
Revisiting Cerebrospinal Fluid Flow Direction and Rate in Physiologically Based Pharmacokinetic Model


Abstract
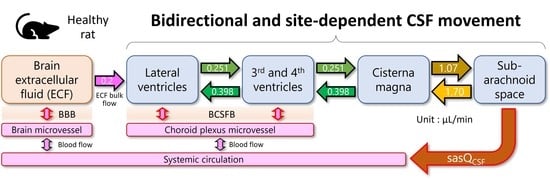
:
1. Introduction
2. Materials and Methods
2.1. In Vivo PK Profiles in Rats after Intra-CSF Administration
2.2. Data Analysis and Software
2.3. Empirical Plasma PK Models
2.4. Drug-Specific Physicochemical Parameters
2.5. Consideration of Intra-CSF Administration Volume
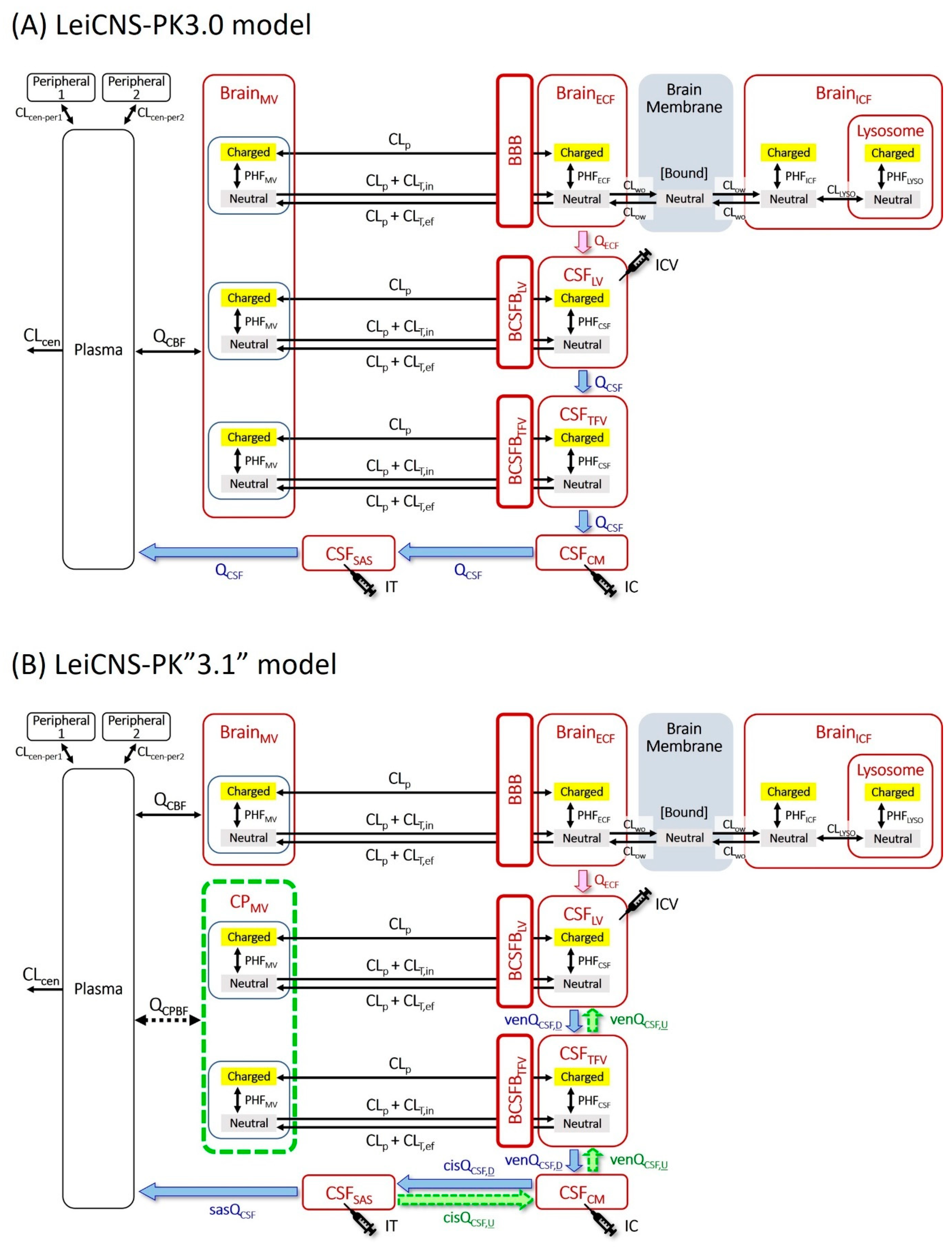
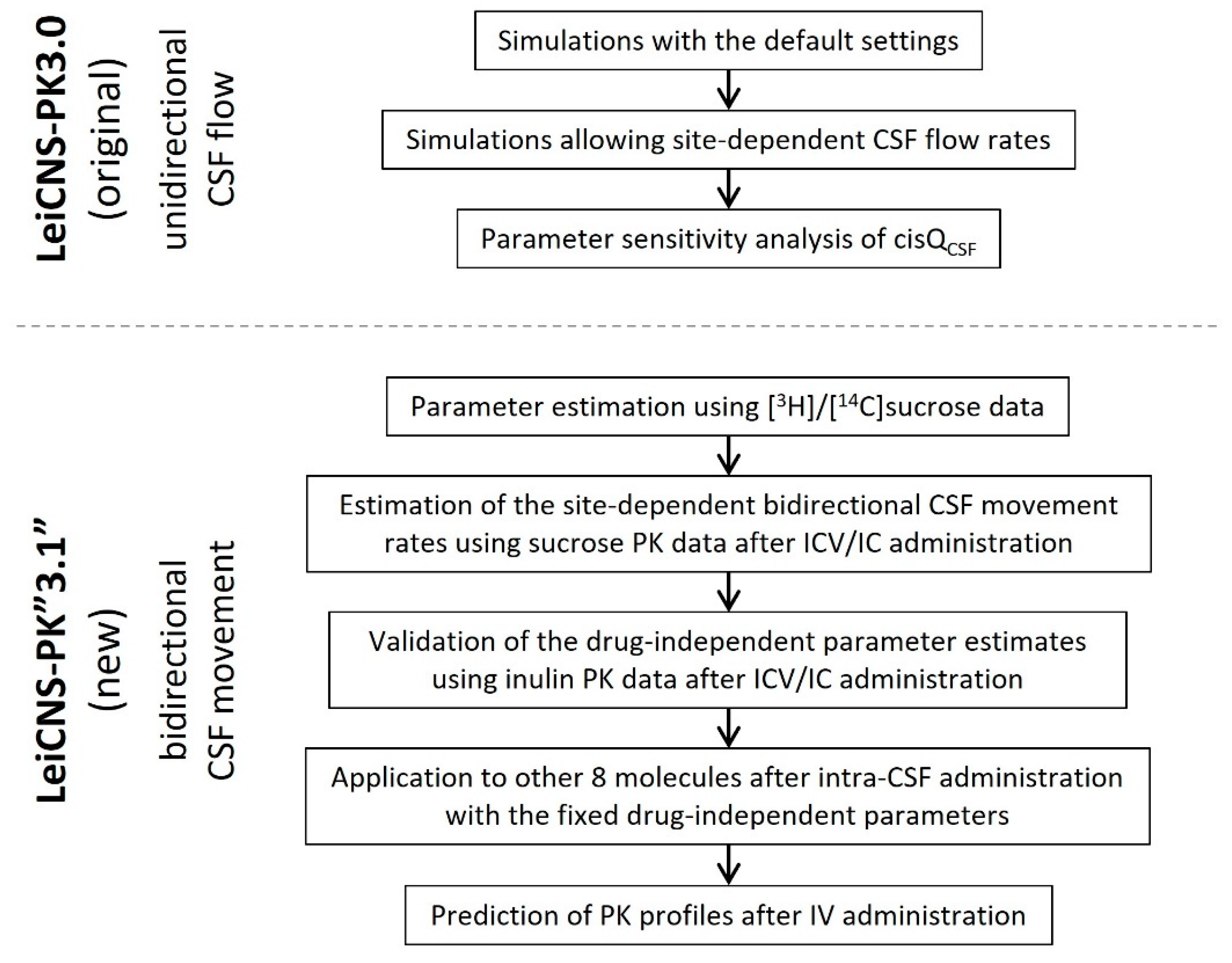
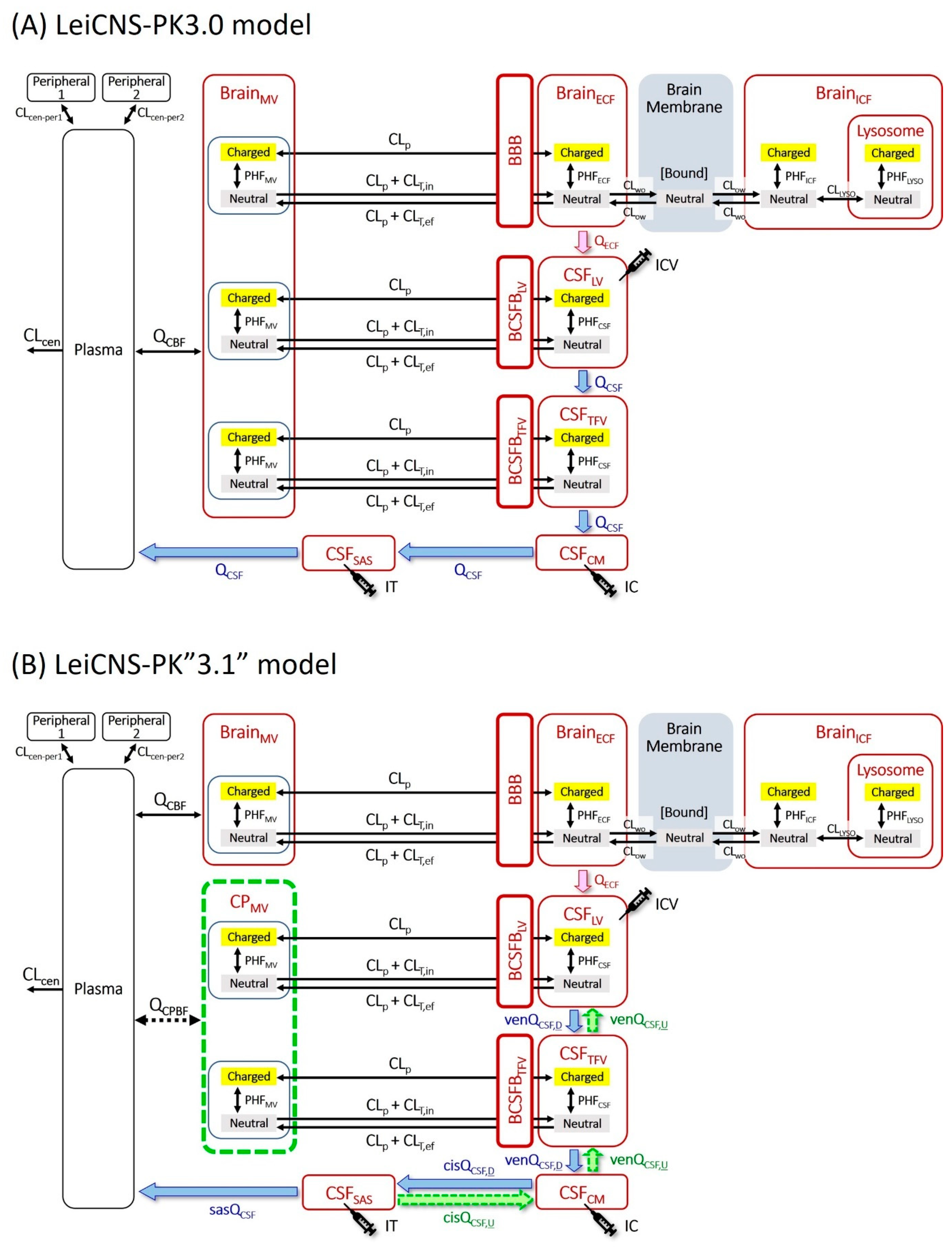
2.6. Simulation with the Original LeiCNS-PK3.0 Model
2.7. Introduction of the Choroid Plexus Microvessel Compartment
2.8. Estimation of the Site-Dependent Bidirectional CSF Movement Rates
2.9. Application of the LeiCNS-PK3.1 Model to Other Molecules
2.10. Application to IV Administration
2.11. Further Modification of Model Structure
2.12. Model Evaluation
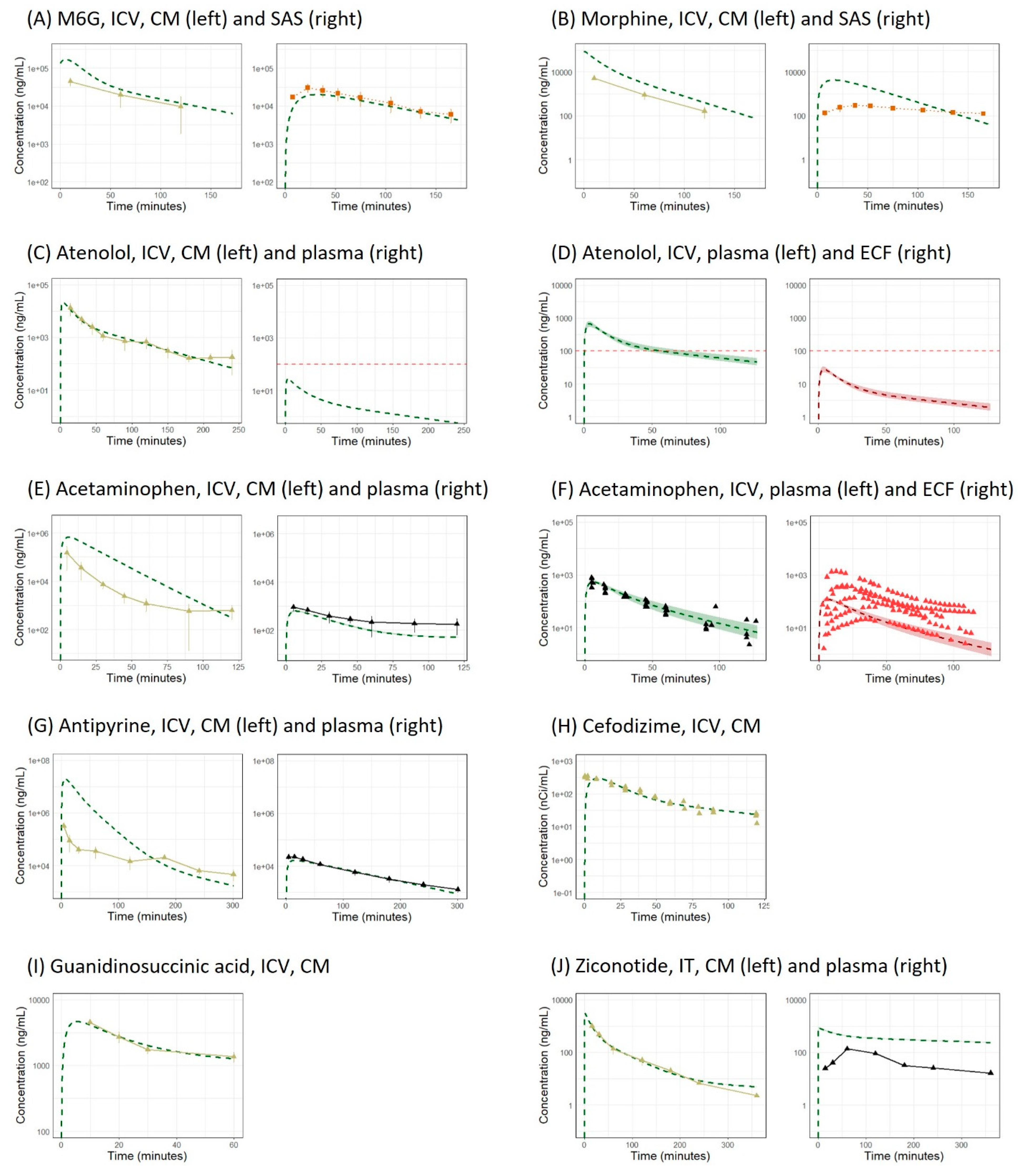
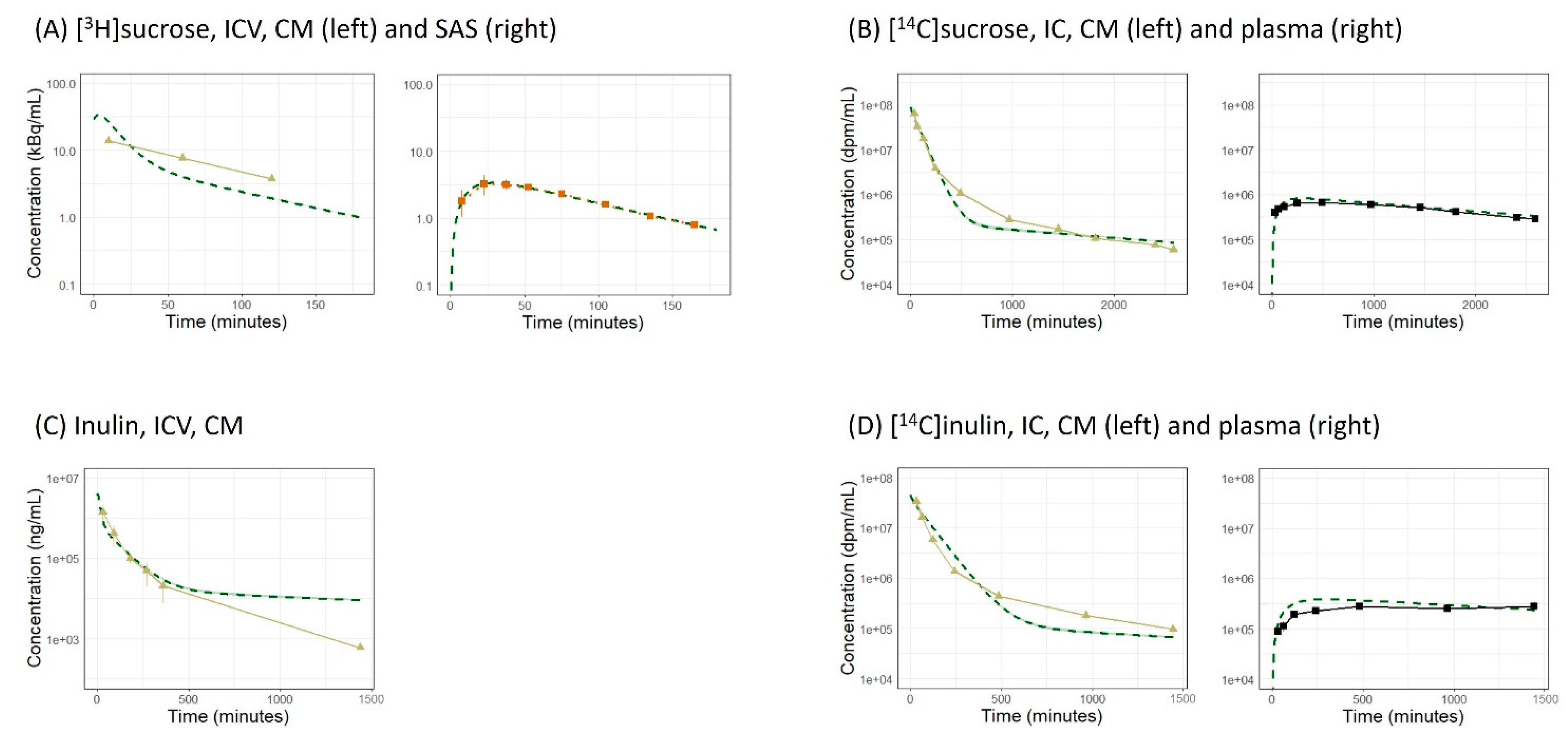
3. Results
3.1. Simulation with the Original LeiCNS-PK3.0 Model
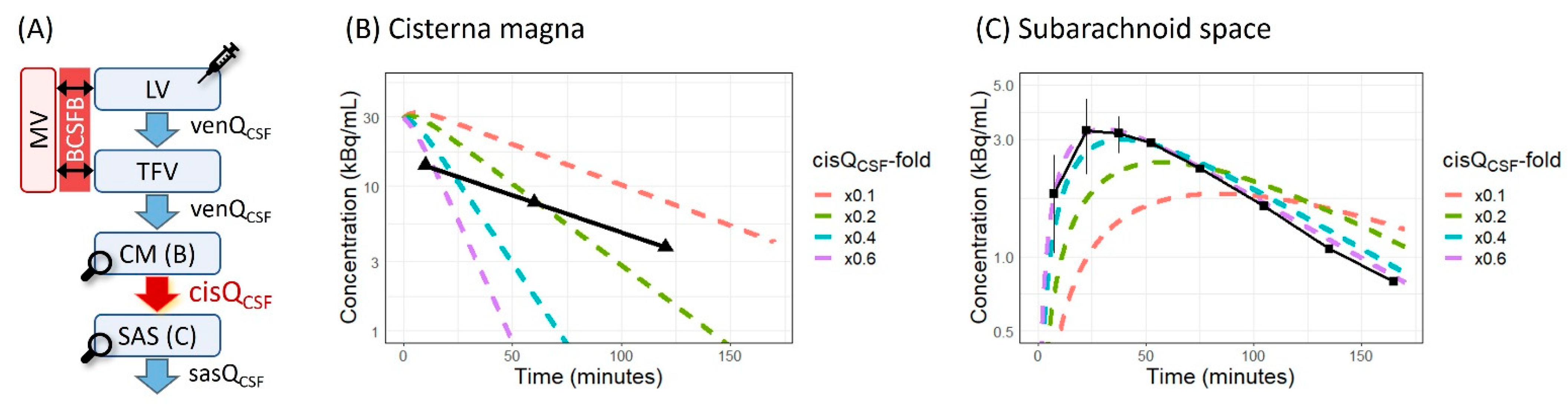
3.2. Parameter Sensitivity Analysis of cisQCSF
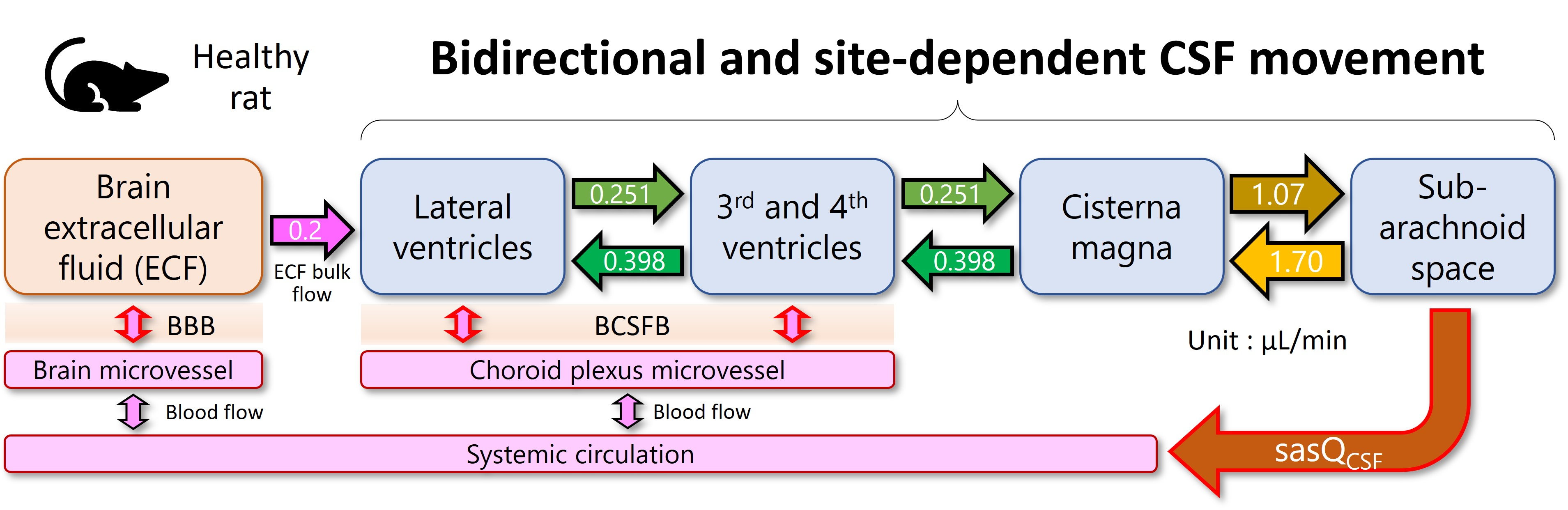
3.3. Estimation of the Site-Dependent Bidirectional CSF Movement Rates
3.4. Application of the LeiCNS-PK3.1 Model to Other Molecules
3.5. Application to IV Administration
3.6. Further Modification of Model Structure
4. Discussion
4.1. CSF Compartments and Parameters in CNS-Specific PBPK Models
4.2. Simulation by the Original LeiCNS-PK3.0 Model with the Unidirectional CSF Flow
4.3. Bidirectional and Pulsatile CSF Movement
