
Targeted Nanocarrier Delivery of RNA Therapeutics to Control HIV Infection


Abstract
:1. Introduction
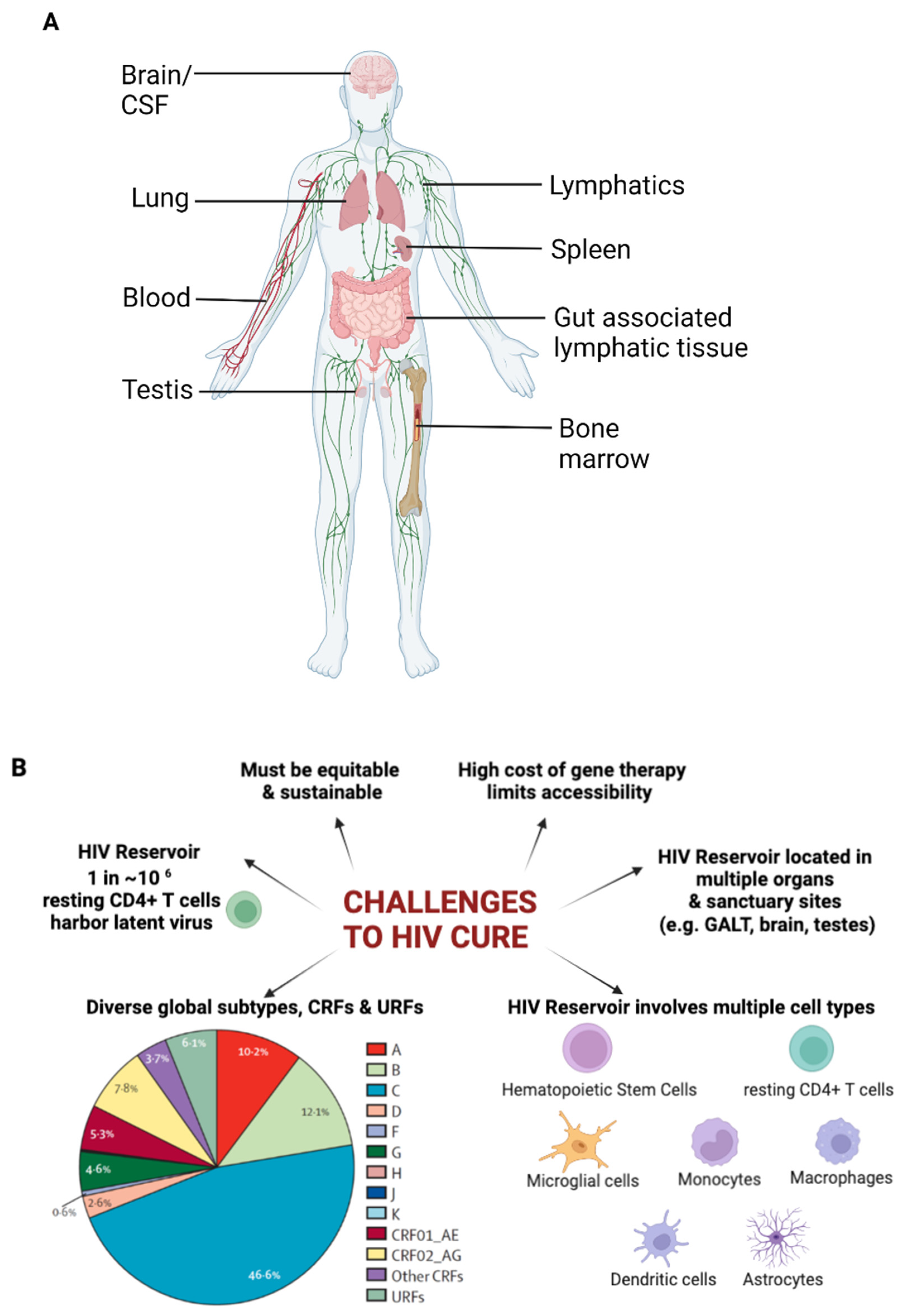
2. HIV Latent Reservoir
3. HIV Cure Strategies
3.1. Hematopoietic Stem Cell Transplant
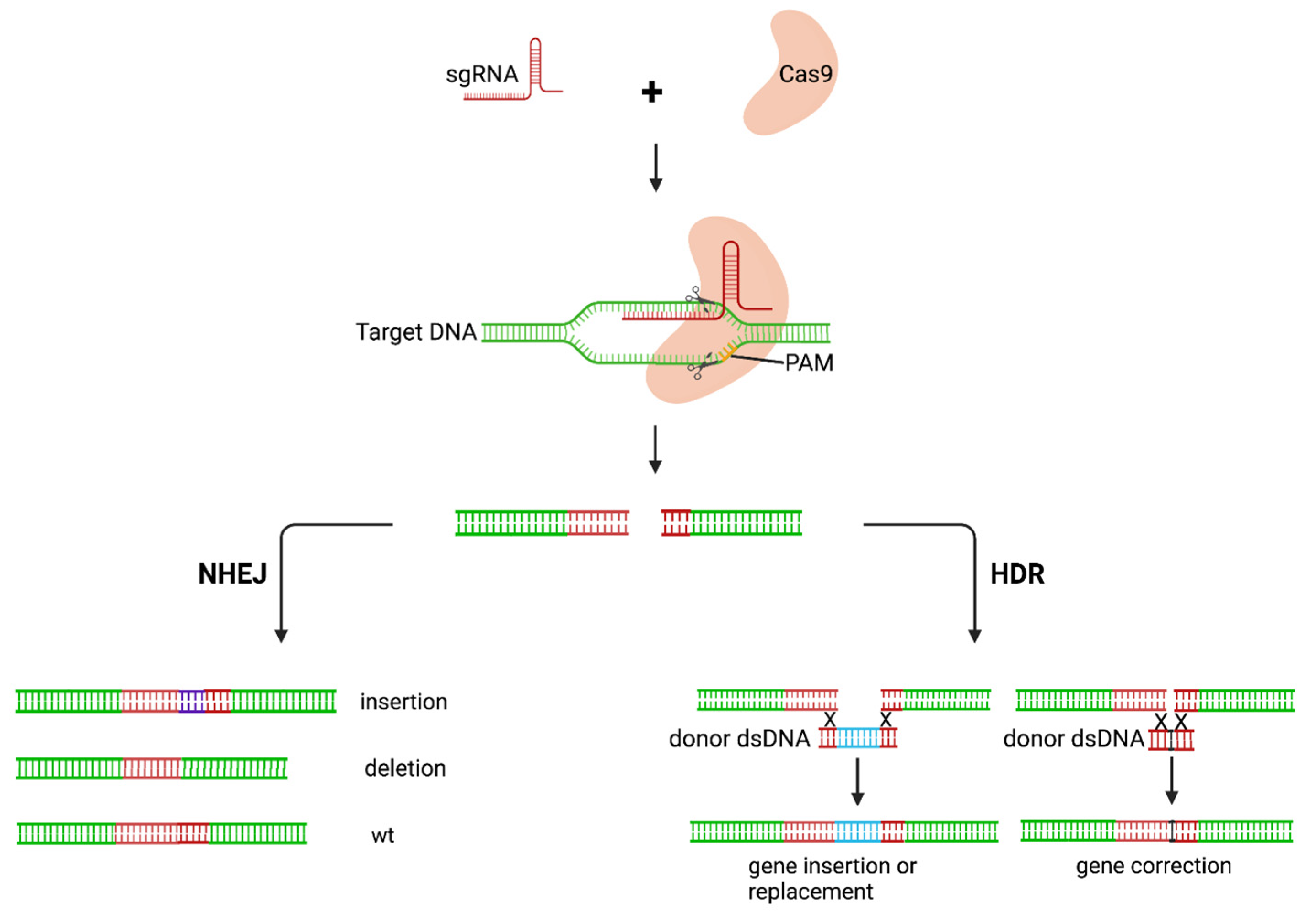
3.2. Gene-Editing of HIV Provirus
3.3. Shock and Kill
3.4. Block and Lock
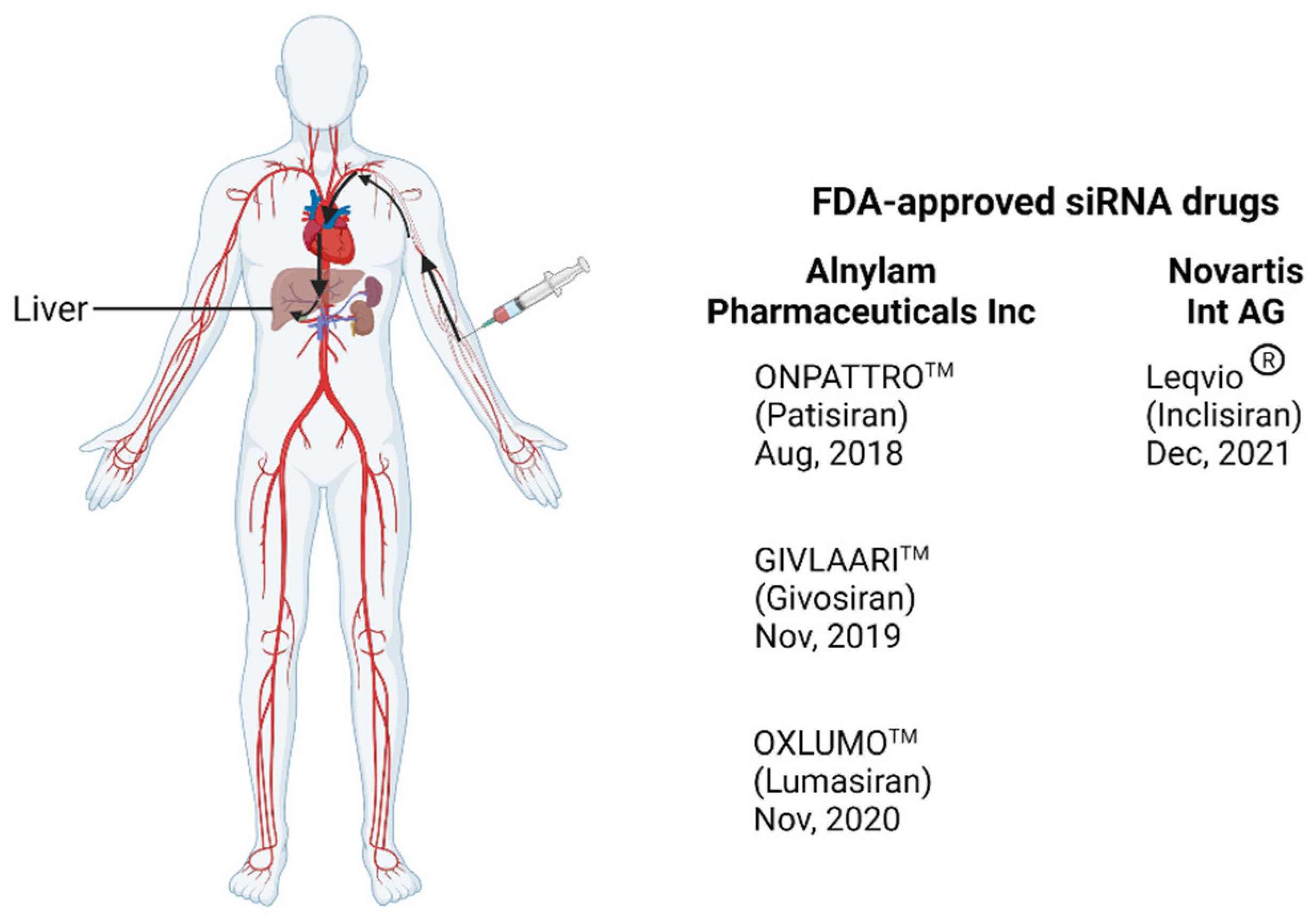
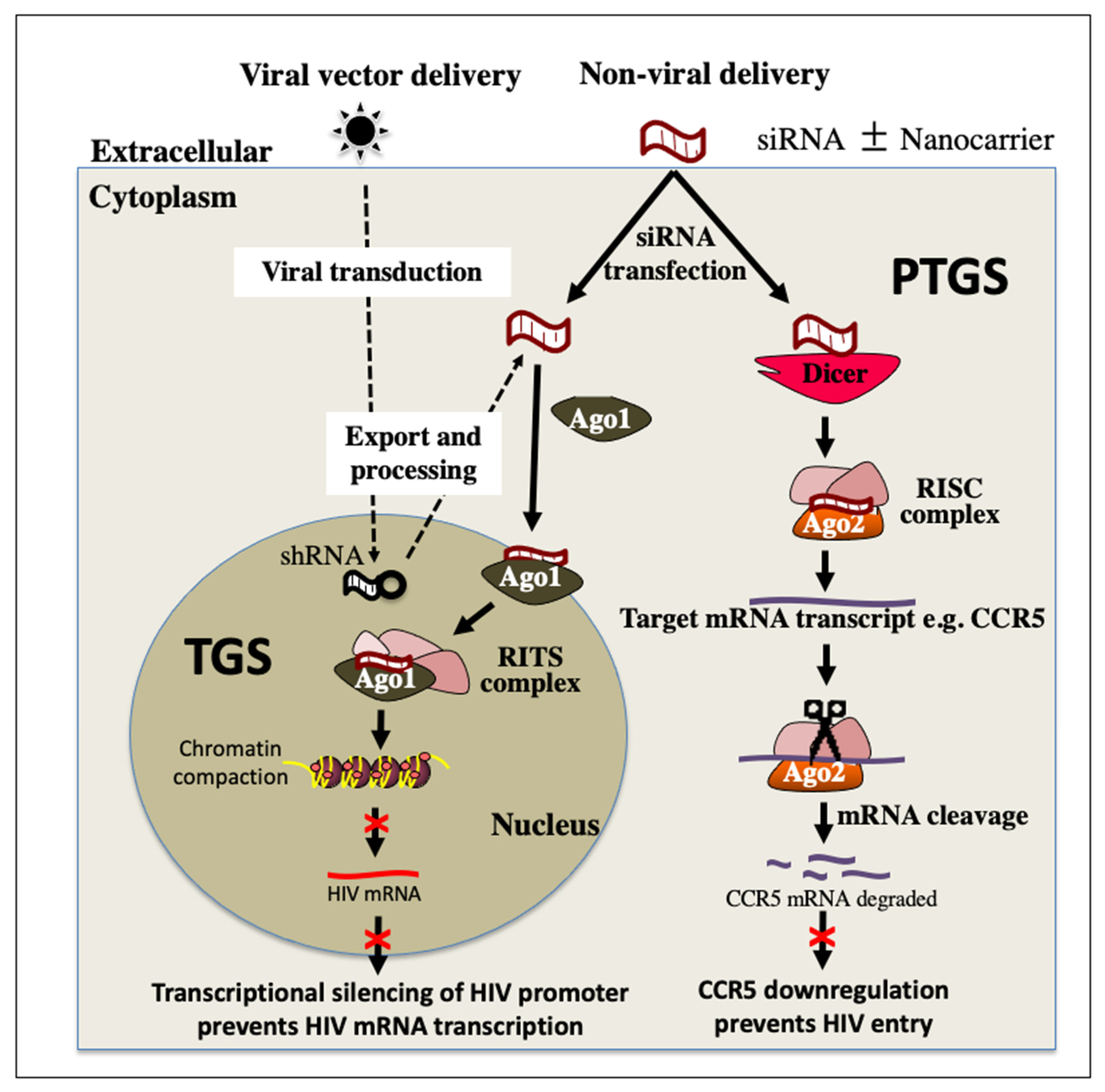
4. RNA Interference (RNAi) Therapeutics for HIV Treatment
5. Non-Viral Delivery Systems
5.1. Nanocarriers
5.1.1. Lipid Nanoparticle
5.1.2. Inorganic Nanocarriers
5.1.3. Polymer-Based Systems
- siRNA-polymer bioconjugates
- Polymeric complexes
5.1.4. N-Acetylgalactosamine Conjugation
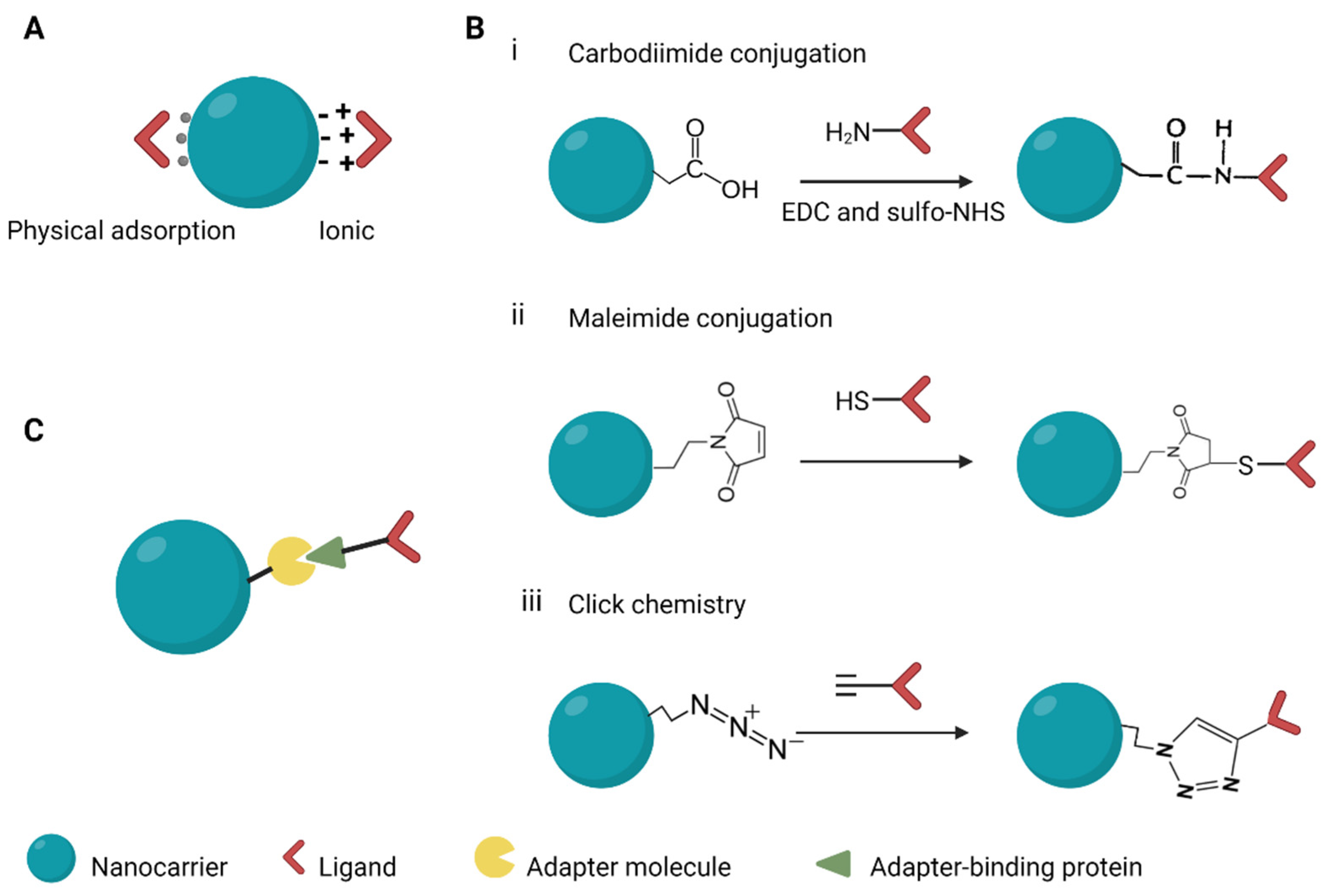
6. Surface Modification of Nanocarriers to Improve Delivery Efficiency
6.1. Targeting Moieties for Specific Cell Types

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Ligand | Targets | Description | Reference |
|---|---|---|---|---|
| Antibodies | Herceptin scAbP-SCA Anti-B2R | HER2Anti-prostate stem cell antigen Bradykinin B2 receptor | High binding affinity High cost of production | [95,141,142] |
| Peptides and proteins | Cilengitide MAdCAM-1 CXCL13 | Integrins Integrin α4β7 + CXCR5 receptor | Low immunogenicity High binding affinity | [146,147] |
| Aptamers | 2′-fluoro-pyridine-RNA aptamer Pegaptanib | Prostate specific membrane antigen VEGF receptor | High specificity and sensitivity Low immunogenicity Low molecular weight High cost of production | [150,151] |
| Small molecules | Folate GalNAc | Folate receptors Asialoglycoprotein receptor | Low molecular weight Ease of production | [140,152] |
6.2. Approaches for Linking Targeting Moieties to NCs
7. Chemical Modifications
7.1. Modification to Nanocarriers
7.2. Modification to siRNA
8. Progression of RNA Therapeutics to the Clinic: Manufacturing Challenges
9. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Damase, T.R.; Sukhovershin, R.; Boada, C.; Taraballi, F.; Pettigrew, R.I.; Cooke, J.P. The Limitless Future of RNA Therapeutics. Front. Bioeng. Biotechnol. 2021, 9, 628137. [Google Scholar] [CrossRef]
- Ibba, M.L.; Ciccone, G.; Esposito, C.L.; Catuogno, S.; Giangrande, P.H. Advances in mRNA non-viral delivery approaches. Adv. Drug Deliv. Rev. 2021, 177, 113930. [Google Scholar] [CrossRef] [PubMed]
