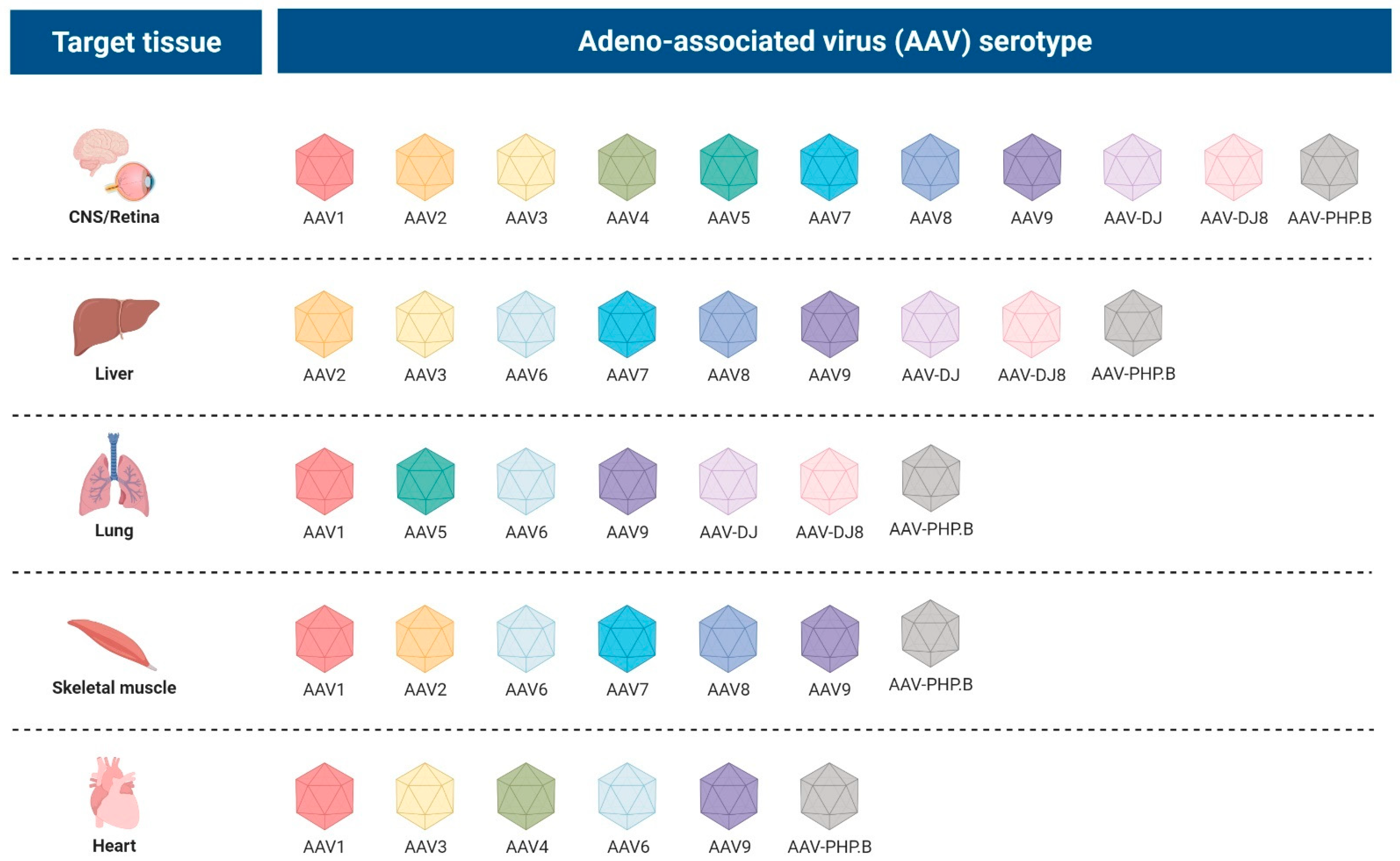
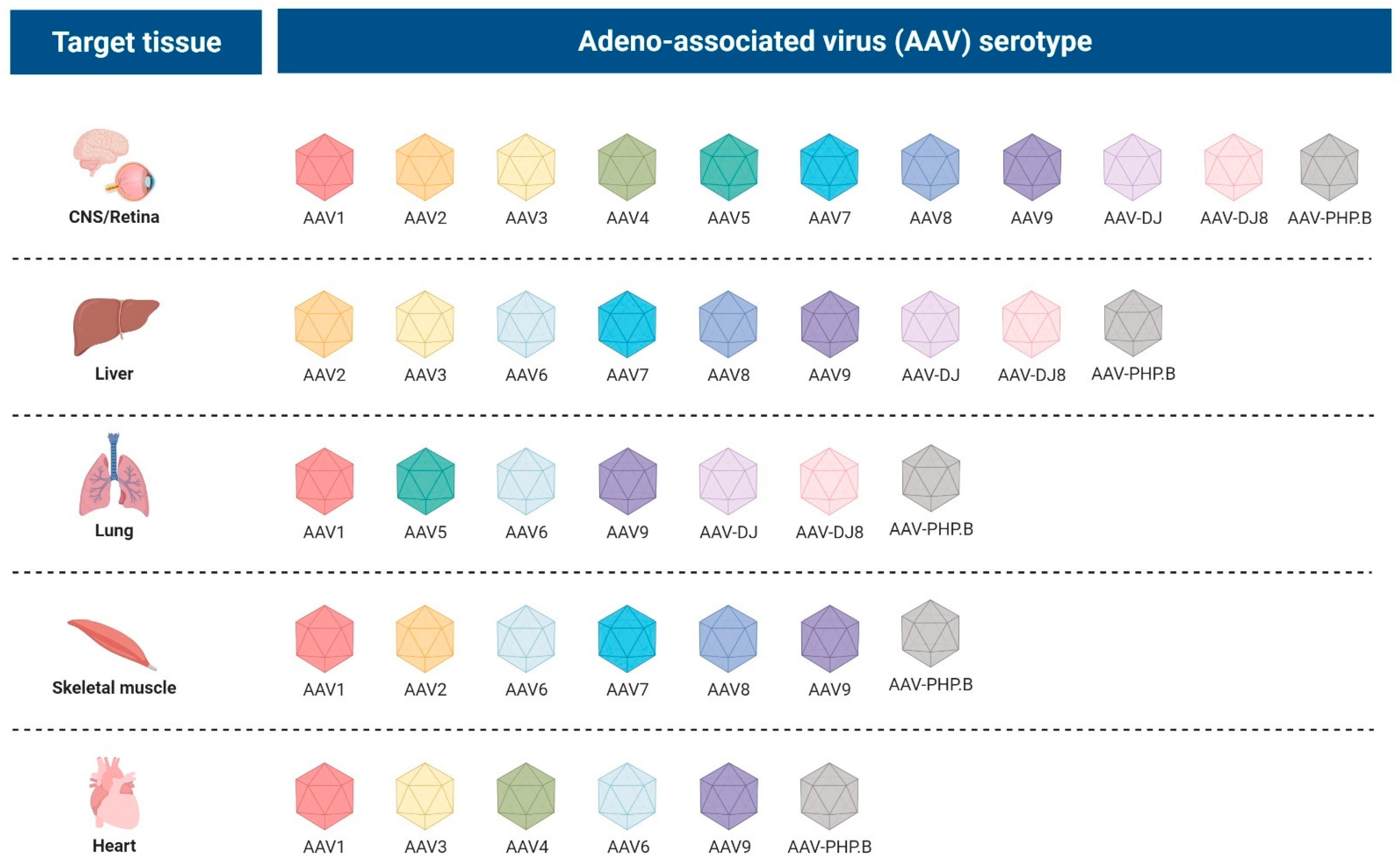
2.2.3. Adeno-Associated Viral Vectors
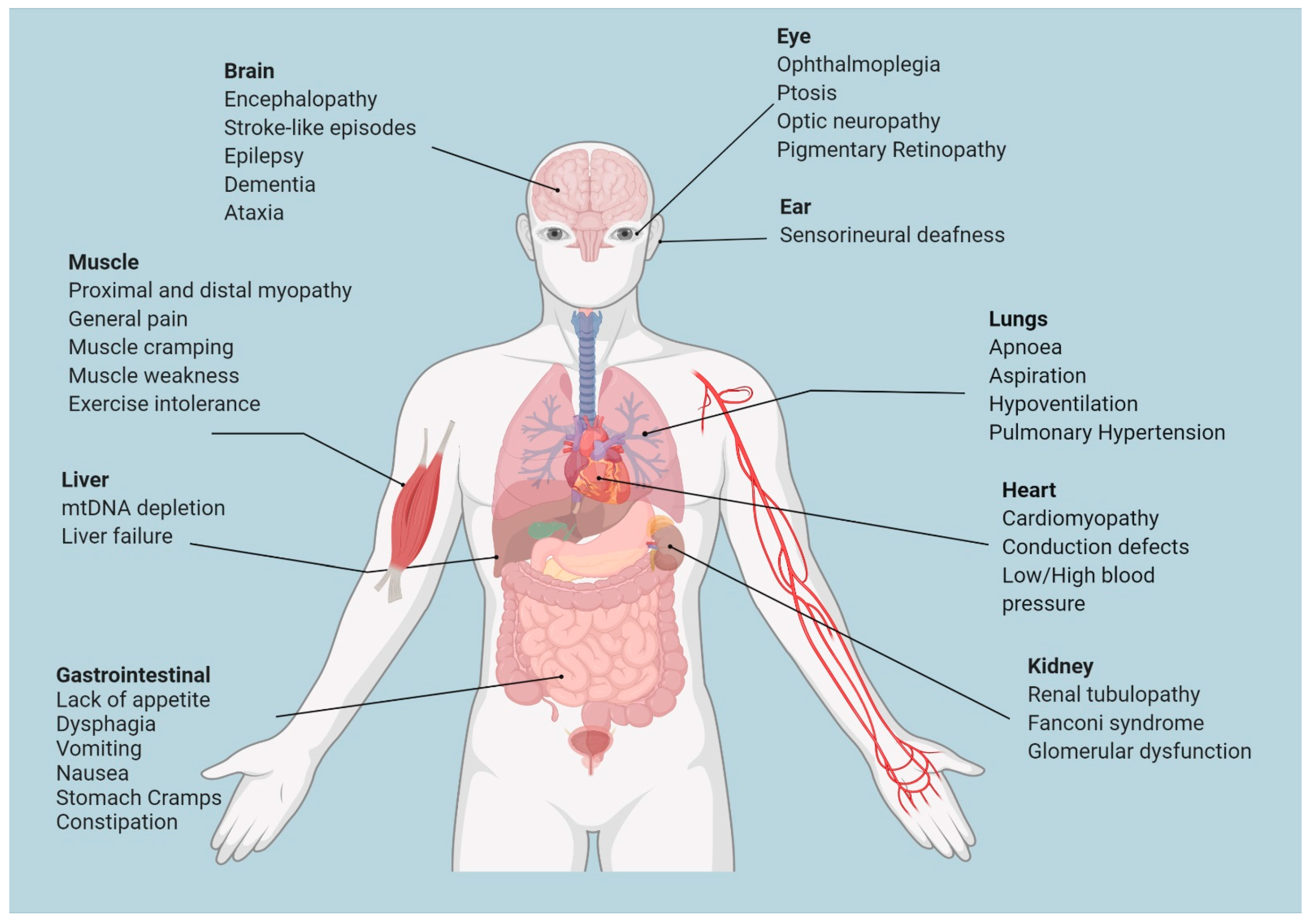
3. Gene Therapy for Mitochondrial Diseases Caused by Mutations in Nuclear Genes
3.1. Mitochondrial Myopathy (MM) and Cardiomyopathy
3.2. Metabolic Disorder Caused by the Accumulation of Toxic Compounds
3.3. Mitochondrial DNA Depletion Syndrome
3.3.1. TYMP
3.3.2. TK2
3.3.3. MPV17
3.4. Leigh Syndrome
3.4.1. NDUFS4
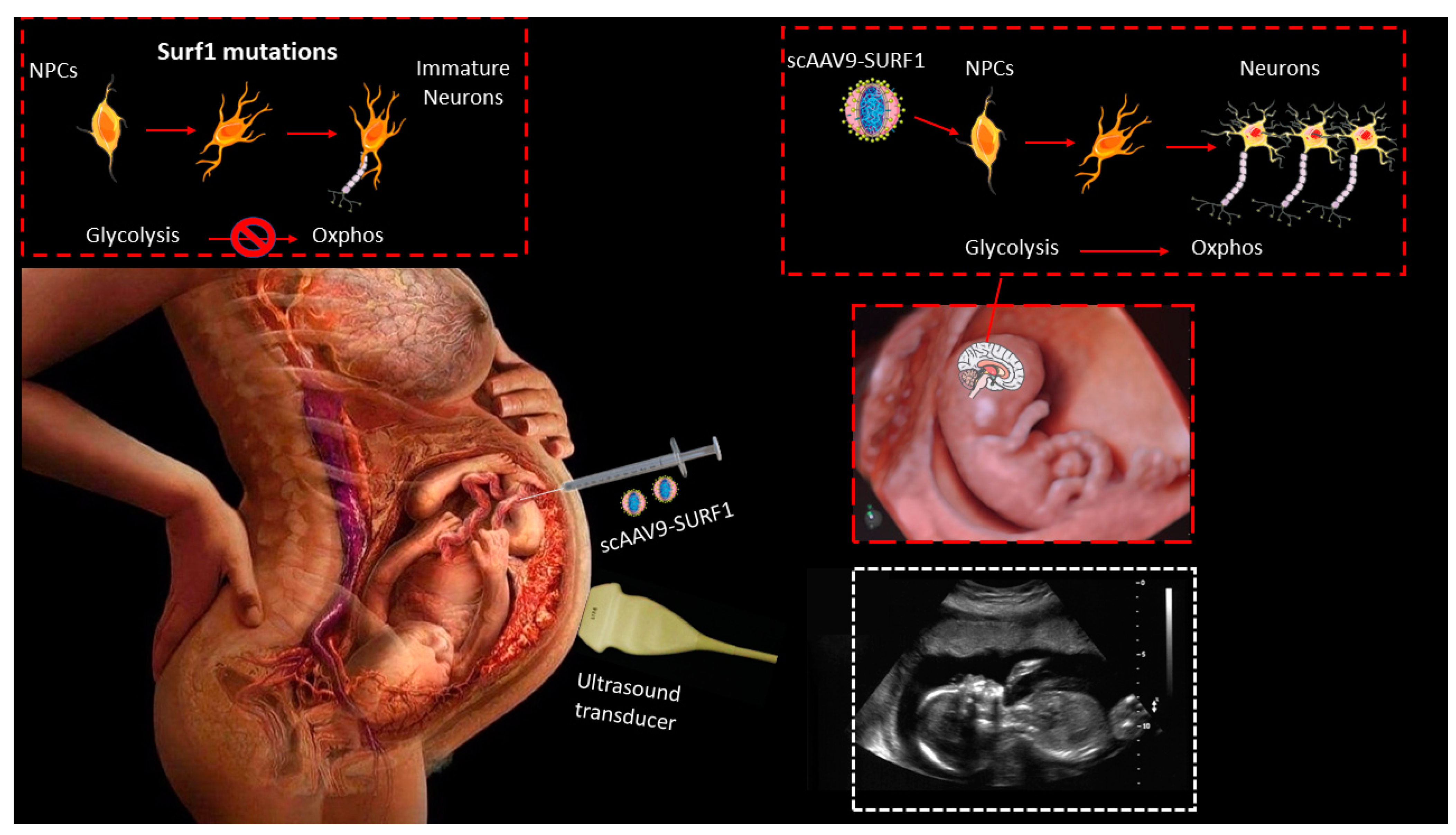
3.4.2. SURF1
3.5. Friedrich Ataxia
4. Gene Therapy for mtDNA-Associated Disorders
4.1. Allotopic Expression
Leber’s Hereditary Optic Neuropathy
4.2. Mitochondrial Delivery of Nucleic Acids
4.3. mtDNA Heteroplasmy Manipulation
4.4. mtDNA Editing
5. Future Perspective
5.1. In Utero Fetal Gene Therapy
5.2. Germline Gene Therapy
5.3. RNA-Based Therapy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Wallace, D.C. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matilainen, O.; Quirós, P.M.; Auwerx, J. Mitochondria and Epigenetics—Crosstalk in Homeostasis and Stress. Trends Cell Biol. 2017, 27, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Dyall, S.D.; Brown, M.T.; Johnson, P.J. Ancient Invasions: From Endosymbionts to Organelles. Science 2004, 304, 253–257. [Google Scholar] [CrossRef] [Green Version]
- Vafai, S.B.; Mootha, V.K. Mitochondrial Disorders as Windows into an Ancient Organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef]
- Taanman, J.-W. The Mitochondrial Genome: Structure, Transcription, Translation and Replication. Biochim. Biophys. Acta BBA-Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef] [Green Version]
- Payne, B.A.I.; Wilson, I.J.; Yu-Wai-Man, P.; Coxhead, J.; Deehan, D.; Horvath, R.; Taylor, R.W.; Samuels, D.C.; Santibanez-Koref, M.; Chinnery, P.F. Universal Heteroplasmy of Human Mitochondrial DNA. Hum. Mol. Genet. 2013, 22, 384–390. [Google Scholar] [CrossRef] [Green Version]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of Nuclear and Mitochondrial DNA Mutations Related to Adult Mitochondrial Disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Goto, Y.; Nonaka, I.; Horai, S. A Mutation in the TRNALeu(UUR) Gene Associated with the MELAS Subgroup of Mitochondrial Encephalomyopathies. Nature 1990, 348, 651–653. [Google Scholar] [CrossRef]
- Rahman, S.; Blok, R.B.; Dahl, H.-H.M.; Danks, D.M.; Kirby, D.M.; Chow, C.W.; Christodoulou, J.; Thorburn, D.R. Leigh Syndrome: Clinical Features and Biochemical and DNA Abnormalities. Ann. Neurol. 1996, 39, 343–351. [Google Scholar] [CrossRef]
- Holt, I.J.; Harding, A.E.; Petty, R.K.; Morgan-Hughes, J.A. A New Mitochondrial Disease Associated with Mitochondrial DNA Heteroplasmy. Am. J. Hum. Genet. 1990, 46, 428–433. [Google Scholar]
- Shoffner, J.M.; Lott, M.T.; Lezza, A.M.S.; Seibel, P.; Ballinger, S.W.; Wallace, D.C. Myoclonic Epilepsy and Ragged-Red Fiber Disease (MERRF) Is Associated with a Mitochondrial DNA TRNALys Mutation. Cell 1990, 61, 931–937. [Google Scholar] [CrossRef]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.S.; Elsas, L.J.; Nikoskelainen, E.K. Mitochondrial DNA Mutation Associated with Leber’s Hereditary Optic Neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Bugiardini, E.; Bottani, E.; Marchet, S.; Poole, O.V.; Beninca, C.; Horga, A.; Woodward, C.; Lam, A.; Hargreaves, I.; Chalasani, A.; et al. Expanding the Molecular and Phenotypic Spectrum of Truncating MT-ATP6 Mutations. Neurol. Genet. 2020, 6, e381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Gallardo, E.; Solano, A.; Herrero-Martin, M.D.; Martinez-Romero, I.; Castano-Perez, M.D.; Andreu, A.L.; Herrera, A.; Lopez-Perez, M.J.; Ruiz-Pesini, E.; Montoya, J. NARP Syndrome in a Patient Harbouring an Insertion in the MT-ATP6 Gene That Results in a Truncated Protein. J. Med. Genet. 2008, 46, 64–67. [Google Scholar] [CrossRef]
- Pitceathly, R.D.S.; Murphy, S.M.; Cottenie, E.; Chalasani, A.; Sweeney, M.G.; Woodward, C.; Mudanohwo, E.E.; Hargreaves, I.; Heales, S.; Land, J.; et al. Genetic Dysfunction of MT-ATP6 Causes Axonal Charcot-Marie-Tooth Disease. Neurology 2012, 79, 1145–1154. [Google Scholar] [CrossRef] [Green Version]
- Moraes, C.T.; DiMauro, S.; Zeviani, M.; Lombes, A.; Shanske, S.; Miranda, A.F.; Nakase, H.; Bonilla, E.; Werneck, L.C.; Servidei, S.; et al. Mitochondrial DNA Deletions in Progressive External Ophthalmoplegia and Kearns-Sayre Syndrome. N. Engl. J. Med. 1989, 320, 1293–1299. [Google Scholar] [CrossRef]
- Pearson, H.A.; Lobel, J.S.; Kocoshis, S.A.; Naiman, J.L.; Windmiller, J.; Lammi, A.T.; Hoffman, R.; Marsh, J.C. A New Syndrome of Refractory Sideroblastic Anemia with Vacuolization of Marrow Precursors and Exocrine Pancreatic Dysfunction. J. Pediatr. 1979, 95, 976–984. [Google Scholar] [CrossRef]
- Koopman, W.J.H.; Willems, P.H.G.M.; Smeitink, J.A.M. Monogenic Mitochondrial Disorders. N. Engl. J. Med. 2012, 366, 1132–1141. [Google Scholar] [CrossRef] [Green Version]
- McFarland, R.; Taylor, R.W.; Turnbull, D.M. A Neurological Perspective on Mitochondrial Disease. Lancet Neurol. 2010, 9, 829–840. [Google Scholar] [CrossRef]
- Bottani, E.; Lamperti, C.; Prigione, A.; Tiranti, V.; Persico, N.; Brunetti, D. Therapeutic Approaches to Treat Mitochondrial Diseases: “One-Size-Fits-All” and “Precision Medicine” Strategies. Pharmaceutics 2020, 12, 1083. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.A.; Rodino-Klapac, L.R.; Goodspeed, K.; Gray, S.J.; Kay, C.N.; Boye, S.L.; Boye, S.E.; George, L.A.; Salabarria, S.; et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther. 2021, 29, 464–488. [Google Scholar] [CrossRef] [PubMed]
