
The Development and Clinical Applications of Oral Arsenic Trioxide for Acute Promyelocytic Leukaemia and Other Diseases


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
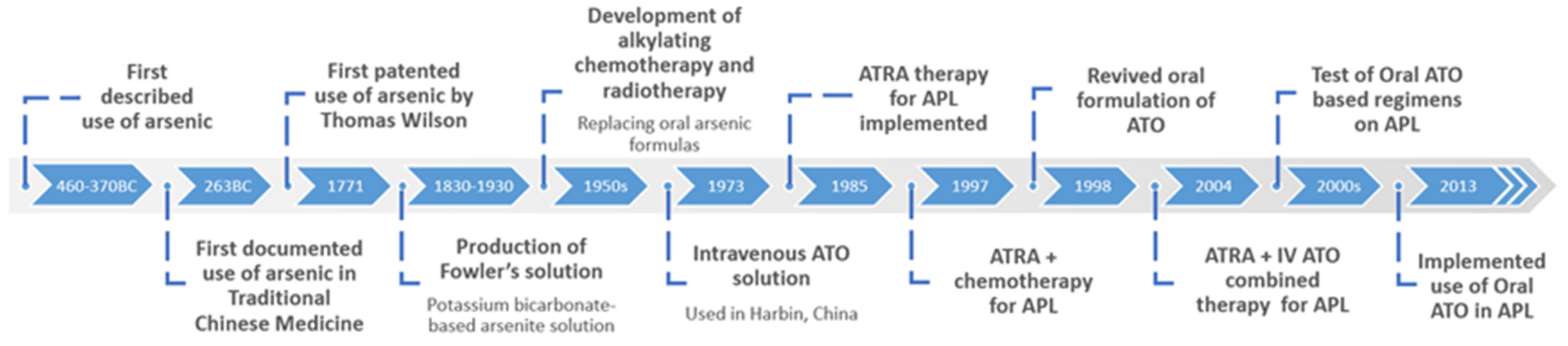
2. The Discovery and History of Arsenic Trioxide
3. Antileukaemic Mechanisms of Arsenic Trioxide
3.1. Caspase Induced Apoptosis
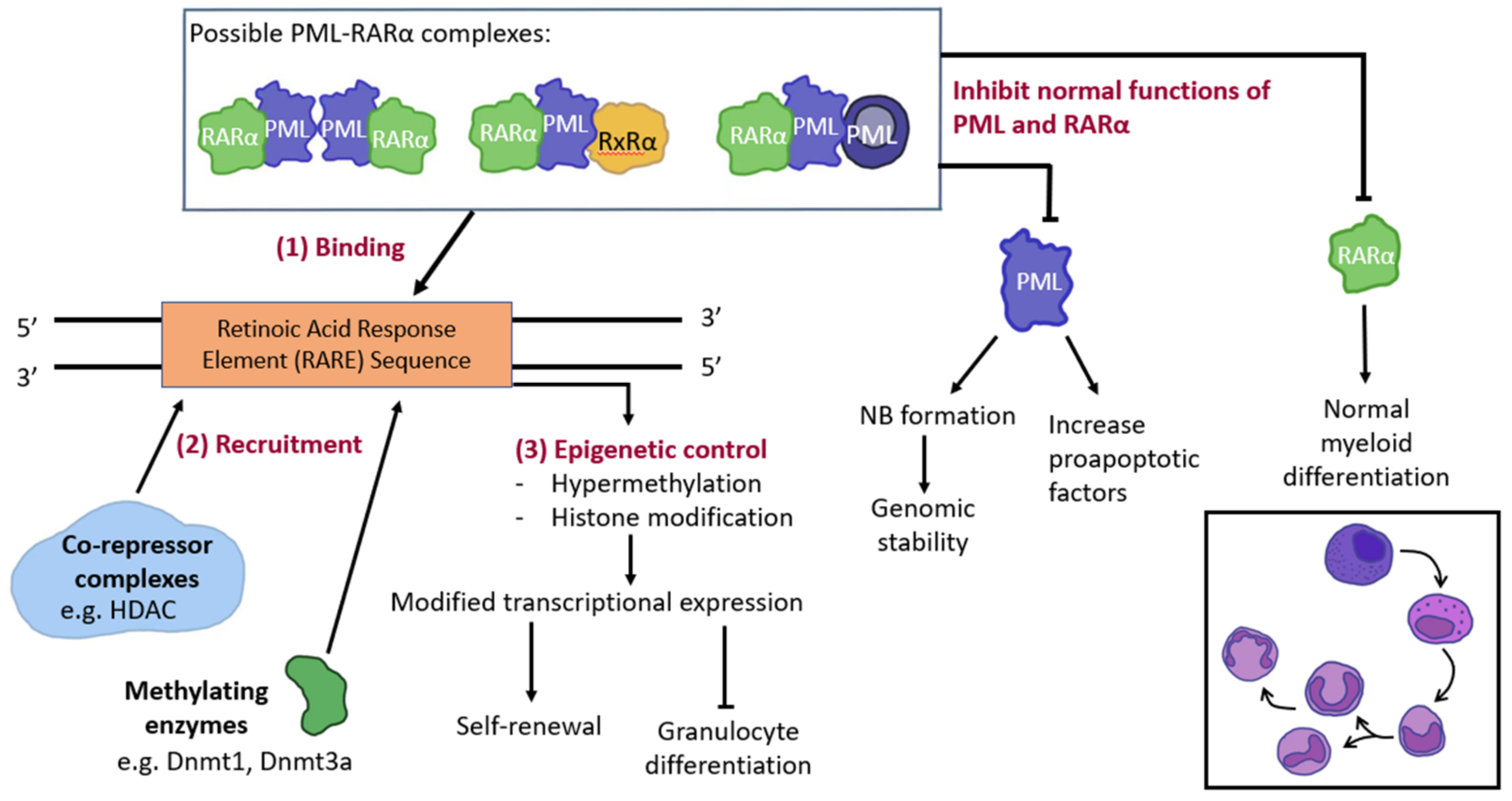
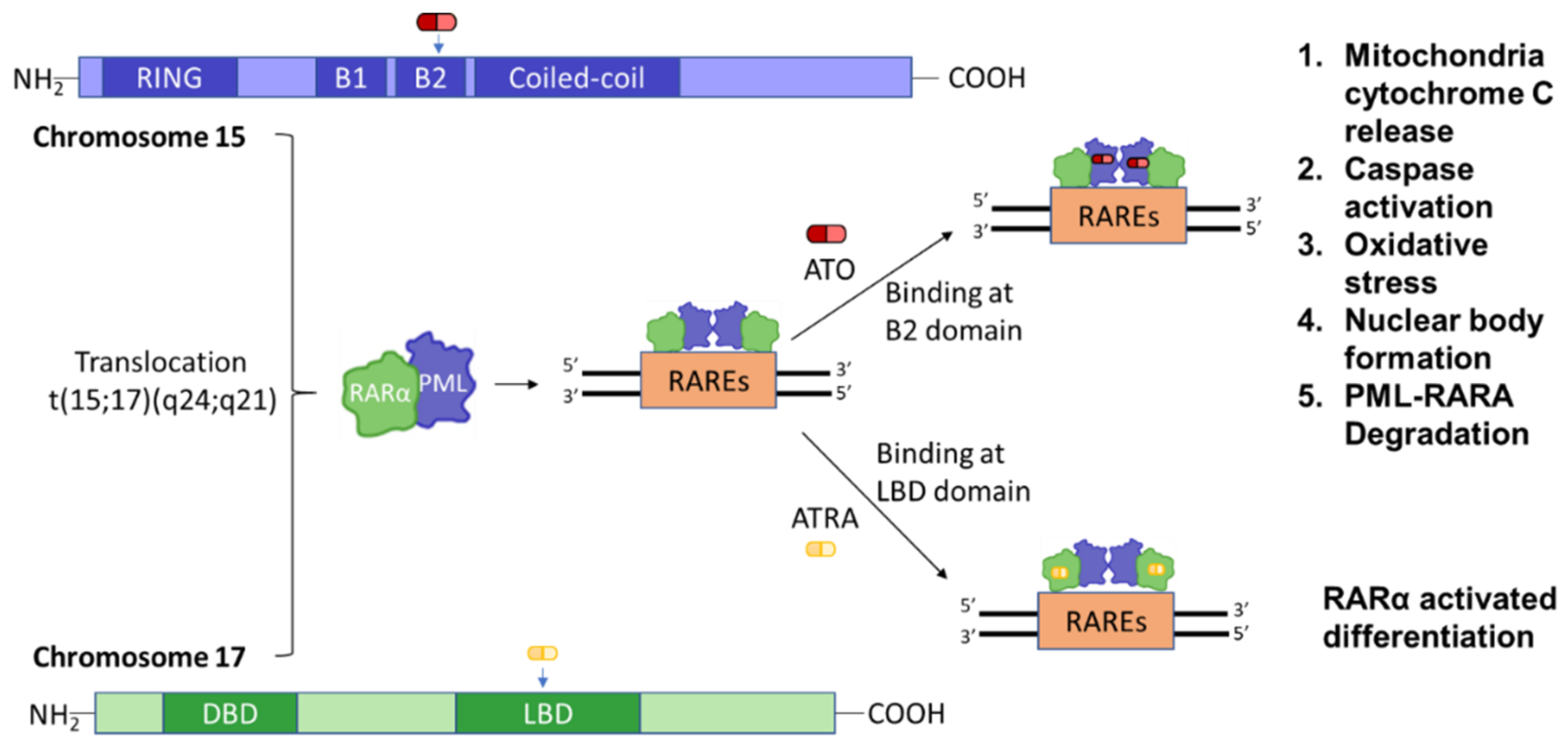
3.2. PML and Gene Modification
3.3. Oxidative Stress
3.4. Other ATO Targeted Protein Interactions
3.5. Myeloid Differentiation
4. Clinical Development of Oral Arsenic Trioxide in the Management of Acute Promyelocytic Leukaemia
5. Applications of Arsenic Trioxide beyond Acute Promyelocytic Leukaemia
5.1. NPM1-Mutated AML
5.2. Lymphoma
5.3. Lung Cancer
5.4. Autoimmune Disorders
5.5. Glioblastoma
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Au, W.Y. A biography of arsenic and medicine in Hong Kong and China. Hong Kong Med. J. 2011, 17, 507–513. [Google Scholar] [PubMed]
- Hoonjan, M.; Jadhav, V.; Bhatt, P. Arsenic trioxide: Insights into its evolution to an anticancer agent. J. Biol. Inorg. Chem. 2018, 23, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Karamanou, M. Arsenic powder in the treatment of cancer: The invention of French physician Pierre Alliot (1610-1685). J. Buon. 2019, 24, 2583–2586. [Google Scholar] [PubMed]
- Yilmaz, M.; Kantarjian, H.; Ravandi, F. Acute promyelocytic leukemia current treatment algorithms. Blood Cancer J. 2021, 11, 123. [Google Scholar] [CrossRef]
- Grimwade, D.; Lo Coco, F. Acute promyelocytic leukemia: A model for the role of molecular diagnosis and residual disease monitoring in directing treatment approach in acute myeloid leukemia. Leukemia 2002, 16, 1959–1973. [Google Scholar] [CrossRef]
- de The, H.; Pandolfi, P.P.; Chen, Z. Acute Promyelocytic Leukemia: A Paradigm for Oncoprotein-Targeted Cure. Cancer Cell 2017, 32, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Gill, H.; Au, W.Y.; Cheung, W.W.; Lee, E.Y.; Kwong, Y.L. Oral arsenic trioxide-based regimen as salvage treatment for relapsed or refractory mantle cell lymphoma. Ann. Oncol. 2014, 25, 1391–1397. [Google Scholar] [CrossRef]
- Antman, K.H. Introduction: The history of arsenic trioxide in cancer therapy. Oncologist 2001, 6, 1–2. [Google Scholar] [CrossRef]
