
Immune Modulatory Effects of Molecularly Targeted Therapy and Its Repurposed Usage in Cancer Immunotherapy


Abstract
:1. Introduction
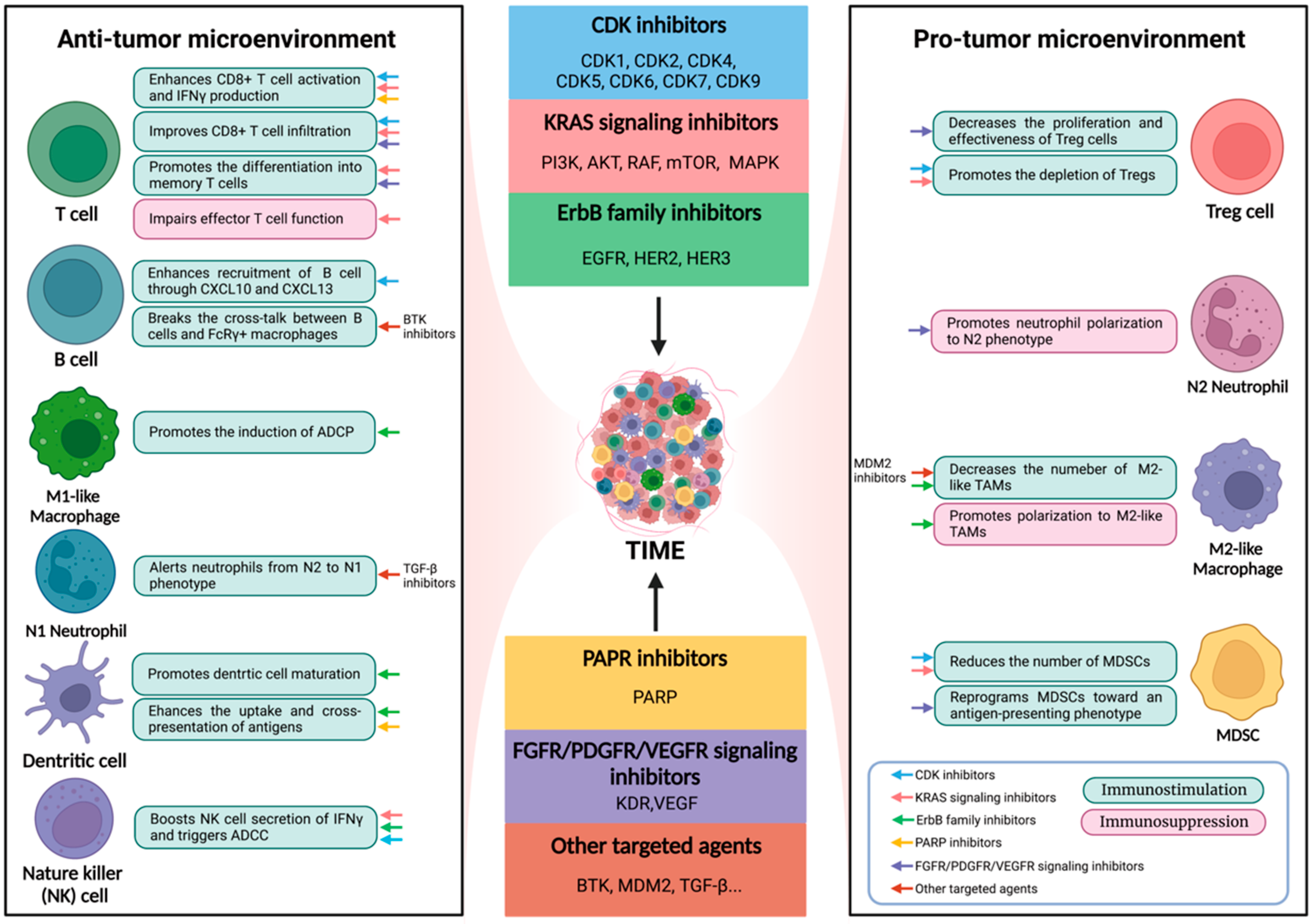
2. Modulatory Effects of Targeted Therapy on Immune Cells
2.1. Effector T Cells
2.2. Regulatory T Cells
2.3. B Cells
2.4. Natural Killer (NK) Cells
2.5. Neutrophils
2.6. Dendritic Cells
2.7. Macrophages
2.8. Myeloid-Derived Suppressor Cells (MDSCs)
3. Modulatory Effects of Targeted Therapy on Immune Checkpoints
4. Potential Combinations of Targeted Therapy and Immunotherapy
4.1. CDK Inhibitors with ICIs
4.2. KRAS Signaling Inhibitors with ICIs
4.3. ErbB Family Inhibitors with ICIs
4.4. PARP Inhibitors with ICIs
4.5. FGFR/PDGFR/VEGFR Signaling Inhibitors with ICIs
4.6. Epigenetic Agents with ICIs
5. Discussion
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADCC | Antibody-Dependent Cell-Mediated Cytotoxicity |
| AML | Acute Myeloid Leukemia |
| BTK | Bruton Tyrosine Kinase |
| CDK | Cycle-dependent Kinase |
| CLL | Chronic Lymphatic Leukemia |
| CRC | Colorectal Cancer |
| DC | Dendritic Cells |
| DDR | DNA Damage Repair |
| DNMT | DNA Methyltransferase |
| DTC | Differentiated Thyroid Cancer |
| HCC | Hepatocellular Carcinoma |
| HDAC | Histone Deacetylases |
| HNSCC | Head and Neck Squamous Cell Carcinoma |
| ICI | Immune Checkpoint Inhibitor |
| MCL | Mantel Cell Lymphoma |
| MDSC | Myeloid Derived Suppressor Cells |
| NK | Nature Killer Cells |
| NSCLC | Non-small Cell Lung Cancer |
| ORR | Objective Response Rate |
| OS | Overall Survival |
| PDAC | Pancreatic Ductal Adenocarcinoma |
| PFS | Progression-free Survival |
| PTC | Papillary Thyroid Carcinoma |
| RCC | Renal Cell Carcinoma |
| TAM | Tumor-associated Macrophages |
| TGF-β | transforming growth factor-β |
| TIB | Tumor-infiltrating B Cells |
| TIME | Tumor Immune Microenvironment |
| TKI | Tyrosine Kinase Inhibitor |
| TNBC | Triple-negative Breast Cancer |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front Pharm. 2018, 9, 1300. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.-M. Targeted therapy in cancer. Cancer Chemother. Pharmacol. 2015, 76, 1113–1132. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Keefe, D.M.K.; Bateman, E.H. Potential Successes and Challenges of Targeted Cancer Therapies. J. Natl. Cancer Inst. Monogr. 2019, 2019, lgz008. [Google Scholar] [CrossRef]
- Petroni, G.; Buque, A.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Immunomodulation by targeted anticancer agents. Cancer Cell 2021, 39, 310–345. [Google Scholar] [CrossRef]
- Tao, L.; Zhu, F.; Xu, F.; Chen, Z.; Jiang, Y.Y.; Chen, Y.Z. Co-targeting cancer drug escape pathways confers clinical advantage for multi-target anticancer drugs. Pharm. Res. 2015, 102, 123–131. [Google Scholar] [CrossRef]
- Gonzalez-Aparicio, M.; Alfaro, C. Implication of Interleukin Family in Cancer Pathogenesis and Treatment. Cancers 2021, 13, 1016. [Google Scholar] [CrossRef]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutierrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Kohlhapp, F.J.; Haribhai, D.; Mathew, R.; Duggan, R.; Ellis, P.A.; Wang, R.; Lasater, E.A.; Shi, Y.; Dave, N.; Riehm, J.J.; et al. Venetoclax Increases Intratumoral Effector T Cells and Antitumor Efficacy in Combination with Immune Checkpoint Blockade. Cancer Discov. 2021, 11, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.-L.; Qin, S.; Ikeda, M.; Galle, P.; Ducreux, M.; Zhu, A.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P. IMbrave150: Efficacy and safety results from a ph III study evaluating atezolizumab (atezo)+ bevacizumab (bev) vs sorafenib (Sor) as first treatment (tx) for patients (pts) with unresectable hepatocellular carcinoma (HCC). Ann. Oncol. 2019, 30, ix186–ix187. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- El Bairi, K.; Haynes, H.R.; Blackley, E.; Fineberg, S.; Shear, J.; Turner, S.; de Freitas, J.R.; Sur, D.; Amendola, L.C.; Gharib, M.; et al. The tale of TILs in breast cancer: A report from The International Immuno-Oncology Biomarker Working Group. NPJ Breast Cancer 2021, 7, 150. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.C.; Vlahovic, G.; et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef]
- Leone, R.D.; Powell, J.D. Metabolism of immune cells in cancer. Nat. Rev. Cancer 2020, 20, 516–531. [Google Scholar] [CrossRef]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef]
- Liu, C.; Peng, W.; Xu, C.; Lou, Y.; Zhang, M.; Wargo, J.A.; Chen, J.Q.; Li, H.S.; Watowich, S.S.; Yang, Y.; et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin. Cancer Res. 2013, 19, 393–403. [Google Scholar] [CrossRef]
- Donia, M.; Fagone, P.; Nicoletti, F.; Andersen, R.S.; Hogdall, E.; Straten, P.T.; Andersen, M.H.; Svane, I.M. BRAF inhibition improves tumor recognition by the immune system: Potential implications for combinatorial therapies against melanoma involving adoptive T-cell transfer. Oncoimmunology 2012, 1, 1476–1483. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandala, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef] [PubMed]
- Erkes, D.A.; Cai, W.; Sanchez, I.M.; Purwin, T.J.; Rogers, C.; Field, C.O.; Berger, A.C.; Hartsough, E.J.; Rodeck, U.; Alnemri, E.S.; et al. Mutant BRAF and MEK Inhibitors Regulate the Tumor Immune Microenvironment via Pyroptosis. Cancer Discov. 2020, 10, 254–269. [Google Scholar] [CrossRef] [PubMed]
- Peiffer, L.; Farahpour, F.; Sriram, A.; Spassova, I.; Hoffmann, D.; Kubat, L.; Stoitzner, P.; Gambichler, T.; Sucker, A.; Ugurel, S.; et al. BRAF and MEK inhibition in melanoma patients enables reprogramming of tumor infiltrating lymphocytes. Cancer Immunol. Immunother. 2021, 70, 1635–1647. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, S.; Eberlein, V.; Schuler, G.; Berking, C.; Heinzerling, L.; Schaft, N.; Dorrie, J. BRAF and MEK Inhibitors Affect Dendritic-Cell Maturation and T-Cell Stimulation. Int. J. Mol. Sci. 2021, 22, 1951. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Singh, M.; Sanz Santos, G.; Guerlavais, V.; Carvajal, L.A.; Aivado, M.; Zhan, Y.; Oliveira, M.M.S.; Westerberg, L.S.; Annis, D.A.; et al. Pharmacologic Activation of p53 Triggers Viral Mimicry Response Thereby Abolishing Tumor Immune Evasion and Promoting Antitumor Immunity. Cancer Discov. 2021, 11, 3090–3105. [Google Scholar] [CrossRef]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Xue, L.; Gao, X.; Zhang, H.; Tang, J.; Wang, Q.; Li, F.; Li, X.; Yu, X.; Lu, Z.; Huang, Y.; et al. Antiangiogenic antibody BD0801 combined with immune checkpoint inhibitors achieves synergistic antitumor activity and affects the tumor microenvironment. BMC Cancer 2021, 21, 1134. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Chen, M.L.; Yan, B.S.; Lu, W.C.; Chen, M.H.; Yu, S.L.; Yang, P.C.; Cheng, A.L. Sorafenib relieves cell-intrinsic and cell-extrinsic inhibitions of effector T cells in tumor microenvironment to augment antitumor immunity. Int. J. Cancer 2014, 134, 319–331. [Google Scholar] [CrossRef]
