
Lentiviral Vectors for Ocular Gene Therapy



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Basics of Lentiviral Vector Technology
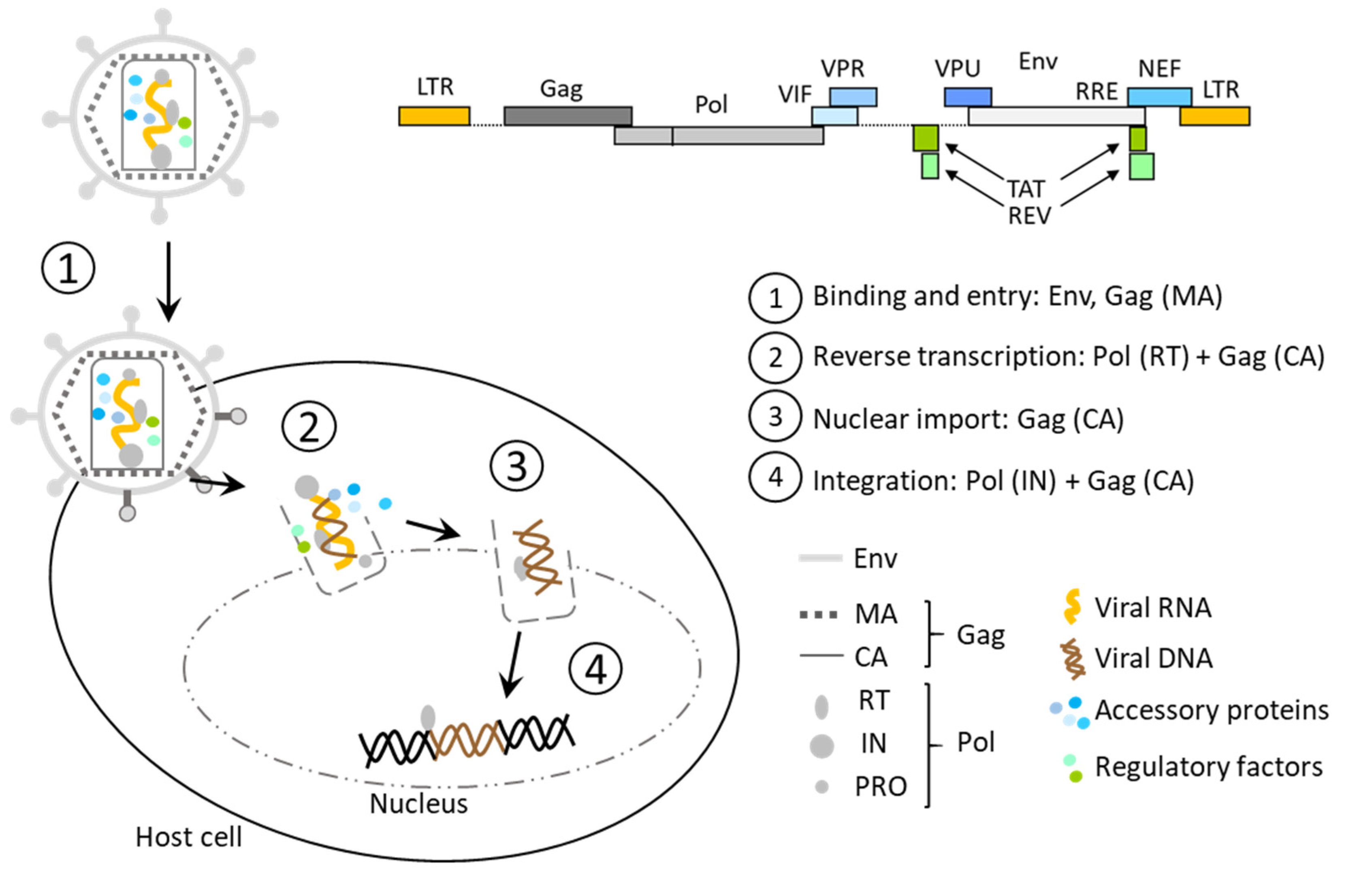
1.1. From Lentivirus to Lentiviral Vector
1.2. Lentivirus and Lentiviral Vector Integration
1.2.1. Components and Functioning of Lentiviral Infection
- Gag encoding for matrix (MA), nucleocapsid (NC), capsid (CA), and p6 proteins required for viral assembly and infection;
- Pol encoding for reverse transcriptase (RT), RNase H, protease (PRO), and integrase (IN), essential for reverse transcription and genomic integration;
- Env encoding for surface glycoproteins (SU, TM) that define tropism of the virus and enable entry into the host cells;
- Tat and Rev, two regulatory genes that activate viral transcription and nuclear export of intron-containing nascent viral RNA, respectively;
- Four accessory genes, Vif, Vpr, Vpu, and Nef;
- Several cis-acting elements are also critical for viral replication, such asψ, the packaging signal;
- RRE, the Rev response element, required for processing and transport of viral RNAs;
- cPPT/CTS, the central polypurine tract, and the central termination sequence, whose role is still questioned, suspected to be linked to nuclear transport and replication;
- TAR, the TAT activation region;
- The splicing donor (SD) and acceptor (SA) sites, which allow the production of all viral proteins and the viral RNA starting from a unique pre-mRNA.
1.2.2. Strategy to Limit the Risks and Increase LV Safety
- The transfer plasmid that contains the transgene and its promoter, the 5′ and ΔU3–3′ LTRs, the ψ sequence, and the RRE sequence;
- One packaging plasmid encoding gag and pol;
- A second packaging plasmid encoding rev;
- The envelope plasmid encoding the envelope proteins, which provide the vector tropism (see Section 1.4).
1.3. Lentiviral Vector Production
1.3.1. Lentiviral Vector Assembly
1.3.2. Recombinant Lentiviral Preparation and Purification
1.3.3. Clinical Preparation
1.4. Envelope
1.4.1. The VSV-G Pseudotyping
1.4.2. Alternative Viral Envelopes
1.4.3. Chimeric Envelopes
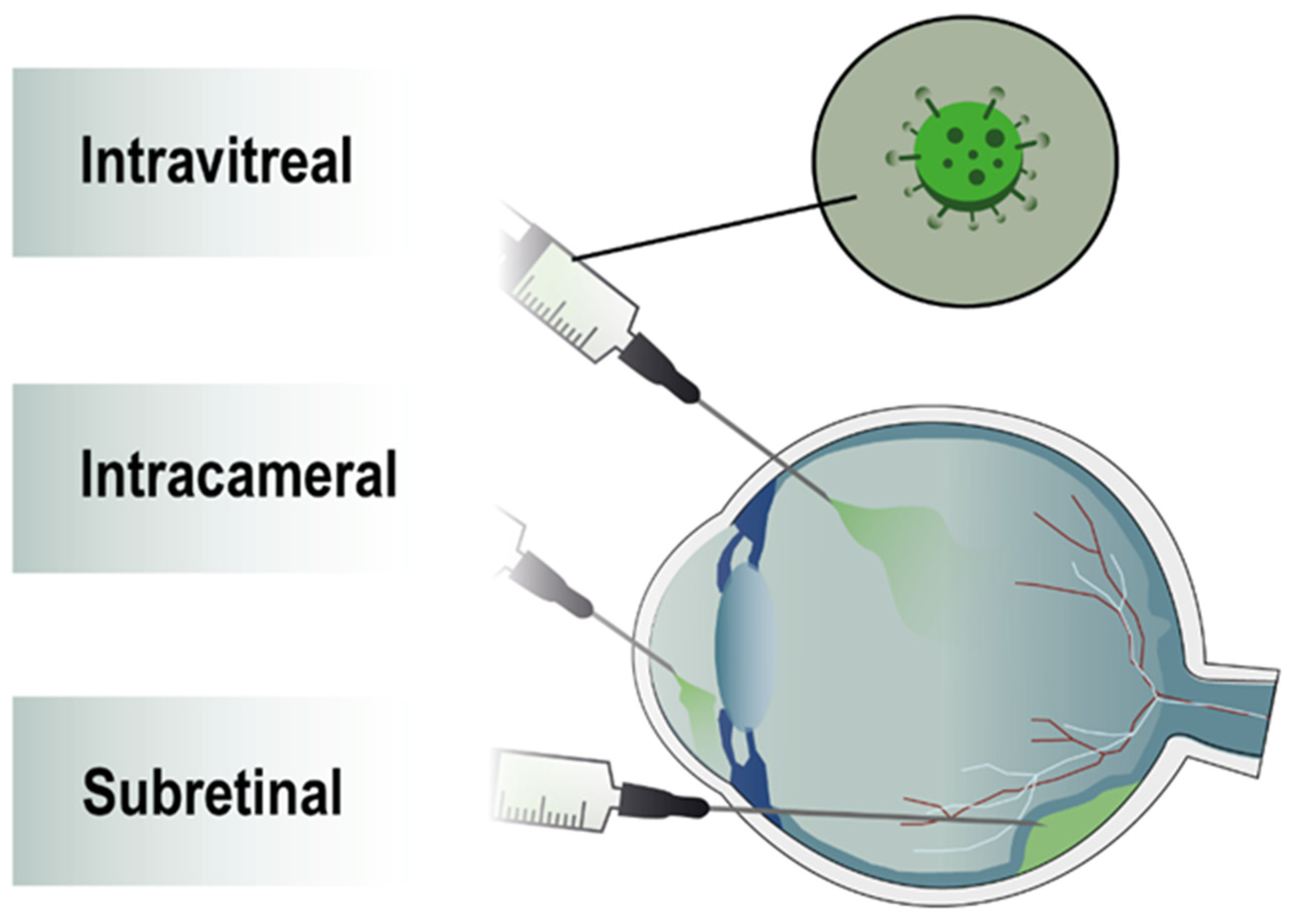
1.5. Biodistribution Safety for Eye Application
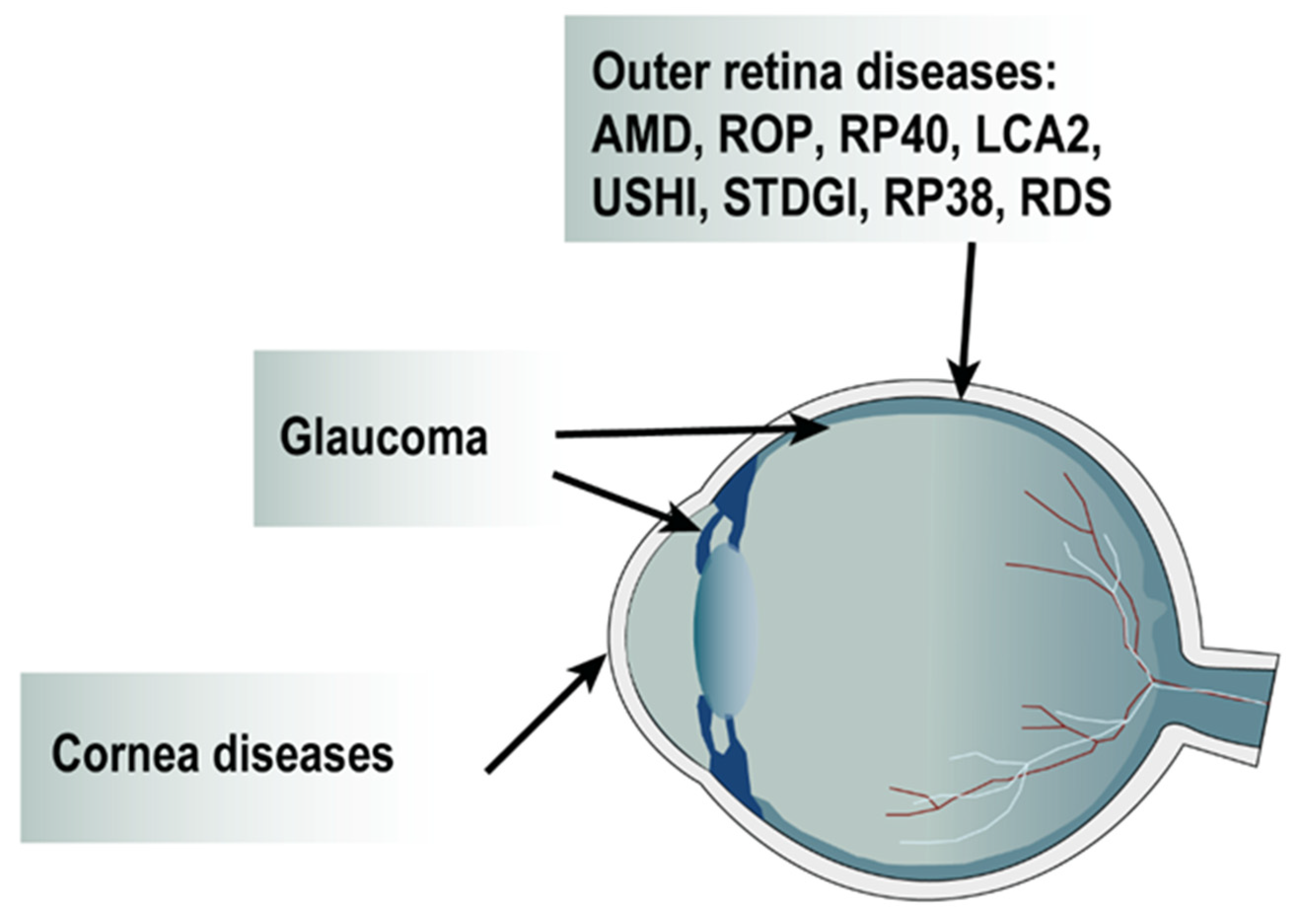
2. Development of Lentiviral Vectors for Ocular Therapeutic Applications
2.1. Gene Transfer Strategy for AMD
2.1.1. Dry and Wet AMD
2.1.2. Gene Therapy for Wet AMD
2.1.3. Anti-Angiogenic Strategy
2.2. Gene Transfer for Retinopathy of Prematurity
2.3. Gene Therapy for Inherited Retinal Dystrophies (IRD)
2.3.1. IRD and Gene Replacement or Correction Strategy
Retinitis Pigmentosa
Leber Congenital Amaurosis
- Gene Replacement
- Gene Editing
Usher Syndrome
Stargardt Disease
2.3.2. IRD and Neuroprotective Strategy
2.4. Gene Therapy Strategies for Glaucoma
2.4.1. Targeting the Trabecular Meshwork
2.4.2. Targeting the Retinal Ganglion Cells for Neuroprotection
2.5. Gene Therapy Strategies to Prevent Corneal Fibrosis and Neovascularization
3. General Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviation
| AAV | Adeno-associated virus |
| AMD | Age-related macular degeneration |
| AE | Adverse effect |
| BDNF | Brain-derived neurotrophic factor |
| BIV | Bovine immunodeficiency virus |
| BCVA | Best-corrected visual acuity |
| C3 | Exoenzyme C3 transferase |
| CA | Capsid |
| cDNA | Complementary DNA |
| CMV | Cytomegalovirus |
| CNTF | Ciliary neurotrophic factor |
| CNV | Choroidal neovascularization |
| cPPT | Central polypurine tract |
| Cas9 | CRISPR-associated protein 9 |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| DNA | Deoxyribonucleic acid |
| EIAV | Equine infectious anemia virus |
| ERG | Electroretinography |
| FAF | Fundus autofluorescence |
| FIV | Feline immunodeficiency virus |
| FGF | Fibroblast growth factor |
| GFP | Green fluorescent protein |
| GMP | Good manufacturing process |
| HIV-1 | Human immunodeficient virus 1 |
| hPEDF | Human pigment epithelium-derived factor |
| IDLV | Integrase-deficient lentiviral vector |
| IN | Integrase |
| IOP | Intraocular pressure |
| iPSC | Induced pluripotent stem cell |
| IRD | Inherited retinal dystrophies |
| IVNV | Intravitreal neovascularization |
| LTR | Long terminal repeat |
| LV | Lentiviral vector |
| MA | Matrix |
| miRNA | microRNA |
| mRNA | Messenger RNA |
| MV | Measles virus |
| NC | Nucleocapsid |
| NHP | Non-human primate |
| OCT | Optical coherence tomography |
| OIR | Oxygen-induced retinopathy |
| OLM | Outer limiting layer |
| ONL | Outer nuclear layer |
| PCR | Polymerase chain reaction |
| p.i. | Post injection |
| PRO | Protease |
| qPCR | Quantitative PCR |
| qRT-PCR | Quantitative RT-PCR |
| rAAV | Recombinant AAV |
| RCL | Recombinant-competent lentivirus |
| RGC | Retinal ganglion cell |
| RPE | Retinal pigment epithelium |
| RNA | Ribonucleic acid |
| ROP | Retinopathy of prematurity |
| RRE | Rev response element |
| RT | Reverse transcriptase |
| RRV | Ross River virus |
| SA | Splicing acceptor |
| scFV | Single-chain antibody |
| shRNA | Short hairpin RNA |
| SIN | Self-inactivating |
| SD | Splicing donor |
| seV | Sendai virus |
| sgRNA | Single guide RNA |
| SIV | Simian immunodeficiency virus |
| SFFV | Spleen focus-forming virus |
| sFlt-1 | Fms-like tyrosine kinase-1 |
| ssRNA | Single-strand RNA |
| SU | Surface |
| TAR | Tat activating region |
| TFF | Tangential flow filtration |
| TM | Transmembrane |
| TU | Transducing unit |
| VEGF | Vascular endothelial growth factor |
| VMD2 | Vitelliform macular dystrophy 2 |
| VSV-G | Vesicular stomatitis virus G |
| WPRE | Woodchuck hepatitis virus post-transcriptional regulatory element |
| WT | Wild type |
| Ψ | Packaging signal sequence |
References
- Bukrinsky, M.I.; Haggerty, S.; Dempsey, M.P.; Sharova, N.; Adzhubel, A.; Spitz, L.; Lewis, P.; Goldfarb, D.; Emerman, M.; Stevenson, M. A nuclear localization signal within HIV-1 matrix protein that governs infection of non-dividing cells. Nature 1993, 365, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Dropulic, B. Lentiviral vectors: Their molecular design, safety, and use in laboratory and preclinical research. Hum. Gene Ther. 2011, 22, 649–657. [Google Scholar] [CrossRef]
- Zennou, V.; Petit, C.; Guetard, D.; Nerhbass, U.; Montagnier, L.; Charneau, P. HIV-1 genome nuclear import is mediated by a central DNA flap. Cell 2000, 101, 173–185. [Google Scholar] [CrossRef] [Green Version]
- Schnittman, S.M.; Lane, H.C.; Roth, J.; Burrows, A.; Folks, T.M.; Kehrl, J.H.; Koenig, S.; Berman, P.; Fauci, A.S. Characterization of GP120 binding to CD4 and an assay that measures ability of sera to inhibit this binding. J. Immunol. 1988, 141, 4181–4186. [Google Scholar] [PubMed]
- Weiss, R.A.; Clapham, P.R.; McClure, M.O.; McKeating, J.A.; McKnight, A.; Dalgleish, A.G.; Sattentau, Q.J.; Weber, J.N. Human immunodeficiency viruses: Neutralization and receptors. J. Acquir. Immune Defic. Syndr. 1988, 1, 536–541. [Google Scholar] [PubMed]
- Naldini, L. Lentiviruses as gene transfer agents for delivery to non-dividing cells. Curr. Opin. Biotechnol. 1998, 9, 457–463. [Google Scholar] [CrossRef]
- Naldini, L.; Blomer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Yee, J.K.; Friedmann, T.; Burns, J.C. Generation of high-titer pseudotyped retroviral vectors with very broad host range. Methods Cell Biol. 1994, 43 Pt A, 99–112. [Google Scholar] [CrossRef]