- Melchiorri, D.; Pani, L.; Gasparini, P.; Cossu, G.; Ancans, J.; Borg, J.J.; Drai, C.; Fiedor, P.; Flory, E.; Hudson, I.; et al. Regulatory Evaluation of Glybera in Europe—Two Committees, One Mission. Nat. Rev. Drug Discov. 2013, 12, 719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and Safety of Voretigene Neparvovec (AAV2-HRPE65v2) in Patients with RPE65 -Mediated Inherited Retinal Dystrophy: A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoy, N.; Fox, T.D. Directed Alteration of Saccharomyces Cerevisiae Mitochondrial DNA by Biolistic Transformation and Homologous Recombination. In Mitochondria; Leister, D., Herrmann, J.M., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2007; Volume 372, pp. 153–166. ISBN 978-1-58829-667-2. [Google Scholar]
- Yasuzaki, Y.; Yamada, Y.; Ishikawa, T.; Harashima, H. Validation of Mitochondrial Gene Delivery in Liver and Skeletal Muscle via Hydrodynamic Injection Using an Artificial Mitochondrial Reporter DNA Vector. Mol. Pharm. 2015, 12, 4311–4320. [Google Scholar] [CrossRef]
- Yamada, Y.; Furukawa, R.; Yasuzaki, Y.; Harashima, H. Dual Function MITO-Porter, a Nano Carrier Integrating Both Efficient Cytoplasmic Delivery and Mitochondrial Macromolecule Delivery. Mol. Ther. 2011, 19, 1449–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, J.; Sousa, F.; Queiroz, J.; Costa, D. Rhodamine Based Plasmid DNA Nanoparticles for Mitochondrial Gene Therapy. Colloids Surf. B Biointerfaces 2014, 121, 129–140. [Google Scholar] [CrossRef]
- Flierl, A.; Jackson, C.; Cottrell, B.; Murdock, D.; Seibel, P.; Wallace, D.C. Targeted Delivery of DNA to the Mitochondrial Compartment via Import Sequence-Conjugated Peptide Nucleic Acid. Mol. Ther. 2003, 7, 550–557. [Google Scholar] [CrossRef]
- Jang, Y.; Lim, K. Recent Advances in Mitochondria-Targeted Gene Delivery. Molecules 2018, 23, 2316. [Google Scholar] [CrossRef] [Green Version]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral Vector Platforms within the Gene Therapy Landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef]
- Neil, J.C. Safety of Retroviral Vectors in Clinical Applications: Lessons from Retroviral Biology and Pathogenesis. In eLS; John Wiley & Sons, Ltd., Ed.; Wiley: Hoboken, NJ, USA, 2017; pp. 1–10. ISBN 978-0-470-01617-6. [Google Scholar]
- Naldini, L.; Blömer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In Vivo Gene Delivery and Stable Transduction of Nondividing Cells by a Lentiviral Vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucci, L.; Lai, M.; Chiuppesi, F.; Ceccherini-Nelli, L.; Pistello, M. Viral Vectors: A Look Back and Ahead on Gene Transfer Technology. New Microbiol. 2013, 36, 1–22. [Google Scholar] [PubMed]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A Third-Generation Lentivirus Vector with a Conditional Packaging System. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohn, D.B.; Booth, C.; Shaw, K.L.; Xu-Bayford, J.; Garabedian, E.; Trevisan, V.; Carbonaro-Sarracino, D.A.; Soni, K.; Terrazas, D.; Snell, K.; et al. Autologous Ex Vivo Lentiviral Gene Therapy for Adenosine Deaminase Deficiency. N. Engl. J. Med. 2021, 384, 2002–2013. [Google Scholar] [CrossRef]
- Fumagalli, F.; Calbi, V.; Natali Sora, M.G.; Sessa, M.; Baldoli, C.; Rancoita, P.M.V.; Ciotti, F.; Sarzana, M.; Fraschini, M.; Zambon, A.A.; et al. Lentiviral Haematopoietic Stem-Cell Gene Therapy for Early-Onset Metachromatic Leukodystrophy: Long-Term Results from a Non-Randomised, Open-Label, Phase 1/2 Trial and Expanded Access. Lancet 2022, 399, 372–383. [Google Scholar] [CrossRef]
- Russell, W.C. Adenoviruses: Update on Structure and Function. J. Gen. Virol. 2009, 90, 1–20. [Google Scholar] [CrossRef]
- Arrand, J.R.; Roberts, R.J. The Nucleotide Sequences at the Termini of Adenovirus-2 DNA. J. Mol. Biol. 1979, 128, 577–594. [Google Scholar] [CrossRef]
- Tomko, R.P.; Xu, R.; Philipson, L. HCAR and MCAR: The Human and Mouse Cellular Receptors for Subgroup C Adenoviruses and Group B Coxsackieviruses. Proc. Natl. Acad. Sci. USA 1997, 94, 3352–3356. [Google Scholar] [CrossRef] [Green Version]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a Common Receptor for Coxsackie B Viruses and Adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Gaggar, A.; Shayakhmetov, D.M.; Lieber, A. CD46 Is a Cellular Receptor for Group B Adenoviruses. Nat. Med. 2003, 9, 1408–1412. [Google Scholar] [CrossRef]
- Nilsson, E.C.; Storm, R.J.; Bauer, J.; Johansson, S.M.C.; Lookene, A.; Ångström, J.; Hedenström, M.; Eriksson, T.L.; Frängsmyr, L.; Rinaldi, S.; et al. The GD1a Glycan Is a Cellular Receptor for Adenoviruses Causing Epidemic Keratoconjunctivitis. Nat. Med. 2011, 17, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Wiethoff, C.M.; Wodrich, H.; Gerace, L.; Nemerow, G.R. Adenovirus Protein VI Mediates Membrane Disruption Following Capsid Disassembly. J. Virol. 2005, 79, 1992–2000. [Google Scholar] [CrossRef] [Green Version]
- Wiethoff, C.M.; Nemerow, G.R. Adenovirus Membrane Penetration: Tickling the Tail of a Sleeping Dragon. Virology 2015, 479–480, 591–599. [Google Scholar] [CrossRef] [Green Version]
- Bremner, K.H.; Scherer, J.; Yi, J.; Vershinin, M.; Gross, S.P.; Vallee, R.B. Adenovirus Transport via Direct Interaction of Cytoplasmic Dynein with the Viral Capsid Hexon Subunit. Cell Host Microbe 2009, 6, 523–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Granelli-Piperno, A.; Choi, Y.; Steinman, R.M. Recombinant Adenovirus Is an Efficient and Non-Perturbing Genetic Vector for Human Dendritic Cells. Eur. J. Immunol. 1999, 29, 964–972. [Google Scholar] [CrossRef]
- Mast, T.C.; Kierstead, L.; Gupta, S.B.; Nikas, A.A.; Kallas, E.G.; Novitsky, V.; Mbewe, B.; Pitisuttithum, P.; Schechter, M.; Vardas, E.; et al. International Epidemiology of Human Pre-Existing Adenovirus (Ad) Type-5, Type-6, Type-26 and Type-36 Neutralizing Antibodies: Correlates of High Ad5 Titers and Implications for Potential HIV Vaccine Trials. Vaccine 2010, 28, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.H.; Kik, S.V.; Weverling, G.J.; Dilan, R.; King, S.L.; Maxfield, L.F.; Clark, S.; Ng’ang’a, D.; Brandariz, K.L.; Abbink, P.; et al. International Seroepidemiology of Adenovirus Serotypes 5, 26, 35, and 48 in Pediatric and Adult Populations. Vaccine 2011, 29, 5203–5209. [Google Scholar] [CrossRef] [Green Version]