- Kalathil, S.G.; Hutson, A.; Barbi, J.; Iyer, R.; Thanavala, Y. Augmentation of IFN-gamma+ CD8+ T cell responses correlates with survival of HCC patients on sorafenib therapy. JCI Insight 2019, 4, e130116. [Google Scholar] [CrossRef]
- Lu, M.; Zhang, X.; Gao, X.; Sun, S.; Wei, X.; Hu, X.; Huang, C.; Xu, H.; Wang, B.; Zhang, W.; et al. Lenvatinib enhances T cell immunity and the efficacy of adoptive chimeric antigen receptor-modified T cells by decreasing myeloid-derived suppressor cells in cancer. Pharm. Res 2021, 174, 105829. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Shaw, A.T.; Sequist, L.V.; Fu, X.; Azzoli, C.G.; Piotrowska, Z.; Huynh, T.G.; Zhao, L.; Fulton, L.; Schultz, K.R.; et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non-Small Cell Lung Cancer: A Retrospective Analysis. Clin. Cancer Res. 2016, 22, 4585–4593. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Li, X.; Jiang, T.; Zhao, S.; Zhao, C.; Zhang, L.; Liu, X.; Shi, J.; Qiao, M.; Luo, J.; et al. EGFR-targeted therapy alters the tumor microenvironment in EGFR-driven lung tumors: Implications for combination therapies. Int. J. Cancer 2019, 145, 1432–1444. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Lama, L.; Galindo-Campos, M.A.; Martinez, C.; Comerma, L.; Vazquez, I.; Vernet-Tomas, M.; Ampurdanes, C.; Lutfi, N.; Martin-Caballero, J.; Dantzer, F.; et al. Coordinated signals from PARP-1 and PARP-2 are required to establish a proper T cell immune response to breast tumors in mice. Oncogene 2020, 39, 2835–2843. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, E.; Togashi, Y.; Takeuchi, Y.; Shinya, S.; Tada, Y.; Kataoka, K.; Tane, K.; Sato, E.; Ishii, G.; Goto, K.; et al. Blockade of EGFR improves responsiveness to PD-1 blockade in EGFR-mutated non-small cell lung cancer. Sci. Immunol. 2020, 5, eaav3937. [Google Scholar] [CrossRef] [PubMed]
- Sharabi, A.; Ghera, N.H. Breaking tolerance in a mouse model of multiple myeloma by chemoimmunotherapy. Adv. Cancer Res. 2010, 107, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Larmonier, N.; Schmitt, E.; Parcellier, A.; Cathelin, D.; Garrido, C.; Chauffert, B.; Solary, E.; Bonnotte, B.; Martin, F. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur. J. Immunol. 2004, 34, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Nizar, S.; Copier, J.; Meyer, B.; Bodman-Smith, M.; Galustian, C.; Kumar, D.; Dalgleish, A. T-regulatory cell modulation: The future of cancer immunotherapy? Br. J. Cancer 2009, 100, 1697–1703. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Peuker, C.A.; Yaghobramzi, S.; Grunert, C.; Keilholz, L.; Gjerga, E.; Hennig, S.; Schaper, S.; Na, I.K.; Keller, U.; Brucker, S.; et al. Treatment with ribociclib shows favourable immunomodulatory effects in patients with hormone receptor-positive breast cancer-findings from the RIBECCA trial. Eur. J. Cancer 2022, 162, 45–55. [Google Scholar] [CrossRef]
- Torrens, L.; Montironi, C.; Puigvehi, M.; Mesropian, A.; Leslie, J.; Haber, P.K.; Maeda, M.; Balaseviciute, U.; Willoughby, C.E.; Abril-Fornaguera, J.; et al. Immunomodulatory Effects of Lenvatinib Plus Anti-Programmed Cell Death Protein 1 in Mice and Rationale for Patient Enrichment in Hepatocellular Carcinoma. Hepatology 2021, 74, 2652–2669. [Google Scholar] [CrossRef]
- Isoyama, S.; Mori, S.; Sugiyama, D.; Kojima, Y.; Tada, Y.; Shitara, K.; Hinohara, K.; Dan, S.; Nishikawa, H. Cancer immunotherapy with PI3K and PD-1 dual-blockade via optimal modulation of T cell activation signal. J. Immunother. Cancer 2021, 9, e002279. [Google Scholar] [CrossRef]
- Eschweiler, S.; Ramirez-Suastegui, C.; Li, Y.; King, E.; Chudley, L.; Thomas, J.; Wood, O.; von Witzleben, A.; Jeffrey, D.; McCann, K.; et al. Intermittent PI3Kdelta inhibition sustains anti-tumour immunity and curbs irAEs. Nature 2022, 605, 741–746. [Google Scholar] [CrossRef]
- Shang, B.; Liu, Y.; Jiang, S.J.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef]
- Frey, D.M.; Droeser, R.A.; Viehl, C.T.; Zlobec, I.; Lugli, A.; Zingg, U.; Oertli, D.; Kettelhack, C.; Terracciano, L.; Tornillo, L. High frequency of tumor-infiltrating FOXP3(+) regulatory T cells predicts improved survival in mismatch repair-proficient colorectal cancer patients. Int. J. Cancer 2010, 126, 2635–2643. [Google Scholar] [CrossRef]
- Lee, W.S.; Park, S.; Lee, W.Y.; Yun, S.H.; Chun, H.K. Clinical impact of tumor-infiltrating lymphocytes for survival in stage II colon cancer. Cancer 2010, 116, 5188–5199. [Google Scholar] [CrossRef]
- Downs-Canner, S.M.; Meier, J.; Vincent, B.G.; Serody, J.S. B Cell Function in the Tumor Microenvironment. Annu. Rev. Immunol. 2022, 40, 169–193. [Google Scholar] [CrossRef]
- Fridman, W.H.; Meylan, M.; Petitprez, F.; Sun, C.M.; Italiano, A.; Sautes-Fridman, C. B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat. Rev. Clin. Oncol. 2022, 19, 441–457. [Google Scholar] [CrossRef]
- Liang, C.; Tian, D.; Ren, X.; Ding, S.; Jia, M.; Xin, M.; Thareja, S. The development of Bruton’s tyrosine kinase (BTK) inhibitors from 2012 to 2017: A mini-review. Eur. J. Med. Chem. 2018, 151, 315–326. [Google Scholar] [CrossRef]