- Manilla, P.; Rebello, T.; Afable, C.; Lu, X.; Slepushkin, V.; Humeau, L.M.; Schonely, K.; Ni, Y.; Binder, G.K.; Levine, B.L.; et al. Regulatory considerations for novel gene therapy products: A review of the process leading to the first clinical lentiviral vector. Hum. Gene Ther. 2005, 16, 17–25. [Google Scholar] [CrossRef]
- Frankel, A.D.; Young, J.A. HIV-1: Fifteen proteins and an RNA. Annu. Rev. Biochem. 1998, 67, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.J.; Stear, J.H.; Jacques, D.A.; Bocking, T. Insights into HIV uncoating from single-particle imaging techniques. Biophys. Rev. 2022, 14, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Hindmarsh, P.; Leis, J. Retroviral DNA integration. Microbiol. Mol. Biol. Rev. 1999, 63, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craigie, R. HIV integrase, a brief overview from chemistry to therapeutics. J. Biol. Chem. 2001, 276, 23213–23216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciuffi, A. Mechanisms governing lentivirus integration site selection. Curr. Gene Ther. 2008, 8, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Busschots, K.; Vercammen, J.; Emiliani, S.; Benarous, R.; Engelborghs, Y.; Christ, F.; Debyser, Z. The interaction of LEDGF/p75 with integrase is lentivirus-specific and promotes DNA binding. J. Biol. Chem. 2005, 280, 17841–17847. [Google Scholar] [CrossRef] [Green Version]
- Engelman, A.; Cherepanov, P. The lentiviral integrase binding protein LEDGF/p75 and HIV-1 replication. PLoS Pathog. 2008, 4, e1000046. [Google Scholar] [CrossRef]
- Ciuffi, A.; Llano, M.; Poeschla, E.; Hoffmann, C.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005, 11, 1287–1289. [Google Scholar] [CrossRef]
- Bukovsky, A.A.; Song, J.P.; Naldini, L. Interaction of human immunodeficiency virus-derived vectors with wild-type virus in transduced cells. J. Virol. 1999, 73, 7087–7092. [Google Scholar] [CrossRef] [Green Version]
- Moiani, A.; Paleari, Y.; Sartori, D.; Mezzadra, R.; Miccio, A.; Cattoglio, C.; Cocchiarella, F.; Lidonnici, M.R.; Ferrari, G.; Mavilio, F. Lentiviral vector integration in the human genome induces alternative splicing and generates aberrant transcripts. J. Clin. Investig. 2012, 122, 1653–1666. [Google Scholar] [CrossRef] [Green Version]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef]
- Bushman, F.D. Retroviral Insertional Mutagenesis in Humans: Evidence for Four Genetic Mechanisms Promoting Expansion of Cell Clones. Mol. Ther. 2020, 28, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.G.; Schmidt, M.; Schwarzwaelder, K.; Stein, S.; Siler, U.; Koehl, U.; Glimm, H.; Kuhlcke, K.; Schilz, A.; Kunkel, H.; et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 2006, 12, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana-Calvo, M.; Hacein-Bey, S.; de Saint Basile, G.; Gross, F.; Yvon, E.; Nusbaum, P.; Selz, F.; Hue, C.; Certain, S.; Casanova, J.L.; et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 2000, 288, 669–672. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef]
- Gabriel, R.; Eckenberg, R.; Paruzynski, A.; Bartholomae, C.C.; Nowrouzi, A.; Arens, A.; Howe, S.J.; Recchia, A.; Cattoglio, C.; Wang, W.; et al. Comprehensive genomic access to vector integration in clinical gene therapy. Nat. Med. 2009, 15, 1431–1436. [Google Scholar] [CrossRef]
- Ryu, J.; Chan, W.; Wettengel, J.M.; Hanna, C.B.; Burwitz, B.J.; Hennebold, J.D.; Bimber, B.N. Rapid, accurate mapping of transgene integration in viable rhesus macaque embryos using enhanced-specificity tagmentation-assisted PCR. Mol. Ther. Methods Clin. Dev. 2022, 24, 241–254. [Google Scholar] [CrossRef]
- Corre, G.; Seye, A.; Frin, S.; Ferrand, M.; Winkler, K.; Luc, C.; Dorange, F.; Rocca, C.J.; Galy, A. Lentiviral standards to determine the sensitivity of assays that quantify lentiviral vector copy numbers and genomic insertion sites in cells. Gene Ther. 2022. [Google Scholar] [CrossRef]
- Balaggan, K.S.; Duran, Y.; Georgiadis, A.; Thaung, C.; Barker, S.E.; Buch, P.K.; MacNeil, A.; Robbie, S.; Bainbridge, J.W.; Smith, A.J.; et al. Absence of ocular malignant transformation after sub-retinal delivery of rAAV2/2 or integrating lentiviral vectors in p53-deficient mice. Gene Ther. 2012, 19, 182–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef]
- Ling, S.; Yang, S.; Hu, X.; Yin, D.; Dai, Y.; Qian, X.; Wang, D.; Pan, X.; Hong, J.; Sun, X.; et al. Lentiviral delivery of co-packaged Cas9 mRNA and a Vegfa-targeting guide RNA prevents wet age-related macular degeneration in mice. Nat. Biomed. Eng. 2021, 5, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Zufferey, R.; Dull, T.; Mandel, R.J.; Bukovsky, A.; Quiroz, D.; Naldini, L.; Trono, D. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 1998, 72, 9873–9880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwakuma, T.; Cui, Y.; Chang, L.J. Self-inactivating lentiviral vectors with U3 and U5 modifications. Virology 1999, 261, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, H.; Takahashi, M.; Gage, F.H.; Verma, I.M. Stable and efficient gene transfer into the retina using an HIV-based lentiviral vector. Proc. Natl. Acad. Sci. USA 1997, 94, 10319–10323. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.F.; von Ruden, T.; Kantoff, P.W.; Garber, C.; Seiberg, M.; Ruther, U.; Anderson, W.F.; Wagner, E.F.; Gilboa, E. Self-inactivating retroviral vectors designed for transfer of whole genes into mammalian cells. Proc. Natl. Acad. Sci. USA 1986, 83, 3194–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zufferey, R.; Donello, J.E.; Trono, D.; Hope, T.J. Woodchuck hepatitis virus posttranscriptional regulatory element enhances expression of transgenes delivered by retroviral vectors. J. Virol. 1999, 73, 2886–2892. [Google Scholar] [CrossRef] [Green Version]
- Binley, K.; Widdowson, P.S.; Kelleher, M.; de Belin, J.; Loader, J.; Ferrige, G.; Carlucci, M.; Esapa, M.; Chipchase, D.; Angell-Manning, D.; et al. Safety and biodistribution of an equine infectious anemia virus-based gene therapy, RetinoStat(®), for age-related macular degeneration. Hum. Gene Ther. 2012, 23, 980–991. [Google Scholar] [CrossRef] [PubMed]
- Verrier, J.D.; Madorsky, I.; Coggin, W.E.; Geesey, M.; Hochman, M.; Walling, E.; Daroszewski, D.; Eccles, K.S.; Ludlow, R.; Semple-Rowland, S.L. Bicistronic lentiviruses containing a viral 2A cleavage sequence reliably co-express two proteins and restore vision to an animal model of LCA1. PLoS ONE 2011, 6, e20553. [Google Scholar] [CrossRef]
- Balaggan, K.S.; Binley, K.; Esapa, M.; Iqball, S.; Askham, Z.; Kan, O.; Tschernutter, M.; Bainbridge, J.W.; Naylor, S.; Ali, R.R. Stable and efficient intraocular gene transfer using pseudotyped EIAV lentiviral vectors. J. Gene Med. 2006, 8, 275–285. [Google Scholar] [CrossRef]
- Cornetta, K.; Yao, J.; Jasti, A.; Koop, S.; Douglas, M.; Hsu, D.; Couture, L.A.; Hawkins, T.; Duffy, L. Replication-competent lentivirus analysis of clinical grade vector products. Mol. Ther. 2011, 19, 557–566. [Google Scholar] [CrossRef]
- Corre, G.; Dessainte, M.; Marteau, J.B.; Dalle, B.; Fenard, D.; Galy, A. “RCL-Pooling Assay”: A Simplified Method for the Detection of Replication-Competent Lentiviruses in Vector Batches Using Sequential Pooling. Hum. Gene Ther. 2016, 27, 202–210. [Google Scholar] [CrossRef] [Green Version]
- Farley, D.C.; McCloskey, L.; Thorne, B.A.; Tareen, S.U.; Nicolai, C.J.; Campbell, D.J.; Bannister, R.; Stewart, H.J.; Pearson, L.J.; Moyer, B.J.; et al. Development of a replication-competent lentivirus assay for dendritic cell-targeting lentiviral vectors. Mol. Ther. Methods Clin. Dev. 2015, 2, 15017. [Google Scholar] [CrossRef]