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef]
- Schlimgen, R.; Howard, J.; Wooley, D.; Thompson, M.; Baden, L.R.; Yang, O.O.; Christiani, D.C.; Mostoslavsky, G.; Diamond, D.V.; Duane, E.G.; et al. Risks Associated with Lentiviral Vector Exposures and Prevention Strategies. J. Occup. Environ. Med. 2016, 58, 1159–1166. [Google Scholar] [CrossRef] [Green Version]
- Kaur, I.P.; Sharma, G.; Singh, M.; Sandhu, S.K.; Deol, P.K.; Yadav, M.; Yakhmi, J.V. Chapter 13—Nanobiomaterials as gene-delivery vehicles. In Nanobiomaterials in Drug Delivery; Grumezescu, A.M., Ed.; William Andrew Publishing: Norwich, NY, USA, 2016; pp. 447–486. [Google Scholar] [CrossRef]
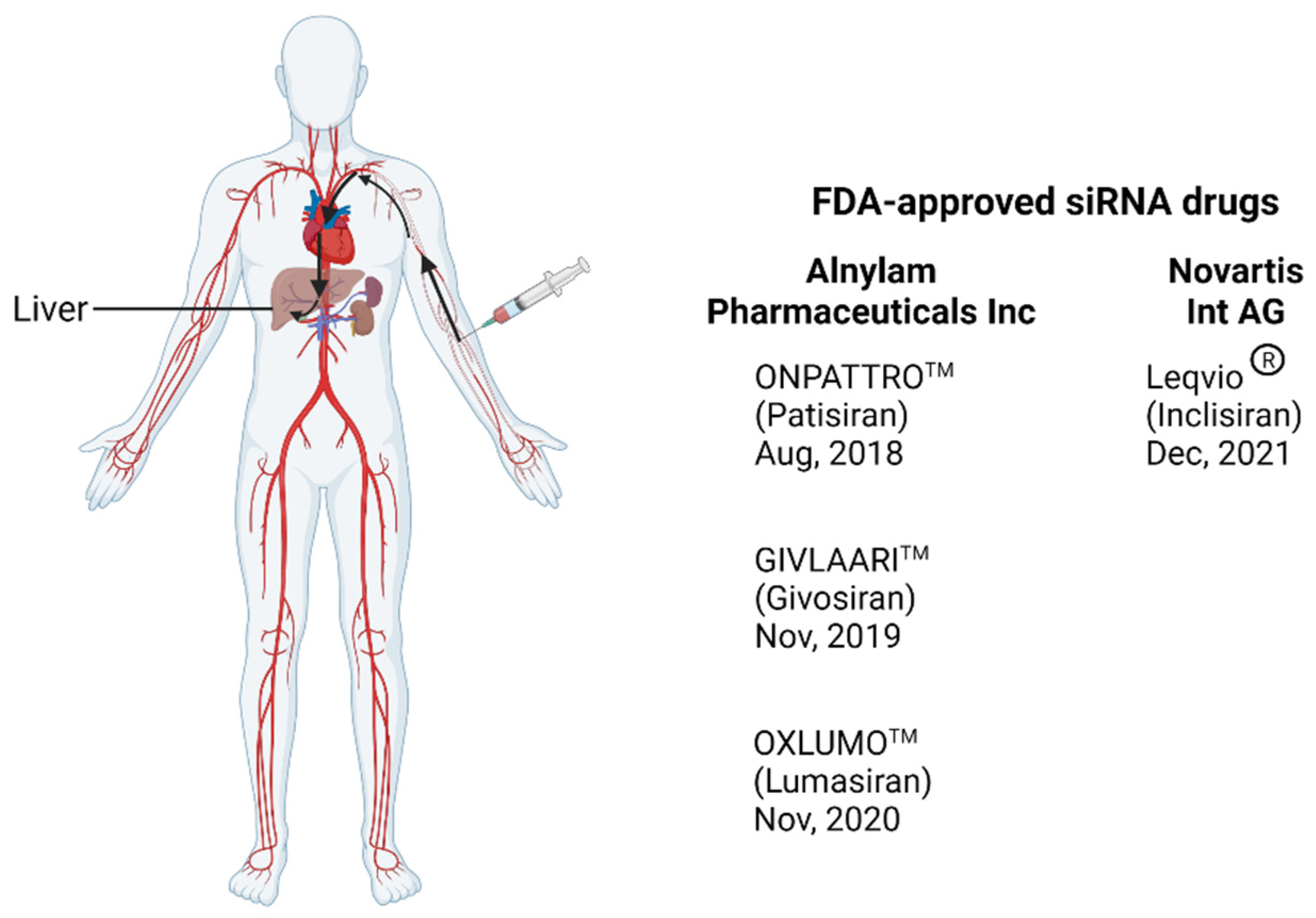
- Kristen, A.V.; Ajroud-Driss, S.; Conceição, I.; Gorevic, P.; Kyriakides, T.; Obici, L. Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener. Dis. Manag. 2019, 9, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Coelho, T.; Adams, D.; Silva, A.; Lozeron, P.; Hawkins, P.N.; Mant, T.; Perez, J.; Chiesa, J.; Warrington, S.; Tranter, E.; et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N. Engl. J. Med. 2013, 369, 819–829. [Google Scholar] [CrossRef]
- Zhang, M.M.; Bahal, R.; Rasmussen, T.P.; Manautou, J.E.; Zhong, X.-b. The growth of siRNA-based therapeutics: Updated clinical studies. Biochem. Pharmacol. 2021, 189, 114432. [Google Scholar] [CrossRef] [PubMed]
- Garrelfs, S.F.; Frishberg, Y.; Hulton, S.A.; Koren, M.J.; O’Riordan, W.D.; Cochat, P.; Deschênes, G.; Shasha-Lavsky, H.; Saland, J.M.; van’t Hoff, W.G.; et al. Lumasiran, an RNAi Therapeutic for Primary Hyperoxaluria Type 1. N. Engl. J. Med. 2021, 384, 1216–1226. [Google Scholar] [CrossRef]
- Cupido, A.J.; Kastelein, J.J.P. Inclisiran for the treatment of hypercholesterolaemia: Implications and unanswered questions from the ORION trials. Cardiovasc. Res. 2020, 116, e136–e139. [Google Scholar] [CrossRef]
- Wong, J.K.; Yukl, S.A. Tissue reservoirs of HIV. Curr. Opin. HIV AIDS 2016, 11, 362–370. [Google Scholar] [CrossRef] [Green Version]
- Ahlenstiel, C.L.; Turville, S.G. Delivery of gene therapy to resting immune cells for an HIV cure. Curr. Opin. HIV AIDS 2019, 14, 129–136. [Google Scholar] [CrossRef]
