
Assessing Drug-Induced Mitochondrial Toxicity in Cardiomyocytes: Implications for Preclinical Cardiac Safety Evaluation

Abstract
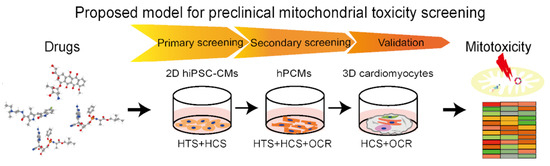
:
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Medicinal Product | Class | Mechanism of Action | Launch Date | Year Withdrawn | Side Effects on Cardiac Function | Mitohondrial Toxicity |
|---|---|---|---|---|---|---|
| Amfepramone | Psychostimulant | Norepinephrine-releasing agent | 1957 | 1975 | - | Unknown |
| Benfluorex | Psychostimulant, anorectic, and hypolipidemic | Blocking of 5-HT2B | 1976 | 2009 | Valvular heart disease | Decrease in CPT I expression [46] |
| Emetine (ipecac syrup) | Emetic | Stimulation of the CTZ, local irritation | 1912 | 1982 | - | Unknown |
| Mephenesin | Muscle relaxant | Spinal reflex inhibition | 1948 | 1976 | - | Unknown |
| Rofecoxib | NSAID | COX-2 inhibitor | 1999 | 2004 | MI, cardiovascular thrombotic events, sudden death | Unknown |
| Adenosine phosphate | Antiarrhythmic | Direct nodal inhibition | 1930 | 1973 | - | Unknown |
| Alphacetylmethadol | Analgesic | OP1 receptor agonist | 1993 | 2003 | - | Unknown |
| Bepridil (Bepridil Hydrochlonde) | Antiarrhythmic, antianginal | Calcium channel blockers | 1981 | 2004 | Prolonged QT, TdP | Unknown |
| Budipine | Antiparkinsonian | Muscarinic and NMDA receptor antagonist | 1979 | 2000 | - | Unknown |
| Cliobutinol | Antitussive | Unclear | 1961 | 2007 | - | Unknown |
| Dofetilide | Antiarrhythmic | Inhibition of KCNH2, KCNK2, KCNJ12 | 1999 | 2004 | QT prolongation, TdP | Unknown |
| Dolansetron | Propulsive | 5-HT3 receptor antagonist | 1997 | 2011 | - | Unknown |
| Encainide | Antiarrhythmic | Na channel blocker | 1985 | 1991 | QT prolongation, TdP | Unknown |
| Grepafloxacin (Grepafloxacin Hydrochloride) | Antimicrobial | Inhibition of DNA gyrase | 1997 | 1999 | QT prolongation | Unknown |
| Indoramin | Vasodilator | Alpha-1 adrenoceptor antagonist | 1981 | 2011 | - | Unknown |
| Isoprenaline | Cardiac stimulant | Non-selective beta-adrenergic agonist | 1949 | 1992 | - | mPTP opening [47] |
| Inhibition of OXPHOS [48] | ||||||
| Levacetylmethadol | Antidote | Mu-opioid receptor agonist, nicotinic acetylcholine receptor antagonist | 1995 | 2001 | - | Unknown |
| Nifedipine (10 mg) | Antihypertensive, antiemetic | Calcium channel blockers | 1975 | 1996 | Hypertension, angina, MI, CHF | Inhibition of ATP synthase [48] |
| Orciprenaline (metaprotenerol) | Bronchodilator | β2 adrenoceptor agonist | 1961 | 2009 | Tachycardia, palpitations | Unknown |
| Pergolide Mesylate | Anti-parkinsonian | Dopamine receptor agonist | 2002 | 2007 | Valvular heart disease | Unknown |
| Rosiglitazone | Hypoglycemic | Gluconeogenesis decrease | 1999 | 2011 | CHF, MI | Inhibition of ETC [48] |
| Increase in mitochondrial oxidative stress, impairment of mitochondrial bioenergetics [13] | ||||||
| Inhibition of complex I; uncoupling of OXPHOS [13] | ||||||
| Sibutramine (Sibutramine Hydrochlonde Hydrate) | Psychostimulant | Serotonin-norepinephrine reuptake inhibitor | 2001 | 2002 | Myocardial infarction | Increase in ROS formation [49] |
| Technetium (99mTc) fanolesomab | Radiography | Radioisotope | 2004 | 2005 | Cardiopulmonary arrest | Unknown |
| Tegaserod (Tegaserod Maleate) | Antispasmodic | 5-HT4 receptor agonist | 2002 | 2007 | HF, ischemia | Unknown |
| Terodiline | Antispasmodic | Calcium channel blockade, blocks cholinergic receptor | 1965 | 1991 | Ventricular tachycardia, cardiac death | Unknown |
| Sertindole | Antipsychotic | 5HT and D2 receptor antagonist/blocking of DRD2,HTR2A, HTR2C, HTR6 | 1996 | 1998 | QT prolongation, TdP, sudden cardiac death | Unknown |
| Cloforex | Psychostimulant | Similar to amphetamine | 1965 | 1967 | - | Unknown |
| Astemizole | Antihistamine | H1-receptor antagonist, inhibition of KCNH2 | 1977 | 1987 | long QT syndrome, TdP | Unknown |
| Cisapride monohydrate | Prokinetic agent | 5-HT4 receptor agonist; inhibition of KCNH2 | 1993 | 2000 | Ventricular arrhythmia, QT prolongation, TdP, cardiac arrest | Unknown |
| Tranylcypromine | Antidepressant | MAOI | 1961 | 1964 | - | Unknown |
| Bromocriptine mesylate | Anti-lactation | D2 and D3 agonist | 1976 | 1989 | - | Swollen mitochondria [50] |
| Domperidone (injectable) | Propulsive | Dopamine receptor antagonist | 1979 | 1985 | - | Unknown |
| Mepazine | Antiepileptic | Unclear | 1955 | 1970 | - | Unknown |
| Clozapine | Antipsychotic | Blocking of DRD2, HTR2A, DRD1, DRD3, DRD4, HTR1A, HTR1B, HTR1D, HTR1E, HTR2C, HTR3A, HTR6, HTR7, HRH1, HRH4, ADRA1A, ADRA1B, ADRA2A, ADRA2B, ADRA2C, CHRM1, CHRM2, CHRM3, CHRM4, CHRM5 | 1972 | 1975 | Cardiomyopathy, MI, myocarditis, arrhythmia, Prolonged QT, TdP, cardiomyopathy | Inhibition of the ETC [51] |
| Increase in ROS formation, GSH depletion, mitochondrial dysfunction, and swelling [52] | ||||||
| Vincamine | Nootropic | Unclear | 1955 | 1980 | - | Unknown |
| Lysine amidotriazoate | Radiography | - | 1975 | 1995 | - | Unknown |
| Terfenadine | Antihistamine | H1-receptor antagonist | 1985 | 1997 | QT prolongation, TdP | Increase in mtROS formation [53] |
| MMP collapse [54] | ||||||
| Naftidrofuryl oxalate (IV) | Vasodilator | 5HT2 receptor antagonist | 1974 | 1992 | - | Unknown |
| Cobalt | Hematinic | As cobalamin | 1951 | 1967 | - | Interruption of TCA and interference with the MRC enzymes [54] |
| MMP collapse [55] | ||||||
| Chloroform (trichloromethane) | Anesthetic | Depression of the respiratory centres | 1847 | 1976 | - | MMP collapse [56] |
| Megamitochondria [57] | ||||||
| Dithiazanine iodide | Antihelminth | Interruption of glucose uptake in cells | 1959 | 1964 | Prolonged QT, TdP | Unknown |
| Epinephrine (topical) | Anesthetic | Vasoconstriction | 1899 | 2004 | - | Unknown |
| Methylhexanamine (DMAA) | Nasal decongestant | Norepinephrine and dopamine transporter blockade | 1948 | 1983 | - | Unknown |
| Dexfenfluramine | Psychostimulant | Serotonin receptor agonist | 1995 | 1997 | Valvular heart disease, cardiac fibrosis | Unknown |
| Fenfluramine | Psychostimulant | Serotonin receptor antagonist | 1973 | 1997 | valvular heart disease | Mitochondrial fragmentation [58] |
| Parecoxib | Analgesic | COX-2 inhibitor | 2002 | 2005 | - | - |
| Prenylamine | Antianginal | Calcium channel blocker | 1960 | 1989 | QT prolongation, sudden cardiac death, ventricular tachycardia, TdP | Inhibition of FAO [59] |
| Probucol | Antioxidant | Inductor of LDL catabolism | 1980 | 1989 | QT prolongation, arrhythmias | Unknown |
| Droperidol | Antipsychotic | Dopamine 2 receptor antagonist | 1970 | 2001 | - | Unknown |
| Valdecoxib | NSAID | COX-2 inhibitor | 2001 | 2005 | Cardiomyopathy, CHF, hypertension, angina, arrhythmia | Inhibition of OXPHOS, mPTP opening [16] |
| Celecoxib (Onsenal) | NSAID | COX-2 inhibitor | 2003 | 2011 | - | Decrease in mitochondrial complex IV activity and induces oxidative stress [14] |
| Increase in ROS formation, MMP collapse, mitochondrial swelling, ATP depletion [60] | ||||||
| Suppression of mitochondrial function [61] | ||||||
| Bismuth salts | Antidyspepsia | Unclear. Forms insoluble complexes | 1875 | 1978 | - | Unknown |
| Levarterenol | Vasopressor | L-norepinephrine analogue | 1904 | 1973 | - | Unknown |
| Pipradrol | Psychostimulant | Norepinephrine-dopamine reuptake inhibitor | 1953 | 1982 | - | Unknown |
| Pseudoephedrine | Sympathomimetic | Direct action on adrenergic receptors | 1959 | 2008 | - | Unknown |
| Gallopamil | Antiarrhythmic | Calcium channel blockers | 1983 | 2001 | - | Decrease in mitochondrial biogenesis and mass [62] |
| Chlorphentermine | Psychostimulant | TAAR1 agonist, blocking of 5-HTs | 1962 | 1969 | Pulmonary heart disease | Inhibition of OXPHOS, uncoupling of OXPHOS [63] |
| Thioridazine | Antipsychotic | 5HT2 receptor antagonist | 1959 | 2000 | QT prolongation, TdP, sudden cardiac death | mPTP opening [64] |
| MMP collapse [65] | ||||||
| Buflomedil | Vasodilator | A-adrenergic blockade | 1970 | 2006 | QT prolongation, cardiac arrest | Unknown |
| Ponatinib Hydrochloride | Antineoplastic | Multi-target kinase inhibitor | 2012 | 2013 | - | Impairment of respiratory chain, increase in ROS formation, MMP collapse, mitochondrial fission [66] |
| Levomethadyl acetate | Analgesic (central nervous system agents) | Activation of OPRM1 | 1993 | 2002 | QT prolongation, TdP | Unknown |
| Mesoridazine Besylate | Antipsychotic | 1970 | - | - | Unknown | |
| Clobutinol Hydrochloride | Antitussive | Inhibition of GABA receptors | 1961 | 2007 | QT prolongation | Unknown |
| Phentermine | Central nervous system agents | Inhibition of SLC6A2, SLC6A3, SLC6A4; blockingof MAOA, MAOB | 1959 | 1997 | Valvular heart disease | Unknown |
| Mibefradil | Antihypertensive | Calcium channel blockers | 1997 | 1998 | QT prolongation | Unknown |
| Sparfloxacin | Antibiotics | Inhibits DNA gyrase | 1997 | 2001 | QT prolongation | MMP collapse [67] |
| Etoricoxib | Anti-inflammatory agents | Inhibition of COX-2 | 2002 | 2007 | thrombotic events | Inhibition of OXPHOS [16] |
| Propoxyphene | Central nervous system agents | Activation of OP1, OP2, OP3 | 1957 | 2010 | QT prolongation, TdP | Unknown |
| Lidoflazine | Cardiovascular agents | Blocking of calcium channels | 1973 | 1989 | QT prolongation | Unknown |
2. Main Properties of Mitochondria and Drug-Induced Mitochondrial Toxicity in Cardiomyocytes
2.1. Morphology, Classification, and Structural Features of Mitochondria
2.2. Substrate Catabolism and OXPHOS
2.3. Mitochondrial ROS (mtROS)
2.4. Replication, Translation, and Transcription of mtDNA
2.5. Mitochondrial Membrane Potential (MMP) and mPTP
| Modules | Alterations | Pharmacology | Drugs | Clinical Manifestations | Cmax | Models | Dose | Time | References |
|---|---|---|---|---|---|---|---|---|---|
| Carrier | Downregulation of CPT I expression | Alkylating agent | Cyclophosphamide | HMC, CMP | 143 μM | Male Wistar rats (IP) | 200 mg/kg | 10 d | [189] |
| Carrier | Downregulation of CPT I expression | Anesthesia | Propofol | HF, arrhythmia | 30.13 μM | HiPSC-CMs | 10 µg/mL | 48 h | [163] |
| Carrier | Downregulation of CPT I expression | TKIs | Sunitinib | Decreased LVEF, QT prolongation, TdP, hypertension, HF, CMP | 0.25 μM | Rats (oral) | 25 mg/kg/d | 28 d | [173] |
| Carrier | Inhibition of CPT1 activity | Anti-arrhythmic drug | Dronedarone | AF, HF | 0.15–0.26 μM | Isolated rat heart mitochondria | IC50 = 40 µM | 20 min | [139] |
| Carrier | loss of carnitine | Co-catalyst | Pivalic acid | CMP | [232] | ||||
| Carrier | Inhibition of ANT | NSAIDs | Diclofenac | Hypertension, arrhythmias | 7.9 µM | Submitochondrial particles | 314 nM/mg protein diminished 76% | [142] | |
| Nimesulide | Submitochondrial particles | 259 nM/mg protein diminished 60% | [142] | ||||||
| Carrier | Inhibition of ANT | NRTIs | Zidovudine | CMP | 4 μM | [233] | |||
| mtDNA | Inhibition of mitochondrial DNA polymerase | NRTIs | Zidovudine | CMP | 4 μM | Cardiac DNA pol-γ | 1 µM | [234] | |
| mtDNA | Inhibition of topoisomerase II | Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP | 15.3 μM | - | - | - | [235] |
| Daunorubicin | CMP, MI, CHF, VA, pericarditis, myocarditis | 89 μM | - | - | - | [207] | |||
| Idarubicin | CMP, MI, CHF, VA, decreased LVEF | 23.22 μM | - | - | - | [207] | |||
| mtDNA | Inhibition of topoisomerase II | Chemotherapeutic agents | Mitoxantrone | CHF, CMP, decreased LVEF, arrhythmia | 3.3 μM | - | - | - | [215] |
| mtDNA | mtDNA content decreasing | Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP | 15.3 μM | Male Wistar rats (IV) | 1 mg/kg/w | 7 w (started at 11 w, observed at 48 w) | [96] |
| mtDNA | mtDNA content decreasing | TKIs | Regorafenib | MI; hypertension | 8.08 μM | H9c2 | 5 μM | 72 h | [90] |
2.6. Mitochondrial Carriers
2.7. Mitochondrial Quality Control (MQC)
2.8. Other Mitotoxicants and Their Targets
3. Limitations of Current Preclinical Models for Assessing Cardiotoxicity
3.1. Limitations in the Current Workflow of Cardiac Safety Testing
3.2. In Vitro Models for Cardiac Toxicity Assessment
3.2.1. H9c2 Cardiomyoblasts
3.2.2. Stem-Cell-Derived Cardiomyocytes
3.2.3. hPCMs
3.2.4. 3D Cardiomyocyte Models
4. Proposed Preclinical Model of Cardiomyocytes for Assessment of Drug-Induced Mitochondria Toxicity
4.1. In Vitro Cell Culture for Cardiotoxicity Assays
4.2. Mitochondrial Target as Readouts in Cardiotoxicity Assays
4.2.1. Mitochondrial Morphology, Structure
4.2.2. Oxygen Consumption Rate (OCR)
4.2.3. ATP
4.2.4. Redox Homeostasis
4.2.5. MMP
4.3. High-Throughput Assessment of Mitochondrial Toxicity
4.4. Proposed Integrated Assays for Drug-Induced Mitochondria Toxicity of Cardiomyocytes
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stummann, T.C.; Beilmann, M.; Duker, G.; Dumotier, B.; Fredriksson, J.M.; Jones, R.L.; Hasiwa, M.; Kang, Y.J.; Mandenius, C.F.; Meyer, T.; et al. Report and recommendations of the workshop of the European Centre for the Validation of Alternative Methods for Drug-Induced Cardiotoxicity. Cardiovasc. Toxicol. 2009, 9, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Albakri, A. Drugs-related cardiomyopathy: A systematic review and pooled analysis of pathophysiology, diagnosis and clinical management. Intern. Med. Care 2019, 3, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Savoji, H.; Mohammadi, M.H.; Rafatian, N.; Toroghi, M.K.; Wang, E.Y.; Zhao, Y.; Korolj, A.; Ahadian, S.; Radisic, M. Cardiovascular disease models: A game changing paradigm in drug discovery and screening. Biomaterials 2019, 198, 3–26. [Google Scholar] [CrossRef]
- Potter, E.; Marwick, T.H. Assessment of Left Ventricular Function by Echocardiography: The Case for Routinely Adding Global Longitudinal Strain to Ejection Fraction. JACC Cardiovasc. Imaging 2018, 11, 260–274. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Yuan, C.; Wang, L.; Chen, R.; Li, X.; Zhang, Y.; Liu, C.; Liu, X.; Liang, W.; Xing, Y. The Beneficial Effects of Saffron Extract on Potential Oxidative Stress in Cardiovascular Diseases. Oxid. Med. Cell. Longev. 2021, 2021, 6699821. [Google Scholar] [CrossRef]
- Cook, D.; Brown, D.; Alexander, R.; March, R.; Morgan, P.; Satterthwaite, G.; Pangalos, M.N. Lessons learned from the fate of AstraZeneca’s drug pipeline: A five-dimensional framework. Nat. Rev. Drug Discov. 2014, 13, 419–431. [Google Scholar] [CrossRef]
- Kuhn, M.; Letunic, I.; Jensen, L.J.; Bork, P. The SIDER database of drugs and side effects. Nucleic Acids Res. 2016, 44, D1075–D1079. [Google Scholar] [CrossRef]
- Magdy, T.; Schuldt, A.J.T.; Wu, J.C.; Bernstein, D.; Burridge, P.W. Human Induced Pluripotent Stem Cell (hiPSC)-Derived Cells to Assess Drug Cardiotoxicity: Opportunities and Problems. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 83–103. [Google Scholar] [CrossRef]
- ICH, S.B. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Steering Committee. The Non Clinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT prolongation) by Human Pharmaceuticals S7B. Available online: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S7B/Step4/S7B_Guideline.pdf (accessed on 12 May 2005).
- International Conference on Harmonisation. Guidance for Industry: E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs. Available online: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E14/E14_Guideline.pdf (accessed on 1 November 2005).