- Kwong, Y.L.; Todd, D. Delicious poison: Arsenic trioxide for the treatment of leukemia. Blood 1997, 89, 3487–3488. [Google Scholar] [CrossRef]
- Kumana, C.; Au, W.; Lee, N.; Kou, M.; Mak, R.; Lam, C.; Kwong, Y. Systemic availability of arsenic from oral arsenic-trioxide used to treat patients with hematological malignancies. Eur. J. Clin. Pharmacol. 2002, 58, 521–526. [Google Scholar]
- Kumana, C.R.; Mak, R.; Kwong, Y.-L.; Gill, H. Resurrection of Oral Arsenic Trioxide for Treating Acute Promyelocytic Leukaemia: A Historical Account from Bedside to Bench to Bedside. Front. Oncol. 2020, 10, 1294. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Yan, H.; Yu, T.; Sun, H.-P.; Liu, J.-X.; Li, X.-S.; Wu, W.; Zhang, F.-Q.; Chen, Y.; Zhou, L.; et al. Studies on Treatment of Acute Promyelocytic Leukemia With Arsenic Trioxide: Remission Induction, Follow-Up, and Molecular Monitoring in 11 Newly Diagnosed and 47 Relapsed Acute Promyelocytic Leukemia Patients. Blood 1999, 94, 3315–3324. [Google Scholar] [CrossRef] [PubMed]
- Carmosino, I.; Latagliata, R.; Avvisati, G.; Breccia, M.; Finolezzi, E.; Lo Coco, F.; Petti, M.C. Arsenic trioxide in the treatment of advanced acute promyelocytic leukemia. Haematologica 2004, 89, 615–617. [Google Scholar]
- Wang, Z.Y.; Chen, Z. Acute promyelocytic leukemia: From highly fatal to highly curable. Blood 2008, 111, 2505–2515. [Google Scholar] [CrossRef]
- Zhang, P. On arsenic trioxide in the clinical treatment of acute promyelocytic leukemia. Leuk. Res. Rep. 2017, 7, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Douer, D.; Hu, W.; Giralt, S.; Lill, M.; DiPersio, J. Arsenic trioxide (trisenox) therapy for acute promyelocytic leukemia in the setting of hematopoietic stem cell transplantation. Oncologist 2003, 8, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Zhang, Y.; Fang, M.; Jehan, S.; Zhou, W. Current Advances of Nanomedicines Delivering Arsenic Trioxide for Enhanced Tumor Therapy. Pharmaceutics 2022, 14, 4. [Google Scholar] [CrossRef]
- Miller, W.H., Jr.; Schipper, H.M.; Lee, J.S.; Singer, J.; Waxman, S. Mechanisms of Action of Arsenic Trioxide1. Cancer Res. 2002, 62, 3893–3903. [Google Scholar]
- Hu, J.; Huang, X.; Hong, X.; Lu, Q.; Zhu, X. Arsenic trioxide inhibits the proliferation of myeloma cell line through notch signaling pathway. Cancer Cell Int. 2013, 13, 25. [Google Scholar] [CrossRef]