- Sastry, L.; Xu, Y.; Johnson, T.; Desai, K.; Rissing, D.; Marsh, J.; Cornetta, K. Certification assays for HIV-1-based vectors: Frequent passage of gag sequences without evidence of replication-competent viruses. Mol. Ther. 2003, 8, 830–839. [Google Scholar] [CrossRef]
- Skrdlant, L.M.; Armstrong, R.J.; Keidaisch, B.M.; Lorente, M.F.; DiGiusto, D.L. Detection of Replication Competent Lentivirus Using a qPCR Assay for VSV-G. Mol. Ther. Methods Clin. Dev. 2018, 8, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tareen, S.U.; Nicolai, C.J.; Campbell, D.J.; Flynn, P.A.; Slough, M.M.; Vin, C.D.; Kelley-Clarke, B.; Odegard, J.M.; Robbins, S.H. A Rev-Independent gag/pol Eliminates Detectable psi-gag Recombination in Lentiviral Vectors. Biores. Open Access 2013, 2, 421–430. [Google Scholar] [CrossRef]
- Kappes, J.C.; Wu, X.; Wakefield, J.K. Production of trans-lentiviral vector with predictable safety. Methods Mol. Med. 2003, 76, 449–465. [Google Scholar] [CrossRef]
- Wu, X.; Wakefield, J.K.; Liu, H.; Xiao, H.; Kralovics, R.; Prchal, J.T.; Kappes, J.C. Development of a novel trans-lentiviral vector that affords predictable safety. Mol. Ther. 2000, 2, 47–55. [Google Scholar] [CrossRef]
- Cornetta, K.; Duffy, L.; Feldman, S.A.; Mackall, C.L.; Davila, M.L.; Curran, K.J.; Junghans, R.P.; Tang, J.Y.; Kochenderfer, J.N.; O’Cearbhaill, R.; et al. Screening Clinical Cell Products for Replication Competent Retrovirus: The National Gene Vector Biorepository Experience. Mol. Ther. Methods Clin. Dev. 2018, 10, 371–378. [Google Scholar] [CrossRef] [Green Version]
- Vink, C.A.; Counsell, J.R.; Perocheau, D.P.; Karda, R.; Buckley, S.M.K.; Brugman, M.H.; Galla, M.; Schambach, A.; McKay, T.R.; Waddington, S.N.; et al. Eliminating HIV-1 Packaging Sequences from Lentiviral Vector Proviruses Enhances Safety and Expedites Gene Transfer for Gene Therapy. Mol. Ther. 2017, 25, 1790–1804. [Google Scholar] [CrossRef] [Green Version]
- Ortinski, P.I.; O’Donovan, B.; Dong, X.; Kantor, B. Integrase-Deficient Lentiviral Vector as an All-in-One Platform for Highly Efficient CRISPR/Cas9-Mediated Gene Editing. Mol. Ther. Methods Clin. Dev. 2017, 5, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Wanisch, K.; Yanez-Munoz, R.J. Integration-deficient lentiviral vectors: A slow coming of age. Mol. Ther. 2009, 17, 1316–1332. [Google Scholar] [CrossRef]
- Ahmed, S.G.; Waddington, S.N.; Boza-Moran, M.G.; Yanez-Munoz, R.J. High-efficiency transduction of spinal cord motor neurons by intrauterine delivery of integration-deficient lentiviral vectors. J. Control. Release 2018, 273, 99–107. [Google Scholar] [CrossRef]
- Yanez-Munoz, R.J.; Balaggan, K.S.; MacNeil, A.; Howe, S.J.; Schmidt, M.; Smith, A.J.; Buch, P.; MacLaren, R.E.; Anderson, P.N.; Barker, S.E.; et al. Effective gene therapy with nonintegrating lentiviral vectors. Nat. Med. 2006, 12, 348–353. [Google Scholar] [CrossRef]
- Brown, H.E.; Chen, H.; Engelman, A. Structure-based mutagenesis of the human immunodeficiency virus type 1 DNA attachment site: Effects on integration and cDNA synthesis. J. Virol. 1999, 73, 9011–9020. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.; Cornetta, K. Design and Potential of Non-Integrating Lentiviral Vectors. Biomedicines 2014, 2, 14–35. [Google Scholar] [CrossRef] [Green Version]
- Koyama, T.; Sun, B.; Tokunaga, K.; Tatsumi, M.; Ishizaka, Y. DNA damage enhances integration of HIV-1 into macrophages by overcoming integrase inhibition. Retrovirology 2013, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- Nightingale, S.J.; Hollis, R.P.; Pepper, K.A.; Petersen, D.; Yu, X.J.; Yang, C.; Bahner, I.; Kohn, D.B. Transient gene expression by nonintegrating lentiviral vectors. Mol. Ther. 2006, 13, 1121–1132. [Google Scholar] [CrossRef]
- Poeschla, E.M.; Wong-Staal, F.; Looney, D.J. Efficient transduction of nondividing human cells by feline immunodeficiency virus lentiviral vectors. Nat. Med. 1998, 4, 354–357. [Google Scholar] [CrossRef]
- Segura, M.M.; Garnier, A.; Durocher, Y.; Coelho, H.; Kamen, A. Production of lentiviral vectors by large-scale transient transfection of suspension cultures and affinity chromatography purification. Biotechnol. Bioeng. 2007, 98, 789–799. [Google Scholar] [CrossRef]
- Tomas, H.A.; Rodrigues, A.F.; Carrondo, M.J.T.; Coroadinha, A.S. LentiPro26: Novel stable cell lines for constitutive lentiviral vector production. Sci. Rep. 2018, 8, 5271. [Google Scholar] [CrossRef] [Green Version]
- Van Craenenbroeck, K.; Vanhoenacker, P.; Haegeman, G. Episomal vectors for gene expression in mammalian cells. Eur. J. Biochem. 2000, 267, 5665–5678. [Google Scholar] [CrossRef]
- Merten, O.W.; Charrier, S.; Laroudie, N.; Fauchille, S.; Dugue, C.; Jenny, C.; Audit, M.; Zanta-Boussif, M.A.; Chautard, H.; Radrizzani, M.; et al. Large-scale manufacture and characterization of a lentiviral vector produced for clinical ex vivo gene therapy application. Hum. Gene Ther. 2011, 22, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Segura, M.M.; Mangion, M.; Gaillet, B.; Garnier, A. New developments in lentiviral vector design, production and purification. Expert Opin. Biol. Ther. 2013, 13, 987–1011. [Google Scholar] [CrossRef] [PubMed]
- Lesch, H.P.; Laitinen, A.; Peixoto, C.; Vicente, T.; Makkonen, K.E.; Laitinen, L.; Pikkarainen, J.T.; Samaranayake, H.; Alves, P.M.; Carrondo, M.J.; et al. Production and purification of lentiviral vectors generated in 293T suspension cells with baculoviral vectors. Gene Ther. 2011, 18, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Lesch, H.P.; Turpeinen, S.; Niskanen, E.A.; Mahonen, A.J.; Airenne, K.J.; Yla-Herttuala, S. Generation of lentivirus vectors using recombinant baculoviruses. Gene Ther. 2008, 15, 1280–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Molina, E.; Chocarro-Wrona, C.; Martinez-Moreno, D.; Marchal, J.A.; Boulaiz, H. Large-Scale Production of Lentiviral Vectors: Current Perspectives and Challenges. Pharmaceutics 2020, 12, 1051. [Google Scholar] [CrossRef] [PubMed]
- Comisel, R.-M.; Kara, B.; Fiesser, F.H.; Farid, S.S. Gene therapy process change evaluation framework: Transient transfection and stable producer cell line comparison. Biochem. Eng. J. 2021, 176, 108202. [Google Scholar] [CrossRef]
- Cooper, A.R.; Patel, S.; Senadheera, S.; Plath, K.; Kohn, D.B.; Hollis, R.P. Highly efficient large-scale lentiviral vector concentration by tandem tangential flow filtration. J. Virol. Methods 2011, 177, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, K.; Scheibe, O.; Kocourek, A.; Muelich, J.; Jurkiewicz, E.; Pfeifer, A. Highly efficient concentration of lenti- and retroviral vector preparations by membrane adsorbers and ultrafiltration. BMC Biotechnol. 2011, 11, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segura, M.M.; Kamen, A.; Trudel, P.; Garnier, A. A novel purification strategy for retrovirus gene therapy vectors using heparin affinity chromatography. Biotechnol. Bioeng. 2005, 90, 391–404. [Google Scholar] [CrossRef]
- Dropulic, B.; Slepushkin, V.; Chang, N.; Gan, Y.; Jiang, B.; Deausen, E.; Berlinger, D.; Binder, G.; Andre, K.; Humeau, L. Large-scale purification of a lentiviral vector by size exclusion chromatography or Mustang Q ion exchange capsule. Bioprocess. J. 2003, 2, 89. [Google Scholar] [CrossRef]