- Buchbinder, S.P.; Mehrotra, D.V.; Duerr, A.; Fitzgerald, D.W.; Mogg, R.; Li, D.; Gilbert, P.B.; Lama, J.R.; Marmor, M.; del Rio, C.; et al. Efficacy Assessment of a Cell-Mediated Immunity HIV-1 Vaccine (the Step Study): A Double-Blind, Randomised, Placebo-Controlled, Test-of-Concept Trial. Lancet 2008, 372, 1881–1893. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zeng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-Mediated Gene Delivery: Potential Applications for Gene and Cell-Based Therapies in the New Era of Personalized Medicine. Genes Dis. 2017, 4, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Cunliffe, T.G.; Bates, E.A.; Parker, A.L. Hitting the Target but Missing the Point: Recent Progress towards Adenovirus-Based Precision Virotherapies. Cancers 2020, 12, 3327. [Google Scholar] [CrossRef]
- Kennedy, S.B.; Bolay, F.; Kieh, M.; Grandits, G.; Badio, M.; Ballou, R.; Eckes, R.; Feinberg, M.; Follmann, D.; Grund, B.; et al. Phase 2 Placebo-Controlled Trial of Two Vaccines to Prevent Ebola in Liberia. N. Engl. J. Med. 2017, 377, 1438–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastian, S.; Lambe, T. Clinical Advances in Viral-Vectored Influenza Vaccines. Vaccines 2018, 6, 29. [Google Scholar] [CrossRef] [Green Version]
- Zhu, F.-C.; Li, Y.-H.; Guan, X.-H.; Hou, L.-H.; Wang, W.-J.; Li, J.-X.; Wu, S.-P.; Wang, B.-S.; Wang, Z.; Wang, L.; et al. Safety, Tolerability, and Immunogenicity of a Recombinant Adenovirus Type-5 Vectored COVID-19 Vaccine: A Dose-Escalation, Open-Label, Non-Randomised, First-in-Human Trial. Lancet 2020, 395, 1845–1854. [Google Scholar] [CrossRef]
- Atchison, R.W.; Casto, B.C.; Hammon, W.M. Adenovirus-Associated Defective Virus Particles. Science 1965, 149, 754–756. [Google Scholar] [CrossRef]
- Linden, R.M.; Berns, K.I. Molecular Biology of Adeno-Associated Viruses. In Contributions to Microbiology; Faisst, S., Rommelaere, J., Eds.; KARGER: Basel, Switzerland, 2000; Volume 4, pp. 68–84. ISBN 978-3-8055-6946-0. [Google Scholar]
- Mietzsch, M.; Broecker, F.; Reinhardt, A.; Seeberger, P.H.; Heilbronn, R. Differential Adeno-Associated Virus Serotype-Specific Interaction Patterns with Synthetic Heparins and Other Glycans. J. Virol. 2014, 88, 2991–3003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somanathan, S.; Breous, E.; Bell, P.; Wilson, J.M. AAV Vectors Avoid Inflammatory Signals Necessary to Render Transduced Hepatocyte Targets for Destructive T Cells. Mol. Ther. 2010, 18, 977–982. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Reiss, U.M.; Tuddenham, E.G.D.; Rosales, C.; Chowdary, P.; McIntosh, J.; Della Peruta, M.; Lheriteau, E.; Patel, N.; Raj, D.; et al. Long-Term Safety and Efficacy of Factor IX Gene Therapy in Hemophilia B. N. Engl. J. Med. 2014, 371, 1994–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, D.; Yue, Y.; Yan, Z.; Engelhardt, J.F. A New Dual-Vector Approach to Enhance Recombinant Adeno-Associated Virus-Mediated Gene Expression through Intermolecular Cis Activation. Nat. Med. 2000, 6, 595–598. [Google Scholar] [CrossRef] [PubMed]
- Nakai, H.; Storm, T.A.; Kay, M.A. Increasing the Size of RAAV-Mediated Expression Cassettes in Vivo by Intermolecular Joining of Two Complementary Vectors. Nat. Biotechnol. 2000, 18, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.; Liu, Y.; Rabinowitz, J.; Li, C.; Samulski, R.J.; Walsh, C.E. Several Log Increase in Therapeutic Transgene Delivery by Distinct Adeno-Associated Viral Serotype Vectors. Mol. Ther. 2000, 2, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Burger, C.; Gorbatyuk, O.S.; Velardo, M.J.; Peden, C.S.; Williams, P.; Zolotukhin, S.; Reier, P.J.; Mandel, R.J.; Muzyczka, N. Recombinant AAV Viral Vectors Pseudotyped with Viral Capsids from Serotypes 1, 2, and 5 Display Differential Efficiency and Cell Tropism after Delivery to Different Regions of the Central Nervous System. Mol. Ther. 2004, 10, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Samulski, R.J. Engineering Adeno-Associated Virus Vectors for Gene Therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Hudry, E.; Vandenberghe, L.H. Therapeutic AAV Gene Transfer to the Nervous System: A Clinical Reality. Neuron 2019, 101, 839–862. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Alvira, M.R.; Somanathan, S.; Lu, Y.; Vandenberghe, L.H.; Rux, J.J.; Calcedo, R.; Sanmiguel, J.; Abbas, Z.; Wilson, J.M. Adeno-Associated Viruses Undergo Substantial Evolution in Primates during Natural Infections. Proc. Natl. Acad. Sci. USA 2003, 100, 6081–6086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, W.P.; Boucher, D.W.; Melnick, J.L.; Taber, L.H.; Yow, M.D. Seroepidemiological and Ecological Studies of the Adenovirus-Associated Satellite Viruses. Infect. Immun. 1970, 2, 716–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, S.; Wang, L.; Takeuchi, T.; Kanda, T. Two Novel Adeno-Associated Viruses from Cynomolgus Monkey: Pseudotyping Characterization of Capsid Protein. Virology 2004, 330, 375–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Martins, I.H.; Chiorini, J.A.; Davidson, B.L. Adeno-Associated Virus Type 4 (AAV4) Targets Ependyma and Astrocytes in the Subventricular Zone and RMS. Gene Ther. 2005, 12, 1503–1508. [Google Scholar] [CrossRef]
- Balaji, S.; King, A.; Dhamija, Y.; Le, L.D.; Shaaban, A.F.; Crombleholme, T.M.; Keswani, S.G. Pseudotyped Adeno-Associated Viral Vectors for Gene Transfer in Dermal Fibroblasts: Implications for Wound-Healing Applications. J. Surg. Res. 2013, 184, 691–698. [Google Scholar] [CrossRef] [Green Version]
- Grimm, D.; Lee, J.S.; Wang, L.; Desai, T.; Akache, B.; Storm, T.A.; Kay, M.A. In Vitro and In Vivo Gene Therapy Vector Evolution via Multispecies Interbreeding and Retargeting of Adeno-Associated Viruses. J. Virol. 2008, 82, 5887–5911. [Google Scholar] [CrossRef] [Green Version]