- UNAIDS. UNAIDS DATA 2021; UNAIDS: Geneva, Switzerland, 2021. [Google Scholar]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.-R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef]
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef]
- Castro-Gonzalez, S.; Colomer-Lluch, M.; Serra-Moreno, R. Barriers for HIV Cure: The Latent Reservoir. AIDS Res. Hum. Retrovir. 2018, 34, 739–759. [Google Scholar] [CrossRef]
- Denton, P.W.; Long, J.M.; Wietgrefe, S.W.; Sykes, C.; Spagnuolo, R.A.; Snyder, O.D.; Perkey, K.; Archin, N.M.; Choudhary, S.K.; Yang, K.; et al. Targeted cytotoxic therapy kills persisting HIV infected cells during ART. PLoS Pathog. 2014, 10, e1003872. [Google Scholar] [CrossRef] [Green Version]
- Pincus, S.H.; Song, K.; Maresh, G.A.; Hamer, D.H.; Dimitrov, D.S.; Chen, W.; Zhang, M.Y.; Ghetie, V.F.; Chan-Hui, P.Y.; Robinson, J.E.; et al. Identification of Human Anti-HIV gp160 Monoclonal Antibodies That Make Effective Immunotoxins. J. Virol. 2017, 91, e01955-16. [Google Scholar] [CrossRef] [Green Version]
- Gaebler, C.; Nogueira, L.; Stoffel, E.; Oliveira, T.Y.; Breton, G.; Millard, K.G.; Turroja, M.; Butler, A.; Ramos, V.; Seaman, M.S.; et al. Prolonged viral suppression with anti-HIV-1 antibody therapy. Nature 2022, 606, 368–374. [Google Scholar] [CrossRef]
- Caskey, M. Broadly neutralizing antibodies for the treatment and prevention of HIV infection. Curr. Opin. HIV AIDS 2020, 15, 49–55. [Google Scholar] [CrossRef]
- Hütter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Müssig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kücherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.K.; Abdul-Jawad, S.; McCoy, L.E.; Mok, H.P.; Peppa, D.; Salgado, M.; Martinez-Picado, J.; Nijhuis, M.; Wensing, A.M.J.; Lee, H.; et al. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic stem-cell transplantation. Nature 2019, 568, 244–248. [Google Scholar] [CrossRef]
- National Insitutes of Health. Researchers Document Third Known Case of HIV Remission Involving Stem Cell Transplant. Available online: https://www.nih.gov/news-events/news-releases/researchers-document-third-known-case-hiv-remission-involving-stem-cell-transplant (accessed on 15 February 2022).