- Shi, R.; Jia, S.; Liu, H.; Nie, H. Clinical grade lentiviral vector purification and quality control requirements. J. Sep. Sci. 2022, 45, 2093–2101. [Google Scholar] [CrossRef] [PubMed]
- Barraza, R.A.; Rasmussen, C.A.; Loewen, N.; Cameron, J.D.; Gabelt, B.T.; Teo, W.L.; Kaufman, P.L.; Poeschla, E.M. Prolonged transgene expression with lentiviral vectors in the aqueous humor outflow pathway of nonhuman primates. Hum. Gene Ther. 2009, 20, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Saenz, D.T.; Barraza, R.; Loewen, N.; Teo, W.; Poeschla, E.M. Production, harvest, and concentration of feline immunodeficiency virus-based lentiviral vector from cells grown in CF10 or CF2 devices. Cold Spring Harb. Protoc. 2012, 2012, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Matet, A.; Kostic, C.; Bemelmans, A.P.; Moulin, A.; Rosolen, S.G.; Martin, S.; Mavilio, F.; Amirjanians, V.; Stieger, K.; Lorenz, B.; et al. Evaluation of tolerance to lentiviral LV-RPE65 gene therapy vector after subretinal delivery in non-human primates. Transl. Res. J. Lab. Clin. Med. 2017, 188, 40–57.e44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, Y.; Yonemitsu, Y.; Miyazaki, M.; Kohno, R.; Murakami, Y.; Murata, T.; Tabata, T.; Ueda, Y.; Ono, F.; Suzuki, T.; et al. Stable retinal gene expression in nonhuman primates via subretinal injection of SIVagm-based lentiviral vectors. Hum. Gene Ther. 2009, 20, 573–579. [Google Scholar] [CrossRef]
- Binley, K.; Widdowson, P.; Loader, J.; Kelleher, M.; Iqball, S.; Ferrige, G.; de Belin, J.; Carlucci, M.; Angell-Manning, D.; Hurst, F.; et al. Transduction of photoreceptors with equine infectious anemia virus lentiviral vectors: Safety and biodistribution of StarGen for Stargardt disease. Investig. Ophthalmol. Vis. Sci. 2013, 54, 4061–4071. [Google Scholar] [CrossRef]
- Zallocchi, M.; Binley, K.; Lad, Y.; Ellis, S.; Widdowson, P.; Iqball, S.; Scripps, V.; Kelleher, M.; Loader, J.; Miskin, J.; et al. EIAV-based retinal gene therapy in the shaker1 mouse model for usher syndrome type 1B: Development of UshStat. PLoS ONE 2014, 9, e94272. [Google Scholar] [CrossRef] [Green Version]
- Miskin, J.; Chipchase, D.; Rohll, J.; Beard, G.; Wardell, T.; Angell, D.; Roehl, H.; Jolly, D.; Kingsman, S.; Mitrophanous, K. A replication competent lentivirus (RCL) assay for equine infectious anaemia virus (EIAV)-based lentiviral vectors. Gene Ther. 2006, 13, 196–205. [Google Scholar] [CrossRef]
- Miyazaki, M.; Ikeda, Y.; Yonemitsu, Y.; Goto, Y.; Kohno, R.; Murakami, Y.; Inoue, M.; Ueda, Y.; Hasegawa, M.; Tobimatsu, S.; et al. Synergistic neuroprotective effect via simian lentiviral vector-mediated simultaneous gene transfer of human pigment epithelium-derived factor and human fibroblast growth factor-2 in rodent models of retinitis pigmentosa. J. Gene Med. 2008, 10, 1273–1281. [Google Scholar] [CrossRef]
- Miyazaki, M.; Ikeda, Y.; Yonemitsu, Y.; Goto, Y.; Sakamoto, T.; Tabata, T.; Ueda, Y.; Hasegawa, M.; Tobimatsu, S.; Ishibashi, T.; et al. Simian lentiviral vector-mediated retinal gene transfer of pigment epithelium-derived factor protects retinal degeneration and electrical defect in Royal College of Surgeons rats. Gene Ther. 2003, 10, 1503–1511. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, Y.; Yonemitsu, Y.; Miyazaki, M.; Kohno, R.; Murakami, Y.; Murata, T.; Goto, Y.; Tabata, T.; Ueda, Y.; Ono, F.; et al. Acute toxicity study of a simian immunodeficiency virus-based lentiviral vector for retinal gene transfer in nonhuman primates. Hum. Gene Ther. 2009, 20, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Fan, N.; Wang, N.; Feng, B.; Yang, M.; Liu, G.; Wang, Y.; Zhu, X.; Kaufman, P.L.; Pang, I.H.; et al. Effects of Lentivirus-Mediated C3 Expression on Trabecular Meshwork Cells and Intraocular Pressure. Invest. Ophthalmol. Vis. Sci. 2018, 59, 4937–4944. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Liu, G.; Zhu, X.; Wu, Z.; Wang, N.; Zhou, L.; Zhang, X.; Fan, N.; Liu, X. Lentiviral Vector-Mediated Expression of Exoenzyme C3 Transferase Lowers Intraocular Pressure in Monkeys. Mol. Ther. 2019, 27, 1327–1338. [Google Scholar] [CrossRef]
- Khare, P.D.; Loewen, N.; Teo, W.; Barraza, R.A.; Saenz, D.T.; Johnson, D.H.; Poeschla, E.M. Durable, safe, multi-gene lentiviral vector expression in feline trabecular meshwork. Mol. Ther. 2008, 16, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Tucci, F.; Galimberti, S.; Naldini, L.; Valsecchi, M.G.; Aiuti, A. A systematic review and meta-analysis of gene therapy with hematopoietic stem and progenitor cells for monogenic disorders. Nat. Commun. 2022, 13, 1315. [Google Scholar] [CrossRef] [PubMed]
- Amirache, F.; Levy, C.; Costa, C.; Mangeot, P.E.; Torbett, B.E.; Wang, C.X.; Negre, D.; Cosset, F.L.; Verhoeyen, E. Mystery solved: VSV-G-LVs do not allow efficient gene transfer into unstimulated T cells, B cells, and HSCs because they lack the LDL receptor. Blood 2014, 123, 1422–1424. [Google Scholar] [CrossRef]
- Finkelshtein, D.; Werman, A.; Novick, D.; Barak, S.; Rubinstein, M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 2013, 110, 7306–7311. [Google Scholar] [CrossRef] [Green Version]
- Nikolic, J.; Belot, L.; Raux, H.; Legrand, P.; Gaudin, Y.; Albertini, A.A. Structural basis for the recognition of LDL-receptor family members by VSV glycoprotein. Nat. Commun. 2018, 9, 1029. [Google Scholar] [CrossRef] [Green Version]
- Kafri, T.; Blomer, U.; Peterson, D.A.; Gage, F.H.; Verma, I.M. Sustained expression of genes delivered directly into liver and muscle by lentiviral vectors. Nat. Genet. 1997, 17, 314–317. [Google Scholar] [CrossRef]
- Kordower, J.H.; Bloch, J.; Ma, S.Y.; Chu, Y.; Palfi, S.; Roitberg, B.Z.; Emborg, M.; Hantraye, P.; Deglon, N.; Aebischer, P. Lentiviral gene transfer to the nonhuman primate brain. Exp. Neurol. 1999, 160, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Blomer, U.; Gage, F.H.; Trono, D.; Verma, I.M. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. USA 1996, 93, 11382–11388. [Google Scholar] [CrossRef] [Green Version]
- Hamaguchi, I.; Woods, N.B.; Panagopoulos, I.; Andersson, E.; Mikkola, H.; Fahlman, C.; Zufferey, R.; Carlsson, L.; Trono, D.; Karlsson, S. Lentivirus vector gene expression during ES cell-derived hematopoietic development in vitro. J. Virol. 2000, 74, 10778–10784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, C.; Rivella, S.; Callegari, J.; Heller, G.; Gaensler, K.M.; Luzzatto, L.; Sadelain, M. Therapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human beta-globin. Nature 2000, 406, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, A.; Ikawa, M.; Dayn, Y.; Verma, I.M. Transgenesis by lentiviral vectors: Lack of gene silencing in mammalian embryonic stem cells and preimplantation embryos. Proc. Natl. Acad. Sci. USA 2002, 99, 2140–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostic, C.; Chiodini, F.; Salmon, P.; Wiznerowicz, M.; Deglon, N.; Hornfeld, D.; Trono, D.; Aebischer, P.; Schorderet, D.F.; Munier, F.L.; et al. Activity analysis of housekeeping promoters using self-inactivating lentiviral vector delivery into the mouse retina. Gene Ther. 2003, 10, 818–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Adel, B.A.; Kostic, C.; Deglon, N.; Ball, A.K.; Arsenijevic, Y. Delivery of Ciliary Neurotrophic Factor via Lentiviral-Mediated Transfer Protects Axotomized Retinal Ganglion Cells for an Extended Period of Time. Hum.Gene Ther. 2003, 14, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Bemelmans, A.P.; Arsenijevic, Y.; Majo, F. Efficient lentiviral gene transfer into corneal stroma cells using a femtosecond laser. Gene Ther. 2009, 16, 933–938. [Google Scholar] [CrossRef] [Green Version]