- Foust, K.D.; Nurre, E.; Montgomery, C.L.; Hernandez, A.; Chan, C.M.; Kaspar, B.K. Intravascular AAV9 Preferentially Targets Neonatal Neurons and Adult Astrocytes. Nat. Biotechnol. 2009, 27, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Bedbrook, C.N.; Deverman, B.E.; Gradinaru, V. Viral Strategies for Targeting the Central and Peripheral Nervous Systems. Annu. Rev. Neurosci. 2018, 41, 323–348. [Google Scholar] [CrossRef]
- Deverman, B.E.; Pravdo, P.L.; Simpson, B.P.; Kumar, S.R.; Chan, K.Y.; Banerjee, A.; Wu, W.-L.; Yang, B.; Huber, N.; Pasca, S.P.; et al. Cre-Dependent Selection Yields AAV Variants for Widespread Gene Transfer to the Adult Brain. Nat. Biotechnol. 2016, 34, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Mathiesen, S.N.; Lock, J.L.; Schoderboeck, L.; Abraham, W.C.; Hughes, S.M. CNS Transduction Benefits of AAV-PHP.EB over AAV9 Are Dependent on Administration Route and Mouse Strain. Mol. Ther.-Methods Clin. Dev. 2020, 19, 447–458. [Google Scholar] [CrossRef]
- Arotcarena, M.-L.; Dovero, S.; Biendon, N.; Dutheil, N.; Planche, V.; Bezard, E.; Dehay, B. Pilot Study Assessing the Impact of Intrathecal Administration of Variants AAV-PHP.B and AAV-PHP.EB on Brain Transduction in Adult Rhesus Macaques. Front. Bioeng. Biotechnol. 2021, 9, 762209. [Google Scholar] [CrossRef] [PubMed]
- McCarty, D.M. Self-Complementary AAV Vectors; Advances and Applications. Mol. Ther. 2008, 16, 1648–1656. [Google Scholar] [CrossRef]
- Wang, Z.; Ma, H.-I.; Li, J.; Sun, L.; Zhang, J.; Xiao, X. Rapid and Highly Efficient Transduction by Double-Stranded Adeno-Associated Virus Vectors in Vitro and in Vivo. Gene Ther. 2003, 10, 2105–2111. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Chan, K.Y.; Tobey, I.G.; Chan, Y.A.; Poterba, T.; Boutros, C.L.; Balazs, A.B.; Daneman, R.; Bloom, J.M.; Seed, C.; et al. Delivering Genes across the Blood-Brain Barrier: LY6A, a Novel Cellular Receptor for AAV-PHP.B Capsids. PLoS ONE 2019, 14, e0225206. [Google Scholar] [CrossRef] [Green Version]
- Kaukonen, J.; Juselius, J.K.; Tiranti, V.; Kyttälä, A.; Zeviani, M.; Comi, G.P.; Keränen, S.; Peltonen, L.; Suomalainen, A. Role of Adenine Nucleotide Translocator 1 in MtDNA Maintenance. Science 2000, 289, 782–785. [Google Scholar] [CrossRef]
- Flierl, A.; Chen, Y.; Coskun, P.E.; Samulski, R.J.; Wallace, D.C. Adeno-Associated Virus-Mediated Gene Transfer of the Heart/Muscle Adenine Nucleotide Translocator (ANT) in Mouse. Gene Ther. 2005, 12, 570–578. [Google Scholar] [CrossRef] [Green Version]
- Di Meo, I.; Lamperti, C.; Tiranti, V. Mitochondrial Diseases Caused by Toxic Compound Accumulation: From Etiopathology to Therapeutic Approaches. EMBO Mol. Med. 2015, 7, 1257–1266. [Google Scholar] [CrossRef]
- Di Meo, I.; Fagiolari, G.; Prelle, A.; Viscomi, C.; Zeviani, M.; Tiranti, V. Chronic Exposure to Sulfide Causes Accelerated Degradation of Cytochrome c Oxidase in Ethylmalonic Encephalopathy. Antioxid. Redox Signal. 2011, 15, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Tiranti, V.; Viscomi, C.; Hildebrandt, T.; Di Meo, I.; Mineri, R.; Tiveron, C.; Levitt, D.M.; Prelle, A.; Fagiolari, G.; Rimoldi, M.; et al. Loss of ETHE1, a Mitochondrial Dioxygenase, Causes Fatal Sulfide Toxicity in Ethylmalonic Encephalopathy. Nat. Med. 2009, 15, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Sun, X.; Wang, R. Hydrogen Sulfide-induced Apoptosis of Human Aorta Smooth Muscle Cells via the Activation of Mitogen-activated Protein Kinases and Caspase-3. FASEB J. 2004, 18, 1782–1784. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, I.; Auricchio, A.; Lamperti, C.; Burlina, A.; Viscomi, C.; Zeviani, M. Effective AAV-mediated Gene Therapy in a Mouse Model of Ethylmalonic Encephalopathy. EMBO Mol. Med. 2012, 4, 1008–1014. [Google Scholar] [CrossRef]
- Dionisi-Vici, C.; Diodato, D.; Torre, G.; Picca, S.; Pariante, R.; Giuseppe Picardo, S.; Di Meo, I.; Rizzo, C.; Tiranti, V.; Zeviani, M.; et al. Liver Transplant in Ethylmalonic Encephalopathy: A New Treatment for an Otherwise Fatal Disease. Brain 2016, 139, 1045–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, A.; AlDhaheri, N.S.; Mysore, K.; Tessier, M.E.; Goss, J.; Fernandez, L.A.; D’Alessandro, A.M.; Schwoerer, J.S.; Rice, G.M.; Elsea, S.H.; et al. Improved Clinical Outcome Following Liver Transplant in Patients with Ethylmalonic Encephalopathy. Am. J. Med. Genet. A. 2019, 179, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, G.; Martinelli, D.; Longo, D.; Grimaldi, C.; Liccardo, D.; Di Meo, I.; Pietrobattista, A.; Sidorina, A.; Semeraro, M.; Dionisi-Vici, C. Ethylmalonic Encephalopathy and Liver Transplantation: Long-Term Outcome of the First Treated Patient. Orphanet J. Rare Dis. 2021, 16, 229. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial DNA Depletion Syndromes: Review and Updates of Genetic Basis, Manifestations, and Therapeutic Options. Neurotherapeutics 2013, 10, 186–198. [Google Scholar] [CrossRef] [Green Version]
- Ronchi, D.; Fassone, E.; Bordoni, A.; Sciacco, M.; Lucchini, V.; Di Fonzo, A.; Rizzuti, M.; Colombo, I.; Napoli, L.; Ciscato, P.; et al. Two Novel Mutations in PEO1 (Twinkle) Gene Associated with Chronic External Ophthalmoplegia. J. Neurol. Sci. 2011, 308, 173–176. [Google Scholar] [CrossRef] [Green Version]
- Spinazzola, A.; Viscomi, C.; Fernandez-Vizarra, E.; Carrara, F.; D’Adamo, P.; Calvo, S.; Marsano, R.M.; Donnini, C.; Weiher, H.; Strisciuglio, P.; et al. MPV17 Encodes an Inner Mitochondrial Membrane Protein and Is Mutated in Infantile Hepatic Mitochondrial DNA Depletion. Nat. Genet. 2006, 38, 570–575. [Google Scholar] [CrossRef]