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. A Combinatorial CRISPR-Cas9 Attack on HIV-1 DNA Extinguishes All Infectious Provirus in Infected T Cell Cultures. Cell Rep. 2016, 17, 2819–2826. [Google Scholar] [CrossRef] [Green Version]
- Ji, H.; Lu, P.; Liu, B.; Qu, X.; Wang, Y.; Jiang, Z.; Yang, X.; Zhong, Y.; Yang, H.; Pan, H.; et al. Zinc-Finger Nucleases Induced by HIV-1 Tat Excise HIV-1 from the Host Genome in Infected and Latently Infected Cells. Mol. Ther. Nucleic Acids 2018, 12, 67–74. [Google Scholar] [CrossRef] [Green Version]
- DiGiusto, D.L.; Cannon, P.M.; Holmes, M.C.; Li, L.; Rao, A.; Wang, J.; Lee, G.; Gregory, P.D.; Kim, K.A.; Hayward, S.B.; et al. Preclinical development and qualification of ZFN-mediated CCR5 disruption in human hematopoietic stem/progenitor cells. Mol. Ther. Methods Clin. Dev. 2016, 3, 16067. [Google Scholar] [CrossRef] [Green Version]
- Stone, D.; Kiem, H.-P.; Jerome, K.R. Targeted gene disruption to cure HIV. Curr. Opin. HIV AIDS 2013, 8, 217–223. [Google Scholar] [CrossRef]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Char, S.N.; Neelakandan, A.K.; Nahampun, H.; Frame, B.; Main, M.; Spalding, M.H.; Becraft, P.W.; Meyers, B.C.; Walbot, V.; Wang, K.; et al. An Agrobacterium-delivered CRISPR/Cas9 system for high-frequency targeted mutagenesis in maize. Plant Biotechnol. J. 2017, 15, 257–268. [Google Scholar] [CrossRef]
- Lai, M.; Maori, E.; Quaranta, P.; Matteoli, G.; Maggi, F.; Sgarbanti, M.; Crucitta, S.; Pacini, S.; Turriziani, O.; Antonelli, G.; et al. CRISPR/Cas9 Ablation of Integrated HIV-1 Accumulates Proviral DNA Circles with Reformed Long Terminal Repeats. J. Virol. 2021, 95, e0135821. [Google Scholar] [CrossRef]
- Dash, P.K.; Kaminski, R.; Bella, R.; Su, H.; Mathews, S.; Ahooyi, T.M.; Chen, C.; Mancuso, P.; Sariyer, R.; Ferrante, P.; et al. Sequential LASER ART and CRISPR Treatments Eliminate HIV-1 in a Subset of Infected Humanized Mice. Nat. Commun. 2019, 10, 2753. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, P.; Chen, C.; Kaminski, R.; Gordon, J.; Liao, S.; Robinson, J.A.; Smith, M.D.; Liu, H.; Sariyer, I.K.; Sariyer, R.; et al. CRISPR based editing of SIV proviral DNA in ART treated non-human primates. Nat. Commun. 2020, 11, 6065. [Google Scholar] [CrossRef]
- Jurczyszak, D.; Manganaro, L.; Buta, S.; Gruber, C.; Martin-Fernandez, M.; Taft, J.; Patel, R.; Cipolla, M.; Alshammary, H.; Mulder, L.C.F.; et al. ISG15 deficiency restricts HIV-1 infection. PLoS Pathog. 2022, 18, e1010405. [Google Scholar] [CrossRef]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [Green Version]
- Manghwar, H.; Lindsey, K.; Zhang, X.; Jin, S. CRISPR/Cas System: Recent Advances and Future Prospects for Genome Editing. Trends Plant Sci. 2019, 24, 1102–1125. [Google Scholar] [CrossRef] [Green Version]
- Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef]
- Freije, C.A.; Myhrvold, C.; Boehm, C.K.; Lin, A.E.; Welch, N.L.; Carter, A.; Metsky, H.C.; Luo, C.Y.; Abudayyeh, O.O.; Gootenberg, J.S.; et al. Programmable Inhibition and Detection of RNA Viruses Using Cas13. Mol. Cell 2019, 76, 826–837.e811. [Google Scholar] [CrossRef] [Green Version]
- Fareh, M.; Zhao, W.; Hu, W.; Casan, J.M.L.; Kumar, A.; Symons, J.; Zerbato, J.M.; Fong, D.; Voskoboinik, I.; Ekert, P.G.; et al. Reprogrammed CRISPR-Cas13b suppresses SARS-CoV-2 replication and circumvents its mutational escape through mismatch tolerance. Nat. Commun. 2021, 12, 4270. [Google Scholar] [CrossRef]
- Ait-Ammar, A.; Kula, A.; Darcis, G.; Verdikt, R.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Rohr, O.; Van Lint, C. Current Status of Latency Reversing Agents Facing the Heterogeneity of HIV-1 Cellular and Tissue Reservoirs. Front. Microbiol. 2019, 10, 3060. [Google Scholar] [CrossRef] [Green Version]
- Spivak, A.M.; Planelles, V. Novel Latency Reversal Agents for HIV-1 Cure. Annu. Rev. Med. 2018, 69, 421–436. [Google Scholar] [CrossRef] [Green Version]
- Ahlenstiel, C.L.; Symonds, G.; Kent, S.J.; Kelleher, A.D. Block and Lock HIV Cure Strategies to Control the Latent Reservoir. Front. Cell. Infect. Microbiol. 2020, 10, 424. [Google Scholar] [CrossRef]
- Acchioni, C.; Palermo, E.; Sandini, S.; Acchioni, M.; Hiscott, J.; Sgarbanti, M. Fighting HIV-1 Persistence: At the Crossroads of “Shoc-K and B-Lock”. Pathogens 2021, 10, 1517. [Google Scholar] [CrossRef]
- Spina, C.A.; Anderson, J.; Archin, N.M.; Bosque, A.; Chan, J.; Famiglietti, M.; Greene, W.C.; Kashuba, A.; Lewin, S.R.; Margolis, D.M.; et al. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog. 2013, 9, e1003834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vansant, G.; Bruggemans, A.; Janssens, J.; Debyser, Z. Block-And-Lock Strategies to Cure HIV Infection. Viruses 2020, 12, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Mori, L.; Valente, S.T. The Block-and-Lock Strategy for Human Immunodeficiency Virus Cure: Lessons Learned from Didehydro-Cortistatin, A. J. Infect. Dis. 2021, 223, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Christ, F.; Voet, A.; Marchand, A.; Nicolet, S.; Desimmie, B.A.; Marchand, D.; Bardiot, D.; Van der Veken, N.J.; Van Remoortel, B.; Strelkov, S.V.; et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 2010, 6, 442–448. [Google Scholar] [CrossRef]
- Vansant, G.; Vranckx, L.S.; Zurnic, I.; Van Looveren, D.; Van de Velde, P.; Nobles, C.; Gijsbers, R.; Christ, F.; Debyser, Z. Impact of LEDGIN treatment during virus production on residual HIV-1 transcription. Retrovirology 2019, 16, 8. [Google Scholar] [CrossRef]
- Ahlenstiel, C.; Mendez, C.; Lim, S.T.; Marks, K.; Turville, S.; Cooper, D.A.; Kelleher, A.D.; Suzuki, K. Novel RNA Duplex Locks HIV-1 in a Latent State via Chromatin-mediated Transcriptional Silencing. Mol. Ther. Nucleic Acids 2015, 4, e261. [Google Scholar] [CrossRef]
- Eekels, J.J.; Berkhout, B. Toward a durable treatment of HIV-1 infection using RNA interference. Prog. Mol. Biol. Transl. Sci. 2011, 102, 141–163. [Google Scholar] [CrossRef]
- Ketting, R.F. The many faces of RNAi. Dev. Cell 2011, 20, 148–161. [Google Scholar] [CrossRef] [Green Version]
- Méndez, C.; Ahlenstiel, C.L.; Kelleher, A.D. Post-transcriptional gene silencing, transcriptional gene silencing and human immunodeficiency virus. World J. Virol. 2015, 4, 219–244. [Google Scholar] [CrossRef] [Green Version]
- Morris, K.V. RNA-mediated transcriptional gene silencing in human cells. Curr. Top. Microbiol. Immunol. 2008, 320, 211–224. [Google Scholar] [CrossRef]
- Weinberg, M.S.; Morris, K.V. Transcriptional gene silencing in humans. Nucleic Acids Res. 2016, 44, 6505–6517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarborough, R.J.; Gatignol, A. RNA Interference Therapies for an HIV-1 Functional Cure. Viruses 2017, 10, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohn, D.B.; Bauer, G.; Rice, C.R.; Rothschild, J.C.; Carbonaro, D.A.; Valdez, P.; Hao, Q.; Zhou, C.; Bahner, I.; Kearns, K.; et al. A clinical trial of retroviral-mediated transfer of a rev-responsive element decoy gene into CD34(+) cells from the bone marrow of human immunodeficiency virus-1-infected children. Blood 1999, 94, 368–371. [Google Scholar] [CrossRef]
- Tebas, P.; Stein, D.; Binder-Scholl, G.; Mukherjee, R.; Brady, T.; Rebello, T.; Humeau, L.; Kalos, M.; Papasavvas, E.; Montaner, L.J.; et al. Antiviral effects of autologous CD4 T cells genetically modified with a conditionally replicating lentiviral vector expressing long antisense to HIV. Blood 2013, 121, 1524–1533. [Google Scholar] [CrossRef] [PubMed]
- Mitsuyasu, R.T.; Merigan, T.C.; Carr, A.; Zack, J.A.; Winters, M.A.; Workman, C.; Bloch, M.; Lalezari, J.; Becker, S.; Thornton, L.; et al. Phase 2 gene therapy trial of an anti-HIV ribozyme in autologous CD34+ cells. Nat. Med. 2009, 15, 285–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolstein, O.; Boyd, M.; Millington, M.; Impey, H.; Boyer, J.; Howe, A.; Delebecque, F.; Cornetta, K.; Rothe, M.; Baum, C.; et al. Preclinical safety and efficacy of an anti-HIV-1 lentiviral vector containing a short hairpin RNA to CCR5 and the C46 fusion inhibitor. Mol. Ther. Methods Clin. Dev. 2014, 1, 11. [Google Scholar] [CrossRef]
- DiGiusto, D.L.; Krishnan, A.; Li, L.; Li, H.; Li, S.; Rao, A.; Mi, S.; Yam, P.; Stinson, S.; Kalos, M.; et al. RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Sci. Transl. Med. 2010, 2, 36ra43. [Google Scholar] [CrossRef] [Green Version]
- Delville, M.; Touzot, F.; Couzin, C.; Hmitou, I.; Djerroudi, L.; Ouedrani, A.; Lefrère, F.; Tuchman-Durand, C.; Mollet, C.; Fabreguettes, J.R.; et al. Safety of CD34(+) Hematopoietic Stem Cells and CD4(+) T Lymphocytes Transduced with LVsh5/C46 in HIV-1 Infected Patients with High-Risk Lymphoma. Mol. Ther. Methods Clin. Dev. 2019, 13, 303–309. [Google Scholar] [CrossRef] [Green Version]
- A Study Evaluating the Safety of Cal-1 (LVsh5/C46) Drug Product in HIV-1 Infected Patient with High Risk Lymphoma (GENHIV) ClinicalTrials.gov Identifier: NCT03593187. Available online: https://clinicaltrials.gov/ct2/show/NCT03593187 (accessed on 12 April 2022).