- Park, E.; Willard, J.; Bi, D.; Fiszman, M.; Kozeli, D.; Koerner, J. The impact of drug-related QT prolongation on FDA regulatory decisions. Int. J. Cardiol. 2013, 168, 4975–4976. [Google Scholar] [CrossRef]
- Geelen, M.J. Mechanisms responsible for the inhibitory effects of benfluorex on hepatic intermediary metabolism. Biochem. Pharmacol. 1983, 32, 1765–1772. [Google Scholar] [CrossRef]
- He, H.; Tao, H.; Xiong, H.; Duan, S.Z.; McGowan, F.X., Jr.; Mortensen, R.M.; Balschi, J.A. Rosiglitazone causes cardiotoxicity via peroxisome proliferator-activated receptor gamma-independent mitochondrial oxidative stress in mouse hearts. Toxicol. Sci. 2014, 138, 468–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atashbar, S.; Jamali, Z.; Khezri, S.; Salimi, A. Celecoxib decreases mitochondrial complex IV activity and induces oxidative stress in isolated rat heart mitochondria: An analysis for its cardiotoxic adverse effect. J. Biochem. Mol. Toxicol. 2021, 36, e22934. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, M.E.; Chun, B.; Moya, R.; Saucerman, J.J. Computational model of cardiomyocyte apoptosis identifies mechanisms of tyrosine kinase inhibitor-induced cardiotoxicity. J. Mol. Cell. Cardiol. 2021, 155, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Syed, M.; Skonberg, C.; Hansen, S.H. Mitochondrial toxicity of selective COX-2 inhibitors via inhibition of oxidative phosphorylation (ATP synthesis) in rat liver mitochondria. Toxicol. In Vitro 2016, 32, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.P.; Zheng, M. Mitochondrial dynamics and inter-mitochondrial communication in the heart. Arch. Biochem. Biophys. 2019, 663, 214–219. [Google Scholar] [CrossRef]
- Bround, M.J.; Wambolt, R.; Luciani, D.S.; Kulpa, J.E.; Rodrigues, B.; Brownsey, R.W.; Allard, M.F.; Johnson, J.D. Cardiomyocyte ATP production, metabolic flexibility, and survival require calcium flux through cardiac ryanodine receptors in vivo. J. Biol. Chem. 2013, 288, 18975–18986. [Google Scholar] [CrossRef] [Green Version]
- Barry, S.P.; Townsend, P.A. What Causes a Broken Heart—Molecular Insights into Heart Failure. Int. Rev. Cell Mol. Biol. 2010, 284, 113–179. [Google Scholar]
- Gintant, G.; Burridge, P.; Gepstein, L.; Harding, S.; Herron, T.; Hong, C.; Jalife, J.; Wu, J.C. Use of Human Induced Pluripotent Stem Cell–Derived Cardiomyocytes in Preclinical Cancer Drug Cardiotoxicity Testing: A Scientific Statement From the American Heart Association. Circ. Res. 2019, 125, e75–e92. [Google Scholar] [CrossRef]
- Varga, Z.V.; Ferdinandy, P.; Liaudet, L.; Pacher, P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1453–H1467. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Liu, F.; Li, J. Mitochondrial Sirtuins and Doxorubicin-induced Cardiotoxicity. Cardiovasc. Toxicol. 2021, 21, 179–191. [Google Scholar] [CrossRef]
- Wallace, K.B.; Sardão, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Yin, Y.; Shen, H. Advances in Cardiotoxicity Induced by Altered Mitochondrial Dynamics and Mitophagy. Front. Cardiovasc. Med. 2021, 8, 739095. [Google Scholar] [CrossRef] [PubMed]
- Hantson, P. Mechanisms of toxic cardiomyopathy. Clin. Toxicol. 2019, 57, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanniah, G.; Kumar, S. Clozapine associated cardiotoxicity: Issues, challenges and way forward. Asian J. Psychiatr. 2020, 50, 101950. [Google Scholar] [CrossRef] [PubMed]
- Arangalage, D.; Pavon, A.G.; Hugelshofer, S.; Desgraz, B.; Tzimas, G.; Delyon, J.; Muller, O.; Obeid, M.; Ribi, C.; Michielin, O.; et al. Cardiotoxicity of immune checkpoint inhibitors used in cancer treatment. Rev. Med. Suisse 2020, 16, 1165–1168. [Google Scholar] [PubMed]
- Grivicich, I.; Regner, A.; da Rocha, A.B.; Grass, L.B.; Alves, P.A.; Kayser, G.B.; Schwartsmann, G.; Henriques, J.A. Irinotecan/5-fluorouracil combination induces alterations in mitochondrial membrane potential and caspases on colon cancer cell lines. Oncol. Res. 2005, 15, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wu, Y. Role of Mitophagy in Coronary Heart Disease: Targeting the Mitochondrial Dysfunction and Inflammatory Regulation. Front. Cardiovasc. Med. 2022, 9, 819454. [Google Scholar] [CrossRef]
- Tantawy, M.; Pamittan, F.G.; Singh, S.; Gong, Y. Epigenetic Changes Associated With Anthracycline-Induced Cardiotoxicity. Clin. Transl. Sci. 2021, 14, 36–46. [Google Scholar] [CrossRef]
- Ma, W.; Liu, M.; Liang, F.; Zhao, L.; Gao, C.; Jiang, X.; Zhang, X.; Zhan, H.; Hu, H.; Zhao, Z. Cardiotoxicity of sorafenib is mediated through elevation of ROS level and CaMKII activity and dysregulation of calcium homoeostasis. Basic Clin. Pharmacol. Toxicol. 2020, 126, 166–180. [Google Scholar] [CrossRef]
- Nishinaka, Y.; Sugiyama, S.; Yokota, M.; Saito, H.; Ozawa, T. The effects of a high dose of ascorbate on ischemia-reperfusion-induced mitochondrial dysfunction in canine hearts. Heart Vessel. 1992, 7, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, L.; Pratt, K.; Gavin, J. Endothelin-3-induced microvascular incompetence and mitochondrial damage in rat myocardium. Clin. Exp. Pharmacol. Physiol. 1992, 19, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Cheng, R.K.; Tian, R. Combat Doxorubicin Cardiotoxicity With the Power of Mitochondria Transfer. JACC CardioOncol. 2021, 3, 441–443. [Google Scholar] [CrossRef]
- Finsterer, J.; Ohnsorge, P. Influence of mitochondrion-toxic agents on the cardiovascular system. Regul. Toxicol. Pharmacol. 2013, 67, 434–445. [Google Scholar] [CrossRef]
- Szendrei, L.; Turoczi, T.; Kovacs, P.; Vecsernyes, M.; Das, D.K.; Tosaki, A. Mitochondrial gene expression and ventricular fibrillation in ischemic/reperfused nondiabetic and diabetic myocardium. Biochem. Pharmacol. 2002, 63, 543–552. [Google Scholar] [CrossRef]
- Aon, M.A.; Cortassa, S.; Akar, F.G.; O’Rourke, B. Mitochondrial criticality: A new concept at the turning point of life or death. Biochim. Biophys. Acta 2006, 1762, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Tosaki, A. ArrhythmoGenoPharmacoTherapy. Front. Pharmacol. 2020, 11, 616. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, W.; Biermans, G.; Callewaert, G.; Vereecke, J.; Opie, L.; Carmeliet, E. The effect of inhibition of mitochondrial energy metabolism on the transient inward current of isolated guinea-pig ventricular myocytes. J. Mol. Cell. Cardiol. 1988, 20, 181–185. [Google Scholar] [CrossRef]
- Yang, K.C.; Bonini, M.G.; Dudley, S.C., Jr. Mitochondria and arrhythmias. Free Radic. Biol. Med. 2014, 71, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Yang, R.; Yang, J.; Zhou, L. Mitochondrial Dysfunction-Associated Arrhythmogenic Substrates in Diabetes Mellitus. Front. Physiol. 2018, 9, 1670. [Google Scholar] [CrossRef] [Green Version]
- Yarmohammadi, F.; Rezaee, R.; Haye, A.W.; Karimi, G. Endoplasmic reticulum stress in doxorubicin-induced cardiotoxicity may be therapeutically targeted by natural and chemical compounds: A review. Pharmacol. Res. 2021, 164, 105383. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Choi, K.C. Effects of anticancer drugs on the cardiac mitochondrial toxicity and their underlying mechanisms for novel cardiac protective strategies. Life Sci. 2021, 277, 119607. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.J.; Zhang, J.; Xiao, M.; Wang, S.; Wang, B.J.; Guo, Y.; Tang, Y.; Gu, J. Molecular mechanisms of doxorubicin-induced cardiotoxicity: Novel roles of sirtuin 1-mediated signaling pathways. Cell. Mol. Life Sci. 2021, 78, 3105–3125. [Google Scholar] [CrossRef] [PubMed]
- Kohl, C.; Ravel, D.; Girard, J.; Pegorier, J.P. Effects of benfluorex on fatty acid and glucose metabolism in isolated rat hepatocytes: From metabolic fluxes to gene expression. Diabetes 2002, 51, 2363–2368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, M.J.; Khaliulin, I.; Hall, K.; Suleiman, M.S. Cardioprotection of Immature Heart by Simultaneous Activation of PKA and Epac: A Role for the Mitochondrial Permeability Transition Pore. Int. J. Mol. Sci. 2022, 23, 1720. [Google Scholar] [CrossRef]
- Krestinin, R.; Baburina, Y.; Odinokova, I.; Kruglov, A.; Fadeeva, I.; Zvyagina, A.; Sotnikova, L.; Krestinina, O. Isoproterenol-Induced Permeability Transition Pore-Related Dysfunction of Heart Mitochondria Is Attenuated by Astaxanthin. Biomedicines 2020, 8, 437. [Google Scholar] [CrossRef]
- Morikawa, Y.; Shibata, A.; Sasajima, Y.; Suenami, K.; Sato, K.; Takekoshi, Y.; Endo, S.; Ikari, A.; Matsunaga, T. Sibutramine facilitates apoptosis and contraction of aortic smooth muscle cells through elevating production of reactive oxygen species. Eur. J. Pharmacol. 2018, 841, 113–121. [Google Scholar] [CrossRef]
- Zhang, S.L.; Tang, H.B.; Hu, J.T.; Zang, Z.L.; Ding, X.; Li, S.; Yang, H. PGAM5-CypD pathway is involved in bromocriptine-induced RIP3/MLKL-dependent necroptosis of prolactinoma cells. Biomed. Pharm. 2019, 111, 638–648. [Google Scholar] [CrossRef]
- Lin, Y.T.; Lin, K.H.; Huang, C.J.; Wei, A.C. MitoTox: A comprehensive mitochondrial toxicity database. BMC Bioinform. 2021, 22, 369. [Google Scholar] [CrossRef]
- Hafez, A.A.; Jamali, Z.; Khezri, S.; Salimi, A. Thymoquinone reduces mitochondrial damage and death of cardiomyocytes induced by clozapine. Naunyn Schmiedebergs Arch. Pharm. 2021, 394, 1675–1684. [Google Scholar] [CrossRef]
- Nicolau-Galmes, F.; Asumendi, A.; Alonso-Tejerina, E.; Perez-Yarza, G.; Jangi, S.M.; Gardeazabal, J.; Arroyo-Berdugo, Y.; Careaga, J.M.; Diaz-Ramon, J.L.; Apraiz, A.; et al. Terfenadine induces apoptosis and autophagy in melanoma cells through ROS-dependent and -independent mechanisms. Apoptosis 2011, 16, 1253–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jangi, S.M.; Diaz-Perez, J.L.; Ochoa-Lizarralde, B.; Martin-Ruiz, I.; Asumendi, A.; Perez-Yarza, G.; Gardeazabal, J.; Diaz-Ramon, J.L.; Boyano, M.D. H1 histamine receptor antagonists induce genotoxic and caspase-2-dependent apoptosis in human melanoma cells. Carcinogenesis 2006, 27, 1787–1796. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.L.; Li, Z.Q.; Zhao, Y.J.; Zhao, S.M.; Zhu, L.; Li, T.; Fu, Y.; Li, H.J. Ginsenoside Rb1 protects cardiomyocytes against CoCl2-induced apoptosis in neonatal rats by inhibiting mitochondria permeability transition pore opening. Acta Pharmacol. Sin. 2010, 31, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Hartig, S.; Fries, S.; Balcarcel, R.R. Reduced mitochondrial membrane potential and metabolism correspond to acute chloroform toxicity of in vitro hepatocytes. J. Appl. Toxicol. 2005, 25, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Guastadisegni, C.; Balduzzi, M.; Mancuso, M.T.; Di Consiglio, E. Liver mitochondria alterations in chloroform-treated Sprague-Dawley rats. J. Toxicol. Environ. Health A 1999, 57, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Mu, W.; Zeifman, A.; Lofti, M.; Remillard, C.V.; Makino, A.; Perkins, D.L.; Garcia, J.G.; Yuan, J.X.; Zhang, W. Fenfluramine-induced gene dysregulation in human pulmonary artery smooth muscle and endothelial cells. Pulm. Circ. 2011, 1, 405–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, G.; Berlin, E.; Smith, J.T. Inhibition of mitochondrial palmitate oxidation by calmodulin antagonists. Int. J. Biochem. 1990, 22, 631–634. [Google Scholar] [CrossRef]
- Salimi, A.; Neshat, M.R.; Naserzadeh, P.; Pourahmad, J. Mitochondrial Permeability Transition Pore Sealing Agents and Antioxidants Protect Oxidative Stress and Mitochondrial Dysfunction Induced by Naproxen, Diclofenac and Celecoxib. Drug Res. 2019, 69, 598–605. [Google Scholar] [CrossRef]
- Tatematsu, Y.; Fujita, H.; Hayashi, H.; Yamamoto, A.; Tabata, A.; Nagamune, H.; Ohkura, K. Effects of the Nonsteroidal Anti-inflammatory Drug Celecoxib on Mitochondrial Function. Biol. Pharm. Bull. 2018, 41, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Beaufils, F.; Esteves, P.; Enaud, R.; Germande, O.; Celle, A.; Marthan, R.; Trian, T.; Fayon, M.; Berger, P. Mitochondria are involved in bronchial smooth muscle remodeling in severe preschool wheezers. J. Allergy Clin. Immunol. 2021, 148, 645–651.e11. [Google Scholar] [CrossRef]
- Zychlinski, L. Mitochondrial alterations in the brain of the rat caused by chlorphentermine. Neuropharmacology 1986, 25, 1111–1117. [Google Scholar] [CrossRef]
- Rana, P.; Aleo, M.D.; Gosink, M.; Will, Y. Evaluation of in Vitro Mitochondrial Toxicity Assays and Physicochemical Properties for Prediction of Organ Toxicity Using 228 Pharmaceutical Drugs. Chem. Res. Toxicol. 2019, 32, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Eftekhari, A.; Ahmadian, E.; Azarmi, Y.; Parvizpur, A.; Fard, J.K.; Eghbal, M.A. Mechanistic Approach for Thioridazine-Induced Hepatotoxicity and Potential Benefits of Melatonin and/or Coenzyme Q10 on Freshly Isolated Rat Hepatocytes. Iran J. Pharm. Res. 2018, 17, 1465–1475. [Google Scholar]
- Paech, F.; Mingard, C.; Grunig, D.; Abegg, V.F.; Bouitbir, J.; Krahenbuhl, S. Mechanisms of mitochondrial toxicity of the kinase inhibitors ponatinib, regorafenib and sorafenib in human hepatic HepG2 cells. Toxicology 2018, 395, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Dwivedi, A.; Ray, L.; Chopra, D.; Dubey, D.; Srivastva, A.K.; Kumari, S.; Yadav, R.K.; Amar, S.K.; Haldar, C.; et al. PLGA nanoformulation of sparfloxacin enhanced antibacterial activity with photoprotective potential under ambient UV-R exposure. Int. J. Pharm. 2018, 541, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C. The importance of drug discovery for treatment of cardiovascular diseases. Future Med. Chem. 2013, 5, 355–357. [Google Scholar] [CrossRef] [Green Version]
- Barth, E.; Stammler, G.; Speiser, B.; Schaper, J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J. Mol. Cell. Cardiol. 1992, 24, 669–681. [Google Scholar] [CrossRef]
- Croston, T.L.; Thapa, D.; Holden, A.A.; Tveter, K.J.; Lewis, S.E.; Shepherd, D.L.; Nichols, C.E.; Long, D.M.; Olfert, I.M.; Jagannathan, R.; et al. Functional deficiencies of subsarcolemmal mitochondria in the type 2 diabetic human heart. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H54–H65. [Google Scholar] [CrossRef] [Green Version]
- Dabkowski, E.R.; Williamson, C.L.; Bukowski, V.C.; Chapman, R.S.; Leonard, S.S.; Peer, C.J.; Callery, P.S.; Hollander, J.M. Diabetic cardiomyopathy-associated dysfunction in spatially distinct mitochondrial subpopulations. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H359–H369. [Google Scholar] [CrossRef] [Green Version]
- Lesnefsky, E.J.; Chen, Q.; Hoppel, C.L. Mitochondrial Metabolism in Aging Heart. Circ. Res. 2016, 118, 1593–1611. [Google Scholar] [CrossRef] [Green Version]
- Shimada, T.; Horita, K.; Murakami, M.; Ogura, R. Morphological studies of different mitochondrial populations in monkey myocardial cells. Cell. Tissue Res. 1984, 238, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Fernandez-Sanz, C.; Sheu, S.S. Regulation of mitochondrial bioenergetics by the non-canonical roles of mitochondrial dynamics proteins in the heart. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1991–2001. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Lochnit, G.; Schulz, R. Mitochondria “THE” target of myocardial conditioning. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1215–H1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, S.B.; Kalkhoran, S.B.; Hernandez-Resendiz, S.; Samangouei, P.; Ong, S.G.; Hausenloy, D.J. Mitochondrial-Shaping Proteins in Cardiac Health and Disease—The Long and the Short of It! Cardiovasc. Drugs Ther. 2017, 31, 87–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobs, S.; Stephan, T.; Ilgen, P.; Bruser, C. Light Microscopy of Mitochondria at the Nanoscale. Annu. Rev. Biophys. 2020, 49, 289–308. [Google Scholar] [CrossRef] [Green Version]
- Chipuk, J.E.; Mohammed, J.N.; Gelles, J.D.; Chen, Y. Mechanistic connections between mitochondrial biology and regulated cell death. Dev. Cell. 2021, 56, 1221–1233. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef]
- Vogel, F.; Bornhovd, C.; Neupert, W.; Reichert, A.S. Dynamic subcompartmentalization of the mitochondrial inner membrane. J. Cell. Biol. 2006, 175, 237–247. [Google Scholar] [CrossRef]
- Portella, D.C.N.; Rossi, E.A.; Paredes, B.D.; Bastos, T.M.; Meira, C.S.; Nonaka, C.V.K.; Silva, D.N.; Improta-Caria, A.; Moreira, D.R.M.; Leite, A.C.L.; et al. A Novel High-Content Screening-Based Method for Anti-Trypanosoma cruzi Drug Discovery Using Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cells Int. 2021, 2021, 2642807. [Google Scholar] [CrossRef]
- Punithavathi, V.R.; Shanmugapriya, K.; Prince, P.S. Protective effects of rutin on mitochondrial damage in isoproterenol-induced cardiotoxic rats: An in vivo and in vitro study. Cardiovasc. Toxicol. 2010, 10, 181–189. [Google Scholar] [CrossRef]
- Devika, P.T.; Stanely Mainzen Prince, P. (-)Epigallocatechin-gallate (EGCG) prevents mitochondrial damage in isoproterenol-induced cardiac toxicity in albino Wistar rats: A transmission electron microscopic and in vitro study. Pharmacol. Res. 2008, 57, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Wang, M.; Wang, R.Y.; Sun, X.; Du, Y.Y.; Ye, J.X.; Sun, G.B.; Sun, X.B. Salvianolic Acid A Ameliorates Arsenic Trioxide-Induced Cardiotoxicity Through Decreasing Cardiac Mitochondrial Injury and Promotes Its Anticancer Activity. Front. Pharmacol. 2018, 9, 487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandolini, L.; Antonosante, A.; Giorgio, C.; Bagnasco, M.; d’Angelo, M.; Castelli, V.; Benedetti, E.; Cimini, A.; Allegretti, M. NSAIDs-dependent adaption of the mitochondria-proteasome system in immortalized human cardiomyocytes. Sci. Rep. 2020, 10, 18337. [Google Scholar] [CrossRef] [PubMed]
- Kwok, M.; Lee, C.; Li, H.S.; Deng, R.; Tsoi, C.; Ding, Q.; Tsang, S.Y.; Leung, K.T.; Yan, B.P.; Poon, E.N. Remdesivir induces persistent mitochondrial and structural damage in human induced pluripotent stem cell derived cardiomyocytes. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef]
- Antonucci, S.; Di Sante, M.; Tonolo, F.; Pontarollo, L.; Scalcon, V.; Alanova, P.; Menabo, R.; Carpi, A.; Bindoli, A.; Rigobello, M.P.; et al. The Determining Role of Mitochondrial Reactive Oxygen Species Generation and Monoamine Oxidase Activity in Doxorubicin-Induced Cardiotoxicity. Antioxid. Redox. Signal 2021, 34, 531–550. [Google Scholar] [CrossRef]
- Jia, G.; Meng, Z.; Liu, C.; Ma, X.; Gao, J.; Liu, J.; Guo, R.; Yan, Z.; Christopher, T.; Lopez, B.; et al. Nicotine induces cardiac toxicity through blocking mitophagic clearance in young adult rat. Life Sci. 2020, 257, 118084. [Google Scholar] [CrossRef]
- French, K.J.; Coatney, R.W.; Renninger, J.P.; Hu, C.X.; Gales, T.L.; Zhao, S.; Storck, L.M.; Davis, C.B.; McSurdy-Freed, J.; Chen, E.; et al. Differences in effects on myocardium and mitochondria by angiogenic inhibitors suggest separate mechanisms of cardiotoxicity. Toxicol. Pathol. 2010, 38, 691–702. [Google Scholar] [CrossRef] [Green Version]
- Boran, T.; Akyildiz, A.G.; Jannuzzi, A.T.; Alpertunga, B. Extended regorafenib treatment can be linked with mitochondrial damage leading to cardiotoxicity. Toxicol. Lett. 2021, 336, 39–49. [Google Scholar] [CrossRef]
- Kerkela, R.; Grazette, L.; Yacobi, R.; Iliescu, C.; Patten, R.; Beahm, C.; Walters, B.; Shevtsov, S.; Pesant, S.; Clubb, F.J.; et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat. Med. 2006, 12, 908–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.M.; Kim, H.; Yeon, J.H.; Lee, J.H.; Park, H.O. Identification of a Mitochondrial DNA Polymerase Affecting Cardiotoxicity of Sunitinib Using a Genome-Wide Screening on S. pombe Deletion Library. Toxicol. Sci. 2016, 149, 4–14. [Google Scholar] [CrossRef] [Green Version]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef] [PubMed]