- Bottani, E.; Giordano, C.; Civiletto, G.; Di Meo, I.; Auricchio, A.; Ciusani, E.; Marchet, S.; Lamperti, C.; d’Amati, G.; Viscomi, C.; et al. AAV-Mediated Liver-Specific MPV17 Expression Restores MtDNA Levels and Prevents Diet-Induced Liver Failure. Mol. Ther. 2014, 22, 10–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Torronteras, J.; Gómez, A.; Eixarch, H.; Palenzuela, L.; Pizzorno, G.; Hirano, M.; Andreu, A.L.; Barquinero, J.; Martí, R. Hematopoietic Gene Therapy Restores Thymidine Phosphorylase Activity in a Cell Culture and a Murine Model of MNGIE. Gene Ther. 2011, 18, 795–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Torronteras, J.; Cabrera-Pérez, R.; Barba, I.; Costa, C.; de Luna, N.; Andreu, A.L.; Barquinero, J.; Hirano, M.; Cámara, Y.; Martí, R. Long-Term Restoration of Thymidine Phosphorylase Function and Nucleoside Homeostasis Using Hematopoietic Gene Therapy in a Murine Model of Mitochondrial Neurogastrointestinal Encephalomyopathy. Hum. Gene Ther. 2016, 27, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Torres-Torronteras, J.; Viscomi, C.; Cabrera-Pérez, R.; Cámara, Y.; Di Meo, I.; Barquinero, J.; Auricchio, A.; Pizzorno, G.; Hirano, M.; Zeviani, M.; et al. Gene Therapy Using a Liver-Targeted AAV Vector Restores Nucleoside and Nucleotide Homeostasis in a Murine Model of MNGIE. Mol. Ther. 2014, 22, 901–907. [Google Scholar] [CrossRef] [Green Version]
- Torres-Torronteras, J.; Cabrera-Pérez, R.; Vila-Julià, F.; Viscomi, C.; Cámara, Y.; Hirano, M.; Zeviani, M.; Martí, R. Long-Term Sustained Effect of Liver-Targeted Adeno-Associated Virus Gene Therapy for Mitochondrial Neurogastrointestinal Encephalomyopathy. Hum. Gene Ther. 2018, 29, 708–718. [Google Scholar] [CrossRef]
- Garcia-Diaz, B.; Garone, C.; Barca, E.; Mojahed, H.; Gutierrez, P.; Pizzorno, G.; Tanji, K.; Arias-Mendoza, F.; Quinzii, C.M.; Hirano, M. Deoxynucleoside Stress Exacerbates the Phenotype of a Mouse Model of Mitochondrial Neurogastrointestinal Encephalopathy. Brain 2014, 137, 1337–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vila-Julià, F.; Cabrera-Pérez, R.; Cámara, Y.; Molina-Berenguer, M.; Lope-Piedrafita, S.; Hirano, M.; Mingozzi, F.; Torres-Torronteras, J.; Martí, R. Efficacy of Adeno-Associated Virus Gene Therapy in a MNGIE Murine Model Enhanced by Chronic Exposure to Nucleosides. eBioMedicine 2020, 62, 103133. [Google Scholar] [CrossRef]
- Parés, M.; Fornaguera, C.; Vila-Julià, F.; Oh, S.; Fan, S.H.Y.; Tam, Y.K.; Comes, N.; Vidal, F.; Martí, R.; Borrós, S.; et al. Preclinical Assessment of a Gene-Editing Approach in a Mouse Model of Mitochondrial Neurogastrointestinal Encephalomyopathy. Hum. Gene Ther. 2021, 32, 1210–1223. [Google Scholar] [CrossRef]
- Wang, L.; Saada, A.; Eriksson, S. Kinetic Properties of Mutant Human Thymidine Kinase 2 Suggest a Mechanism for Mitochondrial DNA Depletion Myopathy. J. Biol. Chem. 2003, 278, 6963–6968. [Google Scholar] [CrossRef] [Green Version]
- Akman, H.O.; Dorado, B.; López, L.C.; García-Cazorla, Á.; Vilà, M.R.; Tanabe, L.M.; Dauer, W.T.; Bonilla, E.; Tanji, K.; Hirano, M. Thymidine Kinase 2 (H126N) Knockin Mice Show the Essential Role of Balanced Deoxynucleotide Pools for Mitochondrial DNA Maintenance. Hum. Mol. Genet. 2008, 17, 2433–2440. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.-J.C.; Brunetti-Pierri, N.; Zhang, Q.; Yazigi, N.; Bove, K.E.; Dahms, B.B.; Puchowicz, M.A.; Gonzalez-Gomez, I.; Schmitt, E.S.; Truong, C.K.; et al. Mutations in TheMPV17 Gene Are Responsible for Rapidly Progressive Liver Failure in Infancy. Hepatology 2007, 46, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Spinazzola, A.; Santer, R.; Akman, O.H.; Tsiakas, K.; Schaefer, H.; Ding, X.; Karadimas, C.L.; Shanske, S.; Ganesh, J.; Di Mauro, S.; et al. Hepatocerebral Form of Mitochondrial DNA Depletion Syndrome: Novel MPV17 Mutations. Arch. Neurol. 2008, 65, 1108–1113. [Google Scholar] [CrossRef] [Green Version]
- Viscomi, C.; Spinazzola, A.; Maggioni, M.; Fernandez-Vizarra, E.; Massa, V.; Pagano, C.; Vettor, R.; Mora, M.; Zeviani, M. Early-Onset Liver MtDNA Depletion and Late-Onset Proteinuric Nephropathy in Mpv17 Knockout Mice. Hum. Mol. Genet. 2009, 18, 12–26. [Google Scholar] [CrossRef] [Green Version]
- Schubert Baldo, M.; Vilarinho, L. Molecular Basis of Leigh Syndrome: A Current Look. Orphanet J. Rare Dis. 2020, 15, 31. [Google Scholar] [CrossRef] [PubMed]
- Budde, S.M.S.; van den Heuvel, L.P.W.J.; Smeets, R.J.P.; Skladal, D.; Mayr, J.A.; Boelen, C.; Petruzzella, V.; Papa, S.; Smeitink, J.A.M. Clinical Heterogeneity in Patients with Mutations in the NDUFS4 Gene of Mitochondrial Complex I. J. Inherit. Metab. Dis. 2003, 26, 813–815. [Google Scholar] [CrossRef]
- Ortigoza-Escobar, J.D.; Oyarzabal, A.; Montero, R.; Artuch, R.; Jou, C.; Jiménez, C.; Gort, L.; Briones, P.; Muchart, J.; López-Gallardo, E.; et al. Ndufs4 Related Leigh Syndrome: A Case Report and Review of the Literature. Mitochondrion 2016, 28, 73–78. [Google Scholar] [CrossRef]
- van de Wal, M.A.E.; Adjobo-Hermans, M.J.W.; Keijer, J.; Schirris, T.J.J.; Homberg, J.R.; Wieckowski, M.R.; Grefte, S.; van Schothorst, E.M.; van Karnebeek, C.; Quintana, A.; et al. Ndufs4 Knockout Mouse Models of Leigh Syndrome: Pathophysiology and Intervention. Brain 2022, 145, 45–63. [Google Scholar] [CrossRef]
- Di Meo, I.; Marchet, S.; Lamperti, C.; Zeviani, M.; Viscomi, C. AAV9-Based Gene Therapy Partially Ameliorates the Clinical Phenotype of a Mouse Model of Leigh Syndrome. Gene Ther. 2017, 24, 661–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Pinheiro, P.; Cerutti, R.; Luna-Sanchez, M.; Zeviani, M.; Viscomi, C. A Single Intravenous Injection of AAV-PHP.B-HNDUFS4 Ameliorates the Phenotype of Ndufs4 Mice. Mol. Ther.-Methods Clin. Dev. 2020, 17, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Reynaud-Dulaurier, R.; Benegiamo, G.; Marrocco, E.; Al-Tannir, R.; Surace, E.M.; Auwerx, J.; Decressac, M. Gene Replacement Therapy Provides Benefit in an Adult Mouse Model of Leigh Syndrome. Brain 2020, 143, 1686–1696. [Google Scholar] [CrossRef]
- Tiranti, V.; Jaksch, M.; Hofmann, S.; Galimberti, C.; Hoertnagel, K.; Lulli, L.; Freisinger, P.; Bindoff, L.; Gerbitz, K.D.; Comi, G.-P.; et al. Loss-of-Function Mutations of SURF-1 Are Specifically Associated with Leigh Syndrome with Cytochromec Oxidase Deficiency. Ann. Neurol. 1999, 46, 161–166. [Google Scholar] [CrossRef]