- Weng, Y.; Huang, Q.; Li, C.; Yang, Y.; Wang, X.; Yu, J.; Huang, Y.; Liang, X.J. Improved Nucleic Acid Therapy with Advanced Nanoscale Biotechnology. Mol. Ther. Nucleic Acids 2020, 19, 581–601. [Google Scholar] [CrossRef]
- Bobbin, M.L.; Burnett, J.C.; Rossi, J.J. RNA interference approaches for treatment of HIV-1 infection. Genome Med. 2015, 7, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenkwein, D.; Afzal, S.; Nousiainen, A.; Schmidt, M.; Ylä-Herttuala, S. Efficient Nuclease-Directed Integration of Lentivirus Vectors into the Human Ribosomal DNA Locus. Mol. Ther. J. Am. Soc. Gene Ther. 2020, 28, 1858–1875. [Google Scholar] [CrossRef] [PubMed]
- Singha, K.; Namgung, R.; Kim, W.J. Polymers in small-interfering RNA delivery. Nucleic Acid Ther. 2011, 21, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Wilczewska, A.Z.; Niemirowicz, K.; Markiewicz, K.H.; Car, H. Nanoparticles as drug delivery systems. Pharmacol. Rep. 2012, 64, 1020–1037. [Google Scholar] [CrossRef]
- Gwinn, M.R.; Vallyathan, V. Nanoparticles: Health effects—Pros and cons. Environ. Health Perspect. 2006, 114, 1818–1825. [Google Scholar] [CrossRef] [Green Version]
- De Jong, W.H.; Borm, P.J. Drug delivery and nanoparticles: Applications and hazards. Int. J. Nanomed. 2008, 3, 133–149. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Du, J.; Gu, Z.; Liang, M.; Hu, Y.; Zhang, W.; Priceman, S.; Wu, L.; Zhou, Z.H.; Liu, Z.; et al. A novel intracellular protein delivery platform based on single-protein nanocapsules. Nat. Nanotechnol. 2010, 5, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Yetisgin, A.A.; Cetinel, S.; Zuvin, M.; Kosar, A.; Kutlu, O. Therapeutic Nanoparticles and Their Targeted Delivery Applications. Molecules 2020, 25, 2193. [Google Scholar] [CrossRef]
- Schluep, T.; Hwang, J.; Hildebrandt, I.J.; Czernin, J.; Choi, C.H.; Alabi, C.A.; Mack, B.C.; Davis, M.E. Pharmacokinetics and tumor dynamics of the nanoparticle IT-101 from PET imaging and tumor histological measurements. Proc. Natl. Acad. Sci. USA 2009, 106, 11394–11399. [Google Scholar] [CrossRef] [Green Version]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Arvizo, R.R.; Miranda, O.R.; Moyano, D.F.; Walden, C.A.; Giri, K.; Bhattacharya, R.; Robertson, J.D.; Rotello, V.M.; Reid, J.M.; Mukherjee, P. Modulating pharmacokinetics, tumor uptake and biodistribution by engineered nanoparticles. PLoS ONE 2011, 6, e24374. [Google Scholar] [CrossRef] [Green Version]
- Gratton, S.E.; Ropp, P.A.; Pohlhaus, P.D.; Luft, J.C.; Madden, V.J.; Napier, M.E.; DeSimone, J.M. The effect of particle design on cellular internalization pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 11613–11618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonezawa, S.; Koide, H.; Asai, T. Recent advances in siRNA delivery mediated by lipid-based nanoparticles. Adv. Drug Deliv. Rev. 2020, 154–155, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.J.; Kulkarni, J.A.; van der Meel, R.; Cullis, P.R.; Vader, P.; Schiffelers, R.M. State-of-the-Art Design and Rapid-Mixing Production Techniques of Lipid Nanoparticles for Nucleic Acid Delivery. Small Methods 2018, 2, 1700375. [Google Scholar] [CrossRef]
- Verbeke, R.; Lentacker, I.; De Smedt, S.C.; Dewitte, H. The dawn of mRNA vaccines: The COVID-19 case. J. Control. Release 2021, 333, 511–520. [Google Scholar] [CrossRef]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.Y.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef]
- Lytton-Jean, A.K.; Langer, R.; Anderson, D.G. Five years of siRNA delivery: Spotlight on gold nanoparticles. Small 2011, 7, 1932–1937. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-S.; Green, J.J.; Love, K.T.; Sunshine, J.; Langer, R.; Anderson, D.G. Gold, Poly(β-amino ester) Nanoparticles for Small Interfering RNA Delivery. Nano Lett. 2009, 9, 2402–2406. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Giljohann, D.A.; Chen, D.L.; Massich, M.D.; Wang, X.Q.; Iordanov, H.; Mirkin, C.A.; Paller, A.S. Topical delivery of siRNA-based spherical nucleic acid nanoparticle conjugates for gene regulation. Proc. Natl. Acad. Sci. USA 2012, 109, 11975–11980. [Google Scholar] [CrossRef] [Green Version]
- Charbe, N.B.; Amnerkar, N.D.; Ramesh, B.; Tambuwala, M.M.; Bakshi, H.A.; Aljabali, A.A.A.; Khadse, S.C.; Satheeshkumar, R.; Satija, S.; Metha, M.; et al. Small interfering RNA for cancer treatment: Overcoming hurdles in delivery. Acta Pharm. Sin. B 2020, 10, 2075–2109. [Google Scholar] [CrossRef]
- Davis, M.E. The First Targeted Delivery of siRNA in Humans via a Self-Assembling, Cyclodextrin Polymer-Based Nanoparticle: From Concept to Clinic. Mol. Pharm. 2009, 6, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, H.; Zhang, X. Design, preparation and application of nucleic acid delivery carriers. Biotechnol. Adv. 2014, 32, 804–817. [Google Scholar] [CrossRef]
- Qi, R.; Liu, S.; Chen, J.; Xiao, H.; Yan, L.; Huang, Y.; Jing, X. Biodegradable copolymers with identical cationic segments and their performance in siRNA delivery. J. Control. Release 2012, 159, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Gabrielson, N.P.; Lu, H.; Yin, L.; Kim, K.H.; Cheng, J. A Cell-penetrating Helical Polymer For siRNA Delivery to Mammalian Cells. Mol. Ther. 2012, 20, 1599–1609. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Mok, H.; Lee, Y.; Park, T.G. Self-assembled siRNA–PLGA conjugate micelles for gene silencing. J. Control. Release 2011, 152, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Iversen, F.; Yang, C.; Dagnæs-Hansen, F.; Schaffert, D.H.; Kjems, J.; Gao, S. Optimized siRNA-PEG conjugates for extended blood circulation and reduced urine excretion in mice. Theranostics 2013, 3, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; van Ee, R.J.; Timmer, K.; Craenmehr, E.G.M.; Huang, J.H.; Öner, F.C.; Dhert, W.J.A.; Kragten, A.H.M.; Willems, N.; Grinwis, G.C.M.; et al. A novel injectable thermoresponsive and cytocompatible gel of poly(N-isopropylacrylamide) with layered double hydroxides facilitates siRNA delivery into chondrocytes in 3D culture. Acta Biomater. 2015, 23, 214–228. [Google Scholar] [CrossRef]
- Rozema, D.B.; Lewis, D.L.; Wakefield, D.H.; Wong, S.C.; Klein, J.J.; Roesch, P.L.; Bertin, S.L.; Reppen, T.W.; Chu, Q.; Blokhin, A.V.; et al. Dynamic PolyConjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 12982–12987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.; Liang, M.; Wen, J.; Liu, Y.; Lu, Y.; Chen, I.S.Y. Single siRNA nanocapsules for enhanced RNAi delivery. J. Am. Chem. Soc. 2012, 134, 13542–13545. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Wen, J.; Liang, M.; Lu, Y.; Kamata, M.; Chen, I.S. Modulation of Gene Expression by Polymer Nanocapsule Delivery of DNA Cassettes Encoding Small RNAs. PLoS ONE 2015, 10, e0127986. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Yan, M.; Liu, Y.; Li, J.; Xie, Y.; Lu, Y.; Kamata, M.; Chen, I.S. Specific Elimination of Latently HIV-1 Infected Cells Using HIV-1 Protease-Sensitive Toxin Nanocapsules. PLoS ONE 2016, 11, e0151572. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Wu, D.; Qin, M.; Liu, C.; Wang, L.; Xu, D.; Vinters, H.V.; Liu, Y.; Kranz, E.; Guan, X.; et al. Sustained delivery and molecular targeting of a therapeutic monoclonal antibody to metastases in the central nervous system of mice. Nat. Biomed. Eng. 2019, 3, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Cheever, T.; Wang, L.; Wu, D.; Reed, J.; Mascola, J.; Chen, X.; Liu, C.; Pegu, A.; Sacha, J.B.; et al. Improved delivery of broadly neutralizing antibodies by nanocapsules suppresses SHIV infection in the CNS of infant rhesus macaques. PLoS Pathog. 2021, 17, e1009738. [Google Scholar] [CrossRef] [PubMed]