- Babaei, H.; Razmaraii, N.; Assadnassab, G.; Mohajjel Nayebi, A.; Azarmi, Y.; Mohammadnejad, D.; Azami, A. Ultrastructural and Echocardiographic Assessment of Chronic Doxorubicin-Induced Cardiotoxicity in Rats. Arch. Razi. Inst. 2020, 75, 55–62. [Google Scholar] [CrossRef]
- Gnanapragasam, A.; Yogeeta, S.; Subhashini, R.; Ebenezar, K.K.; Sathish, V.; Devaki, T. Adriamycin induced myocardial failure in rats: Protective role of Centella asiatica. Mol. Cell. Biochem. 2007, 294, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Lebrecht, D.; Kirschner, J.; Geist, A.; Haberstroh, J.; Walker, U.A. Respiratory chain deficiency precedes the disrupted calcium homeostasis in chronic doxorubicin cardiomyopathy. Cardiovasc. Pathol. 2010, 19, e167–e174. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Zhong, L.; Han, X.; Wang, H.; Zhong, J.; Xuan, Z. Astragalus membranaceus prevents daunorubicin-induced apoptosis of cultured neonatal cardiomyocytes: Role of free radical effect of Astragalus membranaceus on daunorubicin cardiotoxicity. Phytother. Res. 2009, 23, 761–767. [Google Scholar] [CrossRef]
- Jean, S.R.; Tulumello, D.V.; Riganti, C.; Liyanage, S.U.; Schimmer, A.D.; Kelley, S.O. Mitochondrial Targeting of Doxorubicin Eliminates Nuclear Effects Associated with Cardiotoxicity. ACS Chem. Biol. 2015, 10, 2007–2015. [Google Scholar] [CrossRef]
- Sun, J.; Sun, G.; Meng, X.; Wang, H.; Luo, Y.; Qin, M.; Ma, B.; Wang, M.; Cai, D.; Guo, P.; et al. Isorhamnetin protects against doxorubicin-induced cardiotoxicity in vivo and in vitro. PLoS ONE 2013, 8, e64526. [Google Scholar] [CrossRef]
- Brandao, S.R.; Reis-Mendes, A.; Domingues, P.; Duarte, J.A.; Bastos, M.L.; Carvalho, F.; Ferreira, R.; Costa, V.M. Exploring the aging effect of the anticancer drugs doxorubicin and mitoxantrone on cardiac mitochondrial proteome using a murine model. Toxicology 2021, 459, 152852. [Google Scholar] [CrossRef]
- Khuanjing, T.; Ongnok, B.; Maneechote, C.; Siri-Angkul, N.; Prathumsap, N.; Arinno, A.; Chunchai, T.; Arunsak, B.; Chattipakorn, S.C.; Chattipakorn, N. Acetylcholinesterase inhibitor ameliorates doxorubicin-induced cardiotoxicity through reducing RIP1-mediated necroptosis. Pharmacol. Res. 2021, 173, 105882. [Google Scholar] [CrossRef]
- Yao, Y.F.; Liu, X.; Li, W.J.; Shi, Z.W.; Yan, Y.X.; Wang, L.F.; Chen, M.; Xie, M.Y. (-)-Epigallocatechin-3-gallate alleviates doxorubicin-induced cardiotoxicity in sarcoma 180 tumor-bearing mice. Life Sci. 2017, 180, 151–159. [Google Scholar] [CrossRef]
- Bose, C.; Awasthi, S.; Sharma, R.; Benes, H.; Hauer-Jensen, M.; Boerma, M.; Singh, S.P. Sulforaphane potentiates anticancer effects of doxorubicin and attenuates its cardiotoxicity in a breast cancer model. PLoS ONE 2018, 13, e0193918. [Google Scholar] [CrossRef]
- Liu, D.; Ma, Z.; Di, S.; Yang, Y.; Yang, J.; Xu, L.; Reiter, R.J.; Qiao, S.; Yuan, J. AMPK/PGC1alpha activation by melatonin attenuates acute doxorubicin cardiotoxicity via alleviating mitochondrial oxidative damage and apoptosis. Free Radic. Biol. Med. 2018, 129, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Reis-Mendes, A.; Dores-Sousa, J.L.; Padrao, A.I.; Duarte-Araujo, M.; Duarte, J.A.; Seabra, V.; Goncalves-Monteiro, S.; Remiao, F.; Carvalho, F.; Sousa, E.; et al. Inflammation as a Possible Trigger for Mitoxantrone-Induced Cardiotoxicity: An In Vivo Study in Adult and Infant Mice. Pharmaceuticals 2021, 14, 510. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.R.; Sharma, A.; Lytwyn, M.; Bohonis, S.; Thliveris, J.; Singal, P.K.; Jassal, D.S. The cardioprotective role of probucol against anthracycline and trastuzumab-mediated cardiotoxicity. J. Am. Soc. Echocardiogr. 2011, 24, 699–705. [Google Scholar] [CrossRef]
- Al-Harthi, S.E.; Alarabi, O.M.; Ramadan, W.S.; Alaama, M.N.; Al-Kreathy, H.M.; Damanhouri, Z.A.; Khan, L.M.; Osman, A.M. Amelioration of doxorubicininduced cardiotoxicity by resveratrol. Mol. Med. Rep. 2014, 10, 1455–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Ni, J.; Li, M.; Chen, J.; Han, L.; Zhu, Y.; Kong, D.; Mao, J.; Wang, Y.; Zhang, B.; et al. Ginsenoside Rg3 micelles mitigate doxorubicin-induced cardiotoxicity and enhance its anticancer efficacy. Drug Deliv. 2017, 24, 1617–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henninger, C.; Huelsenbeck, S.; Wenzel, P.; Brand, M.; Huelsenbeck, J.; Schad, A.; Fritz, G. Chronic heart damage following doxorubicin treatment is alleviated by lovastatin. Pharmacol. Res. 2015, 91, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Kalender, S.; Kalender, Y.; Ates, A.; Yel, M.; Olcay, E.; Candan, S. Protective role of antioxidant vitamin E and catechin on idarubicin-induced cardiotoxicity in rats. Braz. J. Med. Biol. Res. 2002, 35, 1379–1387. [Google Scholar] [CrossRef] [Green Version]
- Sudharsan, P.T.; Mythili, Y.; Selvakumar, E.; Varalakshmi, P. Lupeol and its ester exhibit protective role against cyclophosphamide-induced cardiac mitochondrial toxicity. J. Cardiovasc. Pharmacol. 2006, 47, 205–210. [Google Scholar] [CrossRef]
- Ma, H.; Jones, K.R.; Guo, R.; Xu, P.; Shen, Y.; Ren, J. Cisplatin compromises myocardial contractile function and mitochondrial ultrastructure: Role of endoplasmic reticulum stress. Clin. Exp. Pharmacol. Physiol. 2010, 37, 460–465. [Google Scholar] [CrossRef]
- Laird-Fick, H.S.; Tokala, H.; Kandola, S.; Kehdi, M.; Pelosi, A.; Wang, L.; Grondahl, B. Early morphological changes in cardiac mitochondria after subcutaneous administration of trastuzumab in rabbits: Possible prevention with oral selenium supplementation. Cardiovasc. Pathol. 2020, 44, 107159. [Google Scholar] [CrossRef] [PubMed]
- Force, T.; Kolaja, K.L. Cardiotoxicity of kinase inhibitors: The prediction and translation of preclinical models to clinical outcomes. Nat. Rev. Drug Discov. 2011, 10, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Khezri, S.; Atashbar, S.; Azizian, S.; Shaikhgermchi, Z.; Kurdpour, P.; Salimi, A. Calcitriol Reduces Adverse Effects of Diclofenac on Mitochondrial Function in Isolated Rat Heart Mitochondria. Drug Res. 2020, 70, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Zhu, Z.N.; Wang, J.Z.; Huang, S.M.; Feng, X.M.; Li, A.Y.; Yang, D.L.; Wang, B.J. Assessment of mitochondrial toxicity induced by zidovudine and adefovir dipivoxil in rats. Chin. J. Hepatol. 2012, 20, 794–797. [Google Scholar] [CrossRef]
- Botelho, A.F.M.; Santos-Miranda, A.; Joca, H.C.; Mattoso, C.R.S.; de Oliveira, M.S.; Pierezan, F.; Cruz, J.S.; Soto-Blanco, B.; Melo, M.M. Hydroalcoholic extract from Nerium oleander L. (Apocynaceae) elicits arrhythmogenic activity. J. Ethnopharmacol. 2017, 206, 170–177. [Google Scholar] [CrossRef]
- Sharmila Queenthy, S.; Stanely Mainzen Prince, P.; John, B. Diosmin Prevents Isoproterenol-Induced Heart Mitochondrial Oxidative Stress in Rats. Cardiovasc. Toxicol. 2018, 18, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Seydi, E.; Tabbati, Y.; Pourahmad, J. Toxicity of Atenolol and Propranolol on Rat Heart Mitochondria. Drug Res. 2020, 70, 151–157. [Google Scholar] [CrossRef]
- Salimi, A.; Eybagi, S.; Seydi, E.; Naserzadeh, P.; Kazerouni, N.P.; Pourahmad, J. Toxicity of macrolide antibiotics on isolated heart mitochondria: A justification for their cardiotoxic adverse effect. Xenobiotica 2016, 46, 82–93. [Google Scholar] [CrossRef]
- Peng, F.; Zhang, N.; Wang, C.; Wang, X.; Huang, W.; Peng, C.; He, G.; Han, B. Aconitine induces cardiomyocyte damage by mitigating BNIP3-dependent mitophagy and the TNFalpha-NLRP3 signalling axis. Cell. Prolif. 2020, 53, e12701. [Google Scholar] [CrossRef] [Green Version]
- Seydi, E.; Servati, T.; Samiei, F.; Naserzadeh, P.; Pourahmad, J. Toxicity of Pioglitazone on Mitochondria Isolated from Brain and Heart: An Analysis for Probable Drug-Induced Neurotoxicity and Cardiotoxicity. Drug Res. 2020, 70, 112–118. [Google Scholar] [CrossRef]
- Liu, Y.; Nguyen, P.; Baris, T.Z.; Poirier, M.C. Molecular analysis of mitochondrial compromise in rodent cardiomyocytes exposed long term to nucleoside reverse transcriptase inhibitors (NRTIs). Cardiovasc. Toxicol. 2012, 12, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Mythili, Y.; Sudharsan, P.T.; Varalakshmi, P. dl-alpha-lipoic acid ameliorates cyclophosphamide induced cardiac mitochondrial injury. Toxicology 2005, 215, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Kusumoto, S.; Kawano, H.; Hayashi, T.; Satoh, O.; Yonekura, T.; Eguchi, M.; Takeno, M.; Tsuneto, A.; Koide, Y.; Jo, T.; et al. Cyclophosphamide-induced cardiotoxicity with a prolonged clinical course diagnosed on an endomyocardial biopsy. Intern. Med. 2013, 52, 2311–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, J.B.; Tani, Y.; Witt, K.; Johnson, J.A.; Peddada, S.; Dunnick, J.; Nyska, A. Mitochondrial damage revealed by morphometric and semiquantitative analysis of mouse pup cardiomyocytes following in utero and postnatal exposure to zidovudine and lamivudine. Toxicol. Sci. 2004, 81, 512–517. [Google Scholar] [CrossRef]
- Yin, J.; Guo, J.; Zhang, Q.; Cui, L.; Zhang, L.; Zhang, T.; Zhao, J.; Li, J.; Middleton, A.; Carmichael, P.L.; et al. Doxorubicin-induced mitophagy and mitochondrial damage is associated with dysregulation of the PINK1/parkin pathway. Toxicol. In Vitro 2018, 51, 1–10. [Google Scholar] [CrossRef]
- Gharanei, M.; Hussain, A.; Janneh, O.; Maddock, H. Attenuation of doxorubicin-induced cardiotoxicity by mdivi-1: A mitochondrial division/mitophagy inhibitor. PLoS ONE 2013, 8, e77713. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Funakoshi, T.; Unuma, K.; Aki, T.; Uemura, K. Activation of the ubiquitin-proteasome system against arsenic trioxide cardiotoxicity involves ubiquitin ligase Parkin for mitochondrial homeostasis. Toxicology 2014, 322, 43–50. [Google Scholar] [CrossRef]
- Mamoshina, P.; Rodriguez, B.; Bueno-Orovio, A. Toward a broader view of mechanisms of drug cardiotoxicity. Cell. Rep. Med. 2021, 2, 100216. [Google Scholar] [CrossRef]
- Nomura, R.; Sato, T.; Sato, Y.; Medin, J.A.; Kushimoto, S.; Yanagisawa, T. Azidothymidine-triphosphate impairs mitochondrial dynamics by disrupting the quality control system. Redox Biol. 2017, 13, 407–417. [Google Scholar] [CrossRef]
- Sivakumar, A.; Shanmugarajan, S.; Subbiah, R.; Balakrishnan, R. Cardiac Mitochondrial PTEN-L determines cell fate between apoptosis and survival during chronic alcohol consumption. Apoptosis 2020, 25, 590–604. [Google Scholar] [CrossRef]
- Sorrentino, V.; Menzies, K.J.; Auwerx, J. Repairing Mitochondrial Dysfunction in Disease. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 353–389. [Google Scholar] [CrossRef] [PubMed]
- Martin-Fernandez, B.; Gredilla, R. Mitochondria and oxidative stress in heart aging. Age 2016, 38, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Alford, J.; Qiu, H. Structural and Functional Remodeling of Mitochondria in Cardiac Diseases. Int. J. Mol. Sci. 2021, 22, 4167. [Google Scholar] [CrossRef]
- Marin, W.; Marin, D.; Ao, X.; Liu, Y. Mitochondria as a therapeutic target for cardiac ischemiareperfusion injury (Review). Int. J. Mol. Med. 2021, 47, 485–499. [Google Scholar] [CrossRef]
- Yehualashet, A.S.; Belachew, T.F.; Kifle, Z.D.; Abebe, A.M. Targeting Cardiac Metabolic Pathways: A Role in Ischemic Management. Vasc. Health Risk Manag. 2020, 16, 353–365. [Google Scholar] [CrossRef]
- Ghosh, R.; Hwang, S.M.; Cui, Z.; Gilda, J.E.; Gomes, A.V. Different effects of the nonsteroidal anti-inflammatory drugs meclofenamate sodium and naproxen sodium on proteasome activity in cardiac cells. J. Mol. Cell. Cardiol. 2016, 94, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Karkhanis, A.; Leow, J.W.H.; Hagen, T.; Chan, E.C.Y. Dronedarone-Induced Cardiac Mitochondrial Dysfunction and Its Mitigation by Epoxyeicosatrienoic Acids. Toxicol. Sci. 2018, 163, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, Z.A.; Harvey, R.F.; Pryde, K.R.; Mistry, S.; Hardy, R.E.; Serreli, R.; Chung, I.; Allen, T.E.; Stoneley, M.; MacFarlane, M.; et al. Identification of a novel toxicophore in anti-cancer chemotherapeutics that targets mitochondrial respiratory complex I. Elife 2020, 9, e55845. [Google Scholar] [CrossRef] [PubMed]
- Vineetha, V.P.; Soumya, R.S.; Raghu, K.G. Phloretin ameliorates arsenic trioxide induced mitochondrial dysfunction in H9c2 cardiomyoblasts mediated via alterations in membrane permeability and ETC complexes. Eur. J. Pharmacol. 2015, 754, 162–172. [Google Scholar] [CrossRef]
- Moreno-Sanchez, R.; Bravo, C.; Vasquez, C.; Ayala, G.; Silveira, L.H.; Martinez-Lavin, M. Inhibition and uncoupling of oxidative phosphorylation by nonsteroidal anti-inflammatory drugs: Study in mitochondria, submitochondrial particles, cells, and whole heart. Biochem. Pharmacol. 1999, 57, 743–752. [Google Scholar] [CrossRef]
- Hoch, F.L. Cardiolipins and biomembrane function. Biochim. Biophys. Acta 1992, 1113, 71–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petronilli, V.; Penzo, D.; Scorrano, L.; Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J. Biol. Chem. 2001, 276, 12030–12034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.N.; Korge, P.; Honda, H.M.; Ping, P. Role of the mitochondrial permeability transition in myocardial disease. Circ. Res. 2003, 93, 292–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sztark, F.; Nouette-Gaulain, K.; Malgat, M.; Dabadie, P.; Mazat, J.P. Absence of stereospecific effects of bupivacaine isomers on heart mitochondrial bioenergetics. Anesthesiology 2000, 93, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Dykens, J.A.; Will, Y. The significance of mitochondrial toxicity testing in drug development. Drug Discov. Today 2007, 12, 777–785. [Google Scholar] [CrossRef]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar] [CrossRef] [Green Version]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Lin, Y.; Xu, X.; Lin, S.; Chen, X.; Wang, S. The alterations of mitochondrial DNA in coronary heart disease. Exp. Mol. Pathol. 2020, 114, 104412. [Google Scholar] [CrossRef]
- Bonifacio, A.; Mullen, P.J.; Mityko, I.S.; Navegantes, L.C.; Bouitbir, J.; Krahenbuhl, S. Simvastatin induces mitochondrial dysfunction and increased atrogin-1 expression in H9c2 cardiomyocytes and mice in vivo. Arch. Toxicol. 2016, 90, 203–215. [Google Scholar] [CrossRef]
- Liu, Y.; Shim, E.; Nguyen, P.; Gibbons, A.T.; Mitchell, J.B.; Poirier, M.C. Tempol protects cardiomyocytes from nucleoside reverse transcriptase inhibitor-induced mitochondrial toxicity. Toxicol. Sci. 2014, 139, 133–141. [Google Scholar] [CrossRef]
- Grundmanova, M.; Jarkovska, D.; Suss, A.; Tuma, Z.; Markova, M.; Grundman, Z.; El-Kadi, A.; Cedikova, M.; Stengl, M.; Kuncova, J. Propofol-induced mitochondrial and contractile dysfunction of the rat ventricular myocardium. Physiol. Res. 2016, 65, S601–S609. [Google Scholar] [CrossRef] [PubMed]
- Hanley, P.J.; Ray, J.; Brandt, U.; Daut, J. Halothane, isoflurane and sevoflurane inhibit NADH:ubiquinone oxidoreductase (complex I) of cardiac mitochondria. J. Physiol. 2002, 544, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Acosta, D., Jr. Effect of cocaine on mitochondrial electron transport chain evaluated in primary cultures of neonatal rat myocardial cells and in isolated mitochondrial preparations. Drug Chem. Toxicol. 2000, 23, 339–348. [Google Scholar] [CrossRef] [PubMed]
- BB, N.G.; Sanchez, H.; Zoll, J.; Ribera, F.; Dufour, S.; Lampert, E.; Kindo, M.; Geny, B.; Ventura-Clapier, R.; Mettauer, B. Oxidative capacities of cardiac and skeletal muscles of heart transplant recipients: Mitochondrial effects of cyclosporin-A and its vehicle Cremophor-EL. Fundam. Clin. Pharmacol. 2014, 28, 151–160. [Google Scholar] [CrossRef]
- Ghosh, R.; Goswami, S.K.; Feitoza, L.; Hammock, B.; Gomes, A.V. Diclofenac induces proteasome and mitochondrial dysfunction in murine cardiomyocytes and hearts. Int. J. Cardiol. 2016, 223, 923–935. [Google Scholar] [CrossRef] [Green Version]
- Pointon, A.V.; Walker, T.M.; Phillips, K.M.; Luo, J.; Riley, J.; Zhang, S.D.; Parry, J.D.; Lyon, J.J.; Marczylo, E.L.; Gant, T.W. Doxorubicin in vivo rapidly alters expression and translation of myocardial electron transport chain genes, leads to ATP loss and caspase 3 activation. PLoS ONE 2010, 5, e12733. [Google Scholar] [CrossRef] [Green Version]
- Nowis, D.; Maczewski, M.; Mackiewicz, U.; Kujawa, M.; Ratajska, A.; Wieckowski, M.R.; Wilczynski, G.M.; Malinowska, M.; Bil, J.; Salwa, P.; et al. Cardiotoxicity of the anticancer therapeutic agent bortezomib. Am. J. Pathol. 2010, 176, 2658–2668. [Google Scholar] [CrossRef]
- Rossato, L.G.; Costa, V.M.; Dallegrave, E.; Arbo, M.; Silva, R.; Ferreira, R.; Amado, F.; Dinis-Oliveira, R.J.; Duarte, J.A.; de Lourdes Bastos, M.; et al. Mitochondrial cumulative damage induced by mitoxantrone: Late onset cardiac energetic impairment. Cardiovasc. Toxicol. 2014, 14, 30–40. [Google Scholar] [CrossRef] [Green Version]
- Dzimiri, N. Effects of procainamide, tocainide and phenytoin on guinea pig cardiac mitochondrial ATPase activity. Res. Commun. Chem. Pathol. Pharmacol. 1993, 80, 121–124. [Google Scholar]
- Hu, C.; Ge, F.; Hyodo, E.; Arai, K.; Iwata, S.; Lobdell, H.t.; Walewski, J.L.; Zhou, S.; Clugston, R.D.; Jiang, H.; et al. Chronic ethanol consumption increases cardiomyocyte fatty acid uptake and decreases ventricular contractile function in C57BL/6J mice. J. Mol. Cell. Cardiol. 2013, 59, 30–40. [Google Scholar] [CrossRef] [Green Version]
- Kido, K.; Ito, H.; Yamamoto, Y.; Makita, K.; Uchida, T. Cytotoxicity of propofol in human induced pluripotent stem cell-derived cardiomyocytes. J. Anesth. 2018, 32, 120–131. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sheehan, R.P.; Palmer, A.C.; Everley, R.A.; Boswell, S.A.; Ron-Harel, N.; Ringel, A.E.; Holton, K.M.; Jacobson, C.A.; Erickson, A.R.; et al. Adaptation of Human iPSC-Derived Cardiomyocytes to Tyrosine Kinase Inhibitors Reduces Acute Cardiotoxicity via Metabolic Reprogramming. Cell. Syst. 2019, 8, 412–426.e7. [Google Scholar] [CrossRef] [PubMed]
- Nulton-Persson, A.C.; Szweda, L.I.; Sadek, H.A. Inhibition of cardiac mitochondrial respiration by salicylic acid and acetylsalicylate. J. Cardiovasc. Pharmacol. 2004, 44, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Hiller, N.; Mirtschink, P.; Merkel, C.; Knels, L.; Oertel, R.; Christ, T.; Deussen, A.; Koch, T.; Stehr, S.N. Myocardial accumulation of bupivacaine and ropivacaine is associated with reversible effects on mitochondria and reduced myocardial function. Anesth. Analg. 2013, 116, 83–92. [Google Scholar] [CrossRef]
- Branca, D.; Vincenti, E.; Scutari, G. Influence of the anesthetic 2,6-diisopropylphenol (propofol) on isolated rat heart mitochondria. Comp. Biochem. Physiol. C Pharmacol. Toxicol. Endocrinol. 1995, 110, 41–45. [Google Scholar] [CrossRef]
- Graf, B.M. The cardiotoxicity of local anesthetics: The place of ropivacaine. Curr. Top Med. Chem. 2001, 1, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Nemade, H.; Chaudhari, U.; Acharya, A.; Hescheler, J.; Hengstler, J.G.; Papadopoulos, S.; Sachinidis, A. Cell death mechanisms of the anti-cancer drug etoposide on human cardiomyocytes isolated from pluripotent stem cells. Arch. Toxicol. 2018, 92, 1507–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asiri, Y.A. Probucol attenuates cyclophosphamide-induced oxidative apoptosis, p53 and Bax signal expression in rat cardiac tissues. Oxid. Med. Cell. Longev. 2010, 3, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Nagi, M.N.; Al-Shabanah, O.A.; Hafez, M.M.; Sayed-Ahmed, M.M. Thymoquinone supplementation attenuates cyclophosphamide-induced cardiotoxicity in rats. J. Biochem. Mol. Toxicol. 2011, 25, 135–142. [Google Scholar] [CrossRef]