- Kitamura, K.; Minami, Y.; Yamamoto, K.; Akao, Y.; Kiyoi, H.; Saito, H.; Naoe, T. Involvement of CD95-independent caspase 8 activation in arsenic trioxide-induced apoptosis. Leukemia 2000, 14, 1743–1750. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; Jeanne, M.; Benhenda, S.; Nasr, R.; Lei, M.; Peres, L.; Zhou, J.; Zhu, J.; Raught, B.; de Thé, H. Arsenic degrades PML or PML-RARalpha through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat. Cell Biol 2008, 10, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Gurnari, C.; Voso, M.T.; Girardi, K.; Mastronuzzi, A.; Strocchio, L. Acute Promyelocytic Leukemia in Children: A Model of Precision Medicine and Chemotherapy-Free Therapy. Int. J. Mol. Sci. 2021, 22, 2. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Salomoni, P.; Luo, J.; Shih, A.; Zhong, S.; Gu, W.; Paolo Pandolfi, P. The function of PML in p53-dependent apoptosis. Nat. Cell Biol. 2000, 2, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Ishitsuka, K.; Hanada, S.; Uozumi, K.; Utsunomiya, A.; Arima, T. Arsenic trioxide and the growth of human T-cell leukemia virus type I infected T-cell lines. Leuk. Lymphoma 2000, 37, 649–655. [Google Scholar] [CrossRef]
- Torii, S.; Egan, D.A.; Evans, R.A.; Reed, J.C. Human Daxx regulates Fas-induced apoptosis from nuclear PML oncogenic domains (PODs). Embo J. 1999, 18, 6037–6049. [Google Scholar] [CrossRef] [PubMed]
- Weisshaar, S.R.; Keusekotten, K.; Krause, A.; Horst, C.; Springer, H.M.; Göttsche, K.; Dohmen, R.J.; Praefcke, G.J. Arsenic trioxide stimulates SUMO-2/3 modification leading to RNF4-dependent proteolytic targeting of PML. FEBS Lett. 2008, 582, 3174–3178. [Google Scholar] [CrossRef]
- Zhao, B.; Zhang, Z.; Chen, X.; Shen, Y.; Qin, Y.; Yang, X.; Xing, Z.; Zhang, S.; Long, X.; Zhang, Y.; et al. The important roles of protein SUMOylation in the occurrence and development of leukemia and clinical implications. J. Cell. Physiol. 2021, 236, 3466–3480. [Google Scholar] [CrossRef]
- Maroui, M.A.; Kheddache-Atmane, S.; El Asmi, F.; Dianoux, L.; Aubry, M.; Chelbi-Alix, M.K. Requirement of PML SUMO interacting motif for RNF4-or arsenic trioxide-induced degradation of nuclear PML isoforms. PLoS ONE 2012, 7, e44949. [Google Scholar]