- Springer, A.D.; Dowdy, S.F. GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther. 2018, 28, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.K.; Willoughby, J.L.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y. Preclinical and Clinical Advances of GalNAc-Decorated Nucleic Acid Therapeutics. Mol. Ther. Nucleic Acids 2017, 6, 116–132. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.C.; Klein, J.J.; Hamilton, H.L.; Chu, Q.; Frey, C.L.; Trubetskoy, V.S.; Hegge, J.; Wakefield, D.; Rozema, D.B.; Lewis, D.L. Co-injection of a targeted, reversibly masked endosomolytic polymer dramatically improves the efficacy of cholesterol-conjugated small interfering RNAs in vivo. Nucleic Acid Ther. 2012, 22, 380–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef] [PubMed]
- El Dika, I.; Lim, H.Y.; Yong, W.P.; Lin, C.C.; Yoon, J.H.; Modiano, M.; Freilich, B.; Choi, H.J.; Chao, T.Y.; Kelley, R.K.; et al. An Open-Label, Multicenter, Phase I, Dose Escalation Study with Phase II Expansion Cohort to Determine the Safety, Pharmacokinetics, and Preliminary Antitumor Activity of Intravenous TKM-080301 in Subjects with Advanced Hepatocellular Carcinoma. Oncologist 2019, 24, 747-e218. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, J.; Shapiro, G.I.; LoRusso, P.M.; Cervantes, A.; Schwartz, G.K.; Weiss, G.J.; Paz-Ares, L.; Cho, D.C.; Infante, J.R.; Alsina, M.; et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013, 3, 406–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, M.J.; Mitra, R.; McArthur, M.J.; Baze, W.; Barnhart, K.; Wu, S.Y.; Rodriguez-Aguayo, C.; Zhang, X.; Coleman, R.L.; Lopez-Berestein, G.; et al. Preclinical Mammalian Safety Studies of EPHARNA (DOPC Nanoliposomal EphA2-Targeted siRNA). Mol. Cancer Ther. 2017, 16, 1114–1123. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, A.; Sarker, D.; Reebye, V.; Jarvis, S.; Sodergren, M.H.; Kossenkov, A.; Sanseviero, E.; Raulf, N.; Vasara, J.; Andrikakou, P.; et al. Upregulation of C/EBPα Inhibits Suppressive Activity of Myeloid Cells and Potentiates Antitumor Response in Mice and Patients with Cancer. Clin. Cancer Res. 2021, 27, 5961–5978. [Google Scholar] [CrossRef] [PubMed]
- Sarker, D.; Plummer, R.; Meyer, T.; Sodergren, M.H.; Basu, B.; Chee, C.E.; Huang, K.W.; Palmer, D.H.; Ma, Y.T.; Evans, T.R.J.; et al. MTL-CEBPA, a Small Activating RNA Therapeutic Upregulating C/EBP-α, in Patients with Advanced Liver Cancer: A First-in-Human, Multicenter, Open-Label, Phase I Trial. Clin. Cancer Res. 2020, 26, 3936–3946. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Quimbo, A.; Xia, F.; Yao, J.; Clamme, J.P.; Zabludoff, S.; Zhang, J.; Ying, W. Anti-HSP47 siRNA lipid nanoparticle ND-L02-s0201 reverses interstitial pulmonary fibrosis in preclinical rat models. ERJ Open Res. 2021, 7, 00733-2020. [Google Scholar] [CrossRef]
- August, A.; Attarwala, H.Z.; Himansu, S.; Kalidindi, S.; Lu, S.; Pajon, R.; Han, S.; Lecerf, J.M.; Tomassini, J.E.; Hard, M.; et al. A phase 1 trial of lipid-encapsulated mRNA encoding a monoclonal antibody with neutralizing activity against Chikungunya virus. Nat. Med. 2021, 27, 2224–2233. [Google Scholar] [CrossRef] [PubMed]
- Streinu-Cercel, A.; Gane, E.; Cheng, W.; Sievert, W.; Roberts, S.; Ahn, S.H.; Kim, Y.J.; Agarwal, K.; Niforos, B.; Symonds, B.; et al. A phase 2a study evaluating the multi-dose activity of ARB-1467 in HBeAg-positive and negative virally suppressed subjects with hepatitis B. J. Hepatol. 2017, 66, s688–s689. [Google Scholar] [CrossRef]
- ModernaTX Inc. Dose-Finding Trial to Evaluate the Safety and Immunogenicity of Cytomegalovirus Vaccine MRNA-1647 in Healthy Adults; Clinical Trial Registration NCT04232280. Available online: https://clinicaltrials.gov/ct2/show/NCT04232280 (accessed on 15 January 2022).
- ModernaTX Inc. Safety and Immunogenicity of mRNA-1653, a Combined Human Metapneumovirus (hMPV) and Parainfluenza Virus Type 3 (PIV3) Vaccine, in Healthy Adults, and Children 12 to 59 Months of Age with Serologic Evidence of Prior Exposure Clinical Trial Registration NCT04144348. Available online: https://clinicaltrials.gov/ct2/show/NCT04144348?term=NCT04144348&draw=2&rank=1 (accessed on 4 April 2022).
- Businesswire. Moderna Announces Clinical Progress from Its Industry-Leading mRNA Vaccine Franchise and Continues Investments to Accelerate Pipeline Development. Available online: https://www.businesswire.com/news/home/20210414005347/en/ (accessed on 31 January 2022).
- Robinson, E.; MacDonald, K.D.; Slaughter, K.; McKinney, M.; Patel, S.; Sun, C.; Sahay, G. Lipid Nanoparticle-Delivered Chemically Modified mRNA Restores Chloride Secretion in Cystic Fibrosis. Mol. Ther. 2018, 26, 2034–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Translate Bio Inc. Study to Evaluate the Safety & Tolerability of MRT5005 Administered by Nebulization in Adults with Cystic Fibrosis (RESTORE-CF) Identifier: NCT03375047. Available online: https://clinicaltrials.gov/ct2/show/NCT03375047 (accessed on 14 April 2022).
- Jensen, S.A.; Day, E.S.; Ko, C.H.; Hurley, L.A.; Luciano, J.P.; Kouri, F.M.; Merkel, T.J.; Luthi, A.J.; Patel, P.C.; Cutler, J.I. Spherical nucleic acid nanoparticle conjugates as an RNAi-based therapy for glioblastoma. Sci. Transl. Med. 2013, 5, 209ra152. [Google Scholar] [CrossRef] [Green Version]
- Kamalzare, S.; Iranpur Mobarakeh, V.; Mirzazadeh Tekie, F.S.; Hajiramezanali, M.; Riazi-Rad, F.; Yoosefi, S.; Normohammadi, Z.; Irani, S.; Tavakoli, M.; Rahimi, P.; et al. Development of a T Cell-targeted siRNA Delivery System Against HIV-1 Using Modified Superparamagnetic Iron Oxide Nanoparticles: An In Vitro Study. J. Pharm. Sci. 2022, 111, 1463–1469. [Google Scholar] [CrossRef]
- Available online: https://www.clinicaltrials.gov/ct2/show/NCT00689065 (accessed on 20 April 2022).
- A Phase 2 Study of siG12D LODER in Combination with Chemotherapy in Patients with Locally Advanced Pancreatic Cancer (PROTACT). Available online: https://www.clinicaltrials.gov/ct2/show/NCT01676259 (accessed on 20 April 2022).
- Varghese, A.M.; Ang, C.; Dimaio, C.J.; Javle, M.M.; Gutierrez, M.; Yarom, N.; Stemmer, S.M.; Golan, T.; Geva, R.; Semenisty, V.; et al. A phase II study of siG12D-LODER in combination with chemotherapy in patients with locally advanced pancreatic cancer (PROTACT). J. Clin. Oncol. 2020, 38, TPS4672. [Google Scholar] [CrossRef]
- Gu, J.; Al-Bayati, K.; Ho, E.A. Development of antibody-modified chitosan nanoparticles for the targeted delivery of siRNA across the blood-brain barrier as a strategy for inhibiting HIV replication in astrocytes. Drug Deliv. Transl. Res. 2017, 7, 497–506. [Google Scholar] [CrossRef]
- van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Habtemariam, B.A.; Karsten, V.; Attarwala, H.; Goel, V.; Melch, M.; Clausen, V.A.; Garg, P.; Vaishnaw, A.K.; Sweetser, M.T.; Robbie, G.J.; et al. Single-Dose Pharmacokinetics and Pharmacodynamics of Transthyretin Targeting N-acetylgalactosamine-Small Interfering Ribonucleic Acid Conjugate, Vutrisiran, in Healthy Subjects. Clin. Pharmacol. Ther. 2021, 109, 372–382. [Google Scholar] [CrossRef]
- Pharmaceuticals, A. HELIOS-A: A Study of Vutrisiran (ALN-TTRSC02) in Patients with Hereditary Transthyretin Amyloidosis (hATTR Amyloidosis) Identifier: NCT03759379. Available online: https://clinicaltrials.gov/ct2/show/NCT03759379?term=NCT03759379&draw=2&rank=1 (accessed on 4 April 2022).