- Gorini, S.; De Angelis, A.; Berrino, L.; Malara, N.; Rosano, G.; Ferraro, E. Chemotherapeutic Drugs and Mitochondrial Dysfunction: Focus on Doxorubicin, Trastuzumab, and Sunitinib. Oxid. Med. Cell. Longev. 2018, 2018, 7582730. [Google Scholar] [CrossRef] [Green Version]
- Sayed-Ahmed, M.M.; Alrufaiq, B.I.; Alrikabi, A.; Abdullah, M.L.; Hafez, M.M.; Al-Shabanah, O.A. Carnitine Supplementation Attenuates Sunitinib-Induced Inhibition of AMP-Activated Protein Kinase Downstream Signals in Cardiac Tissues. Cardiovasc. Toxicol. 2019, 19, 344–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.D.; Babiarz, J.E.; Abrams, R.M.; Guo, L.; Kameoka, S.; Chiao, E.; Taunton, J.; Kolaja, K.L. Use of human stem cell derived cardiomyocytes to examine sunitinib mediated cardiotoxicity and electrophysiological alterations. Toxicol. Appl. Pharmacol. 2011, 257, 74–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thai, P.N.; Ren, L.; Xu, W.; Overton, J.; Timofeyev, V.; Nader, C.E.; Haddad, M.; Yang, J.; Gomes, A.V.; Hammock, B.D.; et al. Chronic Diclofenac Exposure Increases Mitochondrial Oxidative Stress, Inflammatory Mediators, and Cardiac Dysfunction. Cardiovasc. Drugs Ther. 2021. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.W.; Shin, J.S.; Park, S.J.; Jung, E.; Park, Y.G.; Lee, J.; Kim, S.J.; Park, H.J.; Lee, J.H.; Park, S.M.; et al. Antiviral activity and safety of remdesivir against SARS-CoV-2 infection in human pluripotent stem cell-derived cardiomyocytes. Antivir. Res. 2020, 184, 104955. [Google Scholar] [CrossRef]
- Martins, M.J.; Roque Bravo, R.; Enea, M.; Carmo, H.; Carvalho, F.; Bastos, M.L.; Dinis-Oliveira, R.J.; Dias da Silva, D. Ethanol addictively enhances the in vitro cardiotoxicity of cocaine through oxidative damage, energetic deregulation, and apoptosis. Arch. Toxicol. 2018, 92, 2311–2325. [Google Scholar] [CrossRef] [PubMed]
- Vergeade, A.; Mulder, P.; Vendeville-Dehaudt, C.; Estour, F.; Fortin, D.; Ventura-Clapier, R.; Thuillez, C.; Monteil, C. Mitochondrial impairment contributes to cocaine-induced cardiac dysfunction: Prevention by the targeted antioxidant MitoQ. Free Radic. Biol. Med. 2010, 49, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Vergeade, A.; Mulder, P.; Vendeville, C.; Ventura-Clapier, R.; Thuillez, C.; Monteil, C. Xanthine oxidase contributes to mitochondrial ROS generation in an experimental model of cocaine-induced diastolic dysfunction. J. Cardiovasc. Pharmacol. 2012, 60, 538–543. [Google Scholar] [CrossRef]
- Liu, Y.; Shim, E.; Crespo-Mejias, Y.; Nguyen, P.; Gibbons, A.; Liu, D.; Shide, E.; Poirier, M.C. Cardiomyocytes are Protected from Antiretroviral Nucleoside Analog-Induced Mitochondrial Toxicity by Overexpression of PGC-1alpha. Cardiovasc. Toxicol. 2015, 15, 224–231. [Google Scholar] [CrossRef]
- Lange, L.G.; Sobel, B.E. Mitochondrial dysfunction induced by fatty acid ethyl esters, myocardial metabolites of ethanol. J. Clin. Investig. 1983, 72, 724–731. [Google Scholar] [CrossRef]
- Jyoti, S.; Tandon, S. Disruption of mitochondrial membrane potential coupled with alterations in cardiac biomarker expression as early cardiotoxic signatures in human ES cell-derived cardiac cells. Hum. Exp. Toxicol. 2019, 38, 1111–1124. [Google Scholar] [CrossRef]
- Zhao, L. Protective effects of trimetazidine and coenzyme Q10 on cisplatin-induced cardiotoxicity by alleviating oxidative stress and mitochondrial dysfunction. Anatol. J. Cardiol. 2019, 22, 232–239. [Google Scholar] [CrossRef]
- Vineetha, V.P.; Prathapan, A.; Soumya, R.S.; Raghu, K.G. Arsenic trioxide toxicity in H9c2 myoblasts--damage to cell organelles and possible amelioration with Boerhavia diffusa. Cardiovasc. Toxicol. 2013, 13, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, M.; Pinto, A.; Popolo, A. Trastuzumab-induced cardiotoxicity and role of mitochondrial connexin43 in the adaptive response. Toxicol. In Vitro 2020, 67, 104926. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.Q.; Yu, Y.; Chen, H.; Li, M.; Ihsan, A.; Tong, H.Y.; Huang, X.J.; Gao, Y. Sweroside Alleviated Aconitine-Induced Cardiac Toxicity in H9c2 Cardiomyoblast Cell Line. Front. Pharmacol. 2018, 9, 1138. [Google Scholar] [CrossRef] [PubMed]
- Sudheesh, N.P.; Ajith, T.A.; Janardhanan, K.K. Ganoderma lucidum ameliorate mitochondrial damage in isoproterenol-induced myocardial infarction in rats by enhancing the activities of TCA cycle enzymes and respiratory chain complexes. Int. J. Cardiol. 2013, 165, 117–125. [Google Scholar] [CrossRef]
- Pereira, G.C.; Pereira, S.P.; Tavares, L.C.; Carvalho, F.S.; Magalhaes-Novais, S.; Barbosa, I.A.; Santos, M.S.; Bjork, J.; Moreno, A.J.; Wallace, K.B.; et al. Cardiac cytochrome c and cardiolipin depletion during anthracycline-induced chronic depression of mitochondrial function. Mitochondrion 2016, 30, 95–104. [Google Scholar] [CrossRef]
- Sayed-Ahmed, M.M.; Aldelemy, M.L.; Al-Shabanah, O.A.; Hafez, M.M.; Al-Hosaini, K.A.; Al-Harbi, N.O.; Al-Sharary, S.D.; Al-Harbi, M.M. Inhibition of gene expression of carnitine palmitoyltransferase I and heart fatty acid binding protein in cyclophosphamide and ifosfamide-induced acute cardiotoxic rat models. Cardiovasc. Toxicol. 2014, 14, 232–242. [Google Scholar] [CrossRef]
- Mihailovic, D.; Nikolic, J.; Bjelakovic, B.B.; Stankovic, B.N.; Bjelakovic, G. Morphometric and biochemical characteristics of short-term effects of ethanol on rat cardiac muscle. Exp. Toxicol. Pathol. 1999, 51, 545–547. [Google Scholar] [CrossRef]
- Figueira, T.R.; Barros, M.H.; Camargo, A.A.; Castilho, R.F.; Ferreira, J.C.; Kowaltowski, A.J.; Sluse, F.E.; Souza-Pinto, N.C.; Vercesi, A.E. Mitochondria as a source of reactive oxygen and nitrogen species: From molecular mechanisms to human health. Antioxid. Redox Signal. 2013, 18, 2029–2074. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Sun, Y.; Lu, Y.; Saredy, J.; Wang, X.; Drummer Iv, C.; Shao, Y.; Saaoud, F.; Xu, K.; Liu, M.; Yang, W.Y.; et al. ROS systems are a new integrated network for sensing homeostasis and alarming stresses in organelle metabolic processes. Redox Biol. 2020, 37, 101696. [Google Scholar] [CrossRef] [PubMed]
- Berndt, C.; Lillig, C.H.; Holmgren, A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: Implications for diseases in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1227–H1236. [Google Scholar] [CrossRef] [PubMed]
- Wisnovsky, S.; Lei, E.K.; Jean, S.R.; Kelley, S.O. Mitochondrial Chemical Biology: New Probes Elucidate the Secrets of the Powerhouse of the Cell. Cell. Chem. Biol. 2016, 23, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrealba, N.; Aranguiz, P.; Alonso, C.; Rothermel, B.A.; Lavandero, S. Mitochondria in Structural and Functional Cardiac Remodeling. Adv. Exp. Med. Biol. 2017, 982, 277–306. [Google Scholar] [CrossRef]
- Li, A.; Zheng, N.; Ding, X. Mitochondrial abnormalities: A hub in metabolic syndrome-related cardiac dysfunction caused by oxidative stress. Heart Fail. Rev. 2022, 27, 1387–1394. [Google Scholar] [CrossRef]
- Gao, R.Y.; Mukhopadhyay, P.; Mohanraj, R.; Wang, H.; Horvath, B.; Yin, S.; Pacher, P. Resveratrol attenuates azidothymidine-induced cardiotoxicity by decreasing mitochondrial reactive oxygen species generation in human cardiomyocytes. Mol. Med. Rep. 2011, 4, 151–155. [Google Scholar] [CrossRef] [Green Version]
- Myers, C. The role of iron in doxorubicin-induced cardiomyopathy. Semin. Oncol. 1998, 25, 10–14. [Google Scholar]
- Gustafson, D.L.; Swanson, J.D.; Pritsos, C.A. Modulation of glutathione and glutathione dependent antioxidant enzymes in mouse heart following doxorubicin therapy. Free Radic. Res. Commun. 1993, 19, 111–120. [Google Scholar] [CrossRef]
- Adam-Vizi, V.; Chinopoulos, C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol. Sci. 2006, 27, 639–645. [Google Scholar] [CrossRef]
- Nadanaciva, S.; Will, Y. New insights in drug-induced mitochondrial toxicity. Curr. Pharm. Des. 2011, 17, 2100–2112. [Google Scholar] [CrossRef]
- Oz, E.; Erbas, D.; Surucu, H.S.; Duzgun, E. Prevention of doxorubicin-induced cardiotoxicity by melatonin. Mol. Cell. Biochem. 2006, 282, 31–37. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Li, G.Y.; Liu, Y.; Chai, L.M.; Chen, J.X.; Zhang, Y.; Du, Z.M.; Lu, Y.J.; Yang, B.F. Resveratrol protects against arsenic trioxide-induced cardiotoxicity in vitro and in vivo. Br. J. Pharmacol. 2008, 154, 105–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Asuncion, J.G.; Del Olmo, M.L.; Gomez-Cambronero, L.G.; Sastre, J.; Pallardo, F.V.; Vina, J. AZT induces oxidative damage to cardiac mitochondria: Protective effect of vitamins C and E. Life Sci. 2004, 76, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J.; Doroshow, J.H. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J. Biol. Chem. 1986, 261, 3060–3067. [Google Scholar] [CrossRef]
- Bloom, M.W.; Hamo, C.E.; Cardinale, D.; Ky, B.; Nohria, A.; Baer, L.; Skopicki, H.; Lenihan, D.J.; Gheorghiade, M.; Lyon, A.R.; et al. Cancer Therapy-Related Cardiac Dysfunction and Heart Failure: Part 1: Definitions, Pathophysiology, Risk Factors, and Imaging. Circ. Heart Fail. 2016, 9, e002661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Bai, Z.; Lv, D.; Liu, H.; Li, X.; Chen, X. Rescue effect of lipid emulsion on bupivacaine-induced cardiac toxicity in cardiomyocytes. Mol. Med. Rep. 2015, 12, 3739–3747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guven, A.; Yavuz, O.; Cam, M.; Ercan, F.; Bukan, N.; Comunoglu, C. Melatonin protects against epirubicin-induced cardiotoxicity. Acta Histochem. 2007, 109, 52–60. [Google Scholar] [CrossRef]
- Serrano, J.; Palmeira, C.M.; Kuehl, D.W.; Wallace, K.B. Cardioselective and cumulative oxidation of mitochondrial DNA following subchronic doxorubicin administration. Biochim. Biophys. Acta 1999, 1411, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.J.; Yu, J.; Fang, Q.J.; Lian, J.B.; Wang, R.X.; He, R.L.; Lin, M.J. Sodium ferulate protects against daunorubicin-induced cardiotoxicity by inhibition of mitochondrial apoptosis in juvenile rats. J. Cardiovasc. Pharmacol. 2014, 63, 360–368. [Google Scholar] [CrossRef]
- Mercer, T.R.; Neph, S.; Dinger, M.E.; Crawford, J.; Smith, M.A.; Shearwood, A.M.; Haugen, E.; Bracken, C.P.; Rackham, O.; Stamatoyannopoulos, J.A.; et al. The human mitochondrial transcriptome. Cell 2011, 146, 645–658. [Google Scholar] [CrossRef] [Green Version]
- Barshad, G.; Marom, S.; Cohen, T.; Mishmar, D. Mitochondrial DNA Transcription and Its Regulation: An Evolutionary Perspective. Trends Genet. 2018, 34, 682–692. [Google Scholar] [CrossRef]
- Franci, L.; Tubita, A.; Bertolino, F.M.; Palma, A.; Cannino, G.; Settembre, C.; Rasola, A.; Rovida, E.; Chiariello, M. MAPK15 protects from oxidative stress-dependent cellular senescence by inducing the mitophagic process. Aging Cell 2022, e13620. [Google Scholar] [CrossRef]
- Douarre, C.; Sourbier, C.; Dalla Rosa, I.; Brata Das, B.; Redon, C.E.; Zhang, H.; Neckers, L.; Pommier, Y. Mitochondrial topoisomerase I is critical for mitochondrial integrity and cellular energy metabolism. PLoS ONE 2012, 7, e41094. [Google Scholar] [CrossRef] [Green Version]
- Setzer, B.; Schlesier, M.; Thomas, A.K.; Walker, U.A. Mitochondrial toxicity of nucleoside analogues in primary human lymphocytes. Antivir. Ther. 2005, 10, 327–334. [Google Scholar] [CrossRef] [PubMed]
- McKee, E.E.; Ferguson, M.; Bentley, A.T.; Marks, T.A. Inhibition of mammalian mitochondrial protein synthesis by oxazolidinones. Antimicrob. Agents Chemother. 2006, 50, 2042–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagiec, E.E.; Wu, L.; Swaney, S.M.; Chosay, J.G.; Ross, D.E.; Brieland, J.K.; Leach, K.L. Oxazolidinones inhibit cellular proliferation via inhibition of mitochondrial protein synthesis. Antimicrob. Agents Chemother. 2005, 49, 3896–3902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottger, E.C.; Springer, B.; Prammananan, T.; Kidan, Y.; Sander, P. Structural basis for selectivity and toxicity of ribosomal antibiotics. EMBO Rep. 2001, 2, 318–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dykens, J.A.; Marroquin, L.D.; Will, Y. Strategies to reduce late-stage drug attrition due to mitochondrial toxicity. Expert Rev. Mol. Diagn. 2007, 7, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Herrmann, J.M.; Becker, T. Quality control of the mitochondrial proteome. Nat. Rev. Mol. Cell. Biol. 2021, 22, 54–70. [Google Scholar] [CrossRef] [PubMed]
- Gyulkhandanyan, A.V.; Mutlu, A.; Freedman, J.; Leytin, V. Mitochondrial permeability transition pore (MPTP)-dependent and -independent pathways of mitochondrial membrane depolarization, cell shrinkage and microparticle formation during platelet apoptosis. Br. J. Haematol. 2015, 169, 142–145. [Google Scholar] [CrossRef]
- Strubbe-Rivera, J.O.; Schrad, J.R.; Pavlov, E.V.; Conway, J.F.; Parent, K.N.; Bazil, J.N. The mitochondrial permeability transition phenomenon elucidated by cryo-EM reveals the genuine impact of calcium overload on mitochondrial structure and function. Sci. Rep. 2021, 11, 1037. [Google Scholar] [CrossRef] [PubMed]
- Ramaccini, D.; Montoya-Uribe, V.; Aan, F.J.; Modesti, L.; Potes, Y.; Wieckowski, M.R.; Krga, I.; Glibetic, M.; Pinton, P.; Giorgi, C.; et al. Mitochondrial Function and Dysfunction in Dilated Cardiomyopathy. Front. Cell Dev. Biol. 2020, 8, 624216. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Wieckowski, M.R.; Chinopoulos, C.; Kepp, O.; Kroemer, G.; Galluzzi, L.; Pinton, P. Molecular mechanisms of cell death: Central implication of ATP synthase in mitochondrial permeability transition. Oncogene 2015, 34, 1475–1486. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Morganti, C.; Morciano, G.; Pedriali, G.; Lebiedzinska-Arciszewska, M.; Aquila, G.; Giorgi, C.; Rizzo, P.; Campo, G.; Ferrari, R.; et al. Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C-ring conformation. EMBO Rep. 2017, 18, 1077–1089. [Google Scholar] [CrossRef]
- Morciano, G.; Bonora, M.; Giorgi, C.; Pinton, P. Other bricks for the correct construction of the mitochondrial permeability transition pore complex. Cell Death Dis. 2017, 8, e2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonora, M.; Pinton, P. A New Current for the Mitochondrial Permeability Transition. Trends Biochem. Sci. 2019, 44, 559–561. [Google Scholar] [CrossRef]
- Zhou, J.; Peng, F.; Cao, X.; Xie, X.; Chen, D.; Yang, L.; Rao, C.; Peng, C.; Pan, X. Risk Compounds, Preclinical Toxicity Evaluation, and Potential Mechanisms of Chinese Materia Medica-Induced Cardiotoxicity. Front. Pharmacol. 2021, 12, 578796. [Google Scholar] [CrossRef]
- Marroquin, L.; Swiss, R.; Will, Y. Identifying Compounds that Induce Opening of the Mitochondrial Permeability Transition Pore in Isolated Rat Liver Mitochondria. Curr. Protoc. Toxicol. 2014, 60, 25.4.1–25.4.17. [Google Scholar] [CrossRef]
- Pessayre, D.; Mansouri, A.; Berson, A.; Fromenty, B. Mitochondrial involvement in drug-induced liver injury. Handb. Exp. Pharmacol. 2010, 311–365. [Google Scholar] [CrossRef]
- Broderick, T.L. Hypocarnitinaemia induced by sodium pivalate in the rat is associated with left ventricular dysfunction and impaired energy metabolism. Drugs R D 2006, 7, 153–161. [Google Scholar] [CrossRef]
- Barile, M.; Valenti, D.; Passarella, S.; Quagliariello, E. 3′-Azido-3′-deoxythmidine uptake into isolated rat liver mitochondria and impairment of ADP/ATP translocator. Biochem. Pharmacol. 1997, 53, 913–920. [Google Scholar] [CrossRef]
- Lewis, W.; Simpson, J.F.; Meyer, R.R. Cardiac mitochondrial DNA polymerase-gamma is inhibited competitively and noncompetitively by phosphorylated zidovudine. Circ. Res. 1994, 74, 344–348. [Google Scholar] [CrossRef] [Green Version]
- Mitry, M.A.; Edwards, J.G. Doxorubicin induced heart failure: Phenotype and molecular mechanisms. Int. J. Cardiol. Heart Vasc. 2016, 10, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewton, K.G.; Johal, A.S.; Parker, S.J. Transporters at the Interface between Cytosolic and Mitochondrial Amino Acid Metabolism. Metabolites 2021, 11, 112. [Google Scholar] [CrossRef] [PubMed]
- Aquila, H.; Misra, D.; Eulitz, M.; Klingenberg, M. Complete amino acid sequence of the ADP/ATP carrier from beef heart mitochondria. Hoppe. Seylers Z Physiol. Chem. 1982, 363, 345–349. [Google Scholar] [PubMed]
- Hu, W.J.; Chen, X.M.; Meng, H.D.; Meng, Z.H. Fermented corn flour poisoning in rural areas of China. III. Isolation and identification of main toxin produced by causal microorganisms. Biomed. Environ. Sci. 1989, 2, 65–71. [Google Scholar]
- Stewart, M.J.; Steenkamp, V. The biochemistry and toxicity of atractyloside: A review. Ther. Drug Monit. 2000, 22, 641–649. [Google Scholar] [CrossRef]
- Tahrir, F.G.; Langford, D.; Amini, S.; Mohseni Ahooyi, T.; Khalili, K. Mitochondrial quality control in cardiac cells: Mechanisms and role in cardiac cell injury and disease. J. Cell. Physiol. 2019, 234, 8122–8133. [Google Scholar] [CrossRef]
- Chang, X.; Zhang, W.; Zhao, Z.; Ma, C.; Zhang, T.; Meng, Q.; Yan, P.; Zhang, L.; Zhao, Y. Regulation of Mitochondrial Quality Control by Natural Drugs in the Treatment of Cardiovascular Diseases: Potential and Advantages. Front. Cell Dev. Biol. 2020, 8, 616139. [Google Scholar] [CrossRef]
- Suliman, H.B.; Piantadosi, C.A. Mitochondrial Quality Control as a Therapeutic Target. Pharmacol. Rev. 2016, 68, 20–48. [Google Scholar] [CrossRef]
- Tatsuta, T.; Langer, T. Quality control of mitochondria: Protection against neurodegeneration and ageing. EMBO J. 2008, 27, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Koleini, N.; Kardami, E. Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget 2017, 8, 46663–46680. [Google Scholar] [CrossRef] [Green Version]
- Oh, C.M.; Ryu, D.; Cho, S.; Jang, Y. Mitochondrial Quality Control in the Heart: New Drug Targets for Cardiovascular Disease. Korean Circ. J. 2020, 50, 395–405. [Google Scholar] [CrossRef]
- Chang, C.Y.; Kazmin, D.; Jasper, J.S.; Kunder, R.; Zuercher, W.J.; McDonnell, D.P. The metabolic regulator ERRalpha, a downstream target of HER2/IGF-1R, as a therapeutic target in breast cancer. Cancer Cell 2011, 20, 500–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peugnet, V.; Chwastyniak, M.; Mulder, P.; Lancel, S.; Bultot, L.; Fourny, N.; Renguet, E.; Bugger, H.; Beseme, O.; Loyens, A.; et al. Mitochondrial-Targeted Therapies Require Mitophagy to Prevent Oxidative Stress Induced by SOD2 Inactivation in Hypertrophied Cardiomyocytes. Antioxidants 2022, 11, 723. [Google Scholar] [CrossRef]
- Beak, J.Y.; Kang, H.S.; Huang, W.; Aghajanian, A.; Gerrish, K.; Jetten, A.M.; Jensen, B.C. The nuclear receptor RORα preserves cardiomyocyte mitochondrial function by regulating caveolin-3-mediated mitophagy. J. Biol. Chem. 2020, 297. [Google Scholar] [CrossRef]
- Morales, P.E.; Arias-Duran, C.; Avalos-Guajardo, Y.; Aedo, G.; Verdejo, H.E.; Parra, V.; Lavandero, S. Emerging role of mitophagy in cardiovascular physiology and pathology. Mol. Asp. Med. 2020, 71, 100822. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Ma, Z.; An, D.; Liu, Z.; Cai, W.; Bai, Y.; Zhan, Q.; Lai, W.; Zeng, Q.; Ren, H.; et al. Mitofusin 2 Participates in Mitophagy and Mitochondrial Fusion Against Angiotensin II-Induced Cardiomyocyte Injury. Front. Physiol. 2019, 10, 411. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Wang, L.; Qiao, Y.; Yang, B.; Yin, D.; He, M. Epigallocatechin-3-gallate pretreatment alleviates doxorubicin-induced ferroptosis and cardiotoxicity by upregulating AMPKalpha2 and activating adaptive autophagy. Redox Biol. 2021, 48, 102185. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Wan, K.; Yin, M.; Hu, P.; Que, Y.; Zhou, X.; Zhang, L.; Li, T.; Du, Y.; Xu, G.; et al. RIPK3 Induces Cardiomyocyte Necroptosis via Inhibition of AMPK-Parkin-Mitophagy in Cardiac Remodelling after Myocardial Infarction. Oxid. Med. Cell. Longev. 2021, 2021, 6635955. [Google Scholar] [CrossRef]