- Dell’Agnello, C.; Leo, S.; Agostino, A.; Szabadkai, G.; Tiveron, C.; Zulian, A.; Prelle, A.; Roubertoux, P.; Rizzuto, R.; Zeviani, M. Increased Longevity and Refractoriness to Ca2+-Dependent Neurodegeneration in Surf1 Knockout Mice. Hum. Mol. Genet. 2007, 16, 431–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, Q.; Rioux, M.; Hu, Y.; Lee, M.; Gray, S.J. Adeno-Associated Viral Vector Serotype 9-Based Gene Replacement Therapy for SURF1-Related Leigh Syndrome. Mol. Ther.-Methods Clin. Dev. 2021, 23, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Pandolfo, M. Friedreich Ataxia: The Clinical Picture. J. Neurol. 2009, 256, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s Ataxia: Autosomal Recessive Disease Caused by an Intronic GAA Triplet Repeat Expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Ocana-Santero, G.; Díaz-Nido, J.; Herranz-Martín, S. Future Prospects of Gene Therapy for Friedreich’s Ataxia. Int. J. Mol. Sci. 2021, 22, 1815. [Google Scholar] [CrossRef]
- Piguet, F.; de Montigny, C.; Vaucamps, N.; Reutenauer, L.; Eisenmann, A.; Puccio, H. Rapid and Complete Reversal of Sensory Ataxia by Gene Therapy in a Novel Model of Friedreich Ataxia. Mol. Ther. 2018, 26, 1940–1952. [Google Scholar] [CrossRef] [Green Version]
- Belbellaa, B.; Reutenauer, L.; Messaddeq, N.; Monassier, L.; Puccio, H. High Levels of Frataxin Overexpression Lead to Mitochondrial and Cardiac Toxicity in Mouse Models. Mol. Ther -Methods Clin. Dev. 2020, 19, 120–138. [Google Scholar] [CrossRef]
- Artika, I.M. Allotopic Expression of Mitochondrial Genes: Basic Strategy and Progress. Genes Dis. 2020, 7, 578–584. [Google Scholar] [CrossRef]
- McClelland, C.; Meyerson, C.; Van Stavern, G. Leber Hereditary Optic Neuropathy: Current Perspectives. Clin. Ophthalmol. 2015, 9, 1165–1176. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Caspi, R.R. Ocular Immune Privilege. F1000 Biol. Rep. 2010, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Ellouze, S.; Augustin, S.; Bouaita, A.; Bonnet, C.; Simonutti, M.; Forster, V.; Picaud, S.; Sahel, J.-A.; Corral-Debrinski, M. Optimized Allotopic Expression of the Human Mitochondrial ND4 Prevents Blindness in a Rat Model of Mitochondrial Dysfunction. Am. J. Hum. Genet. 2008, 83, 373–387. [Google Scholar] [CrossRef] [Green Version]
- Koilkonda, R.; Yu, H.; Talla, V.; Porciatti, V.; Feuer, W.J.; Hauswirth, W.W.; Chiodo, V.; Erger, K.E.; Boye, S.L.; Lewin, A.S.; et al. LHON Gene Therapy Vector Prevents Visual Loss and Optic Neuropathy Induced by G11778A Mutant Mitochondrial DNA: Biodistribution and Toxicology Profile. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7739–7753. [Google Scholar] [CrossRef] [Green Version]
- Newman, N.J.; Yu-Wai-Man, P.; Carelli, V.; Biousse, V.; Moster, M.L.; Vignal-Clermont, C.; Sergott, R.C.; Klopstock, T.; Sadun, A.A.; Girmens, J.-F.; et al. Intravitreal Gene Therapy vs. Natural History in Patients With Leber Hereditary Optic Neuropathy Carrying the m.11778G>A ND4 Mutation: Systematic Review and Indirect Comparison. Front. Neurol. 2021, 12, 662838. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Newman, N.J.; Carelli, V.; Moster, M.L.; Biousse, V.; Sadun, A.A.; Klopstock, T.; Vignal-Clermont, C.; Sergott, R.C.; Rudolph, G.; et al. Bilateral Visual Improvement with Unilateral Gene Therapy Injection for Leber Hereditary Optic Neuropathy. Sci. Transl. Med. 2020, 12, eaaz7423. [Google Scholar] [CrossRef]
- Sahel, J.-A.; Newman, N.J.; Yu-Wai-Man, P.; Vignal-Clermont, C.; Carelli, V.; Biousse, V.; Moster, M.L.; Sergott, R.; Klopstock, T.; Sadun, A.A.; et al. Gene Therapies for the Treatment of Leber Hereditary Optic Neuropathy. Int. Ophthalmol. Clin. 2021, 61, 195–208. [Google Scholar] [CrossRef]
- Calkins, D.J.; Yu-Wai-Man, P.; Newman, N.J.; Taiel, M.; Singh, P.; Chalmey, C.; Rogue, A.; Carelli, V.; Ancian, P.; Sahel, J.A. Biodistribution of Intravitreal Lenadogene Nolparvovec Gene Therapy in Nonhuman Primates. Mol. Ther.-Methods Clin. Dev. 2021, 23, 307–318. [Google Scholar] [CrossRef]
- Bouquet, C.; Vignal Clermont, C.; Galy, A.; Fitoussi, S.; Blouin, L.; Munk, M.R.; Valero, S.; Meunier, S.; Katz, B.; Sahel, J.A.; et al. Immune Response and Intraocular Inflammation in Patients With Leber Hereditary Optic Neuropathy Treated With Intravitreal Injection of Recombinant Adeno-Associated Virus 2 Carrying the ND4 Gene: A Secondary Analysis of a Phase 1/2 Clinical Trial. JAMA Ophthalmol. 2019, 137, 399. [Google Scholar] [CrossRef] [Green Version]
- Slone, J.; Huang, T. The Special Considerations of Gene Therapy for Mitochondrial Diseases. Npj Med. 2020, 5, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyrawati, D.; Trounson, A.; Cram, D. Expression of GFP in the Mitochondrial Compartment Using DQAsome-Mediated Delivery of an Artificial Mini-Mitochondrial Genome. Pharm. Res. 2011, 28, 2848–2862. [Google Scholar] [CrossRef]
- Weissig, V. DQAsomes as the Prototype of Mitochondria-Targeted Pharmaceutical Nanocarriers: Preparation, Characterization, and Use. In Mitochondrial Medicine; Weissig, V., Edeas, M., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2015; Volume 1265, pp. 1–11. ISBN 978-1-4939-2287-1. [Google Scholar]
- Yasuzaki, Y.; Yamada, Y.; Harashima, H. Mitochondrial Matrix Delivery Using MITO-Porter, a Liposome-Based Carrier That Specifies Fusion with Mitochondrial Membranes. Biochem. Biophys. Res. Commun. 2010, 397, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, E.; Maruyama, M.; Abe, J.; Sudo, A.; Takeda, A.; Takada, S.; Yokota, T.; Kinugawa, S.; Harashima, H.; Yamada, Y. Validation of Gene Therapy for Mutant Mitochondria by Delivering Mitochondrial RNA Using a MITO-Porter. Mol. Ther.-Nucleic Acids 2020, 20, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Somiya, K.; Miyauchi, A.; Osaka, H.; Harashima, H. Validation of a Mitochondrial RNA Therapeutic Strategy Using Fibroblasts from a Leigh Syndrome Patient with a Mutation in the Mitochondrial ND3 Gene. Sci. Rep. 2020, 10, 7511. [Google Scholar] [CrossRef]
- Mahata, B.; Mukherjee, S.; Mishra, S.; Bandyopadhyay, A.; Adhya, S. Functional Delivery of a Cytosolic TRNA into Mutant Mitochondria of Human Cells. Science 2006, 314, 471–474. [Google Scholar] [CrossRef] [Green Version]