- Hoppe, B.; Koch, A.; Cochat, P.; Garrelfs, S.F.; Baum, M.A.; Groothoff, J.W.; Lipkin, G.; Coenen, M.; Schalk, G.; Amrite, A.; et al. Safety, pharmacodynamics, and exposure-response modeling results from a first-in-human phase 1 study of nedosiran (PHYOX1) in primary hyperoxaluria. Kidney Int. 2022, 101, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Dicerna Pharmaceuticals, I. Safety & Efficacy of DCR-PHXC in Patients with PH1/2 and ESRD (PHYOX7) Identifier NCT04580420. Available online: https://clinicaltrials.gov/ct2/show/NCT04580420?term=nedosiran&draw=2&rank=3 (accessed on 4 April 2022).
- Thielmann, M.; Corteville, D.; Szabo, G.; Swaminathan, M.; Lamy, A.; Lehner, L.J.; Brown, C.D.; Noiseux, N.; Atta, M.G.; Squiers, E.C.; et al. Teprasiran, a Small Interfering RNA, for the Prevention of Acute Kidney Injury in High-Risk Patients Undergoing Cardiac Surgery: A Randomized Clinical Study. Circulation 2021, 144, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Herrera, V.L.; Colby, A.H.; Ruiz-Opazo, N.; Coleman, D.G.; Grinstaff, M.W. Nucleic acid nanomedicines in Phase II/III clinical trials: Translation of nucleic acid therapies for reprogramming cells. Nanomedicine 2018, 13, 2083–2098. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.P.; Look, D.C.; Shi, L.; Hickey, M.; Pewe, L.; Netland, J.; Farzan, M.; Wohlford-Lenane, C.; Perlman, S.; McCray, P.B., Jr. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J. Virol. 2005, 79, 14614–14621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beeraka, N.M.; Sadhu, S.P.; Madhunapantula, S.V.; Rao Pragada, R.; Svistunov, A.A.; Nikolenko, V.N.; Mikhaleva, L.M.; Aliev, G. Strategies for targeting SARS-CoV-2: Small molecule inhibitors—The current status. Front. Immunol. 2020, 11, 552925. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Biswas, A.; Shukla, A.; Maiti, P. Targeted therapy in chronic diseases using nanomaterial-based drug delivery vehicles. Signal Transduct. Target. Ther. 2019, 4, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.S.; Xu, Q.; Boylan, N.J.; Chisholm, J.; Tang, B.C.; Schuster, B.S.; Henning, A.; Ensign, L.M.; Lee, E.; Adstamongkonkul, P.; et al. Nanoparticles that do not adhere to mucus provide uniform and long-lasting drug delivery to airways following inhalation. Sci. Adv. 2017, 3, e1601556. [Google Scholar] [CrossRef] [Green Version]
- Reis, F.J.; Damaceno, N. Cystic fibrosis. J. Pediatr. 1998, 74 (Suppl. S1), S76–S94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maule, G.; Casini, A.; Montagna, C.; Ramalho, A.S.; De Boeck, K.; Zeger, D.; Carlon, M.S.; Petris, G.; Cereseto, A. Allele specific repair of splicing mutations in cystic fibrosis through AsCas12a genome editing. Nat. Commun. 2019, 10, 3556. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, C.; Cantin, A.M. New Therapies to Correct the Cystic Fibrosis Basic Defect. Int. J. Mol. Sci. 2021, 22, 6193. [Google Scholar] [CrossRef]
- Rampado, R.; Crotti, S.; Caliceti, P.; Pucciarelli, S.; Agostini, M. Recent Advances in Understanding the Protein Corona of Nanoparticles and in the Formulation of “Stealthy” Nanomaterials. Front. Bioeng. Biotechnol. 2020, 8, 166. [Google Scholar] [CrossRef]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lincoff, A.M.; Mehran, R.; Povsic, T.J.; Zelenkofske, S.L.; Huang, Z.; Armstrong, P.W.; Steg, P.G.; Bode, C.; Cohen, M.G.; Buller, C.; et al. Effect of the REG1 anticoagulation system versus bivalirudin on outcomes after percutaneous coronary intervention (REGULATE-PCI): A randomised clinical trial. Lancet 2016, 387, 349–356. [Google Scholar] [CrossRef]
- Seth, P.P.; Tanowitz, M.; Bennett, C.F. Selective tissue targeting of synthetic nucleic acid drugs. J. Clin. Investig. 2019, 129, 915–925. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.K.; Park, J.; Jon, S. Targeting strategies for multifunctional nanoparticles in cancer imaging and therapy. Theranostics 2012, 2, 3–44. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Lee, C.H.; Ko, H.J.; Suh, J.S.; Yoon, H.G.; Lee, K.; Huh, Y.M.; Haam, S. Multifunctional magneto-polymeric nanohybrids for targeted detection and synergistic therapeutic effects on breast cancer. Angew. Chem. Int. Ed. Engl. 2007, 46, 8836–8839. [Google Scholar] [CrossRef]
- Ling, Y.; Wei, K.; Luo, Y.; Gao, X.; Zhong, S. Dual docetaxel/superparamagnetic iron oxide loaded nanoparticles for both targeting magnetic resonance imaging and cancer therapy. Biomaterials 2011, 32, 7139–7150. [Google Scholar] [CrossRef]
- Wang, A.Z.; Gu, F.; Zhang, L.; Chan, J.M.; Radovic-Moreno, A.; Shaikh, M.R.; Farokhzad, O.C. Biofunctionalized targeted nanoparticles for therapeutic applications. Expert Opin. Biol. Ther. 2008, 8, 1063–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steichen, S.D.; Caldorera-Moore, M.; Peppas, N.A. A review of current nanoparticle and targeting moieties for the delivery of cancer therapeutics. Eur. J. Pharm. Sci. 2013, 48, 416–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brissette, R.; Prendergast, J.; Goldstein, N.I. Identification of cancer targets and therapeutics using phage display. Curr. Opin. Drug Discov. Dev. 2006, 9, 363–369. [Google Scholar]
- Beekman, K.W.; Colevas, A.D.; Cooney, K.; Dipaola, R.; Dunn, R.L.; Gross, M.; Keller, E.T.; Pienta, K.J.; Ryan, C.J.; Smith, D.; et al. Phase II evaluations of cilengitide in asymptomatic patients with androgen-independent prostate cancer: Scientific rationale and study design. Clin. Genitourin. Cancer 2006, 4, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Dammes, N.; Goldsmith, M.; Ramishetti, S.; Dearling, J.L.J.; Veiga, N.; Packard, A.B.; Peer, D. Conformation-sensitive targeting of lipid nanoparticles for RNA therapeutics. Nat. Nanotechnol. 2021, 16, 1030–1038. [Google Scholar] [CrossRef]
- Gu, F.X.; Karnik, R.; Wang, A.Z.; Alexis, F.; Levy-Nissenbaum, E.; Hong, S.; Langer, R.S.; Farokhzad, O.C. Targeted nanoparticles for cancer therapy. Nano Today 2007, 2, 14–21. [Google Scholar] [CrossRef]
- Schneider, D.; Tuerk, C.; Gold, L. Selection of high affinity RNA ligands to the bacteriophage R17 coat protein. J. Mol. Biol. 1992, 228, 862–869. [Google Scholar] [CrossRef]
- Lupold, S.E.; Hicke, B.J.; Lin, Y.; Coffey, D.S. Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer Res. 2002, 62, 4029–4033. [Google Scholar]
- Shukla, D.; Namperumalsamy, P.; Goldbaum, M.; Cunningham, E.T., Jr. Pegaptanib sodium for ocular vascular disease. Indian J. Ophthalmol. 2007, 55, 427–430. [Google Scholar] [CrossRef]
- Mukherjee, P.; Aksamitiene, E.; Alex, A.; Shi, J.; Bera, K.; Zhang, C.; Spillman, D.R.; Marjanovic, M.; Fazio, M.; Seth, P.P.; et al. Differential Uptake of Antisense Oligonucleotides in Mouse Hepatocytes and Macrophages Revealed by Simultaneous Two-Photon Excited Fluorescence and Coherent Raman Imaging. Nucleic Acid Ther. 2022, 32, 163–176. [Google Scholar] [CrossRef]
- Juan, A.; Cimas, F.J.; Bravo, I.; Pandiella, A.; Ocaña, A.; Alonso-Moreno, C. An Overview of Antibody Conjugated Polymeric Nanoparticles for Breast Cancer Therapy. Pharmaceutics 2020, 12, 802. [Google Scholar] [CrossRef] [PubMed]
- Jahan, S.T.; Sadat, S.M.A.; Walliser, M.; Haddadi, A. Targeted Therapeutic Nanoparticles: An Immense Promise to Fight against Cancer. J. Drug Deliv. 2017, 2017, 9090325. [Google Scholar] [CrossRef] [PubMed]
- Roncato, F.; Rruga, F.; Porcù, E.; Casarin, E.; Ronca, R.; Maccarinelli, F.; Realdon, N.; Basso, G.; Alon, R.; Viola, G.; et al. Improvement and extension of anti-EGFR targeting in breast cancer therapy by integration with the Avidin-Nucleic-Acid-Nano-Assemblies. Nat. Commun. 2018, 9, 4070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Gulati, N.M.; Stewart, P.L.; Steinmetz, N.F. Bioinspired Shielding Strategies for Nanoparticle Drug Delivery Applications. Mol. Pharm. 2018, 15, 2900–2909. [Google Scholar] [CrossRef]