- Ramirez-Sagredo, A.; Quiroga, C.; Garrido-Moreno, V.; Lopez-Crisosto, C.; Leiva-Navarrete, S.; Norambuena-Soto, I.; Ortiz-Quintero, J.; Diaz-Vesga, M.C.; Perez, W.; Hendrickson, T.; et al. Polycystin-1 regulates cardiomyocyte mitophagy. FASEB J. 2021, 35, e21796. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, B.; Cortassa, S.; Aon, M.A. Mitochondrial ion channels: Gatekeepers of life and death. Physiology 2005, 20, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P. Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol. Rev. 1999, 79, 1127–1155. [Google Scholar] [CrossRef] [PubMed]
- Beavis, A.D. Properties of the inner membrane anion channel in intact mitochondria. J. Bioenerg. Biomembr. 1992, 24, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Smyrnias, I.; Gray, S.P.; Okonko, D.O.; Sawyer, G.; Zoccarato, A.; Catibog, N.; Lopez, B.; Gonzalez, A.; Ravassa, S.; Diez, J.; et al. Cardioprotective Effect of the Mitochondrial Unfolded Protein Response During Chronic Pressure Overload. J. Am. Coll. Cardiol. 2019, 73, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Rauthan, M.; Ranji, P.; Aguilera Pradenas, N.; Pitot, C.; Pilon, M. The mitochondrial unfolded protein response activator ATFS-1 protects cells from inhibition of the mevalonate pathway. Proc. Natl. Acad. Sci. USA 2013, 110, 5981–5986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquez, J.; Lee, S.R.; Kim, N.; Han, J. Post-Translational Modifications of Cardiac Mitochondrial Proteins in Cardiovascular Disease: Not Lost in Translation. Korean Circ. J. 2016, 46, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves-Figueiredo, H.; Silva-Platas, C.; Lozano, O.; Vazquez-Garza, E.; Guerrero-Beltran, C.E.; Zarain-Herzberg, A.; Garcia-Rivas, G. A systematic review of post-translational modifications in the mitochondrial permeability transition pore complex associated with cardiac diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 165992. [Google Scholar] [CrossRef]
- DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G. The price of innovation: New estimates of drug development costs. J. Health Econ. 2003, 22, 151–185. [Google Scholar] [CrossRef] [Green Version]
- Ferdinandy, P.; Baczko, I.; Bencsik, P.; Giricz, Z.; Gorbe, A.; Pacher, P.; Varga, Z.V.; Varro, A.; Schulz, R. Definition of hidden drug cardiotoxicity: Paradigm change in cardiac safety testing and its clinical implications. Eur. Heart J. 2019, 40, 1771–1777. [Google Scholar] [CrossRef]
- Kramer, J.A.; Sagartz, J.E.; Morris, D.L. The application of discovery toxicology and pathology towards the design of safer pharmaceutical lead candidates. Nat. Rev. Drug. Discov. 2007, 6, 636–649. [Google Scholar] [CrossRef]
- Lin, X.; Tang, J.; Lou, Y.R. Human Pluripotent Stem-Cell-Derived Models as a Missing Link in Drug Discovery and Development. Pharmaceuticals 2021, 14, 525. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, J.; Miller, P. Trial watch: Phase II and phase III attrition rates 2011–2012. Nat. Rev. Drug Discov. 2013, 12, 569. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Gwathmey, J.K.; Tsaioun, K.; Hajjar, R.J. Cardionomics: A new integrative approach for screening cardiotoxicity of drug candidates. Expert Opin. Drug Metab. Toxicol. 2009, 5, 647–660. [Google Scholar] [CrossRef]
- Sager, P.T.; Gintant, G.; Turner, J.R.; Pettit, S.; Stockbridge, N. Rechanneling the cardiac proarrhythmia safety paradigm: A meeting report from the Cardiac Safety Research Consortium. Am. Heart J. 2014, 167, 292–300. [Google Scholar] [CrossRef]
- Clark, M. Prediction of clinical risks by analysis of preclinical and clinical adverse events. J. Biomed. Inform. 2015, 54, 167–173. [Google Scholar] [CrossRef] [Green Version]
- Brenner, G.B.; Makkos, A.; Nagy, C.T.; Onodi, Z.; Sayour, N.V.; Gergely, T.G.; Kiss, B.; Gorbe, A.; Saghy, E.; Zadori, Z.S.; et al. Hidden Cardiotoxicity of Rofecoxib Can be Revealed in Experimental Models of Ischemia/Reperfusion. Cells 2020, 9, 551. [Google Scholar] [CrossRef] [Green Version]
- Piper, H.M.; Sezer, O.; Schleyer, M.; Schwartz, P.; Hutter, J.F.; Spieckermann, P.G. Development of ischemia-induced damage in defined mitochondrial subpopulations. J. Mol. Cell. Cardiol. 1985, 17, 885–896. [Google Scholar] [CrossRef]
- Palmer, J.W.; Tandler, B.; Hoppel, C.L. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J. Biol. Chem. 1977, 252, 8731–8739. [Google Scholar] [CrossRef]
- O’Shea, K.M.; Khairallah, R.J.; Sparagna, G.C.; Xu, W.; Hecker, P.A.; Robillard-Frayne, I.; Des Rosiers, C.; Kristian, T.; Murphy, R.C.; Fiskum, G.; et al. Dietary omega−3 fatty acids alter cardiac mitochondrial phospholipid composition and delay Ca2+-induced permeability transition. J. Mol. Cell. Cardiol. 2009, 47, 819–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimes, B.W.; Brandt, B.L. Properties of a clonal muscle cell line from rat heart. Exp. Cell. Res. 1976, 98, 367–381. [Google Scholar] [CrossRef]
- Hescheler, J.; Meyer, R.; Plant, S.; Krautwurst, D.; Rosenthal, W.; Schultz, G. Morphological, biochemical, and electrophysiological characterization of a clonal cell (H9c2) line from rat heart. Circ. Res. 1991, 69, 1476–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menard, C.; Pupier, S.; Mornet, D.; Kitzmann, M.; Nargeot, J.; Lory, P. Modulation of L-type calcium channel expression during retinoic acid-induced differentiation of H9C2 cardiac cells. J. Biol. Chem. 1999, 274, 29063–29070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branco, A.F.; Pereira, S.P.; Gonzalez, S.; Gusev, O.; Rizvanov, A.A.; Oliveira, P.J. Gene Expression Profiling of H9c2 Myoblast Differentiation towards a Cardiac-Like Phenotype. PLoS ONE 2015, 10, e0129303. [Google Scholar] [CrossRef] [Green Version]
- Sardao, V.A.; Oliveira, P.J.; Holy, J.; Oliveira, C.R.; Wallace, K.B. Morphological alterations induced by doxorubicin on H9c2 myoblasts: Nuclear, mitochondrial, and cytoskeletal targets. Cell Biol. Toxicol. 2009, 25, 227–243. [Google Scholar] [CrossRef] [Green Version]
- Sacks, B.; Onal, H.; Martorana, R.; Sehgal, A.; Harvey, A.; Wastella, C.; Ahmad, H.; Ross, E.; Pjetergjoka, A.; Prasad, S.; et al. Mitochondrial targeted antioxidants, mitoquinone and SKQ1, not vitamin C, mitigate doxorubicin-induced damage in H9c2 myoblast: Pretreatment vs. co-treatment. BMC Pharmacol. Toxicol. 2021, 22, 49. [Google Scholar] [CrossRef]
- Shi, Y.; Li, F.; Shen, M.; Sun, C.; Hao, W.; Wu, C.; Xie, Y.; Zhang, S.; Gao, H.; Yang, J.; et al. Luteolin Prevents Cardiac Dysfunction and Improves the Chemotherapeutic Efficacy of Doxorubicin in Breast Cancer. Front. Cardiovasc. Med. 2021, 8, 750186. [Google Scholar] [CrossRef]
- Helal, M.; Alcorn, J.; Bandy, B. Doxorubicin Cytotoxicity in Differentiated H9c2 Cardiomyocytes: Evidence for Acute Mitochondrial Superoxide Generation. Cardiovasc. Toxicol. 2021, 21, 152–161. [Google Scholar] [CrossRef]
- Zhang, P.; Chen, Z.; Lu, D.; Wu, Y.; Fan, M.; Qian, J.; Ge, J. Overexpression of COX5A protects H9c2 cells against doxorubicin-induced cardiotoxicity. Biochem. Biophys. Res. Commun. 2020, 524, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Bouitbir, J.; Alshaikhali, A.; Panajatovic, M.; Abegg, V.; Paech, F.; Krahenbuhl, S. Mechanisms of Cardiotoxicity Associated with Tyrosine Kinase Inhibitors in H9c2 Cells and Mice. Eur. Cardiol. 2020, 15, e33. [Google Scholar] [CrossRef] [PubMed]
- Bouitbir, J.; Alshaikhali, A.; Panajatovic, M.V.; Abegg, V.F.; Paech, F.; Krahenbuhl, S. Mitochondrial oxidative stress plays a critical role in the cardiotoxicity of sunitinib: Running title: Sunitinib and oxidative stress in hearts. Toxicology 2019, 426, 152281. [Google Scholar] [CrossRef] [PubMed]
- Will, Y.; Dykens, J.A.; Nadanaciva, S.; Hirakawa, B.; Jamieson, J.; Marroquin, L.D.; Hynes, J.; Patyna, S.; Jessen, B.A. Effect of the multitargeted tyrosine kinase inhibitors imatinib, dasatinib, sunitinib, and sorafenib on mitochondrial function in isolated rat heart mitochondria and H9c2 cells. Toxicol. Sci. 2008, 106, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Vineetha, R.C.; Binu, P.; Arathi, P.; Nair, R.H. L-ascorbic acid and alpha-tocopherol attenuate arsenic trioxide-induced toxicity in H9c2 cardiomyocytes by the activation of Nrf2 and Bcl2 transcription factors. Toxicol. Mech. Methods 2018, 28, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Gergely, S.; Hegedus, C.; Lakatos, P.; Kovacs, K.; Gaspar, R.; Csont, T.; Virag, L. High Throughput Screening Identifies a Novel Compound Protecting Cardiomyocytes from Doxorubicin-Induced Damage. Oxid. Med. Cell. Longev. 2015, 2015, 178513. [Google Scholar] [CrossRef]
- Pointon, A.; Abi-Gerges, N.; Cross, M.J.; Sidaway, J.E. Phenotypic profiling of structural cardiotoxins in vitro reveals dependency on multiple mechanisms of toxicity. Toxicol. Sci. 2013, 132, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branco, A.F.; Pereira, S.L.; Moreira, A.C.; Holy, J.; Sardao, V.A.; Oliveira, P.J. Isoproterenol cytotoxicity is dependent on the differentiation state of the cardiomyoblast H9c2 cell line. Cardiovasc. Toxicol. 2011, 11, 191–203. [Google Scholar] [CrossRef]
- Branco, A.F.; Sampaio, S.F.; Moreira, A.C.; Holy, J.; Wallace, K.B.; Baldeiras, I.; Oliveira, P.J.; Sardao, V.A. Differentiation-dependent doxorubicin toxicity on H9c2 cardiomyoblasts. Cardiovasc. Toxicol. 2012, 12, 326–340. [Google Scholar] [CrossRef]
- Gherghiceanu, M.; Barad, L.; Novak, A.; Reiter, I.; Itskovitz-Eldor, J.; Binah, O.; Popescu, L.M. Cardiomyocytes derived from human embryonic and induced pluripotent stem cells: Comparative ultrastructure. J. Cell. Mol. Med. 2011, 15, 2539–2551. [Google Scholar] [CrossRef] [Green Version]
- Mummery, C.L.; Zhang, J.; Ng, E.S.; Elliott, D.A.; Elefanty, A.G.; Kamp, T.J. Differentiation of human embryonic stem cells and induced pluripotent stem cells to cardiomyocytes: A methods overview. Circ. Res. 2012, 111, 344–358. [Google Scholar] [CrossRef]
- Pang, L.; Sager, P.; Yang, X.; Shi, H.; Sannajust, F.; Brock, M.; Wu, J.C.; Abi-Gerges, N.; Lyn-Cook, B.; Berridge, B.R.; et al. Workshop Report: FDA Workshop on Improving Cardiotoxicity Assessment With Human-Relevant Platforms. Circ. Res. 2019, 125, 855–867. [Google Scholar] [CrossRef]
- Beauchamp, P.; Jackson, C.B.; Ozhathil, L.C.; Agarkova, I.; Galindo, C.L.; Sawyer, D.B.; Suter, T.M.; Zuppinger, C. 3D Co-culture of hiPSC-Derived Cardiomyocytes With Cardiac Fibroblasts Improves Tissue-Like Features of Cardiac Spheroids. Front. Mol. Biosci. 2020, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.C.; Ting, S.; Lee, Y.K.; Ng, K.M.; Zhang, J.; Chen, Z.; Siu, C.W.; Oh, S.K.; Tse, H.F. Electrical stimulation promotes maturation of cardiomyocytes derived from human embryonic stem cells. J. Cardiovasc. Transl. Res. 2013, 6, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Feyen, D.A.M.; McKeithan, W.L.; Bruyneel, A.A.N.; Spiering, S.; Hormann, L.; Ulmer, B.; Zhang, H.; Briganti, F.; Schweizer, M.; Hegyi, B.; et al. Metabolic Maturation Media Improve Physiological Function of Human iPSC-Derived Cardiomyocytes. Cell. Rep. 2020, 32, 107925. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.S.; Blackwell, D.J.; Gomez-Hurtado, N.; Frisk, M.; Wang, L.; Kim, K.; Dahl, C.P.; Fiane, A.; Tonnessen, T.; Kryshtal, D.O.; et al. Thyroid and Glucocorticoid Hormones Promote Functional T-Tubule Development in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Circ. Res. 2017, 121, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson-Bouchard, K.; Ma, S.P.; Yeager, K.; Chen, T.; Song, L.; Sirabella, D.; Morikawa, K.; Teles, D.; Yazawa, M.; Vunjak-Novakovic, G. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 2018, 556, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Karbassi, E.; Fenix, A.; Marchiano, S.; Muraoka, N.; Nakamura, K.; Yang, X.; Murry, C.E. Cardiomyocyte maturation: Advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 2020, 17, 341–359. [Google Scholar] [CrossRef]
- Archer, C.R.; Sargeant, R.; Basak, J.; Pilling, J.; Barnes, J.R.; Pointon, A. Characterization and Validation of a Human 3D Cardiac Microtissue for the Assessment of Changes in Cardiac Pathology. Sci. Rep. 2018, 8, 10160. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Shenoy, S.; Sayed, N. Building Multi-Dimensional Induced Pluripotent Stem Cells-Based Model Platforms to Assess Cardiotoxicity in Cancer Therapies. Front. Pharmacol. 2021, 12, 607364. [Google Scholar] [CrossRef] [PubMed]
- Correia, C.; Serra, M.; Espinha, N.; Sousa, M.; Brito, C.; Burkert, K.; Zheng, Y.; Hescheler, J.; Carrondo, M.J.; Saric, T.; et al. Combining hypoxia and bioreactor hydrodynamics boosts induced pluripotent stem cell differentiation towards cardiomyocytes. Stem Cell Rev. Rep. 2014, 10, 786–801. [Google Scholar] [CrossRef] [Green Version]
- Tohyama, S.; Fujita, J.; Fujita, C.; Yamaguchi, M.; Kanaami, S.; Ohno, R.; Sakamoto, K.; Kodama, M.; Kurokawa, J.; Kanazawa, H.; et al. Efficient Large-Scale 2D Culture System for Human Induced Pluripotent Stem Cells and Differentiated Cardiomyocytes. Stem Cell Rep. 2017, 9, 1406–1414. [Google Scholar] [CrossRef] [Green Version]
- Paik, D.T.; Chandy, M.; Wu, J.C. Patient and Disease-Specific Induced Pluripotent Stem Cells for Discovery of Personalized Cardiovascular Drugs and Therapeutics. Pharmacol. Rev. 2020, 72, 320–342. [Google Scholar] [CrossRef] [Green Version]
- Foldes, G.; Mioulane, M.; Wright, J.S.; Liu, A.Q.; Novak, P.; Merkely, B.; Gorelik, J.; Schneider, M.D.; Ali, N.N.; Harding, S.E. Modulation of human embryonic stem cell-derived cardiomyocyte growth: A testbed for studying human cardiac hypertrophy? J. Mol. Cell. Cardiol. 2011, 50, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Juhola, M.; Joutsijoki, H.; Penttinen, K.; Aalto-Setala, K. Detection of genetic cardiac diseases by Ca(2+) transient profiles using machine learning methods. Sci. Rep. 2018, 8, 9355. [Google Scholar] [CrossRef] [PubMed]
- Grafton, F.; Ho, J.; Ranjbarvaziri, S.; Farshidfar, F.; Budan, A.; Steltzer, S.; Maddah, M.; Loewke, K.E.; Green, K.; Patel, S.; et al. Deep learning detects cardiotoxicity in a high-content screen with induced pluripotent stem cell-derived cardiomyocytes. Elife 2021, 10, e68714. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.W.; Zhang, X.; Abi-Gerges, N.; Lamore, S.D.; Abassi, Y.A.; Peters, M.F. An impedance-based cellular assay using human iPSC-derived cardiomyocytes to quantify modulators of cardiac contractility. Toxicol. Sci. 2014, 142, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Palmer, J.A.; Smith, A.M.; Gryshkova, V.; Donley, E.L.R.; Valentin, J.P.; Burrier, R.E. A Targeted Metabolomics-Based Assay Using Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes Identifies Structural and Functional Cardiotoxicity Potential. Toxicol. Sci. 2020, 174, 218–240. [Google Scholar] [CrossRef] [PubMed]
- Louisse, J.; Wust, R.C.I.; Pistollato, F.; Palosaari, T.; Barilari, M.; Macko, P.; Bremer, S.; Prieto, P. Assessment of acute and chronic toxicity of doxorubicin in human induced pluripotent stem cell-derived cardiomyocytes. Toxicol. In Vitro 2017, 42, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Habeler, W.; Pouillot, S.; Plancheron, A.; Puceat, M.; Peschanski, M.; Monville, C. An in vitro beating heart model for long-term assessment of experimental therapeutics. Cardiovasc. Res. 2009, 81, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Kopljar, I.; De Bondt, A.; Vinken, P.; Teisman, A.; Damiano, B.; Goeminne, N.; Van den Wyngaert, I.; Gallacher, D.J.; Lu, H.R. Chronic drug-induced effects on contractile motion properties and cardiac biomarkers in human induced pluripotent stem cell-derived cardiomyocytes. Br. J. Pharmacol. 2017, 174, 3766–3779. [Google Scholar] [CrossRef] [Green Version]
- Dias, T.P.; Pinto, S.N.; Santos, J.I.; Fernandes, T.G.; Fernandes, F.; Diogo, M.M.; Prieto, M.; Cabral, J.M.S. Biophysical study of human induced Pluripotent Stem Cell-Derived cardiomyocyte structural maturation during long-term culture. Biochem. Biophys. Res. Commun. 2018, 499, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hua, Y.; Miyagawa, S.; Zhang, J.; Li, L.; Liu, L.; Sawa, Y. hiPSC-Derived Cardiac Tissue for Disease Modeling and Drug Discovery. Int. J. Mol. Sci. 2020, 21, 8893. [Google Scholar] [CrossRef]
- Clements, M.; Millar, V.; Williams, A.S.; Kalinka, S. Bridging Functional and Structural Cardiotoxicity Assays Using Human Embryonic Stem Cell-Derived Cardiomyocytes for a More Comprehensive Risk Assessment. Toxicol. Sci. 2015, 148, 241–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilsbach, R.; Preissl, S.; Gruning, B.A.; Schnick, T.; Burger, L.; Benes, V.; Wurch, A.; Bonisch, U.; Gunther, S.; Backofen, R.; et al. Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat. Commun. 2014, 5, 5288. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.G.; Kho, C.; Hajjar, R.J.; Ishikawa, K. Experimental models of cardiac physiology and pathology. Heart Fail. Rev. 2019, 24, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Kistamas, K.; Hezso, T.; Horvath, B.; Nanasi, P.P. Late sodium current and calcium homeostasis in arrhythmogenesis. Channels 2021, 15, 1–19. [Google Scholar] [CrossRef]
- Sala, L.; van Meer, B.J.; Tertoolen, L.G.J.; Bakkers, J.; Bellin, M.; Davis, R.P.; Denning, C.; Dieben, M.A.E.; Eschenhagen, T.; Giacomelli, E.; et al. MUSCLEMOTION: A Versatile Open Software Tool to Quantify Cardiomyocyte and Cardiac Muscle Contraction In Vitro and In Vivo. Circ. Res. 2018, 122, e5–e16. [Google Scholar] [CrossRef]
- Abi-Gerges, N.; Indersmitten, T.; Truong, K.; Nguyen, W.; Ratchada, P.; Nguyen, N.; Page, G.; Miller, P.E.; Ghetti, A. Multiparametric Mechanistic Profiling of Inotropic Drugs in Adult Human Primary Cardiomyocytes. Sci. Rep. 2020, 10, 7692. [Google Scholar] [CrossRef]
- Nguyen, N.; Nguyen, W.; Nguyenton, B.; Ratchada, P.; Page, G.; Miller, P.E.; Ghetti, A.; Abi-Gerges, N. Adult Human Primary Cardiomyocyte-Based Model for the Simultaneous Prediction of Drug-Induced Inotropic and Pro-arrhythmia Risk. Front. Physiol. 2017, 8, 1073. [Google Scholar] [CrossRef] [Green Version]
- Cui, N.; Wu, F.; Lu, W.J.; Bai, R.; Ke, B.; Liu, T.; Li, L.; Lan, F.; Cui, M. Doxorubicin-induced cardiotoxicity is maturation dependent due to the shift from topoisomerase IIalpha to IIbeta in human stem cell derived cardiomyocytes. J. Cell. Mol. Med. 2019, 23, 4627–4639. [Google Scholar] [CrossRef] [Green Version]
- Guo, G.R.; Chen, L.; Rao, M.; Chen, K.; Song, J.P.; Hu, S.S. A modified method for isolation of human cardiomyocytes to model cardiac diseases. J. Transl. Med. 2018, 16, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Yu, P.; Zhou, B.; Song, J.; Li, Z.; Zhang, M.; Guo, G.; Wang, Y.; Chen, X.; Han, L.; et al. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat. Cell Biol. 2020, 22, 108–119. [Google Scholar] [CrossRef]