- Gunderson, A.J.; Kaneda, M.M.; Tsujikawa, T.; Nguyen, A.V.; Affara, N.I.; Ruffell, B.; Gorjestani, S.; Liudahl, S.M.; Truitt, M.; Olson, P.; et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Discov. 2016, 6, 270–285. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.F.; Li, J.; Jiang, K.; Wang, R.; Ge, J.L.; Yang, H.; Liu, S.J.; Jia, L.T.; Wang, L.; Chen, B.L. CDK4/6 inhibition promotes immune infiltration in ovarian cancer and synergizes with PD-1 blockade in a B cell-dependent manner. Theranostics 2020, 10, 10619–10633. [Google Scholar] [CrossRef]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Van Acker, H.H.; Capsomidis, A.; Smits, E.L.; Van Tendeloo, V.F. CD56 in the Immune System: More Than a Marker for Cytotoxicity? Front. Immunol. 2017, 8, 892. [Google Scholar] [CrossRef]
- Fu, B.; Tian, Z.; Wei, H. Subsets of human natural killer cells and their regulatory effects. Immunology 2014, 141, 483–489. [Google Scholar] [CrossRef]
- Wang, W.; Jiang, J.; Wu, C. CAR-NK for tumor immunotherapy: Clinical transformation and future prospects. Cancer Lett. 2020, 472, 175–180. [Google Scholar] [CrossRef]
- Ruscetti, M.; Leibold, J.; Bott, M.J.; Fennell, M.; Kulick, A.; Salgado, N.R.; Chen, C.C.; Ho, Y.J.; Sanchez-Rivera, F.J.; Feucht, J.; et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 2018, 362, 1416–1422. [Google Scholar] [CrossRef]
- Witalisz-Siepracka, A.; Gotthardt, D.; Prchal-Murphy, M.; Didara, Z.; Menzl, I.; Prinz, D.; Edlinger, L.; Putz, E.M.; Sexl, V. NK Cell-Specific CDK8 Deletion Enhances Antitumor Responses. Cancer Immunol. Res. 2018, 6, 458–466. [Google Scholar] [CrossRef]
- Ke, C.; Hou, H.; Li, J.; Su, K.; Huang, C.; Lin, Y.; Lu, Z.; Du, Z.; Tan, W.; Yuan, Z. Extracellular Vesicle Delivery of TRAIL Eradicates Resistant Tumor Growth in Combination with CDK Inhibition by Dinaciclib. Cancers 2020, 12, 1157. [Google Scholar] [CrossRef]
- Ferrari de Andrade, L.; Ngiow, S.F.; Stannard, K.; Rusakiewicz, S.; Kalimutho, M.; Khanna, K.K.; Tey, S.K.; Takeda, K.; Zitvogel, L.; Martinet, L.; et al. Natural killer cells are essential for the ability of BRAF inhibitors to control BRAFV600E-mutant metastatic melanoma. Cancer Res. 2014, 74, 7298–7308. [Google Scholar] [CrossRef] [Green Version]
- Mallmann-Gottschalk, N.; Sax, Y.; Kimmig, R.; Lang, S.; Brandau, S. EGFR-Specific Tyrosine Kinase Inhibitor Modifies NK Cell-Mediated Antitumoral Activity against Ovarian Cancer Cells. Int. J. Mol. Sci. 2019, 20, 4693. [Google Scholar] [CrossRef]
- Lopez-Cobo, S.; Pieper, N.; Campos-Silva, C.; Garcia-Cuesta, E.M.; Reyburn, H.T.; Paschen, A.; Vales-Gomez, M. Impaired NK cell recognition of vemurafenib-treated melanoma cells is overcome by simultaneous application of histone deacetylase inhibitors. Oncoimmunology 2018, 7, e1392426. [Google Scholar] [CrossRef]
- Rohrbacher, L.; Brauchle, B.; Ogrinc Wagner, A.; von Bergwelt-Baildon, M.; Bucklein, V.L.; Subklewe, M. The PI3K partial differential-Selective Inhibitor Idelalisib Induces T- and NK-Cell Dysfunction Independently of B-Cell Malignancy-Associated Immunosuppression. Front. Immunol. 2021, 12, 608625. [Google Scholar] [CrossRef]
- Rossi, L.E.; Avila, D.E.; Spallanzani, R.G.; Ziblat, A.; Fuertes, M.B.; Lapyckyj, L.; Croci, D.O.; Rabinovich, G.A.; Domaica, C.I.; Zwirner, N.W. Histone deacetylase inhibitors impair NK cell viability and effector functions through inhibition of activation and receptor expression. J. Leukoc. Biol. 2012, 91, 321–331. [Google Scholar] [CrossRef]
- Powell, D.R.; Huttenlocher, A. Neutrophils in the Tumor Microenvironment. Trends Immunol. 2016, 37, 41–52. [Google Scholar] [CrossRef]
- Li, Y.; Hu, Q.; Li, W.; Liu, S.; Li, K.; Li, X.; Du, J.; Yu, Z.; Wang, C.; Zhang, C. Simultaneous blockage of contextual TGF-beta by cyto-pharmaceuticals to suppress breast cancer metastasis. J. Control. Release 2021, 336, 40–53. [Google Scholar] [CrossRef]
- Peng, H.; Shen, J.; Long, X.; Zhou, X.; Zhang, J.; Xu, X.; Huang, T.; Xu, H.; Sun, S.; Li, C.; et al. Local Release of TGF-beta Inhibitor Modulates Tumor-Associated Neutrophils and Enhances Pancreatic Cancer Response to Combined Irreversible Electroporation and Immunotherapy. Adv. Sci. 2022, 9, e2105240. [Google Scholar] [CrossRef]
- Harizi, H. Reciprocal crosstalk between dendritic cells and natural killer cells under the effects of PGE2 in immunity and immunopathology. Cell. Mol. Immunol. 2013, 10, 213–221. [Google Scholar] [CrossRef]
- Shang, K.; Bai, Y.P.; Wang, C.; Wang, Z.; Gu, H.Y.; Du, X.; Zhou, X.Y.; Zheng, C.L.; Chi, Y.Y.; Mukaida, N.; et al. Crucial involvement of tumor-associated neutrophils in the regulation of chronic colitis-associated carcinogenesis in mice. PLoS ONE 2012, 7, e51848. [Google Scholar] [CrossRef]
- Ning, C.; Li, Y.Y.; Wang, Y.; Han, G.C.; Wang, R.X.; Xiao, H.; Li, X.Y.; Hou, C.M.; Ma, Y.F.; Sheng, D.S.; et al. Complement activation promotes colitis-associated carcinogenesis through activating intestinal IL-1beta/IL-17A axis. Mucosal. Immunol. 2015, 8, 1275–1284. [Google Scholar] [CrossRef] [Green Version]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef]
- Gall, V.A.; Philips, A.V.; Qiao, N.; Clise-Dwyer, K.; Perakis, A.A.; Zhang, M.; Clifton, G.T.; Sukhumalchandra, P.; Ma, Q.; Reddy, S.M.; et al. Trastuzumab Increases HER2 Uptake and Cross-Presentation by Dendritic Cells. Cancer Res 2017, 77, 5374–5383. [Google Scholar] [CrossRef]
- Aksoy, E.; Saveanu, L.; Manoury, B. The Isoform Selective Roles of PI3Ks in Dendritic Cell Biology and Function. Front. Immunol. 2018, 9, 2574. [Google Scholar] [CrossRef]
- Zhu, S.; Ma, A.H.; Zhu, Z.; Adib, E.; Rao, T.; Li, N.; Ni, K.; Chittepu, V.; Prabhala, R.; Garisto Risco, J.; et al. Synergistic antitumor activity of pan-PI3K inhibition and immune checkpoint blockade in bladder cancer. J. Immunother. Cancer 2021, 9, e002917. [Google Scholar] [CrossRef]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- De Palma, M.; Lewis, C.E. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef]
- Poon, E.; Mullins, S.; Watkins, A.; Williams, G.S.; Koopmann, J.O.; Di Genova, G.; Cumberbatch, M.; Veldman-Jones, M.; Grosskurth, S.E.; Sah, V.; et al. The MEK inhibitor selumetinib complements CTLA-4 blockade by reprogramming the tumor immune microenvironment. J. Immunother. Cancer 2017, 5, 63. [Google Scholar] [CrossRef]
- Kaneda, M.M.; Cappello, P.; Nguyen, A.V.; Ralainirina, N.; Hardamon, C.R.; Foubert, P.; Schmid, M.C.; Sun, P.; Mose, E.; Bouvet, M.; et al. Macrophage PI3Kgamma Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer Discov. 2016, 6, 870–885. [Google Scholar] [CrossRef]