- Styblo, M.; Serves, S.V.; Cullen, W.R.; Thomas, D.J. Comparative Inhibition of Yeast Glutathione Reductase by Arsenicals and Arsenothiols. Chem. Res. Toxicol. 1997, 10, 27–33. [Google Scholar] [CrossRef]
- Chen, Y.C.; Lin-Shiau, S.Y.; Lin, J.K. Involvement of reactive oxygen species and caspase 3 activation in arsenite-induced apoptosis. J. Cell Physiol. 1998, 177, 324–333. [Google Scholar] [CrossRef]
- Bernstam, L.; Nriagu, J. Molecular aspects of arsenic stress. J. Toxicol. Environ. Health B Crit. Rev. 2000, 3, 293–322. [Google Scholar] [PubMed]
- Nicolussi, A.; D’Inzeo, S.; Capalbo, C.; Giannini, G.; Coppa, A. The role of peroxiredoxins in cancer (Review). Mol. Clin. Oncol. 2017, 6, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Mun, Y.-C.; Ahn, J.Y.; Yoo, E.S.; Lee, K.E.; Nam, E.M.; Huh, J.; Woo, H.A.; Rhee, S.G.; Seong, C.M. Peroxiredoxin 3 Has Important Roles on Arsenic Trioxide Induced Apoptosis in Human Acute Promyelocytic Leukemia Cell Line via Hyperoxidation of Mitochondrial Specific Reactive Oxygen Species. Mol. Cells 2020, 43, 813–820. [Google Scholar] [PubMed]
- Li, X.; He, P.; Wang, X.-L.; Zhang, S.; Devejian, N.; Bennett, E.; Cai, C. Sulfiredoxin-1 enhances cardiac progenitor cell survival against oxidative stress via the upregulation of the ERK/NRF2 signal pathway. Free Radic. Biol. Med. 2018, 123, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zheng, B.; Liu, P.; Zhang, J.; Chu, X.; Dong, C.; Shi, J.; Liang, Y.; Chu, L.; Liu, Y.; et al. Exploration of the hepatoprotective effect and mechanism of magnesium isoglycyrrhizinate in mice with arsenic trioxide-induced acute liver injury. Mol. Med. Rep. 2021, 23, 6. [Google Scholar] [CrossRef]
- Maimaitiyiming, Y.; Zhu, H.-H.; Yang, C.; Naranmandura, H. Biotransformation of arsenic trioxide by AS3MT favors eradication of acute promyelocytic leukemia: Revealing the hidden facts. Drug Metab. Rev. 2020, 52, 425–437. [Google Scholar] [CrossRef]
- Kozbial, P.Z.; Mushegian, A.R. Natural history of S-adenosylmethionine-binding proteins. BMC Struct. Biol. 2005, 5, 19. [Google Scholar] [CrossRef]
- Wang, Q.Q.; Zhou, X.Y.; Zhang, Y.F.; Bu, N.; Zhou, J.; Cao, F.L.; Naranmandura, H. Methylated arsenic metabolites bind to PML protein but do not induce cellular differentiation and PML-RARα protein degradation. Oncotarget 2015, 6, 25646. [Google Scholar] [CrossRef]