- Shamsaldeen, Y.A.; Culliford, L.; Clout, M.; James, A.F.; Ascione, R.; Hancox, J.C.; Marrion, N.V. Role of SK channel activation in determining the action potential configuration in freshly isolated human atrial myocytes from the SKArF study. Biochem. Biophys. Res. Commun. 2019, 512, 684–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benardeau, A.; Hatem, S.N.; Rucker-Martin, C.; Tessier, S.; Dinanian, S.; Samuel, J.L.; Coraboeuf, E.; Mercadier, J.J. Primary culture of human atrial myocytes is associated with the appearance of structural and functional characteristics of immature myocardium. J. Mol. Cell. Cardiol. 1997, 29, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Bistola, V.; Nikolopoulou, M.; Derventzi, A.; Kataki, A.; Sfyras, N.; Nikou, N.; Toutouza, M.; Toutouzas, P.; Stefanadis, C.; Konstadoulakis, M.M. Long-term primary cultures of human adult atrial cardiac myocytes: Cell viability, structural properties and BNP secretion in vitro. Int. J. Cardiol. 2008, 131, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Saleem, U.; van Meer, B.J.; Katili, P.A.; Mohd Yusof, N.A.N.; Mannhardt, I.; Garcia, A.K.; Tertoolen, L.; de Korte, T.; Vlaming, M.L.H.; McGlynn, K.; et al. Blinded, Multicenter Evaluation of Drug-induced Changes in Contractility Using Human-induced Pluripotent Stem Cell-derived Cardiomyocytes. Toxicol. Sci. 2020, 176, 103–123. [Google Scholar] [CrossRef] [PubMed]
- Pointon, A.; Pilling, J.; Dorval, T.; Wang, Y.; Archer, C.; Pollard, C. From the Cover: High-Throughput Imaging of Cardiac Microtissues for the Assessment of Cardiac Contraction during Drug Discovery. Toxicol. Sci. 2017, 155, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Ravenscroft, S.M.; Pointon, A.; Williams, A.W.; Cross, M.J.; Sidaway, J.E. Cardiac Non-myocyte Cells Show Enhanced Pharmacological Function Suggestive of Contractile Maturity in Stem Cell Derived Cardiomyocyte Microtissues. Toxicol. Sci. 2016, 152, 99–112. [Google Scholar] [CrossRef]
- Kerr, C.M.; Richards, D.; Menick, D.R.; Deleon-Pennell, K.Y.; Mei, Y. Multicellular Human Cardiac Organoids Transcriptomically Model Distinct Tissue-Level Features of Adult Myocardium. Int. J. Mol. Sci. 2021, 22, 8482. [Google Scholar] [CrossRef]
- Branco, M.A.; Cabral, J.M.S.; Diogo, M.M. From Human Pluripotent Stem Cells to 3D Cardiac Microtissues: Progress, Applications and Challenges. Bioengineering 2020, 7, 92. [Google Scholar] [CrossRef]
- Cho, S.; Lee, C.; Skylar-Scott, M.A.; Heilshorn, S.C.; Wu, J.C. Reconstructing the heart using iPSCs: Engineering strategies and applications. J. Mol. Cell. Cardiol. 2021, 157, 56–65. [Google Scholar] [CrossRef]
- Fonoudi, H.; Burridge, P.W. Cellular model systems to study cardiovascular injury from chemotherapy. J. Thromb. Thrombolysis 2021, 51, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.F.; Leong, M.F.; Lim, T.C.; Chua, Y.P.; Lim, J.K.; Du, C.; Wan, A.C.A. Engineering a functional three-dimensional human cardiac tissue model for drug toxicity screening. Biofabrication 2017, 9, 025011. [Google Scholar] [CrossRef] [PubMed]
- Mills, R.J.; Parker, B.L.; Quaife-Ryan, G.A.; Voges, H.K.; Needham, E.J.; Bornot, A.; Ding, M.; Andersson, H.; Polla, M.; Elliott, D.A.; et al. Drug Screening in Human PSC-Cardiac Organoids Identifies Pro-proliferative Compounds Acting via the Mevalonate Pathway. Cell Stem Cell 2019, 24, 895–907.e6. [Google Scholar] [CrossRef] [PubMed]
- Kofron, C.M.; Kim, T.Y.; Munarin, F.; Soepriatna, A.H.; Kant, R.J.; Mende, U.; Choi, B.R.; Coulombe, K.L.K. A predictive in vitro risk assessment platform for pro-arrhythmic toxicity using human 3D cardiac microtissues. Sci. Rep. 2021, 11, 10228. [Google Scholar] [CrossRef]
- Sharma, A.; Marceau, C.; Hamaguchi, R.; Burridge, P.W.; Rajarajan, K.; Churko, J.M.; Wu, H.; Sallam, K.I.; Matsa, E.; Sturzu, A.C.; et al. Human induced pluripotent stem cell-derived cardiomyocytes as an in vitro model for coxsackievirus B3-induced myocarditis and antiviral drug screening platform. Circ. Res. 2014, 115, 556–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Burridge, P.W.; McKeithan, W.L.; Serrano, R.; Shukla, P.; Sayed, N.; Churko, J.M.; Kitani, T.; Wu, H.; Holmstrom, A.; et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci. Transl. Med. 2017, 9, eaaf2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mot, A.I.; Liddell, J.R.; White, A.R.; Crouch, P.J. Circumventing the Crabtree Effect: A method to induce lactate consumption and increase oxidative phosphorylation in cell culture. Int. J. Biochem. Cell. Biol. 2016, 79, 128–138. [Google Scholar] [CrossRef] [Green Version]
- Beeson, C.C.; Beeson, G.C.; Schnellmann, R.G. A high-throughput respirometric assay for mitochondrial biogenesis and toxicity. Anal. Biochem. 2010, 404, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Deus, C.M.; Zehowski, C.; Nordgren, K.; Wallace, K.B.; Skildum, A.; Oliveira, P.J. Stimulating basal mitochondrial respiration decreases doxorubicin apoptotic signaling in H9c2 cardiomyoblasts. Toxicology 2015, 334, 1–11. [Google Scholar] [CrossRef]
- Rana, P.; Anson, B.; Engle, S.; Will, Y. Characterization of human-induced pluripotent stem cell-derived cardiomyocytes: Bioenergetics and utilization in safety screening. Toxicol. Sci. 2012, 130, 117–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Sekine, S.; Song, B.; Ito, K. Use of Primary Rat Hepatocytes for Prediction of Drug-Induced Mitochondrial Dysfunction. Curr. Protoc. Toxicol. 2017, 72, 14.16.1–14.16.10. [Google Scholar] [CrossRef] [PubMed]
- Delp, J.; Funke, M.; Rudolf, F.; Cediel, A.; Bennekou, S.H.; van der Stel, W.; Carta, G.; Jennings, P.; Toma, C.; Gardner, I.; et al. Development of a neurotoxicity assay that is tuned to detect mitochondrial toxicants. Arch. Toxicol. 2019, 93, 1585–1608. [Google Scholar] [CrossRef] [Green Version]
- Rana, P.; Nadanaciva, S.; Will, Y. Mitochondrial membrane potential measurement of H9c2 cells grown in high-glucose and galactose-containing media does not provide additional predictivity towards mitochondrial assessment. Toxicol. In Vitro 2011, 25, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Dykens, J.A.; Jamieson, J.D.; Marroquin, L.D.; Nadanaciva, S.; Xu, J.J.; Dunn, M.C.; Smith, A.R.; Will, Y. In vitro assessment of mitochondrial dysfunction and cytotoxicity of nefazodone, trazodone, and buspirone. Toxicol. Sci. 2008, 103, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Hom, J.; Sheu, S.S. Morphological dynamics of mitochondria—A special emphasis on cardiac muscle cells. J. Mol. Cell. Cardiol. 2009, 46, 811–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, J.E.; Beeson, C.C.; Schnellmann, R.G. Characterization of functionally distinct mitochondrial subpopulations. J. Bioenerg. Biomembr. 2013, 45, 87–99. [Google Scholar] [CrossRef] [Green Version]
- Kepiro, M.; Varkuti, B.H.; Davis, R.L. High Content, Phenotypic Assays and Screens for Compounds Modulating Cellular Processes in Primary Neurons. Methods Enzymol. 2018, 610, 219–250. [Google Scholar] [CrossRef]
- Rizzuto, R.; Brini, M.; De Giorgi, F.; Rossi, R.; Heim, R.; Tsien, R.Y.; Pozzan, T. Double labelling of subcellular structures with organelle-targeted GFP mutants in vivo. Curr. Biol. 1996, 6, 183–188. [Google Scholar] [CrossRef]
- Legros, F.; Lombes, A.; Frachon, P.; Rojo, M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol. Biol. Cell. 2002, 13, 4343–4354. [Google Scholar] [CrossRef] [Green Version]
- Fogo, G.M.; Anzell, A.R.; Maheras, K.J.; Raghunayakula, S.; Wider, J.M.; Emaus, K.J.; Bryson, T.D.; Bukowski, M.J.; Neumar, R.W.; Przyklenk, K.; et al. Machine learning-based classification of mitochondrial morphology in primary neurons and brain. Sci. Rep. 2021, 11, 5133. [Google Scholar] [CrossRef] [PubMed]
- Hallinger, D.R.; Lindsay, H.B.; Paul Friedman, K.; Suarez, D.A.; Simmons, S.O. Respirometric Screening and Characterization of Mitochondrial Toxicants Within the ToxCast Phase I and II Chemical Libraries. Toxicol. Sci. 2020, 176, 175–192. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Williams, G.R. The respiratory chain and oxidative phosphorylation. Adv. Enzymol. Relat. Subj. Biochem. 1956, 17, 65–134. [Google Scholar] [CrossRef]
- Lanza, I.R.; Nair, K.S. Functional assessment of isolated mitochondria in vitro. Methods Enzymol. 2009, 457, 349–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attene-Ramos, M.S.; Huang, R.; Sakamuru, S.; Witt, K.L.; Beeson, G.C.; Shou, L.; Schnellmann, R.G.; Beeson, C.C.; Tice, R.R.; Austin, C.P.; et al. Systematic study of mitochondrial toxicity of environmental chemicals using quantitative high throughput screening. Chem. Res. Toxicol. 2013, 26, 1323–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wills, L.P.; Beeson, G.C.; Trager, R.E.; Lindsey, C.C.; Beeson, C.C.; Peterson, Y.K.; Schnellmann, R.G. High-throughput respirometric assay identifies predictive toxicophore of mitochondrial injury. Toxicol. Appl. Pharmacol. 2013, 272, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Wills, L.P.; Beeson, G.C.; Hoover, D.B.; Schnellmann, R.G.; Beeson, C.C. Assessment of ToxCast Phase II for Mitochondrial Liabilities Using a High-Throughput Respirometric Assay. Toxicol. Sci. 2015, 146, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Hynes, J.; Marroquin, L.D.; Ogurtsov, V.I.; Christiansen, K.N.; Stevens, G.J.; Papkovsky, D.B.; Will, Y. Investigation of drug-induced mitochondrial toxicity using fluorescence-based oxygen-sensitive probes. Toxicol. Sci. 2006, 92, 186–200. [Google Scholar] [CrossRef] [Green Version]
- Wagner, B.K.; Kitami, T.; Gilbert, T.J.; Peck, D.; Ramanathan, A.; Schreiber, S.L.; Golub, T.R.; Mootha, V.K. Large-scale chemical dissection of mitochondrial function. Nat. Biotechnol. 2008, 26, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Wills, L.P. The use of high-throughput screening techniques to evaluate mitochondrial toxicity. Toxicology 2017, 391, 34–41. [Google Scholar] [CrossRef]
- Rosenke, K.; Hansen, F.; Schwarz, B.; Feldmann, F.; Haddock, E.; Rosenke, R.; Barbian, K.; Meade-White, K.; Okumura, A.; Leventhal, S.; et al. Orally delivered MK-4482 inhibits SARS-CoV-2 replication in the Syrian hamster model. Nat. Commun. 2021, 12, 2295. [Google Scholar] [CrossRef]
- Nehdi, A.; Samman, N.; Mashhour, A.; Alhallaj, A.; Trivilegio, T.; Gul, S.; Reinshagen, J.; Alaskar, A.; Gmati, G.; Abuelgasim, K.A.; et al. A Drug Repositioning Approach Identifies a Combination of Compounds as a Potential Regimen for Chronic Lymphocytic Leukemia Treatment. Front. Oncol. 2021, 11, 579488. [Google Scholar] [CrossRef]
- Naia, L.; Pinho, C.M.; Dentoni, G.; Liu, J.; Leal, N.S.; Ferreira, D.M.S.; Schreiner, B.; Filadi, R.; Fao, L.; Connolly, N.M.C.; et al. Neuronal cell-based high-throughput screen for enhancers of mitochondrial function reveals luteolin as a modulator of mitochondria-endoplasmic reticulum coupling. BMC Biol. 2021, 19, 57. [Google Scholar] [CrossRef] [PubMed]
- Bakowski, M.A.; Beutler, N.; Wolff, K.C.; Kirkpatrick, M.G.; Chen, E.; Nguyen, T.H.; Riva, L.; Shaabani, N.; Parren, M.; Ricketts, J.; et al. Drug repurposing screens identify chemical entities for the development of COVID-19 interventions. Nat. Commun. 2021, 12, 3309. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente-Herreruela, D.; Gonzalez-Charro, V.; Almendro-Vedia, V.G.; Moran, M.; Martin, M.A.; Lillo, M.P.; Natale, P.; Lopez-Montero, I. Rhodamine-based sensor for real-time imaging of mitochondrial ATP in living fibroblasts. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 999–1006. [Google Scholar] [CrossRef]
- Tolosa, L.; Jimenez, N.; Perez, G.; Castell, J.V.; Gomez-Lechon, M.J.; Donato, M.T. Customised in vitro model to detect human metabolism-dependent idiosyncratic drug-induced liver injury. Arch. Toxicol. 2018, 92, 383–399. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.; Berntsen, H.F.; Zimmer, K.E.; Frizzell, C.; Verhaegen, S.; Ropstad, E.; Connolly, L. Effects of defined mixtures of persistent organic pollutants (POPs) on multiple cellular responses in the human hepatocarcinoma cell line, HepG2, using high content analysis screening. Toxicol. Appl. Pharmacol. 2016, 294, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, A.V.; Kehrer, I.; Kozlov, A.V.; Haller, M.; Redl, H.; Hermann, M.; Grimm, M.; Troppmair, J. Mitochondrial ROS production under cellular stress: Comparison of different detection methods. Anal. Bioanal. Chem. 2011, 400, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Luczak, E.D.; Wu, Y.; Granger, J.M.; Joiner, M.A.; Wilson, N.R.; Gupta, A.; Umapathi, P.; Murphy, K.R.; Reyes Gaido, O.E.; Sabet, A.; et al. Mitochondrial CaMKII causes adverse metabolic reprogramming and dilated cardiomyopathy. Nat. Commun. 2020, 11, 4416. [Google Scholar] [CrossRef] [PubMed]
- Vieira, M.L.; Teixeira, A.F.; Pidde, G.; Ching, A.T.C.; Tambourgi, D.V.; Nascimento, A.; Herwald, H. Leptospira interrogans outer membrane protein LipL21 is a potent inhibitor of neutrophil myeloperoxidase. Virulence 2018, 9, 414–425. [Google Scholar] [CrossRef] [Green Version]
- Cali, T.; Ottolini, D.; Brini, M. Mitochondria, calcium, and endoplasmic reticulum stress in Parkinson’s disease. Biofactors 2011, 37, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Qiu, B.Y.; Turner, N.; Li, Y.Y.; Gu, M.; Huang, M.W.; Wu, F.; Pang, T.; Nan, F.J.; Ye, J.M.; Li, J.Y.; et al. High-throughput assay for modulators of mitochondrial membrane potential identifies a novel compound with beneficial effects on db/db mice. Diabetes 2010, 59, 256–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, G.; Xue, L.; Zhu, Y.; Qian, X.; Zou, L.; Jin, Q.; Wang, D.; Ge, G. Differences in susceptibility of HT-29 and A549 cells to statin-induced toxicity: An investigation using high content screening. J. Biochem. Mol. Toxicol. 2021, 35, e22699. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lu, Q.; Ding, Y.; Wu, Y.; Qiu, Y.; Wang, P.; Mao, X.; Huang, K.; Xie, Z.; Zou, M.H. Hyperglycemia-Driven Inhibition of AMP-Activated Protein Kinase alpha2 Induces Diabetic Cardiomyopathy by Promoting Mitochondria-Associated Endoplasmic Reticulum Membranes In Vivo. Circulation 2019, 139, 1913–1936. [Google Scholar] [CrossRef] [PubMed]
- Chazotte, B. Labeling mitochondria with MitoTracker dyes. Cold Spring Harb. Protoc. 2011, 2011, 990–992. [Google Scholar] [CrossRef]
- Perry, S.W.; Norman, J.P.; Barbieri, J.; Brown, E.B.; Gelbard, H.A. Mitochondrial membrane potential probes and the proton gradient: A practical usage guide. Biotechniques 2011, 50, 98–115. [Google Scholar] [CrossRef]
- Huang, L.; Su, W.; Zhao, Y.; Zhan, J.; Lin, W. Synthesis, molecular docking calculation, fluorescence and bioimaging of mitochondria-targeted ratiometric fluorescent probes for sensing hypochlorite in vivo. J. Mater. Chem. B 2021, 9, 2666–2673. [Google Scholar] [CrossRef]
- Salvatorelli, E.; Guarnieri, S.; Menna, P.; Liberi, G.; Calafiore, A.M.; Mariggio, M.A.; Mordente, A.; Gianni, L.; Minotti, G. Defective one-or two-electron reduction of the anticancer anthracycline epirubicin in human heart. Relative importance of vesicular sequestration and impaired efficiency of electron addition. J. Biol. Chem. 2006, 281, 10990–11001. [Google Scholar] [CrossRef] [Green Version]
- Korga, A.; Jozefczyk, A.; Zgorka, G.; Homa, M.; Ostrowska, M.; Burdan, F.; Dudka, J. Evaluation of the phytochemical composition and protective activities of methanolic extracts of Centaurea borysthenica and Centaurea daghestanica (Lipsky) Wagenitz on cardiomyocytes treated with doxorubicin. Food Nutr. Res. 2017, 61, 1344077. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Yin, W.; Wang, J.; Feng, L.; Kang, Y.J. Mitophagy promotes the stemness of bone marrow-derived mesenchymal stem cells. Exp. Biol. Med. 2021, 246, 97–105. [Google Scholar] [CrossRef]
- O’Brien, P.J.; Irwin, W.; Diaz, D.; Howard-Cofield, E.; Krejsa, C.M.; Slaughter, M.R.; Gao, B.; Kaludercic, N.; Angeline, A.; Bernardi, P.; et al. High concordance of drug-induced human hepatotoxicity with in vitro cytotoxicity measured in a novel cell-based model using high content screening. Arch. Toxicol. 2006, 80, 580–604. [Google Scholar] [CrossRef]
- Tilmant, K.; Gerets, H.; De Ron, P.; Hanon, E.; Bento-Pereira, C.; Atienzar, F.A. In vitro screening of cell bioenergetics to assess mitochondrial dysfunction in drug development. Toxicol. In Vitro 2018, 52, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Biesemann, N.; Ried, J.S.; Ding-Pfennigdorff, D.; Dietrich, A.; Rudolph, C.; Hahn, S.; Hennerici, W.; Asbrand, C.; Leeuw, T.; Strubing, C. High throughput screening of mitochondrial bioenergetics in human differentiated myotubes identifies novel enhancers of muscle performance in aged mice. Sci. Rep. 2018, 8, 9408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddah, M.; Mandegar, M.A.; Dame, K.; Grafton, F.; Loewke, K.; Ribeiro, A.J.S. Quantifying drug-induced structural toxicity in hepatocytes and cardiomyocytes derived from hiPSCs using a deep learning method. J. Pharmacol. Toxicol. Methods 2020, 105, 106895. [Google Scholar] [CrossRef] [PubMed]
- Grimm, F.A.; Iwata, Y.; Sirenko, O.; Bittner, M.; Rusyn, I. High-Content Assay Multiplexing for Toxicity Screening in Induced Pluripotent Stem Cell-Derived Cardiomyocytes and Hepatocytes. Assay Drug. Dev. Technol. 2015, 13, 529–546. [Google Scholar] [CrossRef] [PubMed]
- Sirenko, O.; Grimm, F.A.; Ryan, K.R.; Iwata, Y.; Chiu, W.A.; Parham, F.; Wignall, J.A.; Anson, B.; Cromwell, E.F.; Behl, M.; et al. In vitro cardiotoxicity assessment of environmental chemicals using an organotypic human induced pluripotent stem cell-derived model. Toxicol. Appl. Pharmacol. 2017, 322, 60–74. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, P.J.; Edvardsson, A. Validation of a Multiparametric, High-Content-Screening Assay for Predictive/Investigative Cytotoxicity: Evidence from Technology Transfer Studies and Literature Review. Chem. Res. Toxicol. 2017, 30, 804–829. [Google Scholar] [CrossRef]
- Attene-Ramos, M.S.; Huang, R.; Michael, S.; Witt, K.L.; Richard, A.; Tice, R.R.; Simeonov, A.; Austin, C.P.; Xia, M. Profiling of the Tox21 chemical collection for mitochondrial function to identify compounds that acutely decrease mitochondrial membrane potential. Env. Health Perspect. 2015, 123, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Nabati, M.; Parsaee, H. Potential Cardiotoxic Effects of Remdesivir on Cardiovascular System: A Literature Review. Cardiovasc. Toxicol. 2021, 22, 268–272. [Google Scholar] [CrossRef]
- McComsey, G.; Lonergan, J.T. Mitochondrial dysfunction: Patient monitoring and toxicity management. J. Acquir. Immune Defic. Syndr. 2004, 37 (Suppl. S1), S30–S35. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertero, E.; Maack, C. Metabolic remodelling in heart failure. Nat. Rev. Cardiol. 2018, 15, 457–470. [Google Scholar] [CrossRef]
- Limongelli, G.; Masarone, D.; Pacileo, G. Mitochondrial disease and the heart. Heart 2017, 103, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Renu, K.; Abilash, V.G.; Tirupathi Pichiah, P.B.; Arunachalam, S. Molecular mechanism of doxorubicin-induced cardiomyopathy—An update. Eur. J. Pharmacol. 2018, 818, 241–253. [Google Scholar] [CrossRef]
- Benjanuwattra, J.; Siri-Angkul, N.; Chattipakorn, S.C.; Chattipakorn, N. Doxorubicin and its proarrhythmic effects: A comprehensive review of the evidence from experimental and clinical studies. Pharmacol. Res. 2020, 151, 104542. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.M.; Cho, S.; Jang, J.Y.; Kim, H.; Chun, S.; Choi, M.; Park, S.; Ko, Y.G. Cardioprotective Potential of an SGLT2 Inhibitor Against Doxorubicin-Induced Heart Failure. Korean Circ. J. 2019, 49, 1183–1195. [Google Scholar] [CrossRef] [Green Version]
- Tong, D.; Zaha, V.G. Metabolic Imaging in Cardio-oncology. J. Cardiovasc. Transl. Res. 2020, 13, 357–366. [Google Scholar] [CrossRef]