4.4. Parameter Estimates Possibly Representing the Physiological CSF Values in Healthy Rats
4.5. Possibility of the LeiCNS-PK3.1 Model as a Generic CNS-PBPK Model
4.6. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Segal, M.B. Extracellular and cerebrospinal fluids. J. Inherit. Metab. Dis. 1993, 16, 617–638. [Google Scholar] [CrossRef]
- Wood, J.H. Neuroendocrinology of Cerebrospinal Fluid: Peptides, Steroids, and Other Hormones. Neurosurgery 1982, 11, 293–305. [Google Scholar] [CrossRef]
- Veening, J.G.; Barendregt, H.P. The regulation of brain states by neuroactive substances distributed via the cerebrospinal fluid; A review. Cereb. Fluid Res. 2010, 7, 1. [Google Scholar] [CrossRef]
- De Lange, E.C.M. Utility of CSF in translational neuroscience. J. Pharmacokinet. Pharmacodyn. 2013, 40, 315–326. [Google Scholar] [CrossRef]
- Kouzehgarani, G.N.; Feldsien, T.; Engelhard, H.H.; Mirakhur, K.K.; Phipps, C.; Nimmrich, V.; Clausznitzer, D.; Lefebvre, D.R. Harnessing cerebrospinal fluid circulation for drug delivery to brain tissues. Adv. Drug Deliv. Rev. 2021, 173, 20–59. [Google Scholar] [CrossRef]
- Saleh, M.A.A.; Loo, C.F.; Elassaiss-Schaap, J.; De Lange, E.C.M. Lumbar cerebrospinal fluid-to-brain extracellular fluid surrogacy is context-specific: Insights from LeiCNS-PK3.0 simulations. J. Pharmacokinet. Pharmacodyn. 2021, 48, 725–741. [Google Scholar] [CrossRef]
- Ball, K.; Bouzom, F.; Scherrmann, J.-M.; Walther, B.; Declèves, X. A Physiologically Based Modeling Strategy during Preclinical CNS Drug Development. Mol. Pharm. 2014, 11, 836–848. [Google Scholar] [CrossRef]
- Gaohua, L.; Neuhoff, S.; Johnson, T.N.; Rostami-Hodjegan, A.; Jamei, M. Development of a permeability-limited model of the human brain and cerebrospinal fluid (CSF) to integrate known physiological and biological knowledge: Estimating time varying CSF drug concentrations and their variability using in vitro data. Drug Metab. Pharmacokinet. 2016, 31, 224–233. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug transport in brain via the cerebrospinal fluid. Fluids Barriers CNS 2011, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.; Miller, M.C.; Caralopoulos, I.N.; Worden, M.S.; Brinker, T.; Gordon, Z.N.; Johanson, C.E.; Silverberg, G.D. Temporal course of cerebrospinal fluid dynamics and amyloid accumulation in the aging rat brain from three to thirty months. Fluids Barriers CNS 2012, 9, 3. [Google Scholar] [CrossRef]
- Edsbagge, M.; Tisell, M.; Jacobsson, L.; Wikkelso, C. Spinal CSF absorption in healthy individuals. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R1450–R1455. [Google Scholar] [CrossRef] [PubMed]
- Orešković, D.; Klarica, M. The formation of cerebrospinal fluid: Nearly a hundred years of interpretations and misinterpretations. Brain Res. Rev. 2010, 64, 241–262. [Google Scholar] [CrossRef] [PubMed]
- Cushing, H. Studies on the Cerebro-Spinal Fluid: I. Introduction. J. Med. Res. 1914, 31, 1–19. [Google Scholar]
- Orešković, D.; Radoš, M.; Klarica, M. New Concepts of Cerebrospinal Fluid Physiology and Development of Hydrocephalus. Pediatr. Neurosurg. 2017, 52, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Klarica, M.; Radoš, M.; Orešković, D. The Movement of Cerebrospinal Fluid and Its Relationship with Substances Behavior in Cerebrospinal and Interstitial Fluid. Neuroscience 2019, 414, 28–48. [Google Scholar] [CrossRef]
- Avdeef, A.; Nielsen, P.E.; Tsinman, O. PAMPA—A drug absorption in vitro model: 11. Matching the in vivo unstirred water layer thickness by individual-well stirring in microtitre plates. Eur. J. Pharm. Sci. 2004, 22, 365–374. [Google Scholar] [CrossRef]
- Bhargava, H.N.; Villar, V.; Cortijo, J.; Morcillo, E. Analgesic and Thermic Effects, and Cerebrospinal Fluid and Plasma Pharmacokinetics, of Intracerebroventricularly Administered Morphine in Normal and Sensitized Rats. J. Pharm. Pharmacol. 1998, 50, 197–203. [Google Scholar] [CrossRef]
- Okura, T.; Saito, M.; Nakanishi, M.; Komiyama, N.; Fujii, A.; Yamada, S.; Kimura, R. Different distribution of morphine and morphine-6β-glucuronide after intracerebroventricular injection in rats. Br. J. Pharmacol. 2003, 140, 211–217. [Google Scholar] [CrossRef]
- Reed, D.J.; Woodbury, D.M. Kinetics of movement of iodide, sucrose, inulin and radio-iodinated serum albumin in the central nervous system and cerebrospinal fluid of the rat. J. Physiol. 1963, 169, 816–850. [Google Scholar] [CrossRef]
- Noguchi, Y.; Kato, M.; Ozeki, K.; Ishigai, M. Pharmacokinetics of an intracerebroventricularly administered antibody in rats. mAbs 2017, 9, 1210–1215. [Google Scholar] [CrossRef]
- Van Bree, J.B.M.M.; Baljet, A.V.; Van Geyt, A.; De Boer, A.G.; Danhof, M.; Breimer, D.D. The unit impulse response procedure for the pharmacokinetic evaluation of drug entry into the central nervous system. J. Pharmacokinet. Biopharm. 1989, 17, 441–462. [Google Scholar] [CrossRef] [PubMed]
- de Lange, E.C.; Danhof, M.; de Boer, A.G.; Breimer, D.D. Critical factors of intracerebral microdialysis as a technique to determined the pharmacokinetics of drugs in rat brain. Brain Res. 1994, 666, 1–8. [Google Scholar] [CrossRef]
- Matsushita, H.; Suzuki, H.; Sugiyama, Y.; Sawada, Y.; Iga, T.; Kawaguchi, Y.; Hanano, M. Facilitated transport of cefodizime into the rat central nervous system. J. Pharmacol. Exp. Ther. 1991, 259, 620–625. [Google Scholar] [PubMed]
- Kasai, Y.; Akanuma, S.-I.; Kubo, Y.; Tachikawa, M.; Hosoya, K.-I. Pharmacokinetics of Guanidinosuccinic Acid in Rat Blood and Cerebrospinal Fluid. Drug Metab. Pharmacokinet. 2014, 29, 97–100. [Google Scholar] [CrossRef]
- Manda, P.; Kushwaha, A.S.; Kundu, S.; Shivakumar, H.; Jo, S.B.; Murthy, S.N. Delivery of ziconotide to cerebrospinal fluid via intranasal pathway for the treatment of chronic pain. J. Control. Release 2016, 224, 69–76. [Google Scholar] [CrossRef]
- Rohatgi, A. WebPlotDigitizer, Version 4.5; Pacifica, CA, USA. 2021. Available online: https://automeris.io/WebPlotDigitizer/ (accessed on 7 June 2022).
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 7 June 2022).
- Bauer, R.J. NONMEM Tutorial Part I: Description of Commands and Options, with Simple Examples of Population Analysis. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 525–537. [Google Scholar] [CrossRef]
- Wang, W.; Hallow, K.; James, D. A Tutorial on RxODE: Simulating Differential Equation Pharmacometric Models in R. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 3–10. [Google Scholar] [CrossRef]
- Maxima, a Computer Algebra System. Version 5.44.0. 2020. Available online: https://maxima.sourceforge.io/ (accessed on 7 June 2022).
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef]
- Manchester, J.; Walkup, G.; Rivin, O.; You, Z. Evaluation of pKa Estimation Methods on 211 Druglike Compounds. J. Chem. Inf. Model. 2010, 50, 565–571. [Google Scholar] [CrossRef]
- Alqahtani, F.; Chowdhury, E.A.; Bhattacharya, R.; Noorani, B.; Mehvar, R.; Bickel, U. Brain Uptake of [13C] and [14C] Sucrose Quantified by Microdialysis and Whole Tissue Analysis in Mice. Drug Metab. Dispos. 2018, 46, 1514–1518. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, M.; Saleh, M.A.A.; de Lange, E.C.M. The Extension of the LeiCNS-PK3.0 Model in Combination with the “Handshake” Approach to Understand Brain Tumor Pathophysiology. Pharm. Res. 2022, 39, 1343–1361. [Google Scholar] [CrossRef]
- Kawakami, J.; Yamamoto, K.; Sawada, Y.; Iga, T. Prediction of brain delivery of ofloxacin, a new quinolone, in the human from animal data. J. Pharmacokinet. Biopharm. 1994, 22, 207–227. [Google Scholar] [CrossRef] [PubMed]
- Heisey, S.R. Brain and choroid plexus blood volumes in vertebrates. Comp. Biochem. Physiol. 1968, 26, 489–498. [Google Scholar] [CrossRef]
- Spector, R.; Keep, R.F.; Snodgrass, S.R.; Smith, Q.R.; Johanson, C.E. A balanced view of choroid plexus structure and function: Focus on adult humans. Exp. Neurol. 2015, 267, 78–86. [Google Scholar] [CrossRef]
- Ennis, S.R.; Keep, R.F. The Effects of Cerebral Ischemia on the Rat Choroid Plexus. J. Cereb. Blood Flow Metab. 2006, 26, 675–683. [Google Scholar] [CrossRef]
- Williams, J.L.; Shea, M.; Furlan, A.J.; Little, J.R.; Jones, S.C. Importance of freezing time when iodoantipyrine is used for measurement of cerebral blood flow. Am. J. Physiol. 1991, 261, H252–H256. [Google Scholar] [CrossRef]
- Eide, P.K.; Valnes, L.M.; Lindstrøm, E.K.; Mardal, K.-A.; Ringstad, G. Direction and magnitude of cerebrospinal fluid flow vary substantially across central nervous system diseases. Fluids Barriers CNS 2021, 18, 16. [Google Scholar] [CrossRef]
- Sartoretti, T.; Wyss, M.; Sartoretti, E.; Reischauer, C.; Hainc, N.; Graf, N.; Binkert, C.; Najafi, A.; Sartoretti-Schefer, S. Sex and Age Dependencies of Aqueductal Cerebrospinal Fluid Dynamics Parameters in Healthy Subjects. Front. Aging Neurosci. 2019, 11, 199. [Google Scholar] [CrossRef]
- Dreha-Kulaczewski, S.; Joseph, A.A.; Merboldt, K.-D.; Ludwig, H.-C.; Gärtner, J.; Frahm, J. Identification of the Upward Movement of Human CSF In Vivo and its Relation to the Brain Venous System. J. Neurosci. 2017, 37, 2395–2402. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Kim, Y.; Hong, S.; Choi, H. Cerebrospinal fluid flow in normal beagle dogs analyzed using magnetic resonance imaging. J. Vet. Sci. 2021, 22, e2. [Google Scholar] [CrossRef] [PubMed]
- Dur, A.H.; Tang, T.; Viviano, S.; Sekuri, A.; Willsey, H.R.; Tagare, H.D.; Kahle, K.T.; Deniz, E. In Xenopus ependymal cilia drive embryonic CSF circulation and brain development independently of cardiac pulsatile forces. Fluids Barriers CNS 2020, 17, 72. [Google Scholar] [CrossRef] [PubMed]
- Mestre, H.; Mori, Y.; Nedergaard, M. The Brain’s Glymphatic System: Current Controversies. Trends Neurosci. 2020, 43, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, M.E.; Wolak, D.J.; Kumar, N.N.; Brunette, E.; Brunnquell, C.L.; Hannocks, M.; Abbott, N.J.; Meyerand, M.E.; Sorokin, L.; Stanimirovic, D.B.; et al. Intrathecal antibody distribution in the rat brain: Surface diffusion, perivascular transport and osmotic enhancement of delivery. J. Physiol. 2018, 596, 445–475. [Google Scholar] [CrossRef] [PubMed]
- Cserr, H. Potassium exchange between cerebrospinal fluid, plasma, and brain. Am. J. Physiol. 1965, 209, 1219–1226. [Google Scholar] [CrossRef]
- Zakaria, Z.; Badhan, R. Development of a Region-Specific Physiologically Based Pharmacokinetic Brain Model to Assess Hippocampus and Frontal Cortex Pharmacokinetics. Pharmaceutics 2018, 10, 14. [Google Scholar] [CrossRef]
- Chang, H.-Y.; Wu, S.; Meno-Tetang, G.; Shah, D.K. A translational platform PBPK model for antibody disposition in the brain. J. Pharmacokinet. Pharmacodyn. 2019, 46, 319–338. [Google Scholar] [CrossRef]
- Bloomingdale, P.; Bakshi, S.; Maass, C.; van Maanen, E.; Pichardo-Almarza, C.; Yadav, D.B.; van der Graaf, P.; Mehrotra, N. Minimal brain PBPK model to support the preclinical and clinical development of antibody therapeutics for CNS diseases. J. Pharmacokinet. Pharmacodyn. 2021, 48, 861–871. [Google Scholar] [CrossRef]
- Monine, M.; Norris, D.; Wang, Y.; Nestorov, I. A physiologically-based pharmacokinetic model to describe antisense oligonucleotide distribution after intrathecal administration. J. Pharmacokinet. Pharmacodyn. 2021, 48, 639–654. [Google Scholar] [CrossRef]
- du Boulay, G.H. Pulsatile Movements in the CSF Pathways. Br. J. Radiol. 1966, 39, 255–262. [Google Scholar] [CrossRef]
- Tarumi, T.; Yamabe, T.; Fukuie, M.; Zhu, D.C.; Zhang, R.; Ogoh, S.; Sugawara, J. Brain blood and cerebrospinal fluid flow dynamics during rhythmic handgrip exercise in young healthy men and women. J. Physiol. 2021, 599, 1799–1813. [Google Scholar] [CrossRef] [PubMed]
- Bhadelia, R.A.; Bogdan, A.R.; Kaplan, R.F.; Wolpert, S.M. Cerebrospinal fluid pulsation amplitude and its quantitative relationship to cerebral blood flow pulsations: A phase-contrast MR flow imaging study. Neuroradiology 1997, 39, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.J.; Lee, J.W.; Lee, E.; Yeom, J.S.; Kim, K.-J.; Kang, H.S. Cervical compressive myelopathy: Flow analysis of cerebrospinal fluid using phase-contrast magnetic resonance imaging. Eur. Spine J. 2017, 26, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Quigley, M.F.; Iskandar, B.; Quigley, M.A.; Nicosia, M.; Haughton, V. Cerebrospinal Fluid Flow in Foramen Magnum: Temporal and Spatial Patterns at MR Imaging in Volunteers and in Patients with Chiari I Malformation. Radiology 2004, 232, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Kelly, E. Cerebrospinal Fluid Dynamics and the Pathophysiology of Hydrocephalus: New Concepts. Semin. Ultrasound CT MRI 2016, 37, 84–91. [Google Scholar] [CrossRef]
- Smith, D.; Mosely, R.; Begent, R.; Coakham, H.; Glaser, M.; Dewhurst, S.; Kelly, A.; Bagshawe, K. Quantitative distribution of 131I-labelled monoclonal antibodies administered by the intra-ventricular route. Eur. J. Cancer Clin. Oncol. 1990, 26, 129–136. [Google Scholar] [CrossRef]
- Berger, B.; Ortiz, O.; Gold, A.; Hilal, S.K. Total cerebrospinal fluid enhancement following intravenous Gd-DTPA administration in a case of meningiomatosis. AJNR Am. J. Neuroradiol. 1992, 13, 15–18. [Google Scholar]
- Tangen, K.M.; Hsu, Y.; Zhu, D.C.; Linninger, A.A. CNS wide simulation of flow resistance and drug transport due to spinal microanatomy. J. Biomech. 2015, 48, 2144–2154. [Google Scholar] [CrossRef]
- de Lange, E.C.; Bouw, M.R.; Mandema, J.W.; Danhof, M.; de Boer, A.G.; Breimer, D.D. Application of intracerebral microdialysis to study regional distribution kinetics of drugs in rat brain. Br. J. Pharmacol. 1995, 116, 2538–2544. [Google Scholar] [CrossRef]
- Vendel, E.; Rottschäfer, V.; De Lange, E.C. The 3D Brain Unit Network Model to Study Spatial Brain Drug Exposure under Healthy and Pathological Conditions. Pharm. Res. 2020, 37, 137. [Google Scholar] [CrossRef]
- Magdoom, K.N.; Brown, A.; Rey, J.; Mareci, T.H.; King, M.A.; Sarntinoranont, M. MRI of Whole Rat Brain Perivascular Network Reveals Role for Ventricles in Brain Waste Clearance. Sci. Rep. 2019, 9, 11480. [Google Scholar] [CrossRef] [PubMed]
- Ray, L.A.; Pike, M.; Simon, M.; Iliff, J.J.; Heys, J.J. Quantitative analysis of macroscopic solute transport in the murine brain. Fluids Barriers CNS 2021, 18, 55. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Lam, M.A.; Sial, A.; Hemley, S.J.; Bilston, L.E.; Stoodley, M.A. Fluid outflow in the rat spinal cord: The role of perivascular and paravascular pathways. Fluids Barriers CNS 2018, 15, 13. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Fahmy, L.M.; Davoodi-Bojd, E.; Zhang, L.; Ding, G.; Hu, J.; Zhang, Z.; Chopp, M.; Jiang, Q. Waste Clearance in the Brain. Front. Neuroanat. 2021, 15, 665803. [Google Scholar] [CrossRef] [PubMed]
- Tunblad, K.; Hammarlund-Udenaes, M.; Jonsson, E.N. Influence of probenecid on the delivery of morphine-6-glucuronide to the brain. Eur. J. Pharm. Sci. 2005, 24, 49–57. [Google Scholar] [CrossRef]
- Stain-Texier, F.; Boschi, G.; Sandouk, P.; Scherrmann, J.-M. Elevated concentrations of morphine 6-beta-D-glucuronide in brain extracellular fluid despite low blood-brain barrier permeability. Br. J. Pharmacol. 1999, 128, 917–924. [Google Scholar] [CrossRef]
- Bouw, M.R.; Xie, R.; Tunblad, K.; Hammarlund-Udenaes, M. Blood-brain barrier transport and brain distribution of morphine-6-glucuronide in relation to the antinociceptive effect in rats–Pharmacokinetic/pharmacodynamic modelling. Br. J. Pharmacol. 2001, 134, 1796–1804. [Google Scholar] [CrossRef]
- Wood, F.L.; Houston, J.B.; Hallifax, D. Clearance Prediction Methodology Needs Fundamental Improvement: Trends Common to Rat and Human Hepatocytes/Microsomes and Implications for Experimental Methodology. Drug Metab. Dispos. 2017, 45, 1178–1188. [Google Scholar] [CrossRef]