- Drobek, T.; Spencer, N.D.; Heuberger, M. Compressing PEG brushes. Macromolecules 2005, 38, 5254–5259. [Google Scholar] [CrossRef]
- Garay, R.P.; El-Gewely, R.; Armstrong, J.K.; Garratty, G.; Richette, P. Antibodies against polyethylene glycol in healthy subjects and in patients treated with PEG-conjugated agents. Expert Opin. Drug Deliv. 2012, 9, 1319–1323. [Google Scholar] [CrossRef]
- Rao, L.; Xu, J.H.; Cai, B.; Liu, H.; Li, M.; Jia, Y.; Xiao, L.; Guo, S.S.; Liu, W.; Zhao, X.Z. Synthetic nanoparticles camouflaged with biomimetic erythrocyte membranes for reduced reticuloendothelial system uptake. Nanotechnology 2016, 27, 085106. [Google Scholar] [CrossRef]
- Yuba, E.; Harada, A.; Sakanishi, Y.; Watarai, S.; Kono, K. A liposome-based antigen delivery system using pH-sensitive fusogenic polymers for cancer immunotherapy. Biomaterials 2013, 34, 3042–3052. [Google Scholar] [CrossRef]
- Li, Z.Z.; Wen, L.X.; Shao, L.; Chen, J.F. Fabrication of porous hollow silica nanoparticles and their applications in drug release control. J. Control. Release 2004, 98, 245–254. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J.E.; Lee, J.; Yu, J.H.; Kim, B.C.; An, K.; Hwang, Y.; Shin, C.H.; Park, J.G.; Kim, J.; et al. Magnetic fluorescent delivery vehicle using uniform mesoporous silica spheres embedded with monodisperse magnetic and semiconductor nanocrystals. J. Am. Chem. Soc. 2006, 128, 688–689. [Google Scholar] [CrossRef] [PubMed]
- Rim, H.P.; Min, K.H.; Lee, H.J.; Jeong, S.Y.; Lee, S.C. pH-Tunable calcium phosphate covered mesoporous silica nanocontainers for intracellular controlled release of guest drugs. Angew. Chem. Int. Ed. Engl. 2011, 50, 8853–8857. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zhong, L.; Weng, Y.; Peng, L.; Huang, Y.; Zhao, Y.; Liang, X.J. Therapeutic siRNA: State of the art. Signal Transduct. Target. Ther. 2020, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Hayase, Y.; Imura, A.; Iwai, S.; Miura, K.; Ohtsuka, E. Synthesis and hybridization studies on two complementary nona(2′-O-methyl)ribonucleotides. Nucleic Acids Res. 1987, 15, 6131–6148. [Google Scholar] [CrossRef]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef]
- Wu, S.Y.; Chen, T.M.; Gmeiner, W.H.; Chu, E.; Schmitz, J.C. Development of modified siRNA molecules incorporating 5-fluoro-2′-deoxyuridine residues to enhance cytotoxicity. Nucleic Acids Res. 2013, 41, 4650–4659. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.F. Therapeutic Antisense Oligonucleotides Are Coming of Age. Annu. Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef]
- Forbes, N.; Hussain, M.T.; Briuglia, M.L.; Edwards, D.P.; Horst, J.H.t.; Szita, N.; Perrie, Y. Rapid and scale-independent microfluidic manufacture of liposomes entrapping protein incorporating in-line purification and at-line size monitoring. Int. J. Pharm. 2019, 556, 68–81. [Google Scholar] [CrossRef] [Green Version]
- Precision NanoSystems. NanoAssemblr Platform: A Disruptive Technology Enabling Transformative Medicine. Available online: https://www.precisionnanosystems.com/platform-technologies/product-comparison (accessed on 26 May 2022).





| Non-Viral System | Nucleic Acid | Target | Condition | Clinical Trial Stage | Current Clinical Trials.gov Identifiers | Reference |
|---|---|---|---|---|---|---|
| Lipid nanoparticles | ||||||
| Liposome | Tetravalent RNA drug products | selected malignant melanoma-associated antigens | Melanoma | I | NCT02410733 | [76] |
| Lipid NP | siRNA | Polo-like kinase 1 | Hepatocellular carcinoma | I/II | NCT02191878 complete | [77] |
| Lipid NP | siRNAs | VEGF and kinesin spindle protein | Pancreatic Ductal Adenocarcinoma, Pancreatic Cancer | II | NCT01158079 NCT00882180 | [78] |
| Liposome | siRNA | EphA2 | Solid tumours | I | NCT01591356 | [79] |
| Amphoteric liposomes | dsRNA | CEBPA gene | Advanced liver cancer and solid tumours | I | NCT02716012 NCT04105335 | [80,81] |
| Lipid NP conjugate to vitamin A | siRNA | HSp47 | Hepatic and pulmonary fibrosis | II | NCT03538301 | [82] |
| Proprietary lipid NP technology | 2 mRNAs | encode heavy and light chains of anti-Chikungunya antibody | Chikungunya infections | I | NCT03829384 | [83] |
| Lipid particle | siRNA | Three viral genes | Hepatitis B | II | NCT02631096 | [84] |
| Lipid NP | mRNA | Encode anti-CMV antibodies | Cytomegalovirus vaccine | II | NCT04232280 | [85] |
| Lipid NP | mRNA | Encode anti-hMPV and PIV3 antibodies | human metapneumovirus and parainfluenza virus type 3 | I | NCT04144348 | [86] |
| Lipid NP | mRNA | Encoding a prefusion F glycoprotein | Respiratory Syncytial Virus | I | NCT04528719 | [87] |
| Lipid NP | mRNA | cystic fibrosis transmembrane conductance regulator | Cystic fibrosis | I/II | NCT03375047 | [88,89] |
| Inorganic nanoparticles | ||||||
| Gold NP | siRNA | Bcl2L12 mRNA | Glioblastoma | I | NCT03020017 | [90] |
| SPION | siRNA | HIV nef | HIV | - | [91] | |
| Polymeric | ||||||
| siRNA-polymer bioconjugates | siRNA | RRM2 | Solid Tumor Cancers | I | NCT00689065 complete | [92] |
| Polymeric complexes | siRNA | KRAS G12D | Pancreatic ductal adenocarcinoma, pancreatic cancer | II | NCT01676259 | [93,94] |
| Chitosan NP | 2 siRNAs | SART3 and hCycT1 | HIV | - | [95] | |
| Nanocapsule | siRNA | CCR5 | HIV | Preclinical | [96] | |
| Nanocells | ||||||
| Targeted nonliving bacterial minicells | miRNA | miR-16-based mimic | Malignant Pleural Mesothelioma, NSCLC | I | NCT02369198 complete | [97] |
| GalNAc conjugation | ||||||
| GalNAc | siRNA | RNAi therapeutic targeting transthyretin (vutrisiran) | Amyloidosis | III | NCT03759379 | [98,99] |
| ESC-GalNAc | siRNA | hepatic expression of LDHA (nedosiran) | primary hyperoxaluria | I and II | NCT03392896 NCT04580420 | [100,101] |
| ESC-GalNAc | siRNA | antithrombin (fitusiran) | haemophilia A and B | II and III | NCT03417245 NCT03417102 NCT03974113 | [8] |
| Naked siRNA | siRNA | p53 mRNA (teprasiran) | prophylactic treatment for acute kidney injury (AKI) following kidney transplant or cardiovascular surgery | II/III | NCT02610283 NCT03510897 NCT02610296 | [8,102] |
| Disease | Cell Type | Location |
|---|---|---|
| Hepatitis B | Hepatocytes | Liver |
| COVID-19 disease | Epithelial cells Alveolar type II cells | Nasal cavity (upper respiratory tract) Lungs (lower respiratory tract) |
| Cystic fibrosis | Mucoid-producing cells | Lungs Pancreas Intestines |
| HIV | CD4+ T cells Myeloid cells Astrocytes Epithelial cells Microglia | Lymph nodes Gut associated lymphoid tissue Reproductive tissue Brain Lungs |
| Target Molecule | Modification |
|---|---|
| Nanocarrier | PEGylation |
| Shielding with biodegradable biomolecules | |
| Physicochemical changes to increase sensitivity to external stimuli such as changes in pH, heat and light | |
| siRNA | 2′ OH modification such as 2′-OMe and 2′-F |
| Thermal destabilization with UNA and GNA | |
| Modification of bases and analogue base substitutions | |
| Phosphorothioate linkage | |
| Bioconjugation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agbosu, E.E.; Ledger, S.; Kelleher, A.D.; Wen, J.; Ahlenstiel, C.L. Targeted Nanocarrier Delivery of RNA Therapeutics to Control HIV Infection. Pharmaceutics 2022, 14, 1352. https://doi.org/10.3390/pharmaceutics14071352
Agbosu EE, Ledger S, Kelleher AD, Wen J, Ahlenstiel CL. Targeted Nanocarrier Delivery of RNA Therapeutics to Control HIV Infection. Pharmaceutics. 2022; 14(7):1352. https://doi.org/10.3390/pharmaceutics14071352
Chicago/Turabian StyleAgbosu, Esinam E., Scott Ledger, Anthony D. Kelleher, Jing Wen, and Chantelle L. Ahlenstiel. 2022. "Targeted Nanocarrier Delivery of RNA Therapeutics to Control HIV Infection" Pharmaceutics 14, no. 7: 1352. https://doi.org/10.3390/pharmaceutics14071352
APA StyleAgbosu, E. E., Ledger, S., Kelleher, A. D., Wen, J., & Ahlenstiel, C. L. (2022). Targeted Nanocarrier Delivery of RNA Therapeutics to Control HIV Infection. Pharmaceutics, 14(7), 1352. https://doi.org/10.3390/pharmaceutics14071352