- Sivapackiam, J.; Sharma, M.; Schindler, T.H.; Sharma, V. PET Radiopharmaceuticals for Imaging Chemotherapy-Induced Cardiotoxicity. Curr. Cardiol. Rep. 2020, 22, 62. [Google Scholar] [CrossRef]
- Mason, F.E.; Pronto, J.R.D.; Alhussini, K.; Maack, C.; Voigt, N. Cellular and mitochondrial mechanisms of atrial fibrillation. Basic Res. Cardiol. 2020, 115, 72. [Google Scholar] [CrossRef]
- Yang, Y.; Wei, S.; Zhang, B.; Li, W. Recent Progress in Environmental Toxins-Induced Cardiotoxicity and Protective Potential of Natural Products. Front. Pharmacol. 2021, 12, 699193. [Google Scholar] [CrossRef]
- Behjati, M.; Sabri, M.R.; Etemadi Far, M.; Nejati, M. Cardiac complications in inherited mitochondrial diseases. Heart Fail. Rev. 2020, 26, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Liu, Z.; Cao, N. Human pluripotent stem cell-based cardiovascular disease modeling and drug discovery. Pflugers Arch. 2021, 473, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Couch, L.; Higuchi, M.; Fang, J.L.; Guo, L. Mitochondrial dysfunction induced by sertraline, an antidepressant agent. Toxicol. Sci. 2012, 127, 582–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
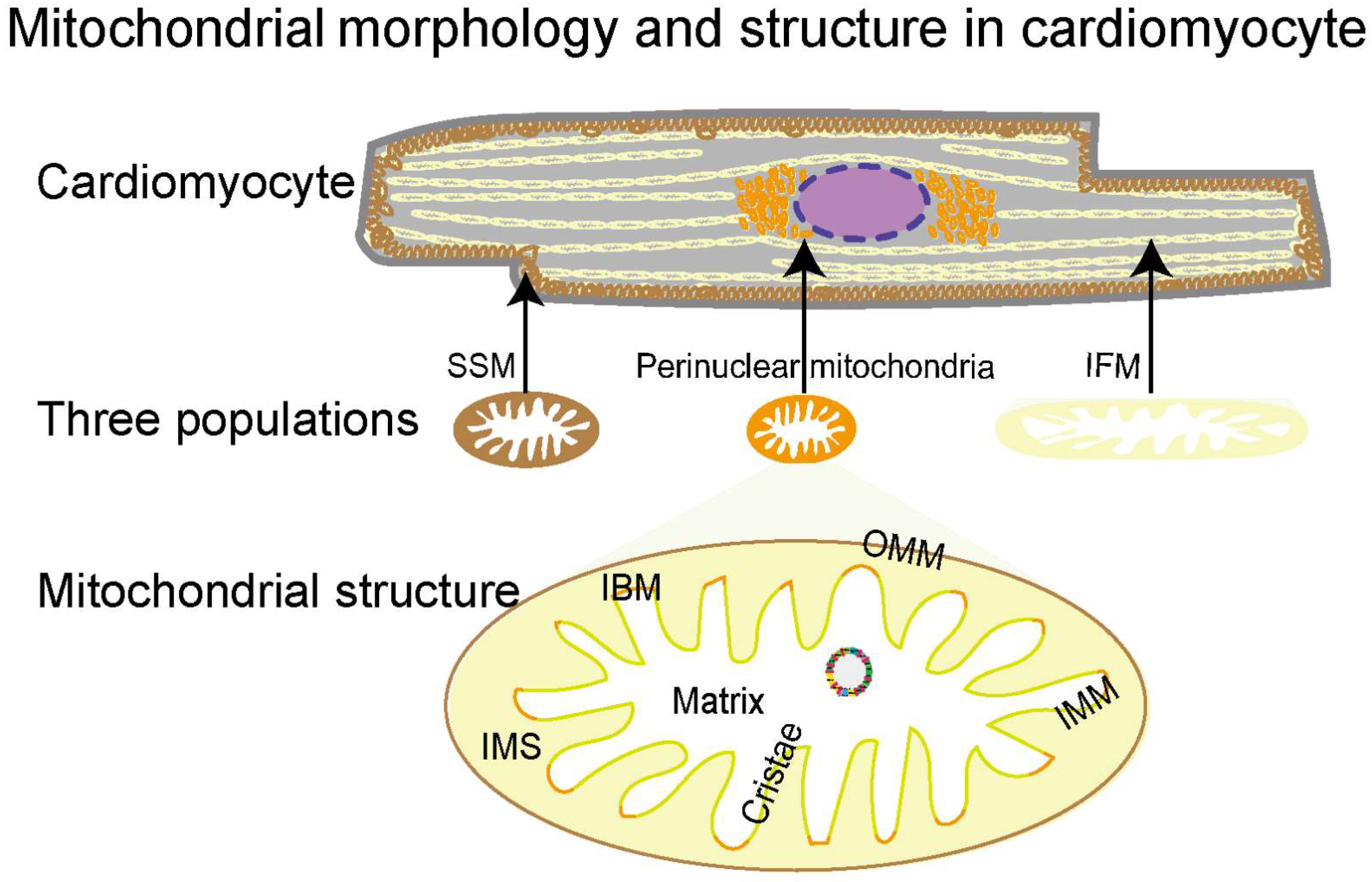
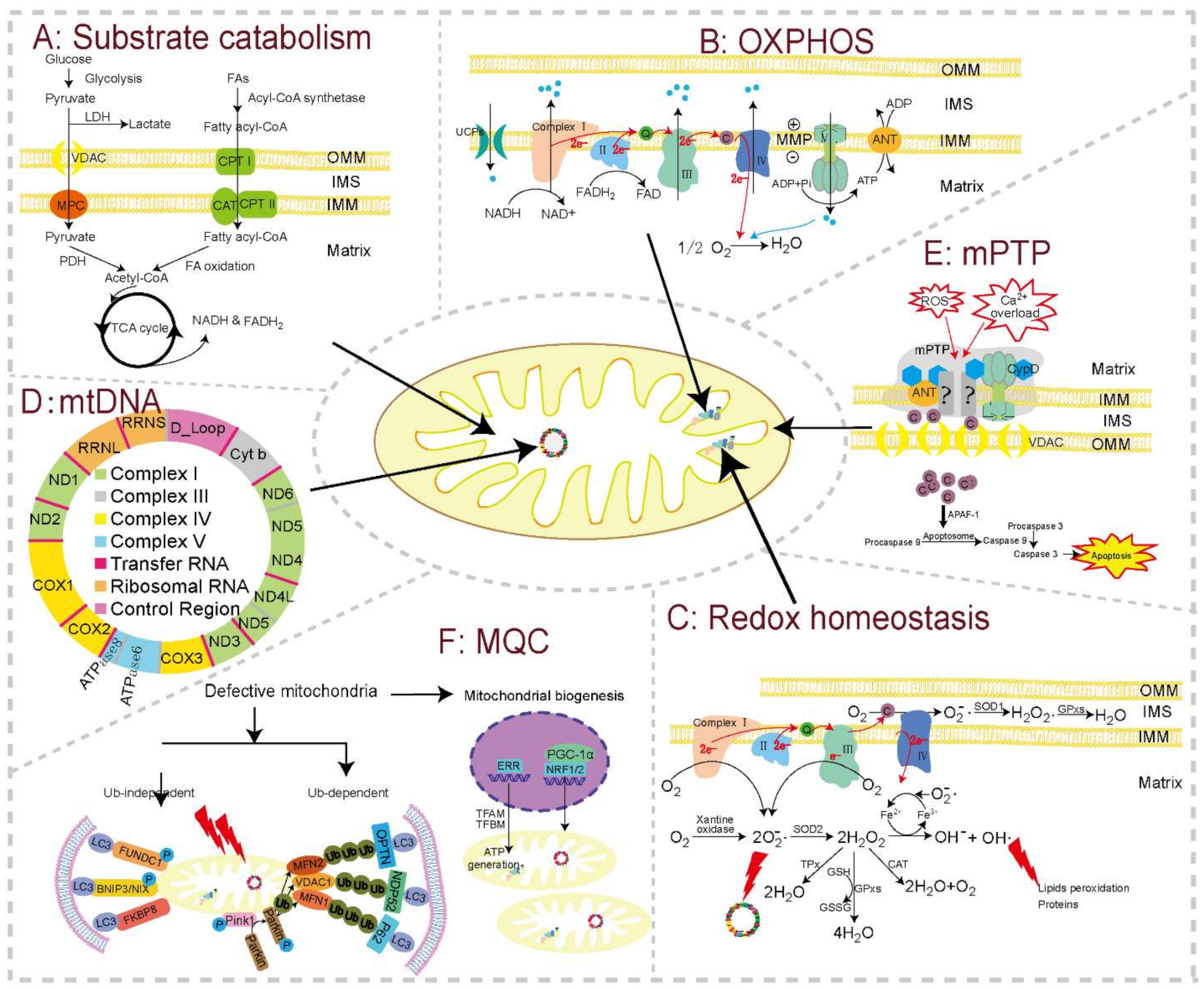
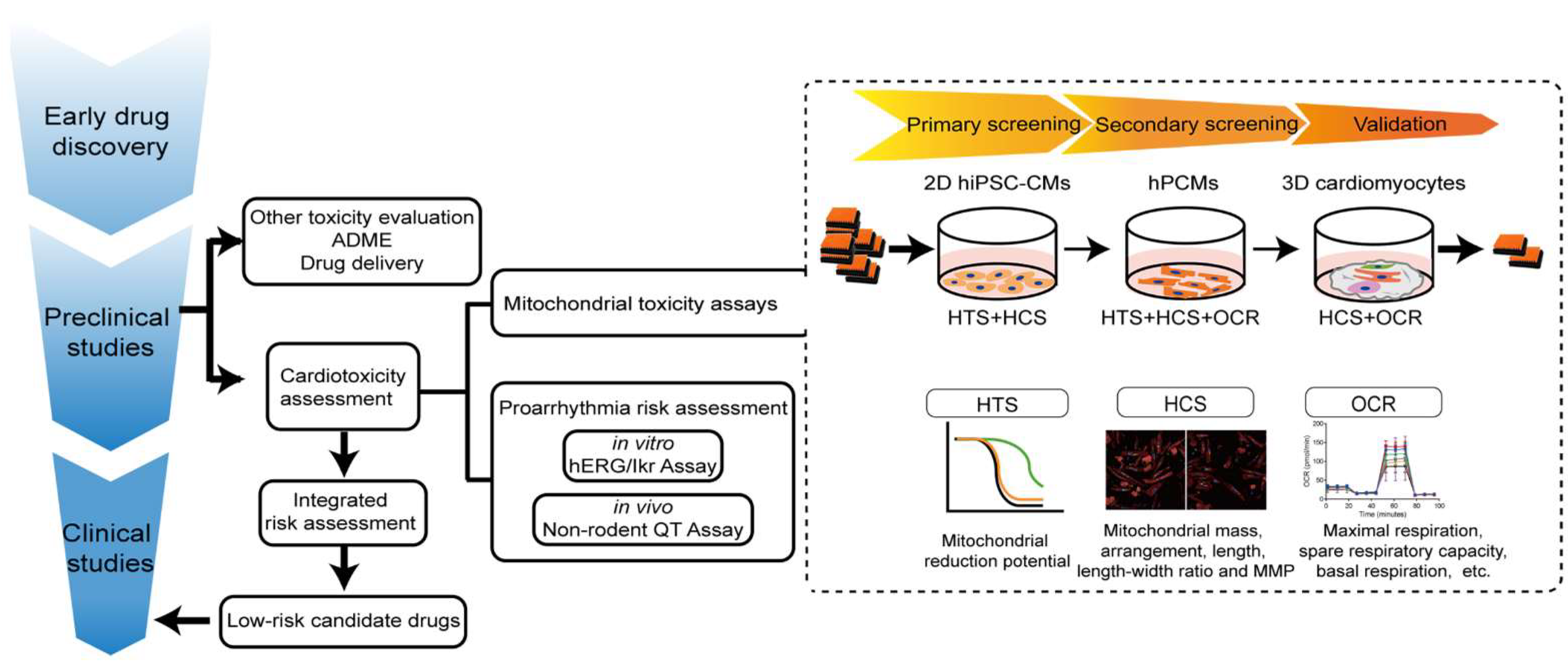
| Modules | Alterations | Pharmacology | Drugs | Clinical Manifestations | Cmax | Models | Dose | Time | References |
|---|---|---|---|---|---|---|---|---|---|
| Morphology | Mitochondrial swelling | Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP | 15.3 μM | Male Wistar rats (IP) | 2&2.5 mg/kg/2 d | 2 w | [94] |
| Male Wistar rats (IP) | 2.5 mg/kg/2 d | 2 w | [95] | ||||||
| Male Wistar rats (IV) | 1 mg/kg/w | 7 w (started at 11 w, observed at 48 w) | [96] | ||||||
| Idarubicin | CMP, MI, CHF, VA, decreased LVEF | 23.22 μM | Male SD rats (IV) | 5 mg/kg/w | 6 w | [110] | |||
| Morphology | Mitochondrial swelling | Alkylating agent | Cyclophosphamide | HMC, CMP | 143 μM | Male Wistar rats (IP) | 200 mg/kg | 10 d | [111] |
| Morphology | Mitochondrial swelling | Chemotherapeutic agents | Cisplatin | Decreased LVEF, arrhythmias, ECA, myocarditis, CMP | 27.54 μM | C57BL mice (IV) | 10 mg/kg/d | 1 w | [112] |
| Morphology | Mitochondrial swelling | Monoclonal antibody | Trastuzumab | CMP, LVD, CHF | 2.59 mM | Female white New Zealand rabbits (SC) | 8 mg/kg, a single dose; 8 mg/kg first w, 6 mg/kg for three additional w | 4 w | [113] |
| Morphology | Mitochondrial swelling | TKIs | Sunitinib | Decreased LVEF, QT prolongation, TdP, hypertension, HF, CMP | 0.25 μM | Patient | [114] | ||
| Male SD rats (oral) | 10 mg/kg/d | 3 w | [89] | ||||||
| Morphology | Mitochondrial swelling | NSAIDs | Diclofenac | Hypertension, arrhythmias | 7.9 µM | Isolated rat heart mitochondria | 10 µg/mL | 1 h | [115] |
| Isolated rat heart mitochondria | 50 μM | 1 h | [60] | ||||||
| Naproxen | - | 100 µM | Isolated rat heart mitochondria | 25 μM | 1 h | [60] | |||
| Celecoxib | Thrombosis, MI, stroke | 3–5 µM | Isolated rat heart mitochondria | 100 μM | 1 h | [60] | |||
| Morphology | Mitochondrial swelling | NRTIs | Zidovudine | CMP | 4 μM | Rats (oral) | 125 mg/kg/d | 4 w | [116] |
| Morphology | Mitochondrial swelling | Cardiac glycosides | Nerium oleander L. | PVB, AVB, VT | - | Guinea pigs (oral) | 150&300 mg/kg | 3 h | [117] |
| Morphology | Mitochondrial swelling | β-adrenoceptor agonists | Isoproterenol | HF | 0.01 μM | Male Wistar rats (SC) | 100 mg/kg, BID | 12 h | [118] |
| Male Wistar rats (SC) | 100 mg/kg, BID | 12 h | [83] | ||||||
| Male Wistar rats (SC) | 100 mg/kg, BID | 12 h | [82] | ||||||
| Propranolol | Cardiotoxicity | 0.22 μM | Isolated rat heart mitochondria | 5 µg/mL | 5 min | [119] | |||
| Atenolol | Cardiotoxicity | 4.99 μM | Isolated rat heart mitochondria | 10 µg/mL | 5 min | [119] | |||
| Morphology | Mitochondrial swelling | Macrolide antibiotics | Azithromycin | Arrhythmia | 0.32–0.87 μM | Isolated rat heart mitochondria | 25 μM | 1 h | [120] |
| Clarithromycin | TdP | 2.67–13.37 μM | Isolated rat heart mitochondria | 50 μM | 1 h | [120] | |||
| Erythromycin | TdP | 11 μM | Isolated rat heart mitochondria | 25 μM | 1 h | [120] | |||
| Morphology | Mitochondrial swelling | Aconitum species | Aconitum sp. | VA | 19.27 μg/ml | H9c2 | 1 μM | [121] | |
| Morphology | Mitochondrial swelling | Diabetes medication | Pioglitazone | HF | 2.6 μM | Isolated rat heart mitochondria | 12.5 µg/mL (30 min), 25 µg/mL (5 min) | [122] | |
| Morphology | Morphological damage | NRTIs | Zidovudine | CMP | 4 μM | H9c2 | 50 μM | 39 passages | [123] |
| Didanosine | CMP | 12 μM | H9c2 | 50 μM | 10 passages | [123] | |||
| Structure | Cristae disappearance | Chemotherapeutic agents | As2O3 | QT prolongation TdP, CMP, tachycardia | 12.1 μM | Male BALB/c mice | 2 mg/kg | 14 d | [84] |
| Structure | Cristae disappearance | Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP | 15.3 μM | Male Wistar rats (IP) | 2&2.5 mg/kg/2 d | 2 w | [94] |
| Male Wistar rats (IP) | 2.5 mg/kg/2 d | 2 w | [95] | ||||||
| Kunming mice (IP) | 2 mg/kg | 10 d | [102] | ||||||
| Male Wistar rats (IV) | 1 mg/kg/w | 7 w (started at 11 w, observed at 48 w) | [96] | ||||||
| Structure | Cristae disappearance | Alkylating agent | Cyclophosphamide | HMC, CMP | 143 μM | Male Wistar rats (IP) | 200 mg/kg | 10 d | [111] |
| Male Wistar rats (IP) | 200 mg/kg | 10 d | [124] | ||||||
| Patient | [125] | ||||||||
| Structure | Cristae disappearance | TKIs | Sorafenib | Bleeding, hypertension,QT prolongation, CHF, CI, MI | 16.6 μM | Male SD rats (oral) | 10 mg/kg/d | 3 w | [89] |
| Structure | Cristae disappearance | NRTIs | Zidovudine | CMP | 4 μM | Rats (oral) | 125 mg/kg/d | 4 w | [116] |
| Pregnant CD-1 mice + pups,oral | 75 mg/kg, BID | 2 w prior to pregnancy to pups postnatal 28 d | [126] | ||||||
| Structure | Cristae disappearance | β-adrenoceptor agonists | Isoproterenol | HF | 0.01 μM | Male Wistar rats (SC) | 100 mg/kg, BID | 12 h | [118] |
| Male Wistar rats (SC) | 100 mg/kg, BID | 12 h | [83] | ||||||
| Male Wistar rats (SC) | 100 mg/kg, BID | 12 h | [82] | ||||||
| Structure | Cristae disorganization | Monoclonal antibody | Trastuzumab | CMP, LVD, CHF | 2.59 mM | Female white New Zealand rabbits (SC) | 8 mg/kg, a single dose; 8 mg/kg first W, 6 mg/kg for three additional w | 4 w | [113] |
| Structure | OMM or/and IMM disruption | NRTIs | Zidovudine | CMP | 4 μM | Rats (oral) | 125 mg/kg/d | 4 w | [116] |
| Monoclonal antibody | Trastuzumab | CMP, LVD, CHF | 2.59 mM | Female white New Zealand rabbits (SC) | 8 mg/kg for first w, 6 mg/kg for three additional w | 4 w | [113] | ||
| Structure | Matrix clearout | Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP | 15.3 μM | Male Wistar rats, intraperitoneal(IP) | 2.5 mg/kg/2 d | 2 w | [95] |
| Structure | Matrix clearout | TKIs | Sunitinib | Decreased LVEF, QT prolongation, TdP, hypertension, HF, CMP | Male SD rats (oral) | 10 mg/kg/d | 3 w | [89] | |
| Regorafenib | MI; hypertension | H9c2 | 10 μM | 72 h | [90] | ||||
| Structure | β-adrenoceptor agonists | Isoproterenol | HF | 0.01 μM | Male Wistar rats (SC) | 100 mg/kg, BID | 12 h | [118] | |
| Matrix clearout | Male Wistar rats (SC) | 100 mg/kg, BID | 12 h | [83] | |||||
| Male Wistar rats (SC) | 100 mg/kg, BID | 12 h | [82] | ||||||
| Structure | Matrix clearout | Chemotherapeutic agents | Cisplatin | Decreased LVEF, arrhythmias, ECA, myocarditis, CMP | 27.54 μM | C57BL mice (IV) | 10 mg/kg/d | 1 w | [112] |
| As2O3 | QT prolongation TdP, CMP, tachycardia | 12.1 μM | Male BALB/c mice | 2 mg/kg | 14 d | [84] | |||
| MQC | Excessive mitophagy | Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP QT prolongation TdP, CMP, tachycardia | 15.3 μM | AC16 cells | 15.625 nM | 24 h | [127] |
| Adult rat cardiac myocytes | 1 μM | 4 h | [128] | ||||||
| MQC | Excessive mitophagy | Chemotherapeutic agents | As2O3 | 12.1 μM | HL-1 | 6 μM | 6 h | [129] | |
| MQC | Inhibition of mitophagy | Aconitum species | Aconitum sp. | VA | 19.27 μg/ml | H9c2 | 2 μM | 24 h | [121] |
| MQC | Inhibition of mitochondrial biogenesis | Monoclonal antibody | Trastuzumab | CMP, LVD, CHF | 2.59 mM | - | - | - | |
| MQC | Mitochondrial dynamics | TKIs | Sunitinib | Decreased LVEF, QT prolongation, TdP, hypertension, HF, CMP | 0.25 μM | - | - | - | [130] |
| Regorafenib | MI; hypertension | 8.08 μM | H9c2 | 20 μM | 48 h | [90] | |||
| MQC | Mitochondrial dynamics | NRTIs | Zidovudine | CMP | 4 μM | Pregnant CD-1 mice + pups, oral | 75 mg/kg, BID | 2 w prior to pregnancy to pups postnatal 28 D | [126] |
| TMPK-overexpressing H9c2 cells | 100 µM | 24 h | [131] | ||||||
| MQC | Mitochondrial dynamics | Nucleoside analogues | Remdesivir | Bradycardia, QT prologation, CA | 9 μM | hiPSC-CMs | 2.5 μM | 3 d | [86] |
| MQC | Mitochondrial dynamics | Addictive drugs | Ethanol | H9c2 | 5 μM | 0.5 h | [132] |
| Modules | Alterations | Pharmacology | Drugs | Clinical Manifestations | Cmax | Models | Dose | Time | References |
|---|---|---|---|---|---|---|---|---|---|
| OXPHOS | Inhibition of complex I | Cholesterol medications | Simvastatin | Cardiac atrophy | 0.02 μM | H9c2 | 10 μM | 24 h | [151] |
| OXPHOS | Inhibition of complex I | β-adrenoceptor agonists | Isoproterenol | HF | 0.01 μM | Male Wistar rats (SC) | 100 mg/kg, BID | 12 h | [83] |
| OXPHOS | Inhibition of complex I | Alkylating agent | Cyclophosphamide | HMC, CMP | 143 μM | Male Wistar rats (IP) | 200 mg/kg | 10 d | [111] |
| Male Wistar rats (IP) | 200 mg/kg | 10 d | [124] | ||||||
| OXPHOS | Inhibition of complex I | NRTIs | Zidovudine | CMP | 4 μM | Isolated mitochondria from H9c2 | 50 μM | 3 passages | [152] |
| Didanosine | CMP | 12 μM | Isolated mitochondria from H9c2 | 50 μM | 3 passages | [152] | |||
| OXPHOS | Inhibition of complex I | Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP | 15.3 μM | Male Wistar rats (IP) | 2.5 mg/kg/2 d | 2 w | [95] |
| OXPHOS | Inhibition of complex I | Chemotherapeutic agents | As2O3 | QT prolongation TdP, CMP, tachycardia | 12.1 μM | Isolated mitochondria from H9c2 | 5 μM | 24 h | [141] |
| OXPHOS | Inhibition of complex I | Anesthesia | Propofol | HF, arrhythmia | 30.13 μM | Cardiac muscle fibers of Wistar male rats | 0.025 mM | [153] | |
| Halothane (fluothane) | - | 10 μM | Pig heart submitochondrial particles | Dose response curve | [154] | ||||
| Inhibition of complex I | TKIs | Mubritinib | - | - | H9c2 | 0.5 μM | [140] | ||
| OXPHOS | Inhibition of complex I | NSAIDs | Nabumetone | - | 2.45 μM | Submitochondrial particles | 55 nmol/mg protein inhibit 50% | [142] | |
| Meclofenamate sodium | - | 3.55 μM | Mitochontria | 100 µM (70% inhibition) | [138] | ||||
| Naproxen | - | 100 µM | Mitochontria | 200 µM (50% inhibition) | [138] | ||||
| OXPHOS | Inhibition of complex I | Addictive drugs | Cocaine | Arrhythmias, angina, MI, HF | 0.76–0.94 µM | Isolated rat heart mitochondria | 1 μM | [155] | |
| OXPHOS | Inhibition of complex I | Anti-arrhythmic drug | Amiodarone | LQT, TdP, Hypotension, AV block, Arrhythmia, heart block, SBC, CHF, VF | 4.65 μM | Isolated rat heart mitochondria | IC50 = 5.24 µM | [139] | |
| Dronedarone | AF, HF | 0.15–0.26 μM | Isolated rat heart mitochondria | IC50 = 3.07 µM | [139] | ||||
| OXPHOS | Inhibition of complex I | Immunosuppressant drug | Cyclosporine A | Cardiotoxicity | 0.5–5 µM | Enzymes and coenzymes | 100 µM | [156] | |
| OXPHOS | Inhibition of complex II | NSAIDs | Diclofenac | Hypertension, arrhythmias | 7.9 µM | Isolated rat heart mitochondria | 10 µg/mL | 1 h | [115] |
| Naproxen | - | 100 µM | Isolated rat heart mitochondria | 50 μM | 1 h | [60] | |||
| OXPHOS | Inhibition of complex II | Alkylating agent | Cyclophosphamide | HMC, CMP Cardiotoxicity | 143 μM 0.22 μM | Male Wistar rats (IP) | 200 mg/kg | 10 d | [124] |
| Male Wistar rats (IP) | 200 mg/kg | 10 d | [111] | ||||||
| OXPHOS | Inhibition of complex II | β receptor blocker drugs | Propranolol | Isolated rat heart mitochondria | 10 µg/mL | 30 min | [119] | ||
| Atenolol | Cardiotoxicity | 4.99 μM | Isolated rat heart mitochondria | 10 µg/mL | 30 min | [119] | |||
| OXPHOS | Inhibition of complex II | Macrolide antibiotics | Azithromycin | Arrhythmia | 0.32–0.87 μM | Isolated rat heart mitochondria | 25 μM | 20 min | [120] |
| Clarithromycin | TdP | 2.67–13.37 μM | Isolated rat heart mitochondria | 50 μM | 20 min | [120] | |||
| Erythromycin | TdP | 11 μM | Isolated rat heart mitochondria | 25 μM | 20 min | [120] | |||
| OXPHOS | Inhibition of complex III | Chemotherapeutic agents | As2O3 | QT prolongation TdP, CMP, tachycardia | 12.1 μM | Isolated mitochondria from H9c2 | 5 μM | 24 h | [141] |
| OXPHOS | Inhibition of complex III | TKIs | Sorafenib | Bleeding, hypertension, QT prolongation, CHF, CI, MI | 16.6 μM | NRVMs | 4.5 µM | 20 min | [32] |
| OXPHOS | Inhibition of complex III | Alkylating agent | Cyclophosphamide | HMC, CMP | 143 μM | Male Wistar rats (IP) | 200 mg/kg | 10 d | [111] |
| 7.9 µM | Male Wistar rats (IP) | 200 mg/kg | 10 d | [124] | |||||
| OXPHOS | Inhibition of complex III | NSAIDs | Diclofenac | Hypertension, arrhythmias | Mitochondria isolated from mouse hearts | 5 µM | [157] | ||
| Meclofenamate sodium | - | 3.55 μM | Mitochontria | 10 µM | [138] | ||||
| Inhibition of complex III | Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP | 15.3 μM | - | 15 mg/kg | - | [158] | |
| OXPHOS | Inhibition of complex IV | Alkylating agent | Cyclophosphamide | HMC, CMP HF | 143 μM 0.01 μM | Male Wistar rats (IP) | 200 mg/kg | 10 d | [111] |
| Male Wistar rats (IP) | 200 mg/kg | 10 d | [124] | ||||||
| OXPHOS | Inhibition of complex IV | β-adrenoceptor agonists | Isoproterenol | Male Wistar rats (SC) | 100 mg/kg, BID | 12 h | [83] | ||
| OXPHOS | Inhibition of complex IV | Cholesterol medications | Simvastatin | Cardiac atrophy | 0.02 μM | H9c2 | 10 μM | 24 h | [151] |
| OXPHOS | Inhibition of complex IV | Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP, QT prolongation TdP, CMP, tachycardia | 15.3 μM 12.1 μM | Male Wistar rats (IP) | 2.5 mg/kg/2 d | 2 w | [95] |
| Male Wistar rats (IV) | 1 mg/kg/w | 7 w(started at 11 w, observed at 48 w) | [96] | ||||||
| OXPHOS | Inhibition of complex IV | Chemotherapeutic agents | As2O3 | Isolated mitochondria from H9c2 | 5 μM | 24 h | [141] | ||
| OXPHOS | Inhibition of complex IV | NSAIDs | Celecoxib | Thrombosis, MI, stroke | 3–5 µM | Isolated rat heart mitochondria | 16 µg/mL | [14] | |
| OXPHOS | Inhibition of complex IV | Proteasome inhibitor | Bortezomib | QT prolongation, hypotension | 0.3 μM | Male Wistar rats | 0.2 mg/kg | 3 w | [159] |
| OXPHOS | Inhibition of complex IV | Immunosuppressant drug | Cyclosporine A | Cardiotoxicity | 0.5–5 µM | Enzymes and coenzymes | 100 µM | [156] | |
| OXPHOS | Inhibition of complex V | Chemotherapeutic agents | Mitoxantrone | CHF, CMP, decreased LVEF, arrhythmia | 3.3 μM | Isolated rat heart mitochondria | 2.5 mg/kg on d 0, 10, and 20 | 22 d | [160] |
| OXPHOS | Inhibition of complex V | Anticonvulsants | Phenytoin | Bradycardia, hypotension | 87.21 μM | guinea pig heart preparations | 1.0 nM | [161] | |
| OXPHOS | Downregulation of complex I expression | TKIs | Regorafenib | MI; hypertension | 8.08 μM | H9c2 | 20 μM | 72 h | [90] |
| OXPHOS | Downregulation of complex I expression | Nucleoside analogues | Remdesivir | Bradycardia, QT prologation, CA | 9 μM | HiPSC-CMs | 2.5 μM | 3 d | [86] |
| OXPHOS | Downregulation of complex I expression | Addictive drugs | Ethanol | Male C57BL/6J mice | 10% (v/v) | 12 w | [162] | ||
| OXPHOS | Downregulation of complex I expression | Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP | 15.3 μM | Male CD-1 mice (IP) | 9 mg/kg | 1 w | [100] |
| Mitoxantrone | CHF, CMP, decreased LVEF, arrhythmia | 3.3 μM | Male CD-1 mice (IP) | 6 mg/kg | 1 w | [100] | |||
| OXPHOS | Downregulation of complexe II expression | Anesthesia | Propofol | HF, arrhythmia | 30.13 μM | HiPSC-CMs | 10 µg/mL | 48 h | [163] |
| Addictive drugs | Ethanol | Male C57BL/6J mice | 10% (v/v) | 12 w | [162] | ||||
| OXPHOS | Downregulation of complex III expression | Addictive drugs | Ethanol | Male C57BL/6J mice | 10% (v/v) | 12 w | [162] | ||
| Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP | 15.3 μM | Male CD-1 mice (IP) | 9 mg/kg | 1 w | [100] | ||
| OXPHOS | Downregulation of complex IV expression | Mitoxantrone | CHF, CMP, decreased LVEF, arrhythmia | 3.3 μM | Male CD-1 mice (IP) | 6 mg/kg | 1 w | [100] | |
| Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP | 15.3 μM | Male Wistar rats (IV) | 1 mg/kg/w | 7 w(started at 11 w, observed at 48 w) | [96] | ||
| Addictive drugs | Ethanol | Male C57BL/6J mice | 10% (v/v) | 12 w | [162] | ||||
| OXPHOS | Downregulation of complex V expression | Nucleoside analogues | Remdesivir | Bradycardia, QT prologation, CA | 9 μM | HiPSC-CMs | 2.5 μM | 3 d | [86] |
| OXPHOS | Downregulation of complex V expression | TKIs | Regorafenib | MI; hypertension | 8.08 μM | H9c2 | 20 μM | 72 h | [90] |
| OXPHOS | Downregulation of complex V expression | Proteasome inhibitor | Bortezomib | QT prolongation, hypotension | 0.3 μM | Male Wistar rats | 0.2 mg/kg | 1 w | [159] |
| OXPHOS | Downregulation of complex V expression | Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP | 15.3 μM | Male CD-1 mice (IP) | 9 mg/kg | 1 w | [100] |
| Mitoxantrone | CHF, CMP, decreased LVEF, arrhythmia | 3.3 μM | Male CD-1 mice (IP) | 6 mg/kg | 1 w | [100] | |||
| OXPHOS | Downregulation of complex V expression | Nucleoside analogues | Remdesivir | Bradycardia, QT prologation, CA | 9 μM | HiPSC-CMs | 2.5 μM | 3 d | [86] |
| OXPHOS | Downregulation of complex V expression | Addictive drugs | Ethanol | Male C57BL/6J mice | 10% (v/v) | 12 w | [162] | ||
| OXPHOS | Inhibition of OXPHOS | Anti-arrhythmic drug | Clofilium | TDP | 1 μM | - | - | - | [64] |
| OXPHOS | Inhibition of OXPHOS | Antipsychotics | Aripiprazole | - | 2.24 μM | - | - | - | [64] |
| OXPHOS | Inhibition of OXPHOS | TKIs | Sorafenib | Bleeding, hypertension, QT prolongation, CHF, CI, MI | 16.6 μM | HiPSC-CMs | 10 µM | 24 h | [164] |
| OXPHOS | OCR reduction | NSAIDs | Acetylsalicylate | - | 0.5–10 mM | Isolated rat heart mitochondria | 5 mM | [165] | |
| OXPHOS | OCR reduction | NRTIs | Zidovudine | CMP | 4 μM | H9c2 | 50 μM | 3 passages | [152] |
| TMPK-overexpressing H9c2 cells | 100 µM | 24 h | [131] | ||||||
| Didanosine | CMP | 12 μM | H9c2 | 50 μM | 3 passages | [152] | |||
| OXPHOS | OCR reduction | Nucleoside analogues | Remdesivir | Bradycardia, QT prologation, CA | 9 μM | HiPSC-CMs | 2.5 μM | 3 d | [86] |
| OXPHOS | OCR reduction | Cholesterol medications | Simvastatin | Cardiac atrophy | 0.02 μM | H9c2 | 10 μM | 24 h | [151] |
| OXPHOS | OCR reduction | Analgesics | Salicylic acid | - | 0.5–10 mM | Isolated rat heart mitochondria | 5 mM | [165] | |
| OXPHOS | OCR reduction | Local anesthetics | Bupivacaine (marcaine) | VF | 0.7 μM | neonatal mouse cardiomyocytes | 5 μM | [166] | |
| OXPHOS | Reduction in ATP content | Anesthesia | Propofol | HF, arrhythmia | 30.13 μM | Isolated rat heart mitochondria | 300 μM | [167] | |
| OXPHOS | Reduction in ATP content | Local anesthetics | Lidocaine | VF | 36 μM | - | - | - | [168] |
| OXPHOS | Reduction in ATP content | Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP | 15.3 μM | - | 15 mg/kg | - | [158] |
| OXPHOS | Reduction in ATP content | Chemotherapeutic agents | Etoposide | Hypotension | 17 μM | hiPSC-CMs | 30 μM | 48 h | [169] |
| OXPHOS | Mitoxantrone | CHF, CMP, decreased LVEF, arrhythmia | 3.3 μM | [160] | |||||