- Adhya, S. Leishmania Mitochondrial TRNA Importers. Int. J. Biochem. Cell Biol. 2008, 40, 2681–2685. [Google Scholar] [CrossRef]
- Yu, H.; Koilkonda, R.D.; Chou, T.-H.; Porciatti, V.; Ozdemir, S.S.; Chiodo, V.; Boye, S.L.; Boye, S.E.; Hauswirth, W.W.; Lewin, A.S.; et al. Gene Delivery to Mitochondria by Targeting Modified Adenoassociated Virus Suppresses Leber’s Hereditary Optic Neuropathy in a Mouse Model. Proc. Natl. Acad. Sci. USA 2012, 109, E1238–E1247. [Google Scholar] [CrossRef] [Green Version]
- Rossignol, R.; Faustin, B.; Rocher, C.; Malgat, M.; Mazat, J.-P.; Letellier, T. Mitochondrial Threshold Effects. Biochem. J. 2003, 370, 751–762. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S. Manipulating Mitochondrial DNA Heteroplasmy by a Mitochondrially Targeted Restriction Endonuclease. Hum. Mol. Genet. 2001, 10, 3093–3099. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Borgeld, H.-J.; Zhang, J.; Muramatsu, S.; Gong, J.-S.; Yoneda, M.; Maruyama, W.; Naoi, M.; Ibi, T.; Sahashi, K.; et al. Gene Therapy for Mitochondrial Disease by Delivering Restriction Endonuclease SmaI into Mitochondria. J. Biomed. Sci. 2002, 9, 534–541. [Google Scholar] [CrossRef]
- Bayona-Bafaluy, M.P.; Blits, B.; Battersby, B.J.; Shoubridge, E.A.; Moraes, C.T. Rapid Directional Shift of Mitochondrial DNA Heteroplasmy in Animal Tissues by a Mitochondrially Targeted Restriction Endonuclease. Proc. Natl. Acad. Sci. USA 2005, 102, 14392–14397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacman, S.R.; Williams, S.L.; Hernandez, D.; Moraes, C.T. Modulating MtDNA Heteroplasmy by Mitochondria-Targeted Restriction Endonucleases in a ‘Differential Multiple Cleavage-Site’ Model. Gene Ther. 2007, 14, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Bacman, S.R.; Williams, S.L.; Garcia, S.; Moraes, C.T. Organ-Specific Shifts in MtDNA Heteroplasmy Following Systemic Delivery of a Mitochondria-Targeted Restriction Endonuclease. Gene Ther. 2010, 17, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Bacman, S.R.; Williams, S.L.; Duan, D.; Moraes, C.T. Manipulation of MtDNA Heteroplasmy in All Striated Muscles of Newborn Mice by AAV9-Mediated Delivery of a Mitochondria-Targeted Restriction Endonuclease. Gene Ther. 2012, 19, 1101–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urnov, F.D.; Miller, J.C.; Lee, Y.-L.; Beausejour, C.M.; Rock, J.M.; Augustus, S.; Jamieson, A.C.; Porteus, M.H.; Gregory, P.D.; Holmes, M.C. Highly Efficient Endogenous Human Gene Correction Using Designed Zinc-Finger Nucleases. Nature 2005, 435, 646–651. [Google Scholar] [CrossRef]
- Hockemeyer, D.; Wang, H.; Kiani, S.; Lai, C.S.; Gao, Q.; Cassady, J.P.; Cost, G.J.; Zhang, L.; Santiago, Y.; Miller, J.C.; et al. Genetic Engineering of Human Pluripotent Cells Using TALE Nucleases. Nat. Biotechnol. 2011, 29, 731–734. [Google Scholar] [CrossRef] [Green Version]
- Minczuk, M.; Papworth, M.A.; Kolasinska, P.; Murphy, M.P.; Klug, A. Sequence-Specific Modification of Mitochondrial DNA Using a Chimeric Zinc Finger Methylase. Proc. Natl. Acad. Sci. USA 2006, 103, 19689–19694. [Google Scholar] [CrossRef] [Green Version]
- Minczuk, M.; Papworth, M.A.; Miller, J.C.; Murphy, M.P.; Klug, A. Development of a Single-Chain, Quasi-Dimeric Zinc-Finger Nuclease for the Selective Degradation of Mutated Human Mitochondrial DNA. Nucleic Acids Res. 2008, 36, 3926–3938. [Google Scholar] [CrossRef] [Green Version]
- Gammage, P.A.; Rorbach, J.; Vincent, A.I.; Rebar, E.J.; Minczuk, M. Mitochondrially Targeted ZFN s for Selective Degradation of Pathogenic Mitochondrial Genomes Bearing Large-scale Deletions or Point Mutations. EMBO Mol. Med. 2014, 6, 458–466. [Google Scholar] [CrossRef]
- Bacman, S.R.; Williams, S.L.; Pinto, M.; Peralta, S.; Moraes, C.T. Specific Elimination of Mutant Mitochondrial Genomes in Patient-Derived Cells by MitoTALENs. Nat. Med. 2013, 19, 1111–1113. [Google Scholar] [CrossRef]
- Kauppila, J.H.K.; Baines, H.L.; Bratic, A.; Simard, M.-L.; Freyer, C.; Mourier, A.; Stamp, C.; Filograna, R.; Larsson, N.-G.; Greaves, L.C.; et al. A Phenotype-Driven Approach to Generate Mouse Models with Pathogenic MtDNA Mutations Causing Mitochondrial Disease. Cell Rep. 2016, 16, 2980–2990. [Google Scholar] [CrossRef] [Green Version]
- Gammage, P.A.; Viscomi, C.; Simard, M.-L.; Costa, A.S.H.; Gaude, E.; Powell, C.A.; Van Haute, L.; McCann, B.J.; Rebelo-Guiomar, P.; Cerutti, R.; et al. Genome Editing in Mitochondria Corrects a Pathogenic MtDNA Mutation in Vivo. Nat. Med. 2018, 24, 1691–1695. [Google Scholar] [CrossRef] [PubMed]
- Bacman, S.R.; Kauppila, J.H.K.; Pereira, C.V.; Nissanka, N.; Miranda, M.; Pinto, M.; Williams, S.L.; Larsson, N.-G.; Stewart, J.B.; Moraes, C.T. MitoTALEN Reduces Mutant MtDNA Load and Restores TRNAAla Levels in a Mouse Model of Heteroplasmic MtDNA Mutation. Nat. Med. 2018, 24, 1696–1700. [Google Scholar] [CrossRef] [PubMed]
- Zekonyte, U.; Bacman, S.R.; Smith, J.; Shoop, W.; Pereira, C.V.; Tomberlin, G.; Stewart, J.; Jantz, D.; Moraes, C.T. Mitochondrial Targeted Meganuclease as a Platform to Eliminate Mutant MtDNA in Vivo. Nat. Commun. 2021, 12, 3210. [Google Scholar] [CrossRef] [PubMed]
- Gammage, P.A.; Moraes, C.T.; Minczuk, M. Mitochondrial Genome Engineering: The Revolution May Not Be CRISPR-Ized. Trends Genet. 2018, 34, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Mok, B.Y.; de Moraes, M.H.; Zeng, J.; Bosch, D.E.; Kotrys, A.V.; Raguram, A.; Hsu, F.; Radey, M.C.; Peterson, S.B.; Mootha, V.K.; et al. A Bacterial Cytidine Deaminase Toxin Enables CRISPR-Free Mitochondrial Base Editing. Nature 2020, 583, 631–637. [Google Scholar] [CrossRef]
- Lee, H.; Lee, S.; Baek, G.; Kim, A.; Kang, B.-C.; Seo, H.; Kim, J.-S. Mitochondrial DNA Editing in Mice with DddA-TALE Fusion Deaminases. Nat. Commun. 2021, 12, 1190. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhang, X.; Chen, X.; Sun, H.; Dai, Y.; Wang, J.; Qian, X.; Tan, L.; Lou, X.; Shen, B. Precision Modeling of Mitochondrial Diseases in Zebrafish via DdCBE-Mediated MtDNA Base Editing. Cell Discov. 2021, 7, 78. [Google Scholar] [CrossRef]