- Calame, M.; Cachafeiro, M.; Philippe, S.; Schouwey, K.; Tekaya, M.; Wanner, D.; Sarkis, C.; Kostic, C.; Arsenijevic, Y. Retinal degeneration progression changes lentiviral vector cell targeting in the retina. PLoS ONE 2011, 6, e23782. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, K.P.; Geller, S.F.; Schaffer, D.V.; Flannery, J.G. Targeted transgene expression in muller glia of normal and diseased retinas using lentiviral vectors. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1844–1852. [Google Scholar] [CrossRef]
- Lipinski, D.M.; Barnard, A.R.; Charbel Issa, P.; Singh, M.S.; De Silva, S.R.; Trabalza, A.; Eleftheriadou, I.; Ellison, S.M.; Mazarakis, N.D.; MacLaren, R.E. Vesicular stomatitis virus glycoprotein- and Venezuelan equine encephalitis virus-derived glycoprotein-pseudotyped lentivirus vectors differentially transduce corneal endothelium, trabecular meshwork, and human photoreceptors. Hum. Gene Ther. 2014, 25, 50–62. [Google Scholar] [CrossRef]
- Loewen, N.; Fautsch, M.P.; Peretz, M.; Bahler, C.K.; Cameron, J.D.; Johnson, D.H.; Poeschla, E.M. Genetic modification of human trabecular meshwork with lentiviral vectors. Hum. Gene Ther. 2001, 12, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Loewen, N.; Fautsch, M.P.; Teo, W.L.; Bahler, C.K.; Johnson, D.H.; Poeschla, E.M. Long-term, targeted genetic modification of the aqueous humor outflow tract coupled with noninvasive imaging of gene expression in vivo. Investig. Ophthalmol. Vis. Sci. 2004, 45, 3091–3098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croyle, M.A.; Callahan, S.M.; Auricchio, A.; Schumer, G.; Linse, K.D.; Wilson, J.M.; Brunner, L.J.; Kobinger, G.P. PEGylation of a vesicular stomatitis virus G pseudotyped lentivirus vector prevents inactivation in serum. J. Virol. 2004, 78, 912–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruter, O.; Kostic, C.; Crippa, S.V.; Perez, M.T.; Zografos, L.; Schorderet, D.F.; Munier, F.L.; Arsenijevic, Y. Lentiviral vector-mediated gene transfer in adult mouse photoreceptors is impaired by the presence of a physical barrier. Gene Ther. 2005, 12, 942–947. [Google Scholar] [CrossRef] [Green Version]
- Busskamp, V.; Duebel, J.; Balya, D.; Fradot, M.; Viney, T.J.; Siegert, S.; Groner, A.C.; Cabuy, E.; Forster, V.; Seeliger, M.; et al. Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science 2010, 329, 413–417. [Google Scholar] [CrossRef] [Green Version]
- Auricchio, A.; Kobinger, G.; Anand, V.; Hildinger, M.; O’Connor, E.; Maguire, A.M.; Wilson, J.M.; Bennett, J. Exchange of surface proteins impacts on viral vector cellular specificity and transduction characteristics: The retina as a model. Hum. Mol. Genet. 2001, 10, 3075–3081. [Google Scholar] [CrossRef] [Green Version]
- Bemelmans, A.P.; Bonnel, S.; Houhou, L.; Dufour, N.; Nandrot, E.; Helmlinger, D.; Sarkis, C.; Abitbol, M.; Mallet, J. Retinal cell type expression specificity of HIV-1-derived gene transfer vectors upon subretinal injection in the adult rat: Influence of pseudotyping and promoter. J. Gene Med. 2005, 7, 1367–1374. [Google Scholar] [CrossRef]
- Puppo, A.; Cesi, G.; Marrocco, E.; Piccolo, P.; Jacca, S.; Shayakhmetov, D.M.; Parks, R.J.; Davidson, B.L.; Colloca, S.; Brunetti-Pierri, N.; et al. Retinal transduction profiles by high-capacity viral vectors. Gene Ther. 2014, 21, 855–865. [Google Scholar] [CrossRef] [Green Version]
- Murakami, Y.; Ikeda, Y.; Yonemitsu, Y.; Miyazaki, M.; Inoue, M.; Hasegawa, M.; Sueishi, K.; Ishibashi, T. Inhibition of choroidal neovascularization via brief subretinal exposure to a newly developed lentiviral vector pseudotyped with Sendai viral envelope proteins. Hum. Gene Ther. 2010, 21, 199–209. [Google Scholar] [CrossRef]
- Thoulouze, M.I.; Lafage, M.; Schachner, M.; Hartmann, U.; Cremer, H.; Lafon, M. The neural cell adhesion molecule is a receptor for rabies virus. J. Virol. 1998, 72, 7181–7190. [Google Scholar] [CrossRef] [Green Version]
- Gastka, M.; Horvath, J.; Lentz, T.L. Rabies virus binding to the nicotinic acetylcholine receptor alpha subunit demonstrated by virus overlay protein binding assay. J. Gen. Virol. 1996, 77 Pt 10, 2437–2440. [Google Scholar] [CrossRef]
- Hanham, C.A.; Zhao, F.; Tignor, G.H. Evidence from the anti-idiotypic network that the acetylcholine receptor is a rabies virus receptor. J. Virol. 1993, 67, 530–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lentz, T.L. Rabies virus binding to an acetylcholine receptor alpha-subunit peptide. J. Mol. Recognit. 1990, 3, 82–88. [Google Scholar] [CrossRef]
- Tuffereau, C.; Benejean, J.; Blondel, D.; Kieffer, B.; Flamand, A. Low-affinity nerve-growth factor receptor (P75NTR) can serve as a receptor for rabies virus. EMBO J. 1998, 17, 7250–7259. [Google Scholar] [CrossRef]
- Mazarakis, N.D.; Azzouz, M.; Rohll, J.B.; Ellard, F.M.; Wilkes, F.J.; Olsen, A.L.; Carter, E.E.; Barber, R.D.; Baban, D.F.; Kingsman, S.M.; et al. Rabies virus glycoprotein pseudotyping of lentiviral vectors enables retrograde axonal transport and access to the nervous system after peripheral delivery. Hum. Mol. Genet. 2001, 10, 2109–2121. [Google Scholar] [CrossRef]
- Mundell, N.A.; Beier, K.T.; Pan, Y.A.; Lapan, S.W.; Goz Ayturk, D.; Berezovskii, V.K.; Wark, A.R.; Drokhlyansky, E.; Bielecki, J.; Born, R.T.; et al. Vesicular stomatitis virus enables gene transfer and transsynaptic tracing in a wide range of organisms. J. Comp. Neurol. 2015, 523, 1639–1663. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Bradow, B.P.; Zimmerberg, J. Large-scale production of pseudotyped lentiviral vectors using baculovirus GP64. Hum. Gene Ther. 2003, 14, 67–77. [Google Scholar] [CrossRef]
- Schauber, C.A.; Tuerk, M.J.; Pacheco, C.D.; Escarpe, P.A.; Veres, G. Lentiviral vectors pseudotyped with baculovirus gp64 efficiently transduce mouse cells in vivo and show tropism restriction against hematopoietic cell types in vitro. Gene Ther. 2004, 11, 266–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guibinga, G.H.; Friedmann, T. Baculovirus GP64-pseudotyped HIV-based lentivirus vectors are stabilized against complement inactivation by codisplay of decay accelerating factor (DAF) or of a GP64-DAF fusion protein. Mol. Ther. 2005, 11, 645–651. [Google Scholar] [CrossRef]
- Hirano, M.; Kato, S.; Kobayashi, K.; Okada, T.; Yaginuma, H.; Kobayashi, K. Highly efficient retrograde gene transfer into motor neurons by a lentiviral vector pseudotyped with fusion glycoprotein. PLoS ONE 2013, 8, e75896. [Google Scholar] [CrossRef]
- Kato, S.; Kobayashi, K.; Kobayashi, K. Improved transduction efficiency of a lentiviral vector for neuron-specific retrograde gene transfer by optimizing the junction of fusion envelope glycoprotein. J. Neurosci. Methods 2014, 227, 151–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funke, S.; Maisner, A.; Muhlebach, M.D.; Koehl, U.; Grez, M.; Cattaneo, R.; Cichutek, K.; Buchholz, C.J. Targeted cell entry of lentiviral vectors. Mol. Ther. 2008, 16, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Morizono, K.; Xie, Y.; Ringpis, G.E.; Johnson, M.; Nassanian, H.; Lee, B.; Wu, L.; Chen, I.S. Lentiviral vector retargeting to P-glycoprotein on metastatic melanoma through intravenous injection. Nat. Med. 2005, 11, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Peng, K.W.; Harvey, M.; Greiner, S.; Lorimer, I.A.; James, C.D.; Russell, S.J. Rescue and propagation of fully retargeted oncolytic measles viruses. Nat. Biotechnol. 2005, 23, 209–214. [Google Scholar] [CrossRef]
- Anliker, B.; Abel, T.; Kneissl, S.; Hlavaty, J.; Caputi, A.; Brynza, J.; Schneider, I.C.; Munch, R.C.; Petznek, H.; Kontermann, R.E.; et al. Specific gene transfer to neurons, endothelial cells and hematopoietic progenitors with lentiviral vectors. Nat. Methods 2010, 7, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Munch, R.C.; Muhlebach, M.D.; Schaser, T.; Kneissl, S.; Jost, C.; Pluckthun, A.; Cichutek, K.; Buchholz, C.J. DARPins: An efficient targeting domain for lentiviral vectors. Mol. Ther. 2011, 19, 686–693. [Google Scholar] [CrossRef]
- Pluckthun, A. Designed ankyrin repeat proteins (DARPins): Binding proteins for research, diagnostics, and therapy. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 489–511. [Google Scholar] [CrossRef]
- Kneissl, S.; Abel, T.; Rasbach, A.; Brynza, J.; Schneider-Schaulies, J.; Buchholz, C.J. Measles virus glycoprotein-based lentiviral targeting vectors that avoid neutralizing antibodies. PLoS ONE 2012, 7, e46667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mettu, P.S.; Allingham, M.J.; Cousins, S.W. Incomplete response to Anti-VEGF therapy in neovascular AMD: Exploring disease mechanisms and therapeutic opportunities. Prog. Retin. Eye Res. 2021, 82, 100906. [Google Scholar] [CrossRef] [PubMed]
- Richard, A.J.; Duker, J.S.; Reichel, E. Geographic atrophy: Where we are now and where we are going. Curr. Opin. Ophthalmol. 2021, 32, 247–252. [Google Scholar] [CrossRef]
- Pugazhendhi, A.; Hubbell, M.; Jairam, P.; Ambati, B. Neovascular Macular Degeneration: A Review of Etiology, Risk Factors, and Recent Advances in Research and Therapy. Int. J. Mol. Sci. 2021, 22, 1170. [Google Scholar] [CrossRef]
- Caballero, B.; Sherman, S.J.; Falk, T. Insights into the Mechanisms Involved in Protective Effects of VEGF-B in Dopaminergic Neurons. Parkinson’s Dis. 2017, 2017, 4263795. [Google Scholar] [CrossRef] [Green Version]
- Semeraro, F.; Morescalchi, F.; Duse, S.; Gambicorti, E.; Cancarini, A.; Costagliola, C. Pharmacokinetic and Pharmacodynamic Properties of Anti-VEGF Drugs After Intravitreal Injection. Curr. Drug. Metab. 2015, 16, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Koponen, S.; Kokki, E.; Kinnunen, K.; Yla-Herttuala, S. Viral-Vector-Delivered Anti-Angiogenic Therapies to the Eye. Pharmaceutics 2021, 13, 219. [Google Scholar] [CrossRef] [PubMed]
- Dawson, D.W.; Volpert, O.V.; Gillis, P.; Crawford, S.E.; Xu, H.; Benedict, W.; Bouck, N.P. Pigment epithelium-derived factor: A potent inhibitor of angiogenesis. Science 1999, 285, 245–248. [Google Scholar] [CrossRef]
- Taniwaki, T.; Hirashima, N.; Becerra, S.P.; Chader, G.J.; Etcheberrigaray, R.; Schwartz, J.P. Pigment epithelium-derived factor protects cultured cerebellar granule cells against glutamate-induced neurotoxicity. J. Neurochem. 1997, 68, 26–32. [Google Scholar] [CrossRef]
- Cachafeiro, M.; Bemelmans, A.P.; Samardzija, M.; Afanasieva, T.; Pournaras, J.A.; Grimm, C.; Kostic, C.; Philippe, S.; Wenzel, A.; Arsenijevic, Y. Hyperactivation of retina by light in mice leads to photoreceptor cell death mediated by VEGF and retinal pigment epithelium permeability. Cell Death Dis. 2013, 4, e781. [Google Scholar] [CrossRef] [PubMed]
- Askou, A.L.; Aagaard, L.; Kostic, C.; Arsenijevic, Y.; Hollensen, A.K.; Bek, T.; Jensen, T.G.; Mikkelsen, J.G.; Corydon, T.J. Multigenic lentiviral vectors for combined and tissue-specific expression of miRNA- and protein-based antiangiogenic factors. Mol. Ther. Methods Clin. Dev. 2015, 2, 14064. [Google Scholar] [CrossRef]
- Askou, A.L.; Benckendorff, J.N.E.; Holmgaard, A.; Storm, T.; Aagaard, L.; Bek, T.; Mikkelsen, J.G.; Corydon, T.J. Suppression of Choroidal Neovascularization in Mice by Subretinal Delivery of Multigenic Lentiviral Vectors Encoding Anti-Angiogenic MicroRNAs. Hum. Gene Ther. Methods 2017, 28, 222–233. [Google Scholar] [CrossRef]
- Holmgaard, A.; Askou, A.L.; Benckendorff, J.N.E.; Thomsen, E.A.; Cai, Y.; Bek, T.; Mikkelsen, J.G.; Corydon, T.J. In vivo Knockout of the Vegfa Gene by Lentiviral Delivery of CRISPR/Cas9 in Mouse Retinal Pigment Epithelium Cells. Mol. Ther. Nucleic Acids 2017, 9, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Kachi, S.; Binley, K.; Yokoi, K.; Umeda, N.; Akiyama, H.; Muramatu, D.; Iqball, S.; Kan, O.; Naylor, S.; Campochiaro, P.A. Equine infectious anemia viral vector-mediated codelivery of endostatin and angiostatin driven by retinal pigmented epithelium-specific VMD2 promoter inhibits choroidal neovascularization. Hum. Gene Ther. 2009, 20, 31–39. [Google Scholar] [CrossRef]
- Campochiaro, P.A.; Lauer, A.K.; Sohn, E.H.; Mir, T.A.; Naylor, S.; Anderton, M.C.; Kelleher, M.; Harrop, R.; Ellis, S.; Mitrophanous, K.A. Lentiviral Vector Gene Transfer of Endostatin/Angiostatin for Macular Degeneration (GEM) Study. Hum. Gene Ther. 2017, 28, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Solebo, A.L.; Teoh, L.; Rahi, J. Epidemiology of blindness in children. Arch. Dis. Child. 2017, 102, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.; Wang, Y.X.; Ma, Y.; Di, Y. The Long-Noncoding RNA TUG1 Regulates Oxygen-Induced Retinal Neovascularization in Mice via MiR-299. Investig. Ophthalmol. Vis. Sci. 2022, 63, 37. [Google Scholar] [CrossRef] [PubMed]
- Simmons, A.B.; Bretz, C.A.; Wang, H.; Kunz, E.; Hajj, K.; Kennedy, C.; Yang, Z.; Suwanmanee, T.; Kafri, T.; Hartnett, M.E. Correction to: Gene therapy knockdown of VEGFR2 in retinal endothelial cells to treat retinopathy. Angiogenesis 2018, 21, 765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Smith, G.W.; Yang, Z.; Jiang, Y.; McCloskey, M.; Greenberg, K.; Geisen, P.; Culp, W.D.; Flannery, J.; Kafri, T.; et al. Short hairpin RNA-mediated knockdown of VEGFA in Muller cells reduces intravitreal neovascularization in a rat model of retinopathy of prematurity. Am. J. Pathol. 2013, 183, 964–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, S.; Wang, H.; Simmons, A.B.; Suwanmanee, T.; Stoddard, G.J.; Kafri, T.; Hartnett, M.E. Targeted Knockdown of Overexpressed VEGFA or VEGF164 in Muller cells maintains retinal function by triggering different signaling mechanisms. Sci. Rep. 2018, 8, 2003. [Google Scholar] [CrossRef] [Green Version]
- Ishida, S.; Usui, T.; Yamashiro, K.; Kaji, Y.; Amano, S.; Ogura, Y.; Hida, T.; Oguchi, Y.; Ambati, J.; Miller, J.W.; et al. VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. J. Exp. Med. 2003, 198, 483–489. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, H.; Culp, D.; Yang, Z.; Fotheringham, L.; Flannery, J.; Hammond, S.; Kafri, T.; Hartnett, M.E. Targeting Muller cell-derived VEGF164 to reduce intravitreal neovascularization in the rat model of retinopathy of prematurity. Investig. Ophthalmol. Vis. Sci. 2014, 55, 824–831. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.; Schorge, S. Gene Therapy for Neurological Disease: State of the Art and Opportunities for Next-generation Approaches. Neuroscience 2022, 490, 309–314. [Google Scholar] [CrossRef]
- Berger, W.; Kloeckener-Gruissem, B.; Neidhardt, J. The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 2010, 29, 335–375. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Miyoshi, H.; Verma, I.M.; Gage, F.H. Rescue from photoreceptor degeneration in the rd mouse by human immunodeficiency virus vector-mediated gene transfer. J. Virol. 1999, 73, 7812–7816. [Google Scholar] [CrossRef] [Green Version]
- Bemelmans, A.P.; Kostic, C.; Crippa, S.V.; Hauswirth, W.W.; Lem, J.; Munier, F.L.; Seeliger, M.W.; Wenzel, A.; Arsenijevic, Y. Lentiviral gene transfer of RPE65 rescues survival and function of cones in a mouse model of Leber congenital amaurosis. PLoS Med. 2006, 3, e347. [Google Scholar] [CrossRef]
- Kostic, C.; Crippa, S.V.; Pignat, V.; Bemelmans, A.P.; Samardzija, M.; Grimm, C.; Wenzel, A.; Arsenijevic, Y. Gene therapy regenerates protein expression in cone photoreceptors in Rpe65(R91W/R91W) mice. PLoS ONE 2011, 6, e16588. [Google Scholar] [CrossRef]
- Udry, F.; Decembrini, S.; Gamm, D.M.; Deglon, N.; Kostic, C.; Arsenijevic, Y. Lentiviral mediated RPE65 gene transfer in healthy hiPSCs-derived retinal pigment epithelial cells markedly increased RPE65 mRNA, but modestly protein level. Sci. Rep. 2020, 10, 8890. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G * C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Suh, S.; Choi, E.H.; Leinonen, H.; Foik, A.T.; Newby, G.A.; Yeh, W.H.; Dong, Z.; Kiser, P.D.; Lyon, D.C.; Liu, D.R.; et al. Restoration of visual function in adult mice with an inherited retinal disease via adenine base editing. Nat. Biomed. Eng. 2021, 5, 169–178. [Google Scholar] [CrossRef]
- Jo, D.H.; Song, D.W.; Cho, C.S.; Kim, U.G.; Lee, K.J.; Lee, K.; Park, S.W.; Kim, D.; Kim, J.H.; Kim, J.S.; et al. CRISPR-Cas9-mediated therapeutic editing of Rpe65 ameliorates the disease phenotypes in a mouse model of Leber congenital amaurosis. Sci. Adv. 2019, 5, eaax1210. [Google Scholar] [CrossRef] [Green Version]
- Weil, D.; Blanchard, S.; Kaplan, J.; Guilford, P.; Gibson, F.; Walsh, J.; Mburu, P.; Varela, A.; Levilliers, J.; Weston, M.D.; et al. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 1995, 374, 60–61. [Google Scholar] [CrossRef]
- Hashimoto, T.; Gibbs, D.; Lillo, C.; Azarian, S.M.; Legacki, E.; Zhang, X.M.; Yang, X.J.; Williams, D.S. Lentiviral gene replacement therapy of retinas in a mouse model for Usher syndrome type 1B. Gene Ther. 2007, 14, 584–594. [Google Scholar] [CrossRef] [Green Version]
- Kong, J.; Kim, S.R.; Binley, K.; Pata, I.; Doi, K.; Mannik, J.; Zernant-Rajang, J.; Kan, O.; Iqball, S.; Naylor, S.; et al. Correction of the disease phenotype in the mouse model of Stargardt disease by lentiviral gene therapy. Gene Ther. 2008, 15, 1311–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, M.A.; Erker, L.R.; Audo, I.; Choi, D.; Mohand-Said, S.; Sestakauskas, K.; Benoit, P.; Appelqvist, T.; Krahmer, M.; Segaut-Prevost, C.; et al. Three-year safety results of SAR422459 (EIAV-ABCA4) gene therapy in patients with ABCA4-associated Stargardt disease: An open-label dose-escalation phase I/IIa clinical trial, cohorts 1-5. Am. J. Ophthalmol. 2022, 240, 285–301. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.A.; Choi, D.; Erker, L.R.; Pennesi, M.E.; Yang, P.; Chegarnov, E.N.; Steinkamp, P.N.; Schlechter, C.L.; Dhaenens, C.M.; Mohand-Said, S.; et al. Test-Retest Variability of Functional and Structural Parameters in Patients with Stargardt Disease Participating in the SAR422459 Gene Therapy Trial. Transl. Vis. Sci. Technol. 2016, 5, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ait-Ali, N.; Fridlich, R.; Millet-Puel, G.; Clerin, E.; Delalande, F.; Jaillard, C.; Blond, F.; Perrocheau, L.; Reichman, S.; Byrne, L.C.; et al. Rod-derived cone viability factor promotes cone survival by stimulating aerobic glycolysis. Cell 2015, 161, 817–832. [Google Scholar] [CrossRef] [Green Version]
- Leveillard, T.; Mohand-Said, S.; Lorentz, O.; Hicks, D.; Fintz, A.C.; Clerin, E.; Simonutti, M.; Forster, V.; Cavusoglu, N.; Chalmel, F.; et al. Identification and characterization of rod-derived cone viability factor. Nat. Genet. 2004, 36, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Punzo, C.; Kornacker, K.; Cepko, C.L. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat. Neurosci. 2009, 12, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Cideciyan, A.V.; Jacobson, S.G.; Beltran, W.A.; Sumaroka, A.; Swider, M.; Iwabe, S.; Roman, A.J.; Olivares, M.B.; Schwartz, S.B.; Komaromy, A.M.; et al. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc. Natl. Acad. Sci. USA 2013, 110, E517–E525. [Google Scholar] [CrossRef] [Green Version]
- Gal, A.; Li, Y.; Thompson, D.A.; Weir, J.; Orth, U.; Jacobson, S.G.; Apfelstedt-Sylla, E.; Vollrath, D. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat. Genet. 2000, 26, 270–271. [Google Scholar] [CrossRef]
- Nandrot, E.; Dufour, E.M.; Provost, A.C.; Pequignot, M.O.; Bonnel, S.; Gogat, K.; Marchant, D.; Rouillac, C.; Sepulchre, d.C.; Bihoreau, M.T.; et al. Homozygous deletion in the coding sequence of the c-mer gene in RCS rats unravels general mechanisms of physiological cell adhesion and apoptosis. Neurobiol. Dis. 2000, 7, 586–599. [Google Scholar] [CrossRef] [Green Version]
- Connell, G.; Bascom, R.; Molday, L.; Reid, D.; McInnes, R.R.; Molday, R.S. Photoreceptor peripherin is the normal product of the gene responsible for retinal degeneration in the rds mouse. Proc. Natl. Acad. Sci. USA 1991, 88, 723–726. [Google Scholar] [CrossRef] [Green Version]
- Travis, G.H.; Sutcliffe, J.G.; Bok, D. The retinal degeneration slow (rds) gene product is a photoreceptor disc membrane-associated glycoprotein. Neuron 1991, 6, 61–70. [Google Scholar] [CrossRef]
- Liu, X.; Hu, Y.; Filla, M.S.; Gabelt, B.T.; Peters, D.M.; Brandt, C.R.; Kaufman, P.L. The effect of C3 transgene expression on actin and cellular adhesions in cultured human trabecular meshwork cells and on outflow facility in organ cultured monkey eyes. Mol. Vis. 2005, 11, 1112–1121. [Google Scholar] [PubMed]
- Slauson, S.R.; Peters, D.M.; Schwinn, M.K.; Kaufman, P.L.; Gabelt, B.T.; Brandt, C.R. Viral Vector Effects on Exoenzyme C3 Transferase-Mediated Actin Disruption and on Outflow Facility. Invest. Ophthalmol. Vis. Sci. 2015, 56, 2431–2438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acott, T.S.; Samples, J.R.; Bradley, J.M.; Bacon, D.R.; Bylsma, S.S.; Van Buskirk, E.M. Trabecular repopulation by anterior trabecular meshwork cells after laser trabeculoplasty. Am. J. Ophthalmol. 1989, 107, 1–6. [Google Scholar] [CrossRef]
- Sundaresan, Y.; Veerappan, M.; Ramasamy, K.S.; Chidambaranathan, G.P. Identification, quantification and age-related changes of human trabecular meshwork stem cells. Eye Vis. 2019, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barraza, R.A.; McLaren, J.W.; Poeschla, E.M. Prostaglandin pathway gene therapy for sustained reduction of intraocular pressure. Mol. Ther. 2010, 18, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Luna, C.; Parker, M.; Challa, P.; Gonzalez, P. Long-Term Decrease of Intraocular Pressure in Rats by Viral Delivery of miR-146a. Transl. Vis. Sci. Technol. 2021, 10, 14. [Google Scholar] [CrossRef]
- Li, G.; Luna, C.; Qiu, J.; Epstein, D.L.; Gonzalez, P. Modulation of inflammatory markers by miR-146a during replicative senescence in trabecular meshwork cells. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2976–2985. [Google Scholar] [CrossRef]
- Luna, C.; Li, G.; Qiu, J.; Epstein, D.L.; Gonzalez, P. MicroRNA-24 regulates the processing of latent TGFbeta1 during cyclic mechanical stress in human trabecular meshwork cells through direct targeting of FURIN. J. Cell Physiol. 2011, 226, 1407–1414. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.S.; Rasmussen, C.A.; Filla, M.S.; Slauson, S.R.; Kolb, A.W.; Peters, D.M.; Kaufman, P.L.; Gabelt, B.T.; Brandt, C.R. Prospects for lentiviral vector mediated prostaglandin F synthase gene delivery in monkey eyes in vivo. Curr. Eye Res. 2014, 39, 859–870. [Google Scholar] [CrossRef] [Green Version]
- Dinslage, S.; Hueber, A.; Diestelhorst, M.; Krieglstein, G. The influence of Latanoprost 0.005% on aqueous humor flow and outflow facility in glaucoma patients: A double-masked placebo-controlled clinical study. Graefe’s Arch. Clin. Exp. Ophthalmol. 2004, 242, 654–660. [Google Scholar] [CrossRef]
- Aktas, Z.; Rao, H.; Slauson, S.R.; Gabelt, B.T.; Larsen, I.V.; Sheridan, R.T.C.; Herrnberger, L.; Tamm, E.R.; Kaufman, P.L.; Brandt, C.R. Proteasome Inhibition Increases the Efficiency of Lentiviral Vector-Mediated Transduction of Trabecular Meshwork. Investig. Ophthalmol. Vis. Sci. 2018, 59, 298–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Q.; Lu, Q.; So, K.F.; Yip, H.K. CNTF, not other trophic factors, promotes axonal regeneration of axotomized retinal ganglion cells in adult hamsters. Investig. Ophthalmol. Vis. Sci. 1999, 40, 760–766. [Google Scholar]
- Koeberle, P.D.; Ball, A.K. Effects of GDNF on retinal ganglion cell survival following axotomy. Vis. Res. 1998, 38, 1505–1515. [Google Scholar] [CrossRef] [Green Version]
- Mey, J.; Thanos, S. Intravitreal injections of neurotrophic factors support the survival of axotomized retinal ganglion cells in adult rats in vivo. Brain Res. 1993, 602, 304–317. [Google Scholar] [CrossRef]
- Peinado-Ramon, P.; Salvador, M.; Villegas-Perez, M.P.; Vidal-Sanz, M. Effects of axotomy and intraocular administration of NT-4, NT-3, and brain-derived neurotrophic factor on the survival of adult rat retinal ganglion cells. A quantitative in vivo study. Investig. Ophthalmol. Vis. Sci. 1996, 37, 489–500. [Google Scholar]
- Deglon, N.; Tseng, J.L.; Bensadoun, J.C.; Zurn, A.D.; Arsenijevic, Y.; Pereira de Almeida, L.; Zufferey, R.; Trono, D.; Aebischer, P. Self-inactivating lentiviral vectors with enhanced transgene expression as potential gene transfer system in Parkinson’s disease. Hum. Gene Ther. 2000, 11, 179–190. [Google Scholar] [CrossRef]
- De Almeida, L.P.; Zala, D.; Aebischer, P.; Deglon, N. Neuroprotective effect of a CNTF-expressing lentiviral vector in the quinolinic acid rat model of Huntington’s disease. Neurobiol. Dis. 2001, 8, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, M.; Ikeda, Y.; Yonemitsu, Y.; Goto, Y.; Murakami, Y.; Yoshida, N.; Tabata, T.; Hasegawa, M.; Tobimatsu, S.; Sueishi, K.; et al. Pigment epithelium-derived factor gene therapy targeting retinal ganglion cell injuries: Neuroprotection against loss of function in two animal models. Hum. Gene Ther. 2011, 22, 559–565. [Google Scholar] [CrossRef] [Green Version]
- Amador, C.; Shah, R.; Ghiam, S.; Kramerov, A.A.; Ljubimov, A.V. Gene Therapy in the Anterior Eye Segment. Curr. Gene Ther. 2022, 22, 104–131. [Google Scholar] [CrossRef]
- Nowell, C.S.; Odermatt, P.D.; Azzolin, L.; Hohnel, S.; Wagner, E.F.; Fantner, G.E.; Lutolf, M.P.; Barrandon, Y.; Piccolo, S.; Radtke, F. Chronic inflammation imposes aberrant cell fate in regenerating epithelia through mechanotransduction. Nat. Cell Biol. 2016, 18, 168–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivas, S.P. Cell signaling in regulation of the barrier integrity of the corneal endothelium. Exp. Eye Res. 2012, 95, 8–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambati, B.K.; Nozaki, M.; Singh, N.; Takeda, A.; Jani, P.D.; Suthar, T.; Albuquerque, R.J.; Richter, E.; Sakurai, E.; Newcomb, M.T.; et al. Corneal avascularity is due to soluble VEGF receptor-1. Nature 2006, 443, 993–997. [Google Scholar] [CrossRef] [Green Version]
- Barcia, R.N.; Dana, M.R.; Kazlauskas, A. Corneal graft rejection is accompanied by apoptosis of the endothelium and is prevented by gene therapy with bcl-xL. Am. J. Transplant. 2007, 7, 2082–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchsluger, T.A.; Jurkunas, U.; Kazlauskas, A.; Dana, R. Corneal endothelial cells are protected from apoptosis by gene therapy. Hum. Gene Ther. 2011, 22, 549–558. [Google Scholar] [CrossRef] [Green Version]
- Catala, P.; Thuret, G.; Skottman, H.; Mehta, J.S.; Parekh, M.; Ni Dhubhghaill, S.; Collin, R.W.J.; Nuijts, R.; Ferrari, S.; LaPointe, V.L.S.; et al. Approaches for corneal endothelium regenerative medicine. Prog. Retin. Eye Res. 2022, 87, 100987. [Google Scholar] [CrossRef]
- Klebe, S.; Coster, D.J.; Sykes, P.J.; Swinburne, S.; Hallsworth, P.; Scheerlinck, J.P.; Krishnan, R.; Williams, K.A. Prolongation of sheep corneal allograft survival by transfer of the gene encoding ovine IL-12-p40 but not IL-4 to donor corneal endothelium. J. Immunol. 2005, 175, 2219–2226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, D.G.; Coster, D.J.; Brereton, H.M.; Hart, P.H.; Koldej, R.; Anson, D.S.; Williams, K.A. Lentivirus-mediated gene transfer of interleukin 10 to the ovine and human cornea. Clin. Exp. Ophthalmol. 2010, 38, 405–413. [Google Scholar] [CrossRef]
- Ghasemi, H.; Ghazanfari, T.; Yaraee, R.; Owlia, P.; Hassan, Z.M.; Faghihzadeh, S. Roles of IL-10 in ocular inflammations: A review. Ocul. Immunol. Inflamm. 2012, 20, 406–418. [Google Scholar] [CrossRef]
- Klebe, S.; Sykes, P.J.; Coster, D.J.; Krishnan, R.; Williams, K.A. Prolongation of sheep corneal allograft survival by ex vivo transfer of the gene encoding interleukin-10. Transplantation 2001, 71, 1214–1220. [Google Scholar] [CrossRef]
- Nosov, M.; Wilk, M.; Morcos, M.; Cregg, M.; O’Flynn, L.; Treacy, O.; Ritter, T. Role of lentivirus-mediated overexpression of programmed death-ligand 1 on corneal allograft survival. Am. J. Transplant. 2012, 12, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Tornabene, P.; Trapani, I. Can Adeno-Associated Viral Vectors Deliver Effectively Large Genes? Hum. Gene Ther. 2020, 31, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Gross, D.A.; Tedesco, N.; Leborgne, C.; Ronzitti, G. Overcoming the Challenges Imposed by Humoral Immunity to AAV Vectors to Achieve Safe and Efficient Gene Transfer in Seropositive Patients. Front. Immunol. 2022, 13, 857276. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Connolly, A.M.; Lehman, K.J.; Griffin, D.A.; Khan, S.Z.; Dharia, S.D.; Quintana-Gallardo, L.; Rodino-Klapac, L.R. Testing preexisting antibodies prior to AAV gene transfer therapy: Rationale, lessons and future considerations. Mol. Ther. Methods Clin. Dev. 2022, 25, 74–83. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arsenijevic, Y.; Berger, A.; Udry, F.; Kostic, C. Lentiviral Vectors for Ocular Gene Therapy. Pharmaceutics 2022, 14, 1605. https://doi.org/10.3390/pharmaceutics14081605
Arsenijevic Y, Berger A, Udry F, Kostic C. Lentiviral Vectors for Ocular Gene Therapy. Pharmaceutics. 2022; 14(8):1605. https://doi.org/10.3390/pharmaceutics14081605
Chicago/Turabian StyleArsenijevic, Yvan, Adeline Berger, Florian Udry, and Corinne Kostic. 2022. "Lentiviral Vectors for Ocular Gene Therapy" Pharmaceutics 14, no. 8: 1605. https://doi.org/10.3390/pharmaceutics14081605
APA StyleArsenijevic, Y., Berger, A., Udry, F., & Kostic, C. (2022). Lentiviral Vectors for Ocular Gene Therapy. Pharmaceutics, 14(8), 1605. https://doi.org/10.3390/pharmaceutics14081605