| OXPHOS | Reduction in ATP content | Alkylating agent | Cyclophosphamide | HMC, CMP CMP, LVD, CHF | 143 μM | Male Wistar rats (IP) | 200 mg/kg | 1 w | [170] |
| Male Wistar rats (IP) | 200 mg/kg | 1 w | [171] | ||||||
| OXPHOS | Reduction in ATP content | Monoclonal antibody | Trastuzumab | 2.59 mM | - | - | - | [172] | |
| OXPHOS | Reduction in ATP content | TKIs | Imatinib mesylate | QT prolongation, CHF, decreased LVEF | 2.71 μM | NRVMs | 5 μM | 24 h | [91] |
| Sunitinib | Decreased LVEF, QT prolongation, TdP, hypertension, HF, CMP Bleeding, hypertension, QT prolongation, CHF, CI, MI | 0.25 μM | Male SD rats (oral) | 10 mg/kg/d | 3 w | [89] | |||
| Male Wistar Rats (oral) | 25 mg/kg/d | 28 d | [173] | ||||||
| NRVMs | 60% of ATP was depleted at 23 µM | 24 h | [174] | ||||||
| Sorafenib | 16.6 μM | Male SD rats (oral) | 10 mg/kg/d | 3 w | [89] | ||||
| Regorafenib | MI; hypertension | 8.08 μM | H9c2 | 5 μM | 48 h | [90] | |||
| OXPHOS | Reduction in ATP content | NSAIDs | Naproxen | - | 100 µM | Isolated rat heart mitochondria | 50 μM | 1 h | [60] |
| Celecoxib | Thrombosis, MI, stroke | 3–5 µM | Isolated rat heart mitochondria | 25 μM | 1 h | [60] | |||
| Diclofenac | Hypertension, arrhythmias - | 7.9 µM | Isolated rat heart mitochondria | 100 μM | 1 h | [60] | |||
| - | - | - | [142] | ||||||
| - | - | - | [175] | ||||||
| Piroxicam | 5 µM | - | - | - | [142] | ||||
| Indomethacin | Hypertension | 6 µM | - | - | - | [142] | |||
| Nimesulide | - | 21.08 µM | - | - | - | [142] | |||
| Meloxicam | HA, stroke | 6.55 µM | - | - | - | [142] | |||
| OXPHOS | Reduction in ATP content | NRTIs | Zidovudine | CMP Bradycardia, QT prologation, CA | 4 μM | Rats (oral) | 125 mg/kg/d | 4 w | [116] |
| TMPK-overexpressing H9c2 cells | Dose response curve(IC50 = 70 μM) | 4 d | [131] | ||||||
| OXPHOS | Reduction in ATP content | Nucleoside analogues | Remdesivir | 9 μM | [176] | ||||
| OXPHOS | Reduction in ATP content | Addictive drugs | Ethanol | Arrhythmias, angina, MI, HF | Male C57BL/6J mice | 10% (v/v) for first w, 14% (v/v) for second w, 18% (v/v) for third w, | 12 w | [162] | |
| H9c2 | 184.34 mM | 24 h | [177] | ||||||
| Cocaine | LQT, TdP, Hypotension, AV block, Arrhythmia, heart block, SBC, CHF, VF | 0.76–0.94 µM | H9c2 | 1.79 mM | 24 h | [177] | |||
| Isolated rat heart mitochondria | 2*7.5 mg/kg/d | 7 d | [178] | ||||||
| Isolated rat heart mitochondria | 2*7.5 mg/kg/d | 7 d | [179] | ||||||
| OXPHOS | Reduction in ATP content | Anti-arrhythmic drug | Amiodarone | 4.65 μM | H9c2 | IC50 = 1.84 µM | 4 h | [139] | |
| Dronedarone | AF, HF | 0.15–0.26 μM | H9c2 | IC50 = 0.49 µM | 4 h | [139] | |||
| OXPHOS | Reduction in ATP content | β-adrenoceptor agonists | Isoproterenol | HF | 0.01 μM | Male Wistar rats (SC) | 100 mg/kg, BID | 12 h | [82] |
| OXPHOS | Reduction in ATP content | Cholesterol medications | Simvastatin | Cardiac atrophy | 0.02 μM | H9c2 | 10 μM (6 h); 100 μM (24 h) | [151] | |
| OXPHOS | Uncoupling of OXPHOS | TKIs | Crizotinib | QT prolongation | 0.73 μM | - | - | - | [64] |
| OXPHOS | Uncoupling of OXPHOS | NSAIDs | Acetylsalicylate | - | 0.5–10 mM | Isolated rat heart mitochondria | 10 mM | [165] | |
| Diclofenac | Hypertension, arrhythmias | 7.9 µM | - | - | - | [142] | |||
| Piroxicam | - | 5 µM | - | - | - | [142] | |||
| Indomethacin | Hypertension | 6 µM | - | - | - | [142] | |||
| Nimesulide | - | 21.08 µM | - | - | - | [142] | |||
| Meloxicam | HA, stroke | 6.55 µM | - | - | - | [142] | |||
| tenidap | - | 8.44 µM (30525499) | - | - | - | [64] | |||
| OXPHOS | Uncoupling of OXPHOS | NRTIs | Zidovudine | CMP | 4 μM | H9c2 | 50 lM | 18 h | [180] |
| Didanosine | CMP | 12 μM | 50 lM | 18 h | [180] | ||||
| OXPHOS | Uncoupling of OXPHOS | Addictive drugs | Ethanol | Isolated mitochondria from rabbit ventricle | 10 µM | 2 h | [181] | ||
| OXPHOS | Uncoupling of OXPHOS | Anti-arrhythmic drug | Amiodarone | LQT, TdP, Hypotension, AV block, Arrhythmia, heart block, SBC, CHF, VF | 4.65 μM | Isolated rat heart mitochondria | 1 µM | [139] | |
| Dronedarone | AF, HF | 0.15–0.26 μM | Isolated rat heart mitochondria | 0.1 µM | [139] | ||||
| OXPHOS | Uncoupling of OXPHOS | Analgesics | Salicylic acid | - | 0.5–10 mM | Isolated rat heart mitochondria | 10 mM | [165] | |
| MMP | Dissipation of MMP | Anthracyclines | DOX | CMP, MI, CHF, VA, pericarditis, myocarditis | 15.3 μM | Kunming mice (IP) | 2 mg/kg | 10 d | [102] |
| KIND-2-derived cardiac cells | 0.24 μM disrupte 48.3% | 48 h | [182] | ||||||
| Daunorubicin | 89 μM | Neonatal rat cardiac cells | 4 μM | 24 h | [97] | ||||
| MMP | Dissipation of MMP | Chemotherapeutic agents | Cisplatin | Decreased LVEF, arrhythmias, ECA, myocarditis, CMP Hypotension | 27.54 μM | C57BL mice (IV) | 10 mg/kg/d | 1 W | [112] |
| NRVMs | 200 μM | 24 h | [183] | ||||||
| Etoposide | 17 μM | HiPSC-CMs | 10 μM | 48 h | [169] | ||||
| As2O3 | QT prolongation TdP, CMP, tachycardia | 12.1 μM | H9c2 | 5 μM | 24 h | [184] | |||
| MMP | Dissipation of MMP | Monoclonal antibody | Trastuzumab | CMP, LVD, CHF | 2.59 mM | H9c2 | 200 nM | 24 h | [185] |
| MMP | Dissipation of MMP | TKIs | Imatinib mesylate | QT prolongation, CHF, decreased LVEF | 2.71 μM | NRVMs | 5 μM | 18 h | [91] |
| Sunitinib | Decreased LVEF, QT prolongation, TdP, hypertension, HF, CMP | 0.25 μM | Male SD rats (oral) | 10 mg/kg/d | 3 W | [89] | |||
| Regorafenib | MI; hypertension | 8.08 μM | H9c2 | 20 μM | 72 h | [90] | |||
| MMP | Dissipation of MMP | NSAIDs | Diclofenac | Hypertension, arrhythmias - | 7.9 µM | - | - | [142] | |
| Isolated rat heart mitochondria | 10 µg/mL | [115] | |||||||
| Mitochondria isolated from mouse hearts | 10 µg/mL | [157] | |||||||
| C57BL/6 mice (oral) | 15 mg/kg/D | [175] | |||||||
| Immortalized human cardiomyocytes | 100 μM | [85] | |||||||
| Piroxicam | 5 µM | - | - | [142] | |||||
| Indomethacin | Hypertension | 6 µM | - | - | [142] | ||||
| Nimesulide | - | 21.08 µM | - | - | [142] | ||||
| Meloxicam | HA, stroke | 6.55 µM | - | - | [142] | ||||
| Meclofenamate sodium | - | 3.55 μM | H9c2 | 5 µM (40% inhibition) | [138] | ||||
| Naproxen | - | 100 µM | Isolated rat heart mitochondria | 25 μM (60 min); 100 μM (30 min) | [60] | ||||
| Diclofenac | Isolated rat heart mitochondria | 50 μM | 5 min | [60] | |||||
| Celecoxib | Thrombosis, MI, stroke | 3–5 µM | Isolated rat heart mitochondria | 25 μM | 5 min | [60] | |||
| MMP | Dissipation of MMP | NRTIs | Zidovudine | CMP | 4 μM | TMPK-overexpressing H9c2 cells | 100 µM | 24 h | [131] |
| MMP | Dissipation of MMP | Anti-arrhythmic drug | Amiodarone | LQT, TdP, Hypotension, AV block, Arrhythmia, heart block, SBC, CHF, VF | 4.65 μM | H9c2 | IC50 = 2.94 μM | 6 h | [139] |
| Dronedarone | AF, HF | 0.15–0.26 μM | H9c2 | IC50 = 0.5 μM | 6 h | [139] | |||
| MMP | Dissipation of MMP | β receptor blocker drugs | Propranolol | Cardiotoxicity | 0.22 μM | Isolated rat heart mitochondria | 5 µg/mL | 5 min | [119] |
| Atenolol | Cardiotoxicity | 4.99 μM | Isolated rat heart mitochondria | 5 µg/mL | 5 min | [119] | |||
| MMP | Dissipation of MMP | Aconitum species | Aconitum sp. | VA | 19.27 μg/ml | H9c2 | 10 μM | 24 h | [186] |
| MMP | Dissipation of MMP | Cholesterol medications | Simvastatin | Cardiac atrophy | 0.02 μM | H9c2 | 10 μM | 24 h | [151] |
| MMP | Dissipation of MMP | Diabetes medication | Pioglitazone | HF | 2.6 μM | Isolated rat heart mitochondria | 12.5 µg/mL | 5 min | [122] |
| MMP | Dissipation of MMP | Anesthesia | Propofol | HF, arrhythmia | 30.13 μM | Isolated rat heart mitochondria | 300 μM | [167] | |
| MMP | Dissipation of MMP | β-adrenoceptor agonists | Isoproterenol | HF | 0.01 μM | Isolated rat heart mitochondria | 85 mg/kg/d | 2 d | [187] |
| mPTP | Increases in mPTP opening | NRTIs | Zidovudine | CMP | 4 μM | TMPK-overexpressing H9c2 cells | 100 µM | 24 h | [131] |
| mPTP | Increases in mPTP opening | Chemotherapeutic agents | As2O3 | QT prolongation TdP, CMP, tachycardia | 12.1 μM | Male BALB/c mice | 2 mg/kg (14 d); 4 mg/kg (3 d) | [84] | |
| mPTP | Increases in mPTP opening | Monoclonal antibody | Trastuzumab | CMP, LVD, CHF | 2.59 mM | - | - | - | [22] |
| mPTP | Loss of cytochrome c | Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP | 15.3 μM | Isolated rat heart mitochondria, subcutaneously (SC) | 2 mg/kg/w | 7 w | [188] |
| mPTP | Loss of cytochrome c | Chemotherapeutic agents | Cisplatin | Decreased LVEF, arrhythmias, ECA, myocarditis, CMP | 27.54 μM | NRVMs | 200 μM | 24 h | [183] |
| mPTP | Loss of cytochrome c | TKIs | Imatinib mesylate | QT prolongation, CHF, decreased LVEF | 2.71 μM | NRVMs | 5 μM | 24 h | [91] |
| mPTP | Loss of cytochrome c | NRTIs | Zidovudine | CMP | 4 μM | Rats (oral) | 125 mg/kg/d | [116] | |
| mPTP | Loss of cytochrome c | β receptor blocker drugs | Propranolol | Cardiotoxicity | 0.22 μM | Isolated rat heart mitochondria | 5 µg/mL | 5 min | [119] |
| Atenolol | Cardiotoxicity | 4.99 μM | Isolated rat heart mitochondria | 10 µg/mL | 5 min | [119] | |||
| mPTP | Loss of cytochrome c | Macrolide antibiotics | Azithromycin | Arrhythmia | 0.32–0.87 μM | Isolated rat heart mitochondria | 50 μM | [120] | |
| Clarithromycin | TdP | 2.67–13.37 μM | Isolated rat heart mitochondria | 50 μM | [120] | ||||
| Erythromycin | TdP | 11 μM | Isolated rat heart mitochondria | 50 μM | [120] | ||||
| mPTP | Loss of cytochrome c | Diabetes medication | Pioglitazone | HF | 2.6 μM | Isolated rat heart mitochondria | 12.5 µg/mL | [122] |
| Modules | Alterations | Pharmacology | Drugs | Clinical Manifestations | Cmax | Models | Dose | Time | References |
|---|---|---|---|---|---|---|---|---|---|
| FA oxidation | Downregulation of FA oxidation related proteins expression | Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP | 15.3 μM | Male CD-1 mice (IP) | 9 mg/kg | 1 w | [100] |
| Mitoxantrone | CHF, CMP, decreased LVEF, arrhythmia | 3.3 μM | Male CD-1 mice (IP) | 6 mg/kg | 1 w | [100] | |||
| FA oxidation | Downregulation of FA oxidation related proteins expression | Alkylating agent | Cyclophosphamide | HMC, CMP | 143 μM | Male Wistar rats (IP) | 200 mg/kg | 10 d | [189] |
| TCA cycle | Downregulation of TCA related proteins expression | Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP | 15.3 μM | Male CD-1 mice (IP) | 9 mg/kg | 1 w | [100] |
| Mitoxantrone | CHF, CMP, decreased LVEF, arrhythmia | 3.3 μM | Male CD-1 mice (IP) | 6 mg/kg | 1 w | [100] | |||
| TCA cycle | Inhibition of the Krebs cycle enzyme | NSAIDs | Acetylsalicylate | - | 0.5–10 mM | Isolated rat heart mitochondria | Dose response curve | [165] | |
| TCA cycle | Inhibition of the Krebs cycle enzyme | Analgesics | Salicylic acid | - | 0.5–10 mM | Isolated rat heart mitochondria | Dose response curve | [165] | |
| TCA cycle | Inhibition of the Krebs cycle enzyme | Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP | 15.3 μM | Male Wistar rats (IP) | 2.5 mg/kg/2 d | 2 w | [95] |
| TCA cycle | Inhibition of the Krebs cycle enzyme | Alkylating agent | Cyclophosphamide | HMC, CMP | 143 μM | Male Wistar rats (IP) | 200 mg/kg | 10 d | [124] |
| Male Wistar rats (IP) | 200 mg/kg | 10 d | [111] | ||||||
| TCA cycle | Inhibition of the Krebs cycle enzyme | β-adrenoceptor agonists | Isoproterenol | HF | 0.01 μM | Male Wistar rats (SC) | 100 mg/kg, BID | 12 h | [83] |
| TCA cycle | Loss of Krebs cycle enzymes | Addictive drugs | Ethanol | Wistar male albino rats | 3 g/kg/d | 10 d | [190] |
| Modules | Alterations | Pharmacology | Drugs | Clinical Manifestations | Cmax | Models | Dose | Time | References |
|---|---|---|---|---|---|---|---|---|---|
| Redox | Decrease in antioxidant enzyme level | NSAIDs | Naproxen | - | 100 µM | Isolated rat heart mitochondria | 25 μM | [60] | |
| Celecoxib | Thrombosis, MI, stroke | 3–5 µM | Isolated rat heart mitochondria | 50 μM | [60] | ||||
| Diclofenac | - | 3.55 μM | Isolated rat heart mitochondria | 25 μM | [60] | ||||
| Redox | Decrease in antioxidant enzyme level | β-adrenoceptor agonists | Isoproterenol | HF | 0.01 μM | Male Wistar rats (SC) | 100 mg/kg, BID | 12 h | [118] |
| Redox | Inhibition of antioxidant enzyme | Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP | 15.3 μM | Male Wistar rats (IP) | 2.5 mg/kg/2 d | 2 w | [95] |
| Kunming mice (IP) | 2 mg/kg | 10 d | [102] | ||||||
| - | - | - | [199] | ||||||
| Male BALB/c mice (IP) | 5 mg/kg/w | 2 w | [200] | ||||||
| Male Wistar rats (IV) | 45 mg/kg | 48 h | [203] | ||||||
| Idarubicin | CMP, MI, CHF, VA, decreased LVEF | 23.22 μM | Rats (IV) | 5 mg/kg/w | 6 w | [110] | |||
| Redox | Inhibition of antioxidant enzyme | Alkylating agent | Cyclophosphamide | HMC, CMP | 143 μM | Male Wistar rats | 200 mg/kg | 1 w | [170] |
| Male Wistar rats (IP) | 200 mg/kg | 1 w | [171] | ||||||
| Redox | Inhibition of antioxidant enzyme | Chemotherapeutic agents | Cisplatin | Decreased LVEF, arrhythmias, ECA, myocarditis, CMP | 27.54 μM | NRVMs | 200 μM | 24 h | [183] |
| As2O3 | QT prolongation TdP, CMP, tachycardia | 12.1 μM | BALB/c mice (IV) | 1 mg/kg/2 d | 6 d | [204] | |||
| Isolated mitochondria from H9c2 | 5 μM | 24 h | [141] | ||||||
| Redox | Inhibition of antioxidant enzyme | TKIs | Sunitinib | Decreased LVEF, QT prolongation, TdP, hypertension, HF, CMP | 0.25 μM | NRVMs | 67% of GSH was oxidized at 23 µM | 24 h | [174] |
| Redox | Inhibition of antioxidant enzyme | NRTIs | Zidovudine | CMP | 4 μM | Male OF1 mice (oral) | 10 mg/kg/d | 35 d | [205] |
| Redox | Inhibition of antioxidant enzyme | Addictive drugs | Cocaine | Arrhythmias, angina, MI, HF | 0.76–0.94 µM | H9c2 | 1.79 mM | 24 h | [177] |
| Redox | Inhibition of antioxidant enzyme | β-adrenoceptor agonists | Isoproterenol | HF | 0.01 μM | Male Wistar rats (SC) | 100 mg/kg, BID | 12 h | [83] |
| Redox | ROS elevation | Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP | 15.3 μM | Beef heart submitochondrial preparations | - | - | [206] |
| - | - | - | [199] | ||||||
| Daunorubicin | CMP, MI, CHF, VA, pericarditis, myocarditis | 89 μM | - | - | - | [207] | |||
| Idarubicin | CMP, MI, CHF, VA, decreased LVEF | 23.22 μM | - | - | - | [207] | |||
| Redox | ROS elevation | Chemotherapeutic agents | Cisplatin | Decreased LVEF, arrhythmias, ECA, myocarditis, CMP | 27.54 μM | NRVMs | 200 μM | 24 h | [183] |
| Etoposide | Hypotension | 17 μM | HiPSC-CMs | 10 μM | 48 h | [169] | |||
| As2O3 | QT prolongation TdP, CMP, tachycardia | 12.1 μM | Male BALB/c mice | 2 mg/kg (14 d); 4 mg/kg (7 d) | [84] | ||||
| Isolated mitochondria from H9c2 | 5 μM | 24 h | [141] | ||||||
| H9c2 | 5 μM | 24 h | [184] | ||||||
| Redox | ROS elevation | Monoclonal antibody | Trastuzumab | CMP, LVD, CHF | 2.59 mM | H9c2 | 200 nM | 24 h | [185] |
| Redox | ROS elevation | TKIs | Sorafenib | Bleeding, hypertension, QT prolongation, CHF, CI, MI | 16.6 μM | NRVMs | 4.5 µM | 10 min | [32] |
| Redox | ROS elevation | NSAIDs | Diclofenac | Hypertension, arrhythmias | 3.55 μM 7.9 µM | Isolated rat heart mitochondria | 25 μM | 5 min | [60] |
| H9c2 | 10 µM | 1.5 h | [157] | ||||||
| Isolated rat heart mitochondria | 10 µg/mL | 1 h | [115] | ||||||
| C57BL/6 mice (oral) | 15 mg/kg/d | 4 w | [175] | ||||||
| Immortalized human cardiomyocytes | 100 μM | 24 h | [85] | ||||||
| Naproxen | - | 100 µM | Isolated rat heart mitochondria | 25 μM | 5 min | [60] | |||
| Celecoxib | Thrombosis, MI, stroke | 3–5 µM | Isolated rat heart mitochondria | 25 μM | 5 min | [60] | |||
| Redox | ROS elevation | NRTIs | Zidovudine | CMP | 4 μM | H9c2 | 50 μM | 3 passages | [152] |
| TMPK-overexpressing H9c2 cells | 100 µM | 24 h | [131] | ||||||
| Human cardiomyocytes | 10 µM | 48 h | [198] | ||||||
| Didanosine | CMP | 12 μM | H9c2 | 50 μM | 3 passages | [152] | |||
| Redox | ROS elevation | Addictive drugs | Ethanol | H9c2 | 184.34 mM | 24 h | [177] | ||
| Cocaine | Arrhythmias, angina, MI, HF | 0.76–0.94 µM | H9c2 | 1.79 mM | 24 h | [177] | |||
| Isolated rat heart mitochondria | 2 × 7.5 mg/kg/d | 8 d | [178] | ||||||
| Isolated rat heart mitochondria | 2 × 7.5 mg/kg/d | 7 d | [179] | ||||||
| Redox | ROS elevation | β-adrenoceptor agonists | Isoproterenol | HF | 0.01 μM | Isolated rat heart mitochondria | 85 mg/kg/d | 2 d | [187] |
| Redox | ROS elevation | β receptor blocker drugs | Propranolol | Cardiotoxicity | 0.22 μM | Isolated rat heart mitochondria | 5 µg/mL | 5 min | [119] |
| Atenolol | Cardiotoxicity | 4.99 μM | Isolated rat heart mitochondria | 5 µg/mL | 30 min | [119] | |||
| Macrolide antibiotics | Azithromycin | Arrhythmia | 0.32–0.87 μM | Isolated rat heart mitochondria | 25 μM | 15 min | [120] | ||
| Clarithromycin | TdP | 2.67–13.37 μM | Isolated rat heart mitochondria | 25 μM | 15 min | [120] | |||
| Erythromycin | TdP | 11 μM | Isolated rat heart mitochondria | 25 μM | 15 min | [120] | |||
| Redox | ROS elevation | Diabetes medication | Pioglitazone | HF | 2.6 μM | Isolated rat heart mitochondria | 12.5 µg/mL | 5 min | [122] |
| Redox | ROS elevation | Local anesthetics | Bupivacaine (marcaine) | VF | 0.7 μM | H9c2 | 1 mM | 24 h | [208] |
| Redox | Nitrozative stress | Anthracyclines | Epirubicin | CHF | 5.68 mM | Male Wistar rats (IP) | 10 mg/kg | 10 d | [209] |
| Redox | Nitrozative stress | Alkylating agent | Cyclophosphamide | HMC, CMP | 143 μM | Male Wistar rats (IP) | 200 mg/kg | 1 w | [171] |
| Redox | 8OHdG adducts in mtDNA | Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP | 15.3 μM | SD rats (IP) | 2 mg/kg/w | 6 w | [210] |
| Redox | Lipid peroxidation | Anthracyclines | DOX | CHF, decreased LVEF, ST, myocarditis, CMP | 15.3 μM | Male Wistar rats (IP) | 2.5 mg/kg/2 d | 2 w | [95] |
| Male Wistar rats (IV) | 45 mg/kg | 48 h | [203] | ||||||
| Daunorubicin | CMP, MI, CHF, VA, pericarditis, myocarditis | 89 μM | Male SD rats | 2.5 mg/kg/w | 5 w | [211] | |||
| Idarubicin | CMP, MI, CHF, VA, decreased LVEF | 23.22 μM | Male SD rats (IV) | 5 mg/kg/w | 6 w | [110] | |||
| Redox | Lipid peroxidation | Alkylating agent | Cyclophosphamide | HMC, CMP | 143 μM | Male Wistar rats | 200 mg/kg | 1 w | [170] |
| Redox | Lipid peroxidation | Chemotherapeutic agents | Cisplatin | Decreased LVEF, arrhythmias, ECA, myocarditis, CMP | 27.54 μM | NRVMs | 200 μM | 24 h | [183] |
| Redox | Lipid peroxidation | NSAIDs | Diclofenac | Hypertension, arrhythmias | 7.9 µM | Isolated rat heart mitochondria | 50 μM | 1 h | [60] |
| Isolated rat heart mitochondria | 10 µg/mL | 1 h | [115] | ||||||
| Naproxen | - | 100 µM | Isolated rat heart mitochondria | 100 μM | 1 h | [60] | |||
| Celecoxib | Thrombosis, MI, stroke | 3–5 µM | Isolated rat heart mitochondria | 100 μM | 1 h | [60] | |||
| Redox | Lipid peroxidation | NRTIs | Zidovudine | CMP | 4 μM | Male OF1 mice (oral) | 10 mg/kg/d | 35 d | [205] |
| Redox | Lipid peroxidation | β-adrenoceptor agonists | Isoproterenol | CHF, decreased LVEF, ST, myocarditis, CMP | 0.01 μM | Rat, subcutaneously (SC) | 100 mg/kg, BID | 12 h | [118] |
| Male Wistar rats (SC) | 100 mg/kg, BID | 12 h | [83] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, X.; Wang, Z.; Hu, S.; Zhou, B. Assessing Drug-Induced Mitochondrial Toxicity in Cardiomyocytes: Implications for Preclinical Cardiac Safety Evaluation. Pharmaceutics 2022, 14, 1313. https://doi.org/10.3390/pharmaceutics14071313
Tang X, Wang Z, Hu S, Zhou B. Assessing Drug-Induced Mitochondrial Toxicity in Cardiomyocytes: Implications for Preclinical Cardiac Safety Evaluation. Pharmaceutics. 2022; 14(7):1313. https://doi.org/10.3390/pharmaceutics14071313
Chicago/Turabian StyleTang, Xiaoli, Zengwu Wang, Shengshou Hu, and Bingying Zhou. 2022. "Assessing Drug-Induced Mitochondrial Toxicity in Cardiomyocytes: Implications for Preclinical Cardiac Safety Evaluation" Pharmaceutics 14, no. 7: 1313. https://doi.org/10.3390/pharmaceutics14071313