- Fang, D.D.; Tang, Q.; Kong, Y.; Wang, Q.; Gu, J.; Fang, X.; Zou, P.; Rong, T.; Wang, J.; Yang, D.; et al. MDM2 inhibitor APG-115 synergizes with PD-1 blockade through enhancing antitumor immunity in the tumor microenvironment. J. Immunother. Cancer 2019, 7, 327. [Google Scholar] [CrossRef]
- Pander, J.; Heusinkveld, M.; van der Straaten, T.; Jordanova, E.S.; Baak-Pablo, R.; Gelderblom, H.; Morreau, H.; van der Burg, S.H.; Guchelaar, H.J.; van Hall, T. Activation of tumor-promoting type 2 macrophages by EGFR-targeting antibody cetuximab. Clin. Cancer Res. 2011, 17, 5668–5673. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.K.; Cheney, E.M.; Hartl, C.A.; Pantelidou, C.; Oliwa, M.; Castrillon, J.A.; Lin, J.R.; Hurst, K.E.; de Oliveira Taveira, M.; Johnson, N.T.; et al. Targeting immunosuppressive macrophages overcomes PARP inhibitor resistance in BRCA1-associated triple-negative breast cancer. Nat. Cancer 2021, 2, 66–82. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, F.; Solito, S.; Ugel, S.; Molon, B.; Bronte, V.; Marigo, I. MDSCs in cancer: Conceiving new prognostic and therapeutic targets. Biochim. Biophys. Acta 2016, 1865, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Teo, Z.L.; Versaci, S.; Dushyanthen, S.; Caramia, F.; Savas, P.; Mintoff, C.P.; Zethoven, M.; Virassamy, B.; Luen, S.J.; McArthur, G.A.; et al. Combined CDK4/6 and PI3Kalpha Inhibition Is Synergistic and Immunogenic in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 6340–6352. [Google Scholar] [CrossRef] [PubMed]
- Allegrezza, M.J.; Rutkowski, M.R.; Stephen, T.L.; Svoronos, N.; Perales-Puchalt, A.; Nguyen, J.M.; Payne, K.K.; Singhal, S.; Eruslanov, E.B.; Tchou, J.; et al. Trametinib Drives T-cell-Dependent Control of KRAS-Mutated Tumors by Inhibiting Pathological Myelopoiesis. Cancer Res. 2016, 76, 6253–6265. [Google Scholar] [CrossRef]
- Lelliott, E.J.; Mangiola, S.; Ramsbottom, K.M.; Zethoven, M.; Lim, L.; Lau, P.K.H.; Oliver, A.J.; Martelotto, L.G.; Kirby, L.; Martin, C.; et al. Combined BRAF, MEK, and CDK4/6 Inhibition Depletes Intratumoral Immune-Potentiating Myeloid Populations in Melanoma. Cancer Immunol. Res. 2021, 9, 136–146. [Google Scholar] [CrossRef]
- Koinis, F.; Vetsika, E.K.; Aggouraki, D.; Skalidaki, E.; Koutoulaki, A.; Gkioulmpasani, M.; Georgoulias, V.; Kotsakis, A. Effect of First-Line Treatment on Myeloid-Derived Suppressor Cells’ Subpopulations in the Peripheral Blood of Patients with Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2016, 11, 1263–1272. [Google Scholar] [CrossRef]
- Du Four, S.; Maenhout, S.K.; De Pierre, K.; Renmans, D.; Niclou, S.P.; Thielemans, K.; Neyns, B.; Aerts, J.L. Axitinib increases the infiltration of immune cells and reduces the suppressive capacity of monocytic MDSCs in an intracranial mouse melanoma model. Oncoimmunology 2015, 4, e998107. [Google Scholar] [CrossRef]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef]
- Li, C.W.; Lim, S.O.; Xia, W.; Lee, H.H.; Chan, L.C.; Kuo, C.W.; Khoo, K.H.; Chang, S.S.; Cha, J.H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun 2016, 7, 12632. [Google Scholar] [CrossRef] [Green Version]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Corte, C.M.D.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Muirhead, G.; Krastev, D.B.; Adam, J.; Morel, D.; Garrido, M.; Lamb, A.; Henon, C.; Dorvault, N.; Rouanne, M.; et al. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J. Clin. Investig. 2019, 129, 1211–1228. [Google Scholar] [CrossRef]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [PubMed]
- Menzies, A.M.; Long, G.V. Dabrafenib and trametinib, alone and in combination for BRAF-mutant metastatic melanoma. Clin. Cancer Res. 2014, 20, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Mayes, P.A.; Eastman, S.; Shi, H.; Yadavilli, S.; Zhang, T.; Yang, J.; Seestaller-Wehr, L.; Zhang, S.Y.; Hopson, C.; et al. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination with Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. 2015, 21, 1639–1651. [Google Scholar] [CrossRef] [PubMed]
- Stutvoet, T.S.; Kol, A.; de Vries, E.G.; de Bruyn, M.; Fehrmann, R.S.; Terwisscha van Scheltinga, A.G.; de Jong, S. MAPK pathway activity plays a key role in PD-L1 expression of lung adenocarcinoma cells. J Pathol 2019, 249, 52–64. [Google Scholar] [CrossRef]
- Xiong, W.; Zhang, B.; Yu, H.; Zhu, L.; Yi, L.; Jin, X. RRM2 Regulates Sensitivity to Sunitinib and PD-1 Blockade in Renal Cancer by Stabilizing ANXA1 and Activating the AKT Pathway. Adv. Sci. 2021, 8, e2100881. [Google Scholar] [CrossRef]
- Stadler, W.M.; Desai, A.A.; Quinn, D.I.; Bukowski, R.; Poiesz, B.; Kardinal, C.G.; Lewis, N.; Makalinao, A.; Murray, P.; Torti, F.M. A Phase I/II study of GTI-2040 and capecitabine in patients with renal cell carcinoma. Cancer Chemother. Pharm. 2008, 61, 689–694. [Google Scholar] [CrossRef]
- Zuckerman, J.E.; Gritli, I.; Tolcher, A.; Heidel, J.D.; Lim, D.; Morgan, R.; Chmielowski, B.; Ribas, A.; Davis, M.E.; Yen, Y. Correlating animal and human phase Ia/Ib clinical data with CALAA-01, a targeted, polymer-based nanoparticle containing siRNA. Proc. Natl. Acad. Sci. USA 2014, 111, 11449–11454. [Google Scholar] [CrossRef]
- Bouattour, M.; Raymond, E.; Qin, S.; Cheng, A.L.; Stammberger, U.; Locatelli, G.; Faivre, S. Recent developments of c-Met as a therapeutic target in hepatocellular carcinoma. Hepatology 2018, 67, 1132–1149. [Google Scholar] [CrossRef]
- Boccaccio, C.; Comoglio, P.M. Invasive growth: A MET-driven genetic programme for cancer and stem cells. Nat. Rev. Cancer 2006, 6, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Rimassa, L.; Assenat, E.; Peck-Radosavljevic, M.; Pracht, M.; Zagonel, V.; Mathurin, P.; Rota Caremoli, E.; Porta, C.; Daniele, B.; Bolondi, L.; et al. Tivantinib for second-line treatment of MET-high, advanced hepatocellular carcinoma (METIV-HCC): A final analysis of a phase 3, randomised, placebo-controlled study. Lancet Oncol. 2018, 19, 682–693. [Google Scholar] [CrossRef]