- Silva-Pinheiro, P.; Nash, P.A.; Van Haute, L.; Mutti, C.D.; Turner, K.; Minczuk, M. In Vivo Mitochondrial Base Editing via Adeno-Associated Viral Delivery to Mouse Post-Mitotic Tissue. Nat. Commun. 2022, 13, 750. [Google Scholar] [CrossRef]
- Wei, Y.; Li, Z.; Xu, K.; Feng, H.; Xie, L.; Li, D.; Zuo, Z.; Zhang, M.; Xu, C.; Yang, H.; et al. Mitochondrial Base Editor DdCBE Causes Substantial DNA Off-Target Editing in Nuclear Genome of Embryos. Cell Discov. 2022, 8, 27. [Google Scholar] [CrossRef]
- Cho, S.-I.; Lee, S.; Mok, Y.G.; Lim, K.; Lee, J.; Lee, J.M.; Chung, E.; Kim, J.-S. Targeted A-to-G Base Editing in Human Mitochondrial DNA with Programmable Deaminases. Cell 2022, 185, 1764–1776.e12. [Google Scholar] [CrossRef]
- Carrabba, G.; Macchini, F.; Fabietti, I.; Schisano, L.; Meccariello, G.; Campanella, R.; Bertani, G.; Locatelli, M.; Boito, S.; Porro, G.A.; et al. Minimally Invasive Fetal Surgery for Myelomeningocele: Preliminary Report from a Single Center. Neurosurg. Focus 2019, 47, E12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, M.E.; MacKenzie, T.C. Prenatal Gene Therapy. Clin. Obstet. Gynecol. 2021, 64, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, D.; Dykstra, W.; Le, S.; Zink, A.; Prigione, A. Mitochondria in Neurogenesis: Implications for Mitochondrial Diseases. Stem Cells 2021, 39, 1289–1297. [Google Scholar] [CrossRef]
- Inak, G.; Rybak-Wolf, A.; Lisowski, P.; Pentimalli, T.M.; Jüttner, R.; Glažar, P.; Uppal, K.; Bottani, E.; Brunetti, D.; Secker, C.; et al. Defective Metabolic Programming Impairs Early Neuronal Morphogenesis in Neural Cultures and an Organoid Model of Leigh Syndrome. Nat. Commun. 2021, 12, 1929. [Google Scholar] [CrossRef] [PubMed]
- Quadalti, C.; Brunetti, D.; Lagutina, I.; Duchi, R.; Perota, A.; Lazzari, G.; Cerutti, R.; Di Meo, I.; Johnson, M.; Bottani, E.; et al. SURF1 Knockout Cloned Pigs: Early Onset of a Severe Lethal Phenotype. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2018, 1864, 2131–2142. [Google Scholar] [CrossRef]
- Massaro, G.; Mattar, C.N.Z.; Wong, A.M.S.; Sirka, E.; Buckley, S.M.K.; Herbert, B.R.; Karlsson, S.; Perocheau, D.P.; Burke, D.; Heales, S.; et al. Fetal Gene Therapy for Neurodegenerative Disease of Infants. Nat. Med. 2018, 24, 1317–1323. [Google Scholar] [CrossRef]
- Wolf, D.P.; Mitalipov, P.A.; Mitalipov, S.M. Principles of and Strategies for Germline Gene Therapy. Nat. Med. 2019, 25, 890–897. [Google Scholar] [CrossRef]
- Cyranoski, D. First CRISPR Babies: Six Questions That Remain. Nature 2018, d41586-018-07607–3. [Google Scholar] [CrossRef]
- Ma, H.; O’Neil, R.C.; Marti Gutierrez, N.; Hariharan, M.; Zhang, Z.Z.; He, Y.; Cinnioglu, C.; Kayali, R.; Kang, E.; Lee, Y.; et al. Functional Human Oocytes Generated by Transfer of Polar Body Genomes. Cell Stem Cell 2017, 20, 112–119. [Google Scholar] [CrossRef] [Green Version]
- Tachibana, M.; Sparman, M.; Sritanaudomchai, H.; Ma, H.; Clepper, L.; Woodward, J.; Li, Y.; Ramsey, C.; Kolotushkina, O.; Mitalipov, S. Mitochondrial Gene Replacement in Primate Offspring and Embryonic Stem Cells. Nature 2009, 461, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Craven, L.; Tuppen, H.A.; Greggains, G.D.; Harbottle, S.J.; Murphy, J.L.; Cree, L.M.; Murdoch, A.P.; Chinnery, P.F.; Taylor, R.W.; Lightowlers, R.N.; et al. Pronuclear Transfer in Human Embryos to Prevent Transmission of Mitochondrial DNA Disease. Nature 2010, 465, 82–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damase, T.R.; Sukhovershin, R.; Boada, C.; Taraballi, F.; Pettigrew, R.I.; Cooke, J.P. The Limitless Future of RNA Therapeutics. Front. Bioeng. Biotechnol. 2021, 9, 628137. [Google Scholar] [CrossRef] [PubMed]
- Garone, C.; Viscomi, C. Towards a Therapy for Mitochondrial Disease: An Update. Biochem. Soc. Trans. 2018, 46, 1247–1261. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Wang, J.; Bonamy, G.M.C.; Meeusen, S.; Brusch, R.G.; Turk, C.; Yang, P.; Schultz, P.G. A Small Molecule Promotes Mitochondrial Fusion in Mammalian Cells. Angew. Chem. Int. Ed. 2012, 51, 9302–9305. [Google Scholar] [CrossRef] [PubMed]
- Jo, A.; Ham, S.; Lee, G.H.; Lee, Y.-I.; Kim, S.; Lee, Y.-S.; Shin, J.-H.; Lee, Y. Efficient Mitochondrial Genome Editing by CRISPR/Cas9. BioMed Res. Int. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valsecchi, M.C. Rare Diseases the next Target for MRNA Therapies. Nat. Italy 2021, d43978-021-00058–x. [Google Scholar] [CrossRef]
- Almási, T.; Guey, L.T.; Lukacs, C.; Csetneki, K.; Vokó, Z.; Zelei, T. Systematic Literature Review and Meta-Analysis on the Epidemiology of Methylmalonic Acidemia (MMA) with a Focus on MMA Caused by Methylmalonyl-CoA Mutase (Mut) Deficiency. Orphanet J. Rare Dis. 2019, 14, 84. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Donfrancesco, A.; Massaro, G.; Di Meo, I.; Tiranti, V.; Bottani, E.; Brunetti, D. Gene Therapy for Mitochondrial Diseases: Current Status and Future Perspective. Pharmaceutics 2022, 14, 1287. https://doi.org/10.3390/pharmaceutics14061287
Di Donfrancesco A, Massaro G, Di Meo I, Tiranti V, Bottani E, Brunetti D. Gene Therapy for Mitochondrial Diseases: Current Status and Future Perspective. Pharmaceutics. 2022; 14(6):1287. https://doi.org/10.3390/pharmaceutics14061287
Chicago/Turabian StyleDi Donfrancesco, Alessia, Giulia Massaro, Ivano Di Meo, Valeria Tiranti, Emanuela Bottani, and Dario Brunetti. 2022. "Gene Therapy for Mitochondrial Diseases: Current Status and Future Perspective" Pharmaceutics 14, no. 6: 1287. https://doi.org/10.3390/pharmaceutics14061287
APA StyleDi Donfrancesco, A., Massaro, G., Di Meo, I., Tiranti, V., Bottani, E., & Brunetti, D. (2022). Gene Therapy for Mitochondrial Diseases: Current Status and Future Perspective. Pharmaceutics, 14(6), 1287. https://doi.org/10.3390/pharmaceutics14061287