- Ajees, A.A.; Marapakala, K.; Packianathan, C.; Sankaran, B.; Rosen, B.P. Structure of an As (III) S-adenosylmethionine methyltransferase: Insights into the mechanism of arsenic biotransformation. Biochemistry 2012, 51, 5476–5485. [Google Scholar] [CrossRef]
- Naranmandura, H.; Xu, S.; Koike, S.; Pan, L.Q.; Chen, B.; Wang, Y.W.; Rehman, K.; Wu, B.; Chen, Z.; Suzuki, N. The endoplasmic reticulum is a target organelle for trivalent dimethylarsinic acid (DMAIII)-induced cytotoxicity. Toxicol. Appl. Pharmacol. 2012, 260, 241–249. [Google Scholar] [CrossRef]
- Dong, H.Y.; Wilkes, S.; Yang, H. CD71 is selectively and ubiquitously expressed at high levels in erythroid precursors of all maturation stages: A comparative immunochemical study with glycophorin A and hemoglobin A. Am. J. Surg. Pathol. 2011, 35, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, W.; He, Y.; Gao, Z.; Liu, L.; Yu, S.; Hu, Y.; Wang, S.; Zhao, C.; Li, H.; et al. Ferritin-based targeted delivery of arsenic to diverse leukaemia types confers strong anti-leukaemia therapeutic effects. Nat. Nanotechnol. 2021, 16, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Hemmati, A.A.; Olapour, S.; Varzi, H.N.; Khodayar, M.J.; Dianat, M.; Mohammadian, B.; Yaghooti, H. Ellagic acid protects against arsenic trioxide–induced cardiotoxicity in rat. Hum. Exp. Toxicol. 2018, 37, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Yedjou, C.G.; Tchounwou, P.B. Arsenic trioxide induces oxidative stress, DNA damage, and mitochondrial pathway of apoptosis in human leukemia (HL-60) cells. J. Exp. Clin. Cancer Res. 2014, 33, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Zhang, H.; Smeester, L.; Zou, F.; Kesic, M.; Jaspers, I.; Pi, J.; Fry, R.C. The NRF2-mediated oxidative stress response pathway is associated with tumor cell resistance to arsenic trioxide across the NCI-60 panel. BMC Med. Genom. 2010, 3, 37. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef]
- Bertero, E.; Maack, C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef]
- dos Santos, G.A.S.; Abreu e Lima, R.S.; Pestana, C.R.; Lima, A.S.G.; Scheucher, P.S.; Thomé, C.H.; Gimenes-Teixeira, H.L.; Santana-Lemos, B.A.A.; Lucena-Araujo, A.R.; Rodrigues, F.P.; et al. (+)α-Tocopheryl succinate inhibits the mitochondrial respiratory chain complex I and is as effective as arsenic trioxide or ATRA against acute promyelocytic leukemia in vivo. Leukemia 2012, 26, 451–460. [Google Scholar] [CrossRef]