- Vanholder, R.; De Smet, R.; Hsu, C.; Vogeleere, P.; Ringoir, S. Uremic toxicity: The middle molecule hypothesis revisited. Semin. Nephrol. 1994, 14, 205–218. [Google Scholar] [PubMed]
- Watanabe, H.; Miyamoto, Y.; Otagiri, M.; Maruyama, T. Update on the Pharmacokinetics and Redox Properties of Protein-Bound Uremic Toxins. J. Pharm. Sci. 2011, 100, 3682–3695. [Google Scholar] [CrossRef]
- De Deyn, P.; Marescau, B.; Cuykens, J.; Van Gorp, L.; Lowenthal, A.; De Potter, W. Guanidino compounds in serum and cerebrospinal fluid of non-dialyzed patients with renal insufficiency. Clin. Chim. Acta 1987, 167, 81–88. [Google Scholar] [CrossRef]
- Bowersox, S.; Mandema, J.; Tarczy-Hornoch, K.; Miljanich, G.; Luther, R.R. Pharmacokinetics of SNX-111, a selective N-type calcium channel blocker, in rats and cynomolgus monkeys. Drug Metab. Dispos. 1997, 25, 379–383. [Google Scholar] [PubMed]
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.; Radchenko, E.; Zefirov, N.S.; Makarenko, A.; et al. Virtual Computational Chemistry Laboratory–Design and Description. J. Comput. -Aided Mol. Des. 2005, 19, 453–463. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
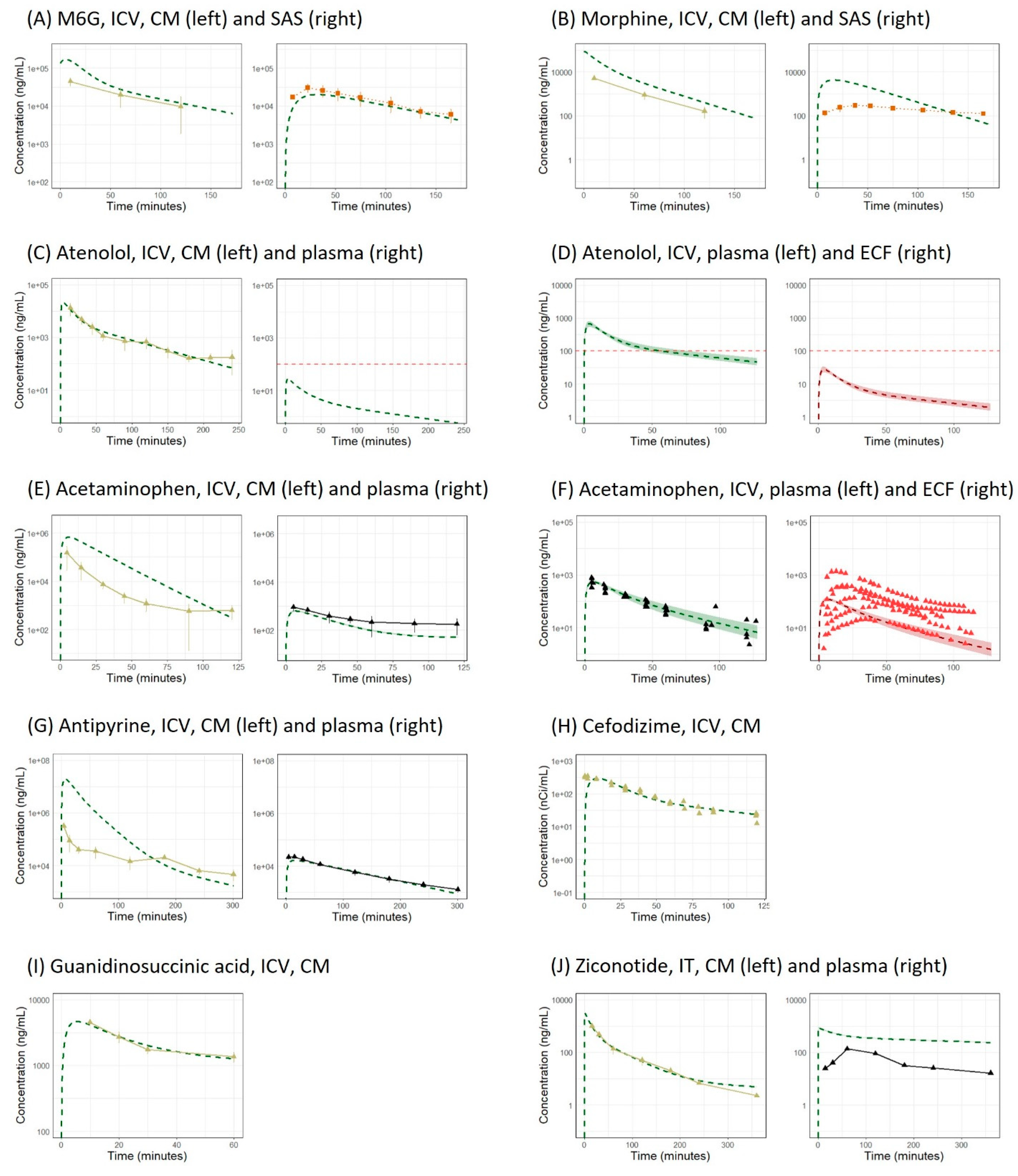{kind=link}
{kind=link}
{kind=link}
| Molecule | Reference | Dosing Route | Dosing Volume (µL) | Dosing Duration (s) | Dose | Reported PK Profile | |||
|---|---|---|---|---|---|---|---|---|---|
| CM | SAS | ECF | PL | ||||||
| [3H]sucrose | [18] | ICV | 12.5 a | NR | 5 kBq | √ | √ | - | - |
| [14C]sucrose | [19] | IC | 100 b | NR | 4 µCi | √ | - | - | √ |
| IV | 100 | NR | 4 µCi | √ | - | - | √ | ||
| Inulin | [20] | ICV | 12.5 c | NR | 2.5 mg/kg | √ | - | - | - |
| [14C]inulin | [19] | IC | 100 b | NR | 2 µCi | √ | - | - | √ |
| IV | 100 | NR | 2 µCi | √ | - | - | √ | ||
| Morphine-6-glucuronide | [18] | ICV | 12.5 a | NR | 50 nmol | √ | √ | - | √ d |
| Morphine | [18] | ICV | 12.5 a | NR | 50 nmol | √ | √ | - | √ e |
| Atenolol | [21] | ICV | 10 | 5–10 | 12 µg | √ | - | - | √ d |
| IV | 500 | 60 | 1 mg | √ | - | - | √ | ||
| [22] | ICV | 15 | 5 | 150 µg | √ | - | √ d | √ d | |
| IV | 500 | 60 | 10 mg | √ | - | √ | √ | ||
| Acetaminophen | [21] | ICV | 10 | 5–10 | 210 µg | √ | - | - | √ |
| IV | 500 | 60 | 825 µg | √ | - | - | √ | ||
| [22] | ICV | 10 | 5 | 210 µg | √ | - | √ | √ | |
| IV | 500 | 60 | 825 µg | √ | - | √ | √ | ||
| Antipyrine | [21] | ICV | 10 | 5–10 | 5 mg | √ | - | - | √ |
| IV | 500 | 60 | 20 mg | √ | - | - | √ | ||
| Cefodizime | [23] | ICV | 10 | NR | 0.074 µCi | √ | - | - | - |
| IV | 1 f | NR | 100 µCi/kg | √ | - | √ | √ | ||
| Guanidinosuccinic acid | [24] | ICV | 10 | NR | 0.05 µmol/kg | √ | - | - | - |
| IV | NR | NR | 1 µmol/kg | √ | - | - | √ | ||
| Ziconotide | [25] | IT | 100 | NR | 100 µg/kg | √ | - | - | √ |
| IV | 100 | NR | 100 µg/kg | √ | - | - | √ | ||
| Molecule | Reference | Dosing Route | Parameter Estimates | ||||
|---|---|---|---|---|---|---|---|
| venQCSF,D (µL/min) | cisQCSF,D (µL/min) | U/D Ratio | sasQCSF (µL/min) | CFPPA | |||
| [3H]sucrose | [18] | ICV | 0.251 | 1.07 | 1.59 | 1.41 | 0.0387 |
| [14C]sucrose | [19] | IC | |||||
| Inulin | [20] | ICV | - a | - a | - a | 1.22 | 0.262 |
| [14C]inulin | [19] | IC | |||||
| Morphine-6-glucuronide | [18] | ICV | - a | - a | - a | 1.52 | 0.00268 |
| Morphine | [18] | ICV | - a | - a | - a | 4.75 b | 95.2 b |
| Atenolol | [21] | ICV | - a | - a | - a | 2.22 | 0.685 |
| Acetaminophen | [21] | ICV | - a | - a | - a | 15.0 c | 529 c |
| Antipyrine | [21] | ICV | - a | - a | - a | 7.18 c | 48.0 c |
| Cefodizime | [23] | ICV | - a | - a | - a | 1.38 | 0.00308 |
| Guanidinosuccinic acid | [24] | ICV | - a | - a | - a | −0.411 | 0.000239 |
| Ziconotide | [25] | IT | - a | - a | - a | 763 | 0.00121 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirasawa, M.; de Lange, E.C.M. Revisiting Cerebrospinal Fluid Flow Direction and Rate in Physiologically Based Pharmacokinetic Model. Pharmaceutics 2022, 14, 1764. https://doi.org/10.3390/pharmaceutics14091764
Hirasawa M, de Lange ECM. Revisiting Cerebrospinal Fluid Flow Direction and Rate in Physiologically Based Pharmacokinetic Model. Pharmaceutics. 2022; 14(9):1764. https://doi.org/10.3390/pharmaceutics14091764
Chicago/Turabian StyleHirasawa, Makoto, and Elizabeth C. M. de Lange. 2022. "Revisiting Cerebrospinal Fluid Flow Direction and Rate in Physiologically Based Pharmacokinetic Model" Pharmaceutics 14, no. 9: 1764. https://doi.org/10.3390/pharmaceutics14091764
APA StyleHirasawa, M., & de Lange, E. C. M. (2022). Revisiting Cerebrospinal Fluid Flow Direction and Rate in Physiologically Based Pharmacokinetic Model. Pharmaceutics, 14(9), 1764. https://doi.org/10.3390/pharmaceutics14091764