- Li, H.; Li, C.W.; Li, X.; Ding, Q.; Guo, L.; Liu, S.; Liu, C.; Lai, C.C.; Hsu, J.M.; Dong, Q.; et al. MET Inhibitors Promote Liver Tumor Evasion of the Immune Response by Stabilizing PDL1. Gastroenterology 2019, 156, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Deng, J.; Li, S.; Silk, T.; Dong, L.; Brea, E.J.; Houghton, S.; Redmond, D.; Zhong, H.; Boiarsky, J.; et al. Pulsatile MEK Inhibition Improves Anti-tumor Immunity and T Cell Function in Murine Kras Mutant Lung Cancer. Cell Rep. 2019, 27, 806–819.e5. [Google Scholar] [CrossRef]
- Yang, H.; Bueso-Ramos, C.; DiNardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef]
- Wang, L.; Amoozgar, Z.; Huang, J.; Saleh, M.H.; Xing, D.; Orsulic, S.; Goldberg, M.S. Decitabine Enhances Lymphocyte Migration and Function and Synergizes with CTLA-4 Blockade in a Murine Ovarian Cancer Model. Cancer Immunol. Res. 2015, 3, 1030–1041. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, L.; Hei, R.; Li, X.; Cai, H.; Wu, X.; Zheng, Q.; Cai, C. CDK inhibitors in cancer therapy, an overview of recent development. Am. J. Cancer Res. 2021, 11, 1913–1935. [Google Scholar]
- Bonelli, P.; Tuccillo, F.M.; Borrelli, A.; Schiattarella, A.; Buonaguro, F.M. CDK/CCN and CDKI Alterations for Cancer Prognosis and Therapeutic Predictivity. BioMed Res. Int. 2014, 2014, e361020. [Google Scholar] [CrossRef]
- Shan, W.; Yuan, J.; Hu, Z.; Jiang, J.; Wang, Y.; Loo, N.; Fan, L.; Tang, Z.; Zhang, T.; Xu, M.; et al. Systematic Characterization of Recurrent Genomic Alterations in Cyclin-Dependent Kinases Reveals Potential Therapeutic Strategies for Cancer Treatment. Cell Rep. 2020, 32, 107884. [Google Scholar] [CrossRef]
- Yuan, Y.; Lee, J.S.; Yost, S.E.; Frankel, P.H.; Ruel, C.; Egelston, C.A.; Guo, W.; Padam, S.; Tang, A.; Martinez, N.; et al. Phase I/II trial of palbociclib, pembrolizumab and letrozole in patients with hormone receptor-positive metastatic breast cancer. Eur. J. Cancer 2021, 154, 11–20. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Kabos, P.; Dickler, M.N.; Gianni, L.; Jansen, V.; Lu, Y.; Young, S.; Rugo, H.S. Updated efficacy, safety, & PD-L1 status of patients with HR+, HER2- metastatic breast cancer administered abemaciclib plus pembrolizumab. J. Clin. Oncol. 2018, 36, 1059. [Google Scholar] [CrossRef] [Green Version]
- Pujol, J.-L.; Vansteenkiste, J.; Paz-Ares Rodríguez, L.; Gregorc, V.; Mazieres, J.; Awad, M.; Jänne, P.A.; Chisamore, M.; Hossain, A.M.; Chen, Y.; et al. Abemaciclib in Combination With Pembrolizumab for Stage IV KRAS-Mutant or Squamous NSCLC: A Phase 1b Study. JTO Clin. Res. Rep. 2021, 2, 100234. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sinica B 2019, 9, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Grob, J.-J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.-Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef]
- Planchard, D.; Kim, T.M.; Mazieres, J.; Quoix, E.; Riely, G.; Barlesi, F.; Souquet, P.-J.; Smit, E.F.; Groen, H.J.M.; Kelly, R.J.; et al. Dabrafenib in patients with BRAF(V600E)-positive advanced non-small-cell lung cancer: A single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 642–650. [Google Scholar] [CrossRef]
- Robert, C.; Flaherty, K.T.; Hersey, P.; Nathan, P.D.; Garbe, C.; Milhem, M.M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. METRIC phase III study: Efficacy of trametinib (T), a potent and selective MEK inhibitor (MEKi), in progression-free survival (PFS) and overall survival (OS), compared with chemotherapy (C) in patients (pts) with BRAFV600E/K mutant advanced or metastatic melanoma (MM). J. Clin. Oncol. 2012, 30, LBA8509. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.-J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Planchard, D.; Smit, E.F.; Groen, H.J.M.; Mazieres, J.; Besse, B.; Helland, Å.; Giannone, V.; D’Amelio, A.M.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: An open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1307–1316. [Google Scholar] [CrossRef]
- Zhang, Z.; Richmond, A.; Yan, C. Immunomodulatory Properties of PI3K/AKT/mTOR and MAPK/MEK/ERK Inhibition Augment Response to Immune Checkpoint Blockade in Melanoma and Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2022, 23, 7353. [Google Scholar] [CrossRef]
- Candido, S.; Salemi, R.; Piccinin, S.; Falzone, L.; Libra, M. The PIK3CA H1047R Mutation Confers Resistance to BRAF and MEK Inhibitors in A375 Melanoma Cells through the Cross-Activation of MAPK and PI3K–Akt Pathways. Pharmaceutics 2022, 14, 590. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Lee, S.J.; Chmielowski, B.; Ribas, A.; Tarhini, A.A.; Truong, T.-G.; Davar, D.; O’Rourke, M.A.; Curti, B.D.; Brell, J.M.; et al. DREAMseq (Doublet, Randomized Evaluation in Advanced Melanoma Sequencing): A phase III trial—ECOG-ACRIN EA6134. J. Clin. Oncol. 2021, 39, 356154. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Ferrucci, P.F.; Fisher, R.; Del Vecchio, M.; Atkinson, V.; Schmidt, H.; Schachter, J.; Queirolo, P.; Long, G.V.; Di Giacomo, A.M.; et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat. Med. 2019, 25, 941–946. [Google Scholar] [CrossRef]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Gogas, H.; Dréno, B.; Larkin, J.; Demidov, L.; Stroyakovskiy, D.; Eroglu, Z.; Ferrucci, P.F.; Pigozzo, J.; Rutkowski, P.; Mackiewicz, J.; et al. Cobimetinib plus atezolizumab in BRAFV600 wild-type melanoma: Primary results from the randomized phase III IMspire170 study. Ann. Oncol. 2021, 32, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAFV600 mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- Yarchoan, M.; Cope, L.; Ruggieri, A.N.; Anders, R.A.; Noonan, A.M.; Goff, L.W.; Goyal, L.; Lacy, J.; Li, D.; Patel, A.K.; et al. Multicenter randomized phase II trial of atezolizumab with or without cobimetinib in biliary tract cancers. J. Clin. Investig. 2021, 131, e152670. [Google Scholar] [CrossRef]
- Liu, S.V.; Hall, R.D.; Saltos, A.N.; Otterson, G.A.; Tan, M.T.; Gnjatic, S.; Gentzler, R.D.; Tanvetyanon, T.; Chen, H.X.; Sharon, E. A phase II study of atezolizumab and cobimetinib in PD-1/PD-L1 inhibitor resistant or refractory non-small cell lung cancer: ETCTN #10166. J. Clin. Oncol. 2020, 38, TPS9638. [Google Scholar] [CrossRef]
- Subramaniam, D.; He, A.R.; Hwang, J.; Deeken, J.; Pishvaian, M.; Hartley, M.L.; Marshall, J.L. Irreversible multitargeted ErbB family inhibitors for therapy of lung and breast cancer. Curr. Cancer Drug Targets 2015, 14, 775–793. [Google Scholar] [CrossRef]
- Roskoski, R. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol. Res. 2019, 139, 395–411. [Google Scholar] [CrossRef]
- Creelan, B.C.; Yeh, T.C.; Kim, S.-W.; Nogami, N.; Kim, D.-W.; Chow, L.Q.M.; Kanda, S.; Taylor, R.; Tang, W.; Tang, M.; et al. A Phase 1 study of gefitinib combined with durvalumab in EGFR TKI-naive patients with EGFR mutation-positive locally advanced/metastatic non-small-cell lung cancer. Br. J. Cancer 2021, 124, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Riudavets, M.; Naigeon, M.; Texier, M.; Dorta, M.; Barlesi, F.; Mazieres, J.; Varga, A.; Cassard, L.; Boselli, L.; Grivel, J.; et al. Gefitinib plus tremelimumab combination in refractory non-small cell lung cancer patients harbouring EGFR mutations: The GEFTREM phase I trial. Lung Cancer 2022, 166, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.-H.; Gadgeel, S.M.; Sequist, L.V.; Wu, C.-L.; Papadimitrakopoulou, V.A.; Su, W.-C.; Fiore, J.; Saraf, S.; Raftopoulos, H.; Patnaik, A. Pembrolizumab in Combination With Erlotinib or Gefitinib as First-Line Therapy for Advanced NSCLC With Sensitizing EGFR Mutation. J. Thorac. Oncol. 2019, 14, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, O.; Kaira, K.; Kawasaki, T.; Mouri, A.; Hashimoto, K.; Shiono, A.; Shinomiya, S.; Miura, Y.; Nishihara, F.; Murayama, Y.; et al. Severe hepatotoxicity due to osimertinib after nivolumab therapy in patients with non-small cell lung cancer harboring EGFR mutation. Thorac. Cancer 2020, 11, 1045–1051. [Google Scholar] [CrossRef]
- Sacco, A.G.; Chen, R.; Worden, F.P.; Wong, D.J.L.; Adkins, D.; Swiecicki, P.; Chai-Ho, W.; Oppelt, P.; Ghosh, D.; Bykowski, J.; et al. Pembrolizumab plus cetuximab in patients with recurrent or metastatic head and neck squamous cell carcinoma: An open-label, multi-arm, non-randomised, multicentre, phase 2 trial. Lancet Oncol. 2021, 22, 883–892. [Google Scholar] [CrossRef]
- Boland, P.M.; Hutson, A.; Maguire, O.; Minderman, H.; Fountzilas, C.; Iyer, R.V. A phase Ib/II study of cetuximab and pembrolizumab in RAS-wt mCRC. J. Clin. Oncol. 2018, 36, 834. [Google Scholar] [CrossRef]
- Lee, M.S.; Loehrer, P.J.; Imanirad, I.; Cohen, S.; Ciombor, K.K.; Moore, D.T.; Carlson, C.A.; Sanoff, H.K.; McRee, A.J. Phase II study of ipilimumab, nivolumab, and panitumumab in patients with KRAS/NRAS/BRAF wild-type (WT) microsatellite stable (MSS) metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2021, 39, 7. [Google Scholar] [CrossRef]
- Besse, B.; Garrido, P.; Cortot, A.B.; Johnson, M.; Murakami, H.; Gazzah, A.; Gil, M.; Bennouna, J. Efficacy and safety of necitumumab and pembrolizumab combination therapy in patients with Stage IV non-small cell lung cancer. Lung Cancer 2020, 142, 63–69. [Google Scholar] [CrossRef]
- Krishnamurti, U.; Silverman, J.F. HER2 in breast cancer: A review and update. Adv. Anat. Pathol. 2014, 21, 100–107. [Google Scholar] [CrossRef]
- Kao, H.-F.; Liao, B.-C.; Huang, Y.-L.; Huang, H.-C.; Chen, C.-N.; Chen, T.-C.; Hong, Y.-J.; Chan, C.-Y.; Chia, J.-S.; Hong, R.-L. Afatinib and Pembrolizumab for Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma (ALPHA Study): A Phase II Study with Biomarker Analysis. Clin. Cancer Res. 2022, 28, 1560–1571. [Google Scholar] [CrossRef]
- Loi, S.; Giobbie-Hurder, A.; Gombos, A.; Bachelot, T.; Hui, R.; Curigliano, G.; Campone, M.; Biganzoli, L.; Bonnefoi, H.; Jerusalem, G.; et al. Pembrolizumab plus trastuzumab in trastuzumab-resistant, advanced, HER2-positive breast cancer (PANACEA): A single-arm, multicentre, phase 1b–2 trial. Lancet Oncol. 2019, 20, 371–382. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Maron, S.B.; Chatila, W.K.; Millang, B.; Chavan, S.S.; Alterman, C.; Chou, J.F.; Segal, M.F.; Simmons, M.Z.; Momtaz, P.; et al. First-line pembrolizumab and trastuzumab in HER2-positive oesophageal, gastric, or gastro-oesophageal junction cancer: An open-label, single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 821–831. [Google Scholar] [CrossRef]
- Chen, A. PARP inhibitors: Its role in treatment of cancer. Chin. J. Cancer 2011, 30, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef]
- Lampert, E.J.; Zimmer, A.; Padget, M.; Cimino-Mathews, A.; Nair, J.R.; Liu, Y.; Swisher, E.M.; Hodge, J.W.; Nixon, A.B.; Nichols, E.; et al. Combination of PARP Inhibitor Olaparib, and PD-L1 Inhibitor Durvalumab, in Recurrent Ovarian Cancer: A Proof-of-Concept Phase II Study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 4268–4279. [Google Scholar] [CrossRef]
- Karzai, F.; VanderWeele, D.; Madan, R.A.; Owens, H.; Cordes, L.M.; Hankin, A.; Couvillon, A.; Nichols, E.; Bilusic, M.; Beshiri, M.L.; et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J. ImmunoTherapy Cancer 2018, 6, 141. [Google Scholar] [CrossRef]
- Fumet, J.-D.; Limagne, E.; Thibaudin, M.; Truntzer, C.; Bertaut, A.; Rederstorff, E.; Ghiringhelli, F. Precision medicine phase II study evaluating the efficacy of a double immunotherapy by durvalumab and tremelimumab combined with olaparib in patients with solid cancers and carriers of homologous recombination repair genes mutation in response or stable after olaparib treatment. BMC Cancer 2020, 20, 748. [Google Scholar] [CrossRef]
- Domchek, S.M.; Postel-Vinay, S.; Im, S.-A.; Park, Y.H.; Delord, J.-P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): An open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020, 21, 1155–1164. [Google Scholar] [CrossRef]
- Taeger, J.; Moser, C.; Hellerbrand, C.; Mycielska, M.E.; Glockzin, G.; Schlitt, H.J.; Geissler, E.K.; Stoeltzing, O.; Lang, S.A. Targeting FGFR/PDGFR/VEGFR impairs tumor growth, angiogenesis, and metastasis by effects on tumor cells, endothelial cells, and pericytes in pancreatic cancer. Mol. Cancer Ther. 2011, 10, 2157–2167. [Google Scholar] [CrossRef]
- Chu, T.; Zhong, R.; Zhong, H.; Zhang, B.; Zhang, W.; Shi, C.; Qian, J.; Zhang, Y.; Chang, Q.; Zhang, X.; et al. Phase 1b Study of Sintilimab Plus Anlotinib as First-line Therapy in Patients With Advanced NSCLC. J. Thorac. Oncol. 2021, 16, 643–652. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, Z.; Li, Q.; Li, Y.; Liu, Q.; Song, E. Efficacy and safety of anti-PD-1 antibody SHR-1210 combined with apatinib in patients with advanced triple-negative breast cancer. J. Clin. Oncol. 2019, 37, 1066. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y.; Jia, R.; Yue, C.; Chang, L.; Liu, R.; Zhang, G.; Zhao, C.; Zhang, Y.; Chen, C.; et al. Anti-PD-1 Antibody SHR-1210 Combined with Apatinib for Advanced Hepatocellular Carcinoma, Gastric, or Esophagogastric Junction Cancer: An Open-label, Dose Escalation and Expansion Study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Mai, Z.-J.; Jin, Y.; He, M.-M.; Wang, Z.-Q.; Luo, H.-Y.; Zhang, D.-S.; Wu, C.-Y.; Wang, F.; Xu, R.-H. Anti-PD-1 antibody SHR-1210 plus apatinib for metastatic colorectal cancer: A prospective, single-arm, open-label, phase II trial. Am. J. Cancer Res. 2020, 10, 2946–2954. [Google Scholar] [PubMed]
- Powles, T.; Plimack, E.R.; Soulières, D.; Waddell, T.; Stus, V.; Gafanov, R.; Nosov, D.; Pouliot, F.; Melichar, B.; Vynnychenko, I.; et al. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): Extended follow-up from a randomised, open-label, phase 3 trial. Lancet Oncol. 2020, 21, 1563–1573. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Powles, T.; Burotto, M.; Escudier, B.; Bourlon, M.T.; Zurawski, B.; Oyervides Juárez, V.M.; Hsieh, J.J.; Basso, U.; Shah, A.Y.; et al. Nivolumab plus Cabozantinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2021, 384, 829–841. [Google Scholar] [CrossRef]
- Finn, R.S.; Ikeda, M.; Zhu, A.X.; Sung, M.W.; Baron, A.D.; Kudo, M.; Okusaka, T.; Kobayashi, M.; Kumada, H.; Kaneko, S.; et al. Phase Ib Study of Lenvatinib Plus Pembrolizumab in Patients With Unresectable Hepatocellular Carcinoma. J. Clin. Oncol. 2020, 38, 2960–2970. [Google Scholar] [CrossRef]
- Haugen, B.; French, J.; Worden, F.P.; Konda, B.; Sherman, E.J.; Dadu, R.; Gianoukakis, A.G.; Wolfe, E.G.; Foster, N.R.; Bowles, D.W.; et al. Lenvatinib plus pembrolizumab combination therapy in patients with radioiodine-refractory (RAIR), progressive differentiated thyroid cancer (DTC): Results of a multicenter phase II international thyroid oncology group trial. J. Clin. Oncol. 2020, 38, 6512. [Google Scholar] [CrossRef]
- Kawazoe, A.; Fukuoka, S.; Nakamura, Y.; Kuboki, Y.; Wakabayashi, M.; Nomura, S.; Mikamoto, Y.; Shima, H.; Fujishiro, N.; Higuchi, T.; et al. Lenvatinib plus pembrolizumab in patients with advanced gastric cancer in the first-line or second-line setting (EPOC1706): An open-label, single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 1057–1065. [Google Scholar] [CrossRef]
- Lin, J.; Yang, X.; Long, J.; Zhao, S.; Mao, J.; Wang, D.; Bai, Y.; Bian, J.; Zhang, L.; Yang, X.; et al. Pembrolizumab combined with lenvatinib as non-first-line therapy in patients with refractory biliary tract carcinoma. Hepatobiliary Surg. Nutr. 2020, 9, 414–424. [Google Scholar] [CrossRef]
- Llovet, J.; Shepard, K.V.; Finn, R.S.; Ikeda, M.; Sung, M.; Baron, A.D.; Kudo, M.; Okusaka, T.; Kobayashi, M.; Kumada, H.; et al. 747P—A phase Ib trial of lenvatinib (LEN) plus pembrolizumab (PEMBRO) in unresectable hepatocellular carcinoma (uHCC): Updated results. Ann. Oncol. 2019, 30, v286–v287. [Google Scholar] [CrossRef]
- Cousin, S.; Cantarel, C.; Guegan, J.-P.; Gomez-Roca, C.; Metges, J.-P.; Adenis, A.; Pernot, S.; Bellera, C.; Kind, M.; Auzanneau, C.; et al. Regorafenib-Avelumab Combination in Patients with Microsatellite Stable Colorectal Cancer (REGOMUNE): A Single-arm, Open-label, Phase II Trial. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 2139–2147. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Guo, Y.; Huang, W.; Hong, X.; Quan, Y.; Lin, L.; Zhou, J.; Liang, L.; Zhang, Y.; Zhou, J.; et al. Regorafenib Combined with PD-1 Blockade Immunotherapy versus Regorafenib as Second-Line Treatment for Advanced Hepatocellular Carcinoma: A Multicenter Retrospective Study. J. Hepatocell. Carcinoma 2022, 9, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.; Subbiah, V.; Nemunaitis, J.J.; Mettu, N.B.; Papadopoulos, K.P.; Barve, M.A.; Féliz, L.; Lihou, C.F.; Tian, C.; Ji, T.; et al. Safety and efficacy of pemigatinib plus pembrolizumab combination therapy in patients (pts) with advanced malignancies: Results from FIGHT-101, an open-label phase I/II study. J. Clin. Oncol. 2020, 38, 3606. [Google Scholar] [CrossRef]
- Zhu, X.D.; Huang, C.; Shen, Y.H.; Ji, Y.; Ge, N.L.; Qu, X.D.; Chen, L.; Shi, W.K.; Li, M.L.; Zhu, J.J.; et al. Downstaging and Resection of Initially Unresectable Hepatocellular Carcinoma with Tyrosine Kinase Inhibitor and Anti-PD-1 Antibody Combinations. Liver Cancer 2021, 10, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Lee, J.-L.; et al. IMmotion151: A Randomized Phase III Study of Atezolizumab Plus Bevacizumab vs Sunitinib in Untreated Metastatic Renal Cell Carcinoma (mRCC). J. Clin. Oncol. 2018, 36, 578. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Jänne, P.A.; Opyrchal, M.; Hafez, N.; Raez, L.E.; Gabrilovich, D.I.; Wang, F.; Trepel, J.B.; Lee, M.-J.; Yuno, A.; et al. Entinostat plus Pembrolizumab in Patients with Metastatic NSCLC Previously Treated with Anti–PD-(L)1 Therapy. Clin. Cancer Res. 2021, 27, 1019–1028. [Google Scholar] [CrossRef]
- Ny, L.; Jespersen, H.; Karlsson, J.; Alsén, S.; Filges, S.; All-Eriksson, C.; Andersson, B.; Carneiro, A.; Helgadottir, H.; Levin, M.; et al. The PEMDAC phase 2 study of pembrolizumab and entinostat in patients with metastatic uveal melanoma. Nat. Commun. 2021, 12, 5155. [Google Scholar] [CrossRef]
- Wang, C.; Liu, Y.; Dong, L.; Li, X.; Yang, Q.; Brock, M.V.; Mei, Q.; Liu, J.; Chen, M.; Shi, F.; et al. Efficacy of Decitabine plus Anti-PD-1 Camrelizumab in Patients with Hodgkin Lymphoma Who Progressed or Relapsed after PD-1 Blockade Monotherapy. Clin. Cancer Res. 2021, 27, 2782–2791. [Google Scholar] [CrossRef]
- Saxena, K.; Herbrich, S.M.; Pemmaraju, N.; Kadia, T.M.; DiNardo, C.D.; Borthakur, G.; Pierce, S.A.; Jabbour, E.; Wang, S.A.; Bueso-Ramos, C.; et al. A phase 1b/2 study of azacitidine with PD-L1 antibody avelumab in relapsed/refractory acute myeloid leukemia. Cancer 2021, 127, 3761–3771. [Google Scholar] [CrossRef]
- Petroni, G.; Buqué, A.; Coussens, L.M.; Galluzzi, L. Targeting oncogene and non-oncogene addiction to inflame the tumour microenvironment. Nat. Rev. Drug Discov. 2022, 21, 440–462. [Google Scholar] [CrossRef]
- Palmer, A.C.; Sorger, P.K. Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy. Cell 2017, 171, 1678–1691.e1613. [Google Scholar] [CrossRef] [PubMed]
- Van Herck, Y.; Feyaerts, A.; Alibhai, S.; Papamichael, D.; Decoster, L.; Lambrechts, Y.; Pinchuk, M.; Bechter, O.; Herrera-Caceres, J.; Bibeau, F.; et al. Is cancer biology different in older patients? Lancet Healthy Longev. 2021, 2, e663–e677. [Google Scholar] [CrossRef]
- Fane, M.; Weeraratna, A.T. How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 2020, 20, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Howie, L.J.; Singh, H.; Bloomquist, E.; Wedam, S.; Amiri-Kordestani, L.; Tang, S.; Sridhara, R.; Sanchez, J.; Prowell, T.M.; Kluetz, P.G.; et al. Outcomes of Older Women With Hormone Receptor–Positive, Human Epidermal Growth Factor Receptor–Negative Metastatic Breast Cancer Treated With a CDK4/6 Inhibitor and an Aromatase Inhibitor: An FDA Pooled Analysis. J. Clin. Oncol. 2019, 37, 3475–3483. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Cancer Type | Targeted Agents | Combination Agents | Phase | Control | Primary Endpoint | NCT Number |
|---|---|---|---|---|---|---|
| BRAF/MEK Inhibitors | ||||||
| Melanoma * | Cobimetinib + Vemurafenib | Atezolizumab | III | Cobimetinib + Vemurafenib + | PFS (vs. control): 15.1 vs. 10.6 months, HR = 0.78 | NCT02908672 |
| placebo | ||||||
| Melanoma | Trametinib + Dabrafenib | Ipilimumab + Nivolumab | III | Arm A: N/I + D/T | 2-year OS rate (Arm A vs. Arm B): 72% vs. 52% | NCT02224781 |
| Arm B: D/T + N/I | ||||||
| CDK Inhibitors | ||||||
| Breast Cancer | Palbociclib | Pembrolizumab + Letrozole | I/II | - | ORR: 56% | NCT02778685 |
| EGFR Inhibitors | ||||||
| HNSCC | Cetuximab | Pembrolizumab | II | - | ORR: 45% (95% CI 28–62) | NCT03082534 |
| CRC | Panitumumab | Ipilimumab + Nivolumab | II | - | ORR: 35% (95% CI 21–48) | NCT03442569 |
| CRC | Cetuximab | Pembrolizumab | Ib/II | - | A manageable safety profile | NCT02713373 |
| HER2 | ||||||
| HNSCC | Afatinib | Pembrolizumab | II | - | High PD-L1 expression (TPS ≥ 50 ORR: 71%, CPS ≥ 20 ORR: 63%); | NCT03695510 |
| EGFR amplification (ORR: 100%); | ||||||
| MTAP loss or mutation (ORR: 0%) | ||||||
| Oesophageal, G/GEJ Cancer | Trastuzumab | Pembrolizumab | II | - | 6-months PFSR: 70% (95% CI 54–83) | NCT02954536 |
| Breast Cancer | Trastuzumab | Pembrolizumab | Ib/II | - | ORR: 15% (90% CI 7–29) | NCT02129556 |
| Cancer Type | Targeted Agents | Combination Agents | Phase | Control | Primary Endpoint | NCT Number |
|---|---|---|---|---|---|---|
| Endometrial Cancer * | Lenvatinib | Pembrolizumab | III | Chemotherapy | Median OS (vs. control): 17.4 vs. 12.0 months, HR = 0.68 | NCT03517449 |
| Median PFS (vs. control): 6.6 vs. 3.8 months, HR = 0.60 | ||||||
| HCC * | Bevacizumab | Atezolizumab | III | Sorafenib | Median OS (vs. control): 19.2 vs. 13.4 months, HR = 0.66 | NCT03434379 |
| Median PFS (vs. control): 6.9 vs. 4.3 months, HR = 0.65 | ||||||
| RCC * | Lenvatinib | Pembrolizumab | III | Lenvatinib + | PFS (vs. control): 23.9 vs. 9.2 months, HR = 0.39 | NCT02811861 |
| Sunitinib | ||||||
| RCC * | Axitinib | Pembrolizumab | III | Sunitinib | OS (vs. control): median not reached vs. 35·7 months, HR = 0.68 | NCT02853331 |
| RCC * | Cabozantinib | Nivolumab | III | Sunitinib | PFS (vs. control): 16.6 vs. 8.3 months, HR = 0.51 | NCT03141177 |
| RCC | Bevacizumab | Atezolizumab | III | Sunitinib | PD-L1 positive group median PFS (vs. control): 11.2 vs. 7.7 months, HR = 0.74 | NCT02420821 |
| Breast Cancer | Apatinib | Camrelizumab | II | - | ORR: 43.3% (95% CI 25.5–62.6) | NCT03394287 |
| CRC | Regorafenib | Avelumab | II | - | CR, PR: 0; SD: 57.5% | NCT03475953 |
| DTC | Lenvatinib | Pembrolizumab | II | - | ORR: 62% | NCT02973997 |
| Endometrial Cancer | Lenvatinib | Pembrolizumab | II | - | ORR: 38% (95% CI 28.8–47.8) | NCT02501096 |
| GC | Lenvatinib | Pembrolizumab | II | - | ORR: 69% (95% CI 49–85) | NCT03609359 |
| Ovarian Cancer | Bevacizumab | Nivolumab | II | - | ORR: 21% | NCT02873962 |
| CRC | Regorafenib | Toripalimab | Ib/II | - | ORR: 15.2% (95% CI 5.7–32.7) | NCT03946917 |
| Pancancer | Pemigatinib | Pembrolizumab | I/II | - | A manageable safety profile and pharmacodynamic and clinical activity | NCT02393248 |
| Sarcomas | Sunitinib | Nivolumab | Ib/II | - | 6-months PFSR: 48% (95% CI 41–55) | NCT03277924 |
| Gastric Cancer/CRC | Regorafenib | Nivolumab | Ib | - | Safety: Regorafenib 80 mg plus nivolumab | NCT03406871 |
| HCC | Lenvatinib | Pembrolizumab | Ib | - | Promising antitumor activity with a tolerable safety profile | NCT03006926 |
| NSCLC | Anlotinib | Sintilimab | Ib | - | ORR: 72.7% (95% CI 49.8–89.3) | NCT03628521 |
| HCC | Apatinib | Camrelizumab | I | - | ORR: 30.8% (95% CI 17–47.6) | NCT02942329 |
| NSCLC, G/GEJ Cancer, UC | Ramucirumab | Pembrolizumab | I | - | A manageable safety profile with favorable antitumor activity | NCT02443324 |
| Cancer Type | Targeted Agents | Combination Agents | Phase | Control | Primary Endpoint | NCT Number |
|---|---|---|---|---|---|---|
| Uveal Melanoma | Entinostat | Pembrolizumab | II | - | ORR: 14% (95% CI 3.9–31.7) | NCT02697630 |
| AML | Azacitidine | Nivolumab | II | - | ORR: 33% | NCT02397720 |
| Hodgkin Lymphoma | Decitabine | Camrelizumab | II | - | ORR: 60% (95% CI 45–74) | NCT02961101 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Zhang, C.; Fu, Z.; Gao, Q. Immune Modulatory Effects of Molecularly Targeted Therapy and Its Repurposed Usage in Cancer Immunotherapy. Pharmaceutics 2022, 14, 1768. https://doi.org/10.3390/pharmaceutics14091768
Zhang T, Zhang C, Fu Z, Gao Q. Immune Modulatory Effects of Molecularly Targeted Therapy and Its Repurposed Usage in Cancer Immunotherapy. Pharmaceutics. 2022; 14(9):1768. https://doi.org/10.3390/pharmaceutics14091768
Chicago/Turabian StyleZhang, Tiancheng, Chenhao Zhang, Zile Fu, and Qiang Gao. 2022. "Immune Modulatory Effects of Molecularly Targeted Therapy and Its Repurposed Usage in Cancer Immunotherapy" Pharmaceutics 14, no. 9: 1768. https://doi.org/10.3390/pharmaceutics14091768