
Analysis of Biologics Molecular Descriptors towards Predictive Modelling for Protein Drug Development Using Time-Gated Raman Spectroscopy

 , ,
, , 
Abstract
:
1. Introduction
- (i)
- The detection and characterization of (sub)visible particles (e.g., visual inspection, optical microscopy, light obscuration, flow imaging, fluorescence microscopy, conductivity-based particle counter, laser diffraction, dynamic light scattering (DLS), nanoparticle tracking analysis, MALLS, turbidimetry, and nephelometry);
- (ii)
- the use of separation techniques for the detection and characterization of aggregates, i.e., (denaturing/reducing) size exclusion chromatography, SDS/Native PAGE, capillary-SDS electrophoresis, and AF4 [13];
- (iii)
- other techniques, e.g., electron/atomic force microscopy, mass spectrometry, macro-ion mobility spectrometry, and AUC [14]. Label-free methods for evaluating protein folding states, such as infrared spectroscopy, Raman spectroscopy, UV/VIS absorption spectroscopy, fluorescence spectroscopy, and circular dichroism spectroscopy, are relevant to mention as they are utilized as in-line analytical techniques due to their non-invasive nature. A recent review outlines the importance of Raman spectroscopy to biopharmaceuticals in greater detail [15]. However, the sensitivity, robustness, and the ability of these label-free techniques for quantification need to be improved for protein applications.
2. Materials and Methods
2.1. Chemical, Reagents, and Protein Samples
2.2. Dynamic Light Scattering
2.3. Tryptophan Fluorescence
2.4. Circular Dichroism (CD) Spectroscopy
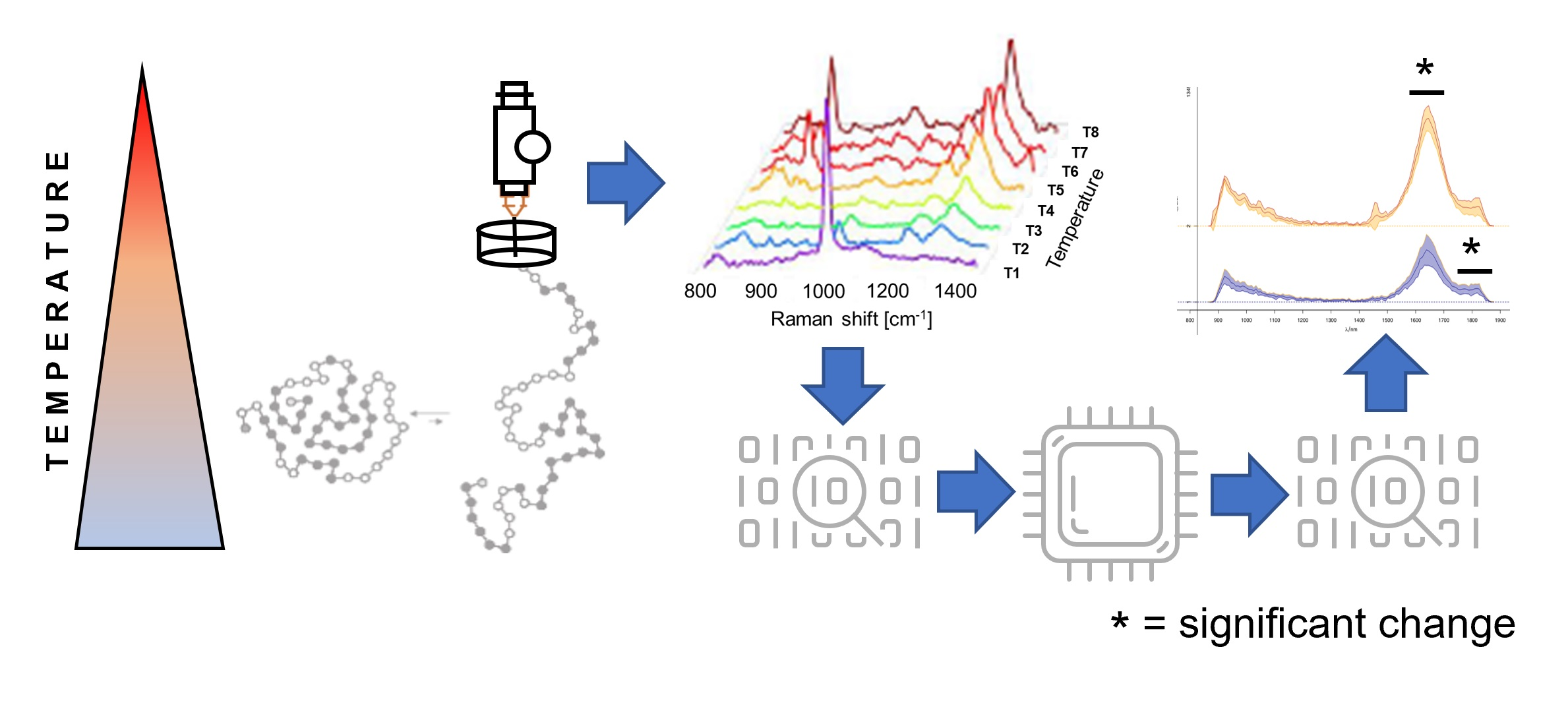
2.5. Time-Gated Raman Spectroscopy
2.6. Data Preprocessing
2.7. Data Analysis
3. Results
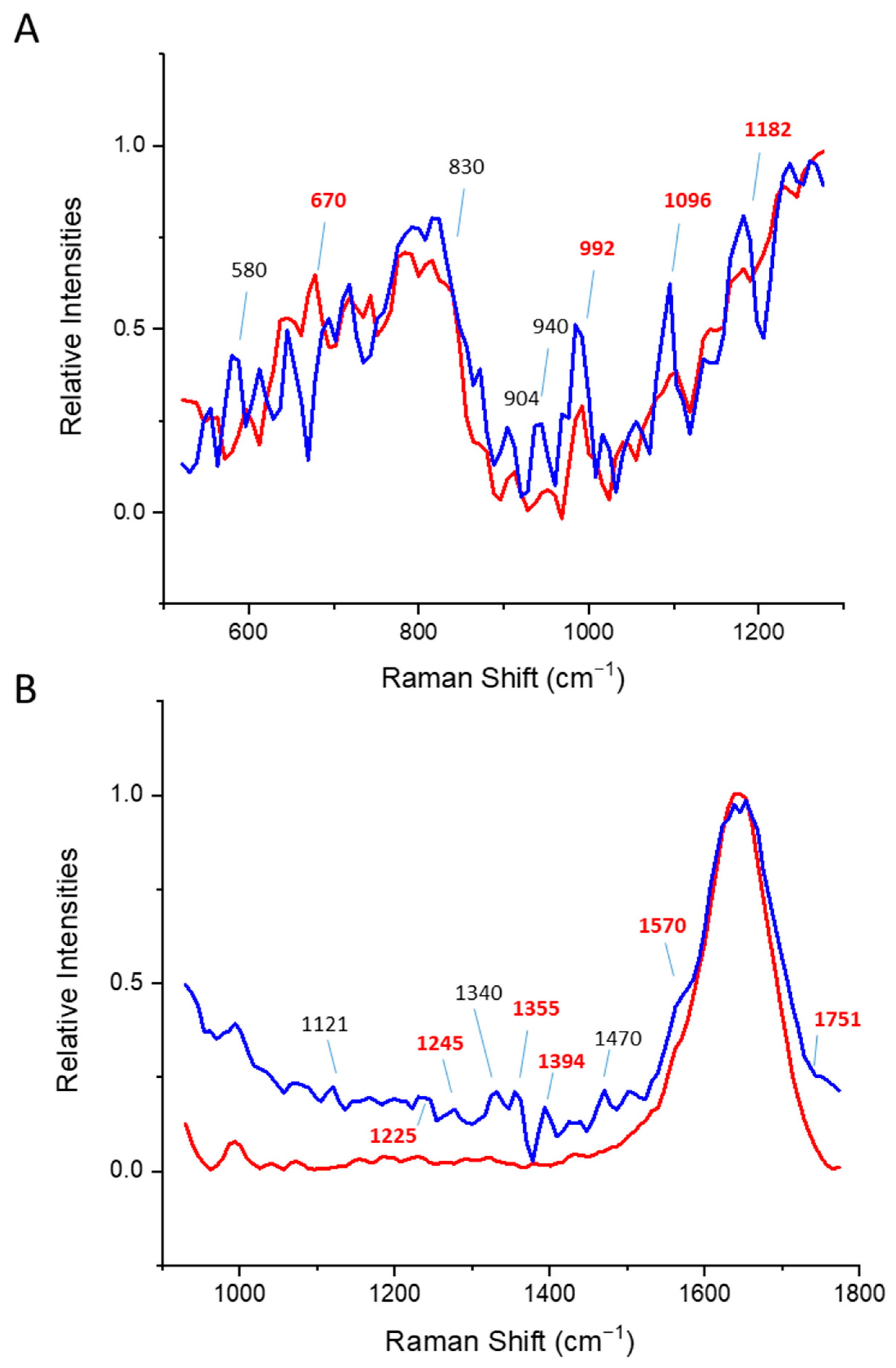
3.1. Alpha Helical Proteins
3.2. Beta Sheet Proteins
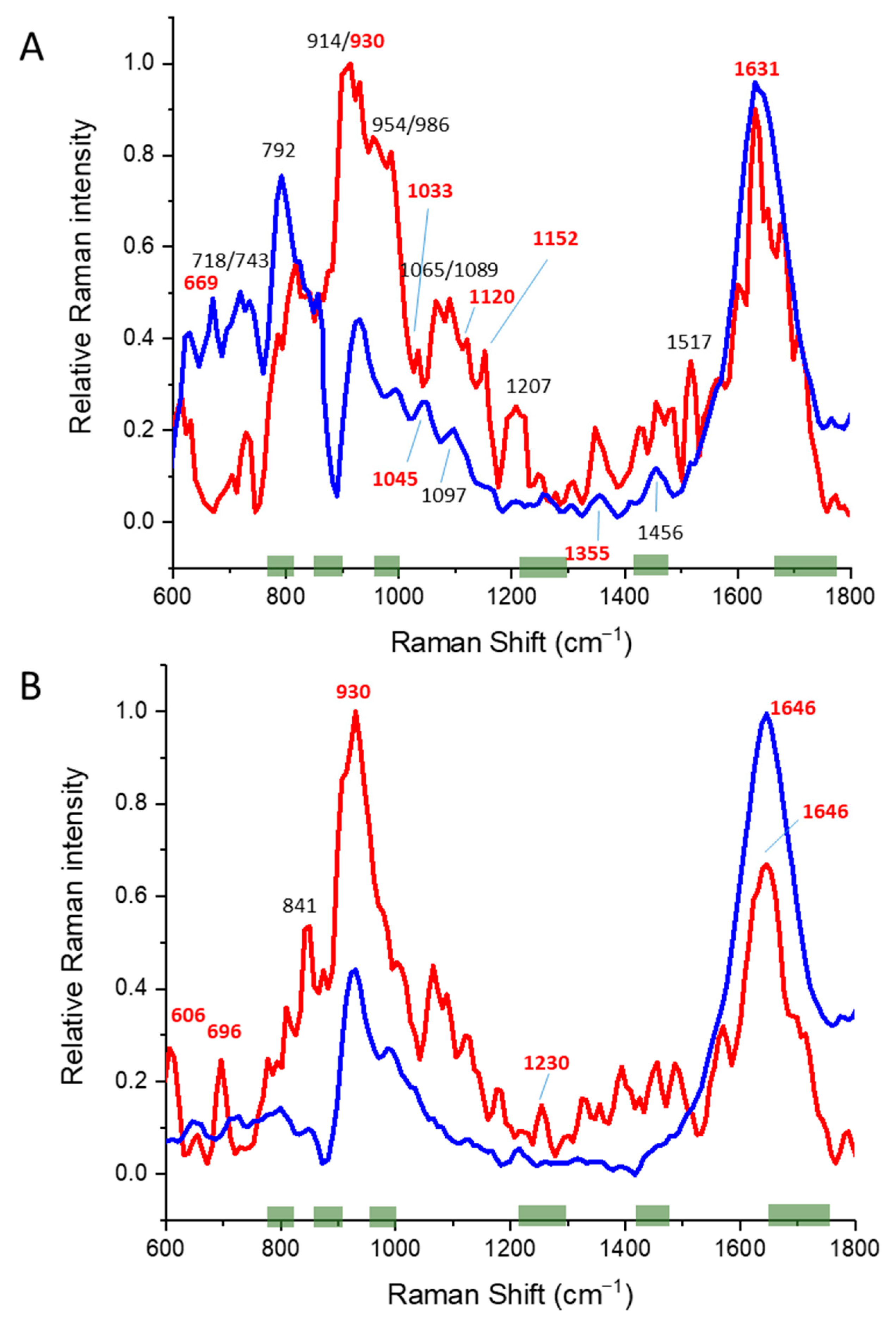
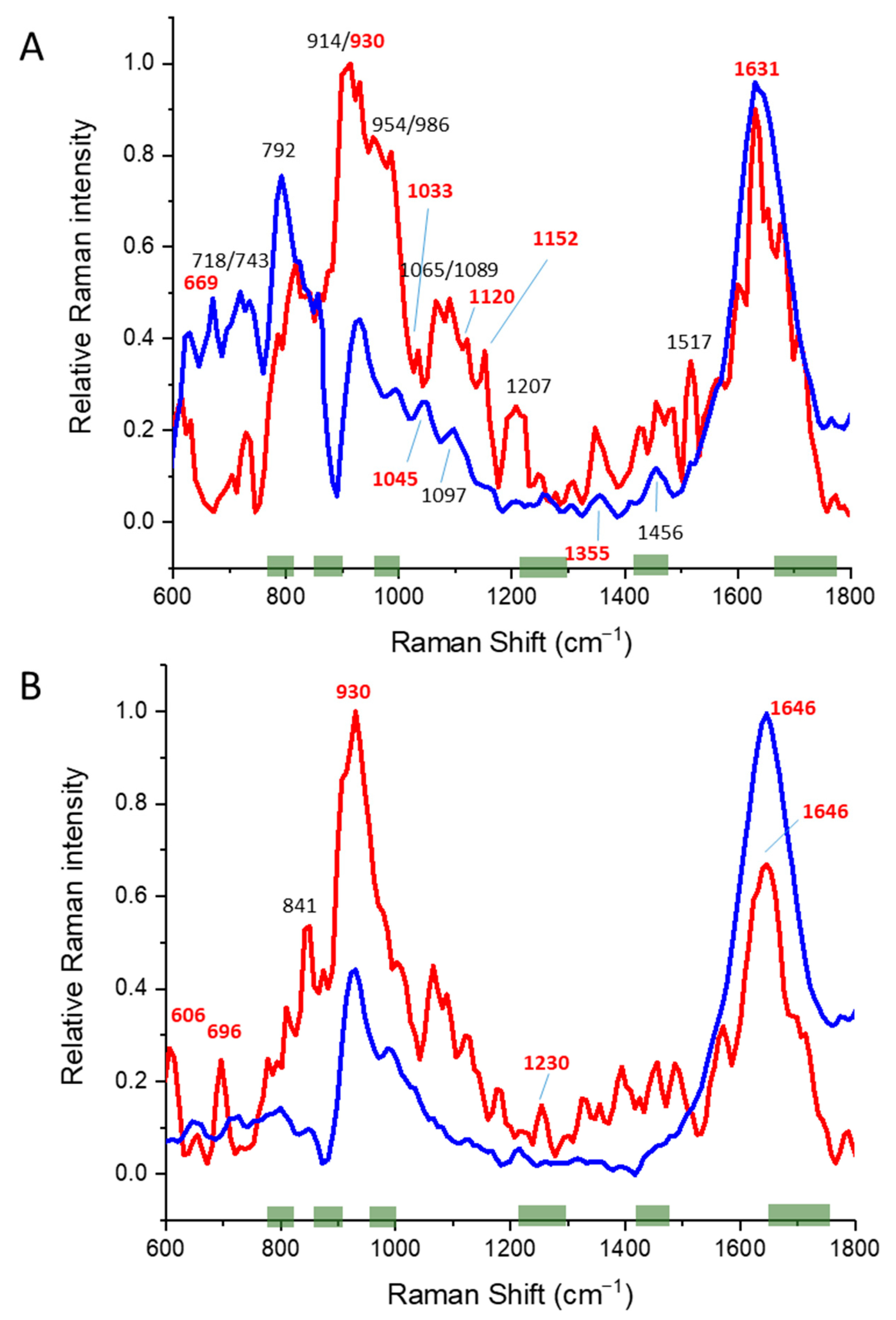
3.3. Alpha/Beta Proteins
4. Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein (α, β, α/β) | Tm (°C) | Van’t Hoff Enthalpy (kJ/mol) | Literature Value Tm (°C) |
|---|---|---|---|
| BSA (α) | 61.2 ± 0.1 a | 256.6 ± 8.4 | 63 [59] |
| CNTF (α) | 55.0 ± 1.9 b | 360.0 ± 34.3 | 53 [27] |
| Fab (β) | 73.9 ± 0.3 | 437.5 ± 17.4 | 61–70 [60,61] |
| IgGglycosylated (β) c | 65.5 ± 0.2 72.1 ± 0.4 | 336.6 ± 7.9 203.9 ± 10.6 | 60–68 71–77 [60,62] |
| IgGnon-glycosylated (β) | 71.5 ± 0.2 | 265.2 ± 12.6 | 62–66 [63] |
| Pepsin (β) | 49.9 ± 0.2 d | 352.1 ± 17.0 | 52 e [64] |
| Ovalbumin (α/β) | 72.3 ± 0.1 | 181.3 ± 2.2 | 71–76 [65] |
| ScTIM (α/β) | 55.3 ± 1.7 | 360.0 ± 13.4 | ~58 [66] |
| LmTIME65Q (α/β) | 81.0 ± 0.3 a | 172.9 ± 12.6 | 83 [26] |
| Protein (α, β, α/β) | Hydrodynamic Diameter at 20 °C [nm] | Taggregation [°C] | Literature Value Taggregation [°C] c |
|---|---|---|---|
| BSA (α) | 8.0 | 58 | ~62 [67] |
| CNTF (α) | ND | ND | 38 a [27] |
| Fab (β) | 205.8 | 60 | - |
| F(ab′)2 (β) | 11.2 | 63 | - |
| IgGglycosylated (β) | 12.3 | 64 | 55–80 [68] |
| IgGnon-glycosylated (β) | ND | ND | - |
| Pepsin (β) | 50.9 | 64 | - |
| Ovalbumin (α/β) | 28.8 | - b | 71 [48] |
| ScTIM (α/β) | 68.4 | 58 | - |
| LmTIME65Q (α/β) | ND | ND | - |
| Protein (α, β, α/β) | Maximum Fluorescence Intensity at Temperature [°C] | Red/Blue Shift a | Tryptophan Oxidation (Peak at 515) |
|---|---|---|---|
| BSA (α) | 45 | Red (10 nm) | No |
| CNTF (α) | 30 | Red (12 nm) | No |
| Fab (β) | 85 | Red (4 nm) | No |
| F(ab′)2 (β) | ND b | ND | ND |
| IgGglycosylated (β) | 85 | Red (5 nm) | No |
| IgGnon-glycosylated (β) | ND | ND | ND |
| Pepsin (β) | 37 | No | No |
| Ovalbumin (α/β) | 37 50 | Blue (5 nm) Blue (5 nm) | No |
| ScTIM (α/β) | 65 | Red (10 nm) | No |
| LmTIME65Q (α/β) | 86 | Red (6 nm) | No |
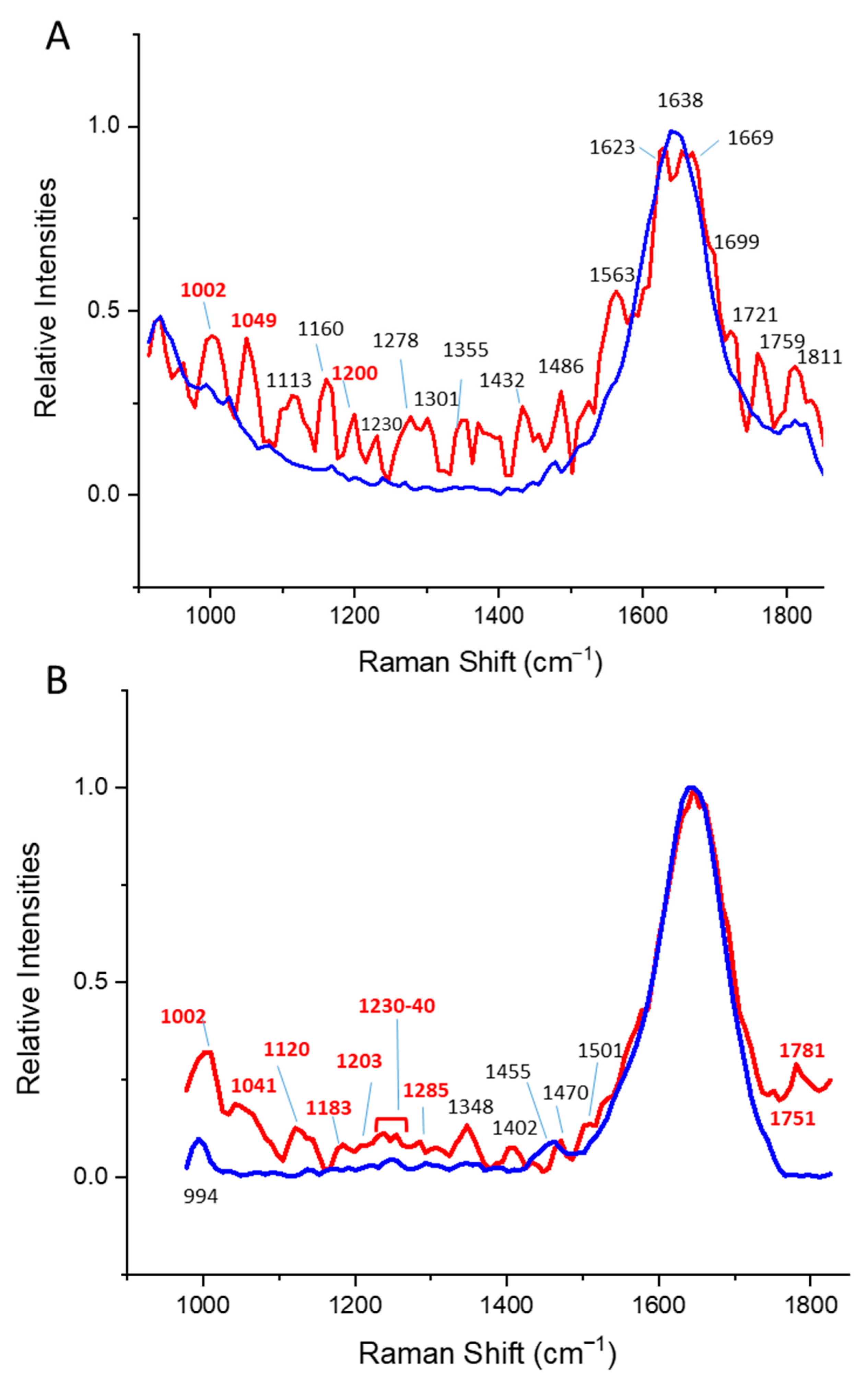
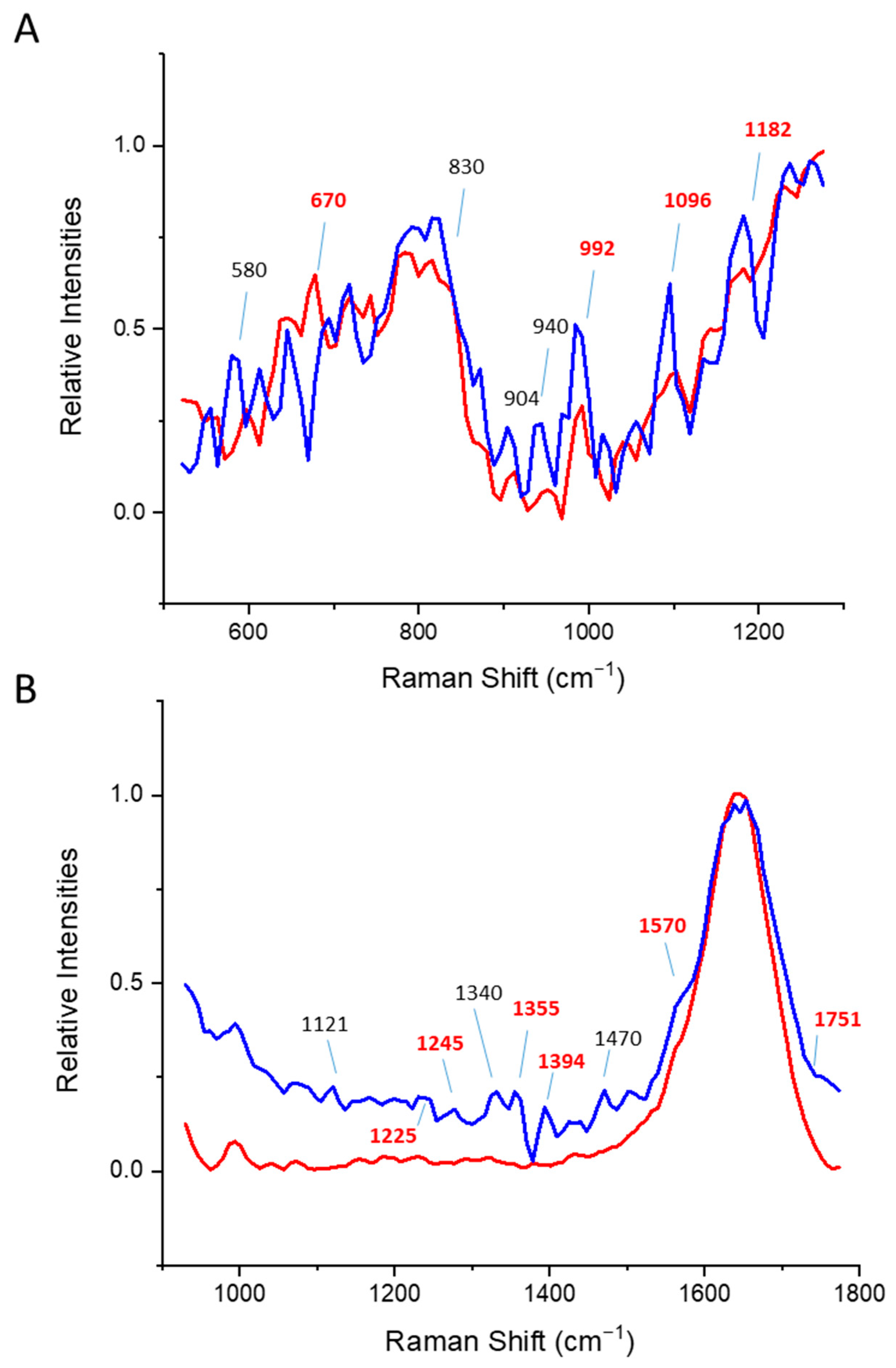
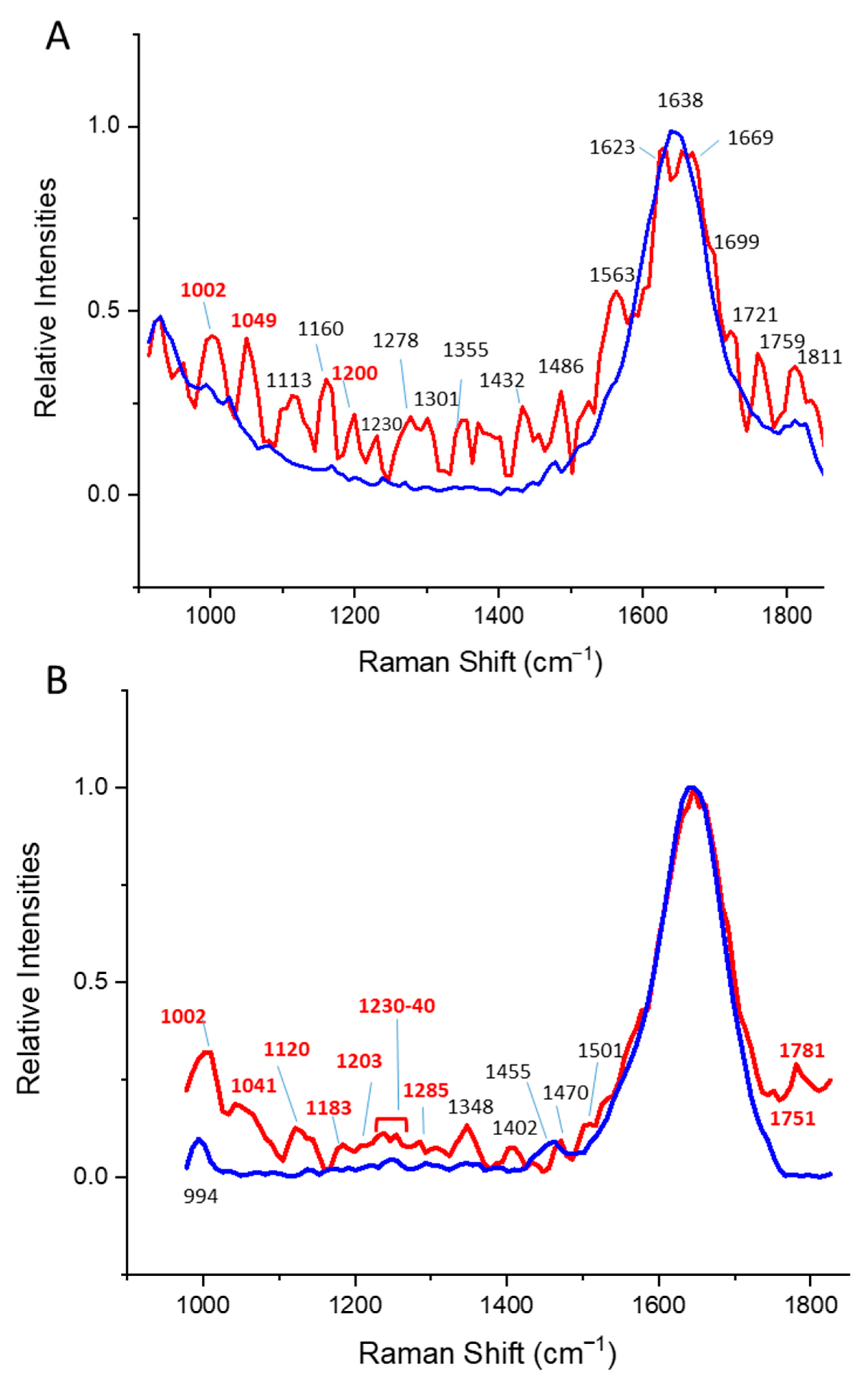
| Protein Structural Class | Most Relevant Changes [cm−1] | Bond Type | Relevant Temperature Change Correlates with |
|---|---|---|---|
| α | 880–900 | Trp | Trp-fluorescence |
| 910 | Ser a | DLS | |
| 940 | N-Ca-C | CD | |
| 950 | N-Ca-C | ||
| 970 | Ser/His a | ||
| 1180 | Val/Arg/other amino acids a | ||
| 1280 | Amide II | ||
| 1310 | Phe, Tyr, Trp a | ||
| β | 1350–1390 | Trp a | Trp-fluorescence |
| 1695–1760 | Amide I/carbonyl stretch | ||
| α/β | 980–1020 | Phe | Trp-fluorescence |
| 1200–1205 | Amide II | DLS | |
| 1820–1860 | C=O | CD |
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Casteleijn, M.G.; Richardson, D. Engineering Cells and Proteins—Creating pharmaceuticals. Eur. Pharm. Rev. 2014, 2014, 4. [Google Scholar]
- Leader, B.; Baca, Q.J.; Golan, D.E. Protein therapeutics: A summary and pharmacological classification. Nat. Rev. Drug Discov. 2008, 7, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Sauna, Z.E.; Lagassé, H.A.D.; Alexaki, A.; Simhadri, V.L.; Katagiri, N.H.; Jankowski, W.; Kimchi-Sarfaty, C. Recent advances in (therapeutic protein) drug development. F1000Research 2017, 6, 113. [Google Scholar] [CrossRef] [Green Version]
- Quianzon, C.C.; Cheikh, I. History of insulin. J. Community Hosp. Intern. Med. Perspect. 2012, 2, 18701. [Google Scholar] [CrossRef] [Green Version]
- Zheng, K.; Bantog, C.; Bayer, R. The impact of glycosylation on monoclonal antibody conformation and stability. mAbs 2011, 3, 568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, S. Biologics: An Update and Challenge of Their Pharmacokinetics. Curr. Drug Metab. 2014, 15, 271–290. [Google Scholar] [CrossRef]
- Frokjaer, S.; Otzen, D.E. Protein drug stability: A formulation challenge. Nat. Rev. Drug Discov. 2005, 4, 298–306. [Google Scholar] [CrossRef]
- Mahler, H.C.; Friess, W.; Grauschopf, U.; Kiese, S. Protein aggregation: Pathways, induction factors and analysis. J. Pharm. Sci. 2009, 98, 2909–2934. [Google Scholar] [CrossRef]
- Lipiäinen, T.; Peltoniemi, M.; Sarkhel, S.; Yrjönen, T.; Vuorela, H.; Urtti, A.; Juppo, A. Formulation and Stability of Cytokine Therapeutics. J. Pharm. Sci. 2015, 104, 307–326. [Google Scholar] [CrossRef]
- Nejadnik, M.R.; Randolph, T.W.; Volkin, D.B.; Schöneich, C.; Carpenter, J.F.; Crommelin, D.J.A.; Jiskoot, W. Postproduction Handling and Administration of Protein Pharmaceuticals and Potential Instability Issues. J. Pharm. Sci. 2018, 107, 2013–2019. [Google Scholar] [CrossRef] [Green Version]
- Rathore, A.S. Roadmap for implementation of quality by design (QbD) for biotechnology products. Trends Biotechnol. 2009, 27, 546–553. [Google Scholar] [CrossRef]
- Den Engelsman, J.; Garidel, P.; Smulders, R.; Koll, H.; Smith, B.; Bassarab, S.; Seidl, A.; Hainzl, O.; Jiskoot, W. Strategies for the assessment of protein aggregates in pharmaceutical biotech product development. Pharm. Res. 2011, 28, 920–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawe, A.; Romeijn, S.; Filipe, V.; Jiskoot, W. Asymmetrical flow field-flow fractionation method for the analysis of submicron protein aggregates. J. Pharm. Sci. 2012, 101, 4129–4139. [Google Scholar] [CrossRef] [PubMed]
- Lebowitz, J.; Lewis, M.S.; Schuck, P. Modern analytical ultracentrifugation in protein science: A tutorial review. Protein Sci. A Publ. Protein Soc. 2002, 11, 2067. [Google Scholar] [CrossRef] [Green Version]
- Esmonde-White, K.A.; Cuellar, M.; Lewis, I.R. The role of Raman spectroscopy in biopharmaceuticals from development to manufacturing. Anal. Bioanal. Chem. 2021, 414, 969–991. [Google Scholar] [CrossRef] [PubMed]
- Oshinbolu, S.; Shah, R.; Finka, G.; Molloy, M.; Uden, M.; Bracewell, D.G. Evaluation of fluorescent dyes to measure protein aggregation within mammalian cell culture supernatants. J. Chem. Technol. Biotechnol. 2018, 93, 909–917. [Google Scholar] [CrossRef]
- Kögler, M.; Itkonen, J.; Viitala, T.; Casteleijn, M.G. Assessment of recombinant protein production in E. coli with Time-Gated Surface Enhanced Raman Spectroscopy (TG-SERS). Sci. Rep. 2020, 10, 2472. [Google Scholar] [CrossRef] [PubMed]
- Varmuza, K.; Filzmoser, P. Introduction to Multivariate Statistical Analysis in Chemometrics, 1st ed.; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]
- Korenius, T.; Laurikkala, J.; Juhola, M. On principal component analysis, cosine and Euclidean measures in information retrieval. Inf. Sci. 2007, 177, 4893–4905. [Google Scholar] [CrossRef]
- Gautam, R.; Vanga, S.; Ariese, F.; Umapathy, S. Review of multidimensional data processing approaches for Raman and infrared spectroscopy. EPJ Tech. Instrum. 2015, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Abdi, H.; Williams, L.J. Principal component analysis. WIREs Comput. Stat. 2010, 2, 433–459. [Google Scholar] [CrossRef]
- Bonnier, F.; Byrne, H.J. Understanding the molecular information contained in principal component analysis of vibrational spectra of biological systems. Analyst 2012, 137, 322–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, J.; Qin, J.; Zeng, H.; Wang, K.; Wang, D.; Wang, S. Confocal Raman microspectroscopic analysis on the time-dependent impact of DAPT, a γ-secretase inhibitor, to osteosarcoma cells. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 239, 118372. [Google Scholar] [CrossRef] [PubMed]
- Itkonen, J.M.; Urtti, A.; Bird, L.E.; Sarkhel, S. Codon optimization and factorial screening for enhanced soluble expression of human ciliary neurotrophic factor in Escherichia coli. BMC Biotechnol. 2014, 14, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, D.; Itkonen, J.; Nievas, J.; Urtti, A.; Casteleijn, M.G. Accelerated pharmaceutical protein development with integrated cell free expression, purification, and bioconjugation. Sci. Rep. 2018, 8, 11967. [Google Scholar] [CrossRef]
- Williams, J.C.; Zeelen, J.P.; Neubauer, G.; Vriend, G.; Backmann, J.; Michels, P.A.; Lambeir, A.M.; Wierenga, R.K. Structural and mutagenesis studies of leishmania triosephosphate isomerase: A point mutation can convert a mesophilic enzyme into a superstable enzyme without losing catalytic power. Protein Eng. 1999, 12, 243–250. [Google Scholar] [CrossRef] [Green Version]
- Itkonen, J.; Annala, A.; Tavakoli, S.; Arango-Gonzalez, B.; Ueffing, M.; Toropainen, E.; Ruponen, M.; Casteleijn, M.G.; Urtti, A. Characterization, Stability, and In Vivo Efficacy Studies of Recombinant Human CNTF and Its Permeation into the Neural Retina in Ex Vivo Organotypic Retinal Explant Culture Models. Pharmaceutics 2020, 12, 611. [Google Scholar] [CrossRef]
- Kostamovaara, J.; Tenhunen, J.; Kögler, M.; Nissinen, I.; Nissinen, J.; Keränen, P. Fluorescence suppression in Raman spectroscopy using a time-gated CMOS SPAD. Opt. Express 2013, 21, 31632. [Google Scholar] [CrossRef]
- Lipiäinen, T.; Pessi, J.; Movahedi, P.; Koivistoinen, J.; Kurki, L.; Tenhunen, M.; Yliruusi, J.; Juppo, A.M.; Heikkonen, J.; Pahikkala, T.; et al. Time-Gated Raman Spectroscopy for Quantitative Determination of Solid-State Forms of Fluorescent Pharmaceuticals. Anal. Chem. 2018, 90, 4832–4839. [Google Scholar] [CrossRef] [Green Version]
- Wen, C.-D.; Mudawar, I. Emissivity characteristics of polished aluminum alloy surfaces and assessment of multispectral radiation thermometry (MRT) emissivity models. Int. J. Heat Mass Transf. 2005, 48, 1316–1329. [Google Scholar] [CrossRef]
- Woody, R.W.; Tinoco, I. Optical rotation of oriented helices. III. Calculation of the rotatory dispersion and circular dichroism of the alpha-and 310-helix. J. Chem. Phys. 1967, 46, 4927–4945. [Google Scholar] [CrossRef]
- Majorek, K.A.; Porebski, P.J.; Dayal, A.; Zimmerman, M.D.; Jablonska, K.; Stewart, A.J.; Chruszcz, M.; Minor, W. Structural and immunologic characterization of bovine, horse, and rabbit serum albumins. Mol. Immunol. 2012, 52, 174–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sielecki, A.R.; Fedorov, A.A.; Boodhoo, A.; Andreeva, N.S.; James, M.N.G. Molecular and crystal structures of monoclinic porcine pepsin refined at 1.8A resolution. J. Mol. Biol. 1990, 214, 143–170. [Google Scholar] [CrossRef]
- Rygula, A.; Majzner, K.; Marzec, K.M.; Kaczor, A.; Pilarczyk, M.; Baranska, M. Raman spectroscopy of proteins: A review. J. Raman Spectrosc. 2013, 44, 1061–1076. [Google Scholar] [CrossRef]
- Charles, A.; Janeway, J.; Travers, P.; Walport, M.; Shlomchik, M.J. The Structure of A Typical Antibody Molecule; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Krapp, S.; Mimura, Y.; Jefferis, R.; Huber, R.; Sondermann, P. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. J. Mol. Biol. 2003, 325, 979–989. [Google Scholar] [CrossRef]
- McConnell, A.; Zhang, X.; Macomber, J.; Chau, B.; Sheffer, J.; Rahmanian, S.; Hare, E.; Spasojevic, V.; Horlick, R.; King, D.; et al. A general approach to antibody thermostabilization. mAbs 2014, 6, 1274–1282. [Google Scholar] [CrossRef] [Green Version]
- Cobb, B.A. The history of IgG glycosylation and where we are now. Glycobiology 2021, 30, 202–213. [Google Scholar] [CrossRef]
- Crispin, M. Therapeutic potential of deglycosylated antibodies. Glycobiology 2013, 30, 202–213. [Google Scholar] [CrossRef] [Green Version]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef] [Green Version]
- Crichton, R.R. 4—Structural and Molecular Biology for Chemists. In Biological Inorganic Chemistry; Crichton, R.R., Ed.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 43–76. [Google Scholar]
- Sadat, A.; Joye, I.J. Peak Fitting Applied to Fourier Transform Infrared and Raman Spectroscopic Analysis of Proteins. Appl. Sci. 2020, 10, 5918. [Google Scholar] [CrossRef]
- Freire, P.T.C.; Barboza, F.M.; Lima, J.A.; Melo, F.E.A.; Filho, J.M. Raman Spectroscopy of Amino Acid Crystals. In Raman Spectroscopy of Amino Acid Crystals; IntechOpen: London, UK, 2017. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; McWilliams, A.; Lui, H.; McLean, D.I.; Lam, S.; Zeng, H. Near-infrared Raman spectroscopy for optical diagnosis of lung cancer. Int. J. Cancer 2003, 107, 1047–1052. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, Q.; Huang, Y.; Li, Z.; Zhang, Z. Contrastive Analysis of the Raman Spectra of Polychlorinated Benzene: Hexachlorobenzene and Benzene. Sensors 2011, 11, 11510–11515. [Google Scholar] [CrossRef] [PubMed]
- Brewster, V.L.; Ashton, L.; Goodacre, R. Monitoring the Glycosylation Status of Proteins Using Raman Spectroscopy. Anal. Chem. 2011, 83, 6074–6081. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.E.; Leslie, A.G.W.; Finch, J.T.; Carrell, R.W. Crystal structure of uncleaved ovalbumin at 1·95 Å resolution. J. Mol. Biol. 1991, 221, 941–959. [Google Scholar] [CrossRef]
- Panalytical, M. Proteins Melting Point Characterization Using The Zetasizer Nano System. Azo Nano 2019. [Google Scholar]
- Vivian, J.T.; Callis, P.R. Mechanisms of Tryptophan Fluorescence Shifts in Proteins. Biophys. J. 2001, 80, 2093–2109. [Google Scholar] [CrossRef] [Green Version]
- Friess, W.; Lee, G. Basic thermoanalytical studies of insoluble collagen matrices. Biomaterials 1996, 17, 2289–2294. [Google Scholar] [CrossRef]
- Casteleijn, M.G. Towards New Enzymes: Protein Engineering versus Bioinformatic Studies; University of Oulu: Finland, UK, 2009. [Google Scholar]
- Lolis, E.; Petsko, G.A. Crystallographic analysis of the complex between triosephosphate isomerase and 2-phosphoglycolate at 2.5-.ANG. resolution: Implications for catalysis. Biochemistry 1990, 29, 6619–6625. [Google Scholar] [CrossRef]
- Lambeir, A.-M.; Backmann, J.; Ruiz-Sanz, J.; Filimonov, V.; Nielsen, J.E.; Kursula, I.; Norledge, B.V.; Wierenga, R.K. The ionization of a buried glutamic acid is thermodynamically linked to the stability of Leishmania mexicana triose phosphate isomerase. Eur. J. Biochem. 2000, 267, 2516–2524. [Google Scholar] [CrossRef] [Green Version]
- Alahuhta, M.; Casteleijn, M.G.; Neubauer, P.; Wierenga, R.K. Structural studies show that the A178L mutation in the C-terminal hinge of the catalytic loop-6 of triosephosphate isomerase (TIM) induces a closed-like conformation in dimeric and monomeric TIM. Acta Crystallogr. Sect. D Biol. Crystallogr. 2008, 64, 178–188. [Google Scholar] [CrossRef]
- Caron, J.; Rochoy, M.; Gaboriau, L.; Gautier, S. The history of pharmacovigilance. Therapies 2016, 71, 129–134. [Google Scholar] [CrossRef]
- Nicoud, L.; Owczarz, M.; Arosio, P.; Morbidelli, M. A multiscale view of therapeutic protein aggregation: A colloid science perspective. Biotechnol. J. 2015, 10, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Möller, M.; Denicola, A. Protein tryptophan accessibility studied by fluorescence quenching. Biochem. Mol. Biol. Educ. 2002, 30, 175–178. [Google Scholar] [CrossRef]
- Luykx, D.M.; Casteleijn, M.G.; Jiskoot, W.; Westdijk, J.; Jongen, P.M. Physicochemical studies on the stability of influenza haemagglutinin in vaccine bulk material. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2004, 23, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Jain, A.; Lu, Y.; Hoag, S.W. Probing Thermal Stability of Proteins with Temperature Scanning Viscometer. Mol. Pharm. 2019, 16, 3687–3693. [Google Scholar] [CrossRef]
- Vermeer, A.W.P.; Norde, W. The Thermal Stability of Immunoglobulin: Unfolding and Aggregation of a Multi-Domain Protein. Biophys. J. 2000, 78, 394–404. [Google Scholar] [CrossRef] [Green Version]
- Zav’yalov, V.; Tishchenko, V. Mechanisms of generation of antibody diversity as a cause for natural selection of homoiothermal animals in the process of evolution. Scand. J. Immunol. 1991, 33, 755–762. [Google Scholar] [CrossRef]
- Vermeer, A.W.P.; Norde, W.; Amerongen, A.V. The Unfolding/Denaturation of Immunogammaglobulin of Isotype 2b and its Fab and Fc Fragments. Biophys. J. 2000, 79, 2150–2154. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, F.; Stevenson, R.; Li, C.; Salimi-Moosavi, H.; Liu, L.; Wen, J.; Luo, Q.; Daris, K.; Buck, L.; Miller, S.; et al. Engineering an IgG Scaffold Lacking Effector Function with Optimized Developability. J. Biol. Chem. 2017, 292, 1865–1875. [Google Scholar] [CrossRef] [Green Version]
- Kamatari, Y.O.; Dobson, C.M.; Konno, T. Structural dissection of alkaline-denatured pepsin. Spectroscopy 2004, 18, 227–236. [Google Scholar] [CrossRef]
- Tani, F.; Shirai, N.; Nakanishi, Y.; Yasumoto, K.; Kitabatake, N. Role of the Carbohydrate Chain and Two Phosphate Moieties in the Heat-Induced Aggregation of Hen Ovalbumin. Biosci. Biotechnol. Biochem. 2004, 68, 2466–2476. [Google Scholar] [CrossRef] [Green Version]
- Benítez-Cardoza, C.G.; Rojo-Domínguez, A.; Hernández-Arana, A. Temperature-Induced Denaturation and Renaturation of Triosephosphate Isomerase from Saccharomyces cerevisiae: Evidence of Dimerization Coupled to Refolding of the Thermally Unfolded Protein. Biochemistry 2001, 40, 9049–9058. [Google Scholar] [CrossRef] [PubMed]
- Panalytical, M. Dynamic Light Scattering as a Method for Understanding the Colloidal Stability of Protein Therapeutics. AZoM 2019. [Google Scholar]
- Berner, C.; Menzen, T.; Winter, G.; Svilenov, H. Combining Unfolding Reversibility Studies and Molecular Dynamics Simulations to Select Aggregation-Resistant Antibodies. Mol. Pharm. 2021, 18, 2242–2253. [Google Scholar] [CrossRef] [PubMed]
- De Gelder, J.; De Gussem, K.; Vandenabeele, P.; Moens, L. Reference database of Raman spectra of biological molecules. J. Raman Spectrosc. 2007, 38, 1133–1147. [Google Scholar] [CrossRef]
- Gremlich, H.-U.B.Y. Infrared and Raman Spectroscopy of Biological Materials; CRC Press: Boca Raton, FL, USA, 2021. [Google Scholar]
- Cattell, R.B. The Scree Test for the Number Of Factors. Multivar. Behav. Res. 1966, 1, 245–276. [Google Scholar] [CrossRef]
| Protein (α, β, α/β) | DLS | Tryptophan Fluorescence | CD | Time-Gated a |
|---|---|---|---|---|
| BSA (α) | yes | yes | yes | yes |
| CNTF (α) | no b | yes | yes | no c |
| Fab (β) | yes | yes | yes | no |
| F(ab′)2 (β) | yes | no | no | no |
| IgGglycosylated (β) | yes | yes | yes | yes |
| IgGnon-glycosylated (β) | yes | no | yes | yes d |
| Pepsin A (EC 3.4.23.1) (α/β) | yes | no | yes | yes |
| Ovalbumin (α/β) | yes | yes | yes | yes |
| ScTIM (EC 5.3.1.1) (α/β) | yes | yes | yes | no |
| LmTIME65Q (EC 5.3.1.1) (α/β) | no | yes | yes | yes |
| Technique | Parameter 1 | Parameter 2 | Parameter 3 |
|---|---|---|---|
| DLS | Z-average | Hydrodynamic diameter | Polydispersity index |
| Tryptophan fluorescence | Fluorescence intensity (internal) quenching | Red/blue shift | Tryptophan oxidation (peak at 515 nm) |
| CD | Melting temperature (°C) | Van’t Hoff enthalpy (kJ/mol) | - |
| Time-gated Raman spectroscopy | Raman spectra similarity clustering according to temperature | Relevant time-gated Raman peaks a | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Itkonen, J.; Ghemtio, L.; Pellegrino, D.; Jokela, P.J.; Xhaard, H.; Casteleijn, M.G. Analysis of Biologics Molecular Descriptors towards Predictive Modelling for Protein Drug Development Using Time-Gated Raman Spectroscopy. Pharmaceutics 2022, 14, 1639. https://doi.org/10.3390/pharmaceutics14081639
Itkonen J, Ghemtio L, Pellegrino D, Jokela PJ, Xhaard H, Casteleijn MG. Analysis of Biologics Molecular Descriptors towards Predictive Modelling for Protein Drug Development Using Time-Gated Raman Spectroscopy. Pharmaceutics. 2022; 14(8):1639. https://doi.org/10.3390/pharmaceutics14081639
Chicago/Turabian StyleItkonen, Jaakko, Leo Ghemtio, Daniela Pellegrino, Pia J. Jokela (née Heinonen), Henri Xhaard, and Marco G. Casteleijn. 2022. "Analysis of Biologics Molecular Descriptors towards Predictive Modelling for Protein Drug Development Using Time-Gated Raman Spectroscopy" Pharmaceutics 14, no. 8: 1639. https://doi.org/10.3390/pharmaceutics14081639
APA StyleItkonen, J., Ghemtio, L., Pellegrino, D., Jokela, P. J., Xhaard, H., & Casteleijn, M. G. (2022). Analysis of Biologics Molecular Descriptors towards Predictive Modelling for Protein Drug Development Using Time-Gated Raman Spectroscopy. Pharmaceutics, 14(8), 1639. https://doi.org/10.3390/pharmaceutics14081639