- Angulo-Molina, A.; Reyes-Leyva, J.; López-Malo, A.; Hernández, J. The role of alpha tocopheryl succinate (α-TOS) as a potential anticancer agent. Nutr. Cancer 2014, 66, 167–176. [Google Scholar] [CrossRef]
- Cavigelli, M.; Li, W.W.; Lin, A.; Su, B.; Yoshioka, K.; Karin, M. The tumor promoter arsenite stimulates AP-1 activity by inhibiting a JNK phosphatase. EMBO J. 1996, 15, 6269–6279. [Google Scholar] [CrossRef]
- Applegate, L.A.; Luscher, P.; Tyrrell, R.M. Induction of heme oxygenase: A general response to oxidant stress in cultured mammalian cells. Cancer Res. 1991, 51, 974–978. [Google Scholar] [PubMed]
- Hu, X.Q.; Li, H.Y.; Ip, T.K.Y.; Cheung, Y.F.; Koohi-Moghadam, M.; Wang, H.B.; Yang, X.M.; Tritton, D.N.; Wang, Y.C.; Wang, Y.; et al. Arsenic trioxide targets Hsp60, triggering degradation of p53 and survivin. Chem. Sci. 2021, 12, 10893–10900. [Google Scholar] [CrossRef] [PubMed]
- Warrier, N.M.; Agarwal, P.; Kumar, P. Emerging Importance of Survivin in Stem Cells and Cancer: The Development of New Cancer Therapeutics. Stem Cell Rev. Rep. 2020, 16, 828–852. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhou, Y.; Fan, S.; Wen, Q. The multiple roles and therapeutic potential of HSP60 in cancer. Biochem. Pharmacol. 2022, 201, 115096. [Google Scholar] [CrossRef]
- Kozono, S.; Lin, Y.-M.; Seo, H.-S.; Pinch, B.; Lian, X.; Qiu, C.; Herbert, M.K.; Chen, C.-H.; Tan, L.; Gao, Z.J.; et al. Arsenic targets Pin1 and cooperates with retinoic acid to inhibit cancer-driving pathways and tumor-initiating cells. Nat. Commun. 2018, 9, 3069. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, Y.-r.; Yang, H.-y.; Li, X.-z.; Jie, M.-m.; Hu, C.-j.; Wu, Y.-y.; Yang, S.-m.; Yang, Y.-b. Prolyl isomerase Pin1: A promoter of cancer and a target for therapy. Cell Death Dis. 2018, 9, 883. [Google Scholar] [CrossRef]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.C.; Peaceman, D.; Ozdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Invest. 2011, 121, 148–160. [Google Scholar] [CrossRef]
- Abele, M.; Müller, S.L.; Schleicher, S.; Hartmann, U.; Döring, M.; Queudeville, M.; Lang, P.; Handgretinger, R.; Ebinger, M. Arsenic trioxide in pediatric cancer—A case series and review of literature. Pediatr. Hematol. Oncol. 2021, 38, 471–485. [Google Scholar] [CrossRef]
- Lainez-González, D.; Serrano-López, J.; Alonso-Domínguez, J.M. Understanding the Hedgehog Signaling Pathway in Acute Myeloid Leukemia Stem Cells: A Necessary Step toward a Cure. Biology 2021, 10, 4. [Google Scholar] [CrossRef]
- Briscoe, J.; Thérond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef]
- Zhang, H.-n.; Yang, L.; Ling, J.-y.; Czajkowsky, D.M.; Wang, J.-F.; Zhang, X.-W.; Zhou, Y.-M.; Ge, F.; Yang, M.-k.; Xiong, Q.; et al. Systematic identification of arsenic-binding proteins reveals that hexokinase-2 is inhibited by arsenic. Proc. Natl. Acad. Sci. USA 2015, 112, 15084–15089. [Google Scholar] [CrossRef] [PubMed]
- Ciscato, F.; Filadi, R.; Masgras, I.; Pizzi, M.; Marin, O.; Damiano, N.; Pizzo, P.; Gori, A.; Frezzato, F.; Chiara, F.; et al. Hexokinase 2 displacement from mitochondria-associated membranes prompts Ca2+-dependent death of cancer cells. EMBO Rep. 2020, 21, e49117. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Zhang, J.; Zhang, J.; Shi, H.; Zhang, Y.; Ji, R.; Mao, F.; Qian, H.; Xu, W.; Zhang, X. SALL4 promotes gastric cancer progression via hexokinase II mediated glycolysis. Cancer Cell Int. 2020, 20, 188. [Google Scholar] [CrossRef] [PubMed]
- Rego, E.M.; De Santis, G.C. Differentiation syndrome in promyelocytic leukemia: Clinical presentation, pathogenesis and treatment. Mediterr. J. Hematol. Infect. Dis. 2011, 3, e2011048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, T.T.; Sultan, M.; Vidovic, D.; Dean, C.A.; Cruickshank, B.M.; Lee, K.; Loung, C.Y.; Holloway, R.W.; Hoskin, D.W.; Waisman, D.M.; et al. Retinoic acid and arsenic trioxide induce lasting differentiation and demethylation of target genes in APL cells. Sci. Rep. 2019, 9, 9414. [Google Scholar] [CrossRef]
- Wang, Z.G.; Ruggero, D.; Ronchetti, S.; Zhong, S.; Gaboli, M.; Rivi, R.; Pandolfi, P.P. PML is essential for multiple apoptotic pathways. Nat. Genet. 1998, 20, 266–272. [Google Scholar] [CrossRef]
- Yih, L.-H.; Wu, Y.-C.; Hsu, N.-C.; Kuo, H.-H. Arsenic Trioxide Induces Abnormal Mitotic Spindles Through a PIP4KIIγ/Rho Pathway. Toxicol. Sci. 2012, 128, 115–125. [Google Scholar] [CrossRef]
- Carré, M.; Carles, G.; André, N.; Douillard, S.; Ciccolini, J.; Briand, C.; Braguer, D. Involvement of microtubules and mitochondria in the antagonism of arsenic trioxide on paclitaxel-induced apoptosis. Biochem. Pharmacol. 2002, 63, 1831–1842. [Google Scholar] [CrossRef]
- Ferreira, L.T.; Figueiredo, A.C.; Orr, B.; Lopes, D.; Maiato, H. Chapter 3—Dissecting the role of the tubulin code in mitosis. In Methods in Cell Biology; Maiato, H., Schuh, M., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 144, pp. 33–74. [Google Scholar]
- Taylor, B.F.; McNeely, S.C.; Miller, H.L.; States, J.C. Arsenite-induced mitotic death involves stress response and is independent of tubulin polymerization. Toxicol. Appl. Pharmacol. 2008, 230, 235–246. [Google Scholar] [CrossRef]
- Hollenbeck, P. Cytoskeleton: Microtubules get the signal. Curr. Biol. 2001, 11, R820–R823. [Google Scholar] [CrossRef]
- Jung, Y.S.; Cheong, H.-J.; Kim, S.-J.; Kim, K.H.; Lee, N.; Park, H.S.; Won, J.-H. Src family kinase inhibitor PP2 enhances differentiation of acute promyelocytic leukemia cell line induced by combination of all-trans-retinoic acid and arsenic trioxide. Leuk. Res. 2014, 38, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Wang, L.J.; Wu, J.; Shi, Y.X.; Li, G.P.; Yang, X.Q. Src kinase inhibitor PP2 regulates the biological characteristics of A549 cells via the PI3K/Akt signaling pathway. Oncol. Lett. 2018, 16, 5059–5065. [Google Scholar] [CrossRef] [PubMed]
- Mandal, K. Review of PIP2 in Cellular Signaling, Functions and Diseases. Int. J. Mol. Sci. 2020, 21, 21. [Google Scholar] [CrossRef]
- Tu, C.L.; Chang, W.; Bikle, D.D. Phospholipase cgamma1 is required for activation of store-operated channels in human keratinocytes. J. Invest. Dermatol. 2005, 124, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neaga, A.; Bagacean, C.; Tempescul, A.; Jimbu, L.; Mesaros, O.; Blag, C.; Tomuleasa, C.; Bocsan, C.; Gaman, M.; Zdrenghea, M. MicroRNAs associated with a good prognosis of acute myeloid leukemia and their effect on macrophage polarization. Front. Immunol. 2021, 11, 582915. [Google Scholar] [CrossRef]
- O’connell, R.M.; Rao, D.S.; Chaudhuri, A.A.; Baltimore, D. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol. 2010, 10, 111–122. [Google Scholar] [CrossRef]
- Liang, H.; Li, X.; Wang, L.; Yu, S.; Xu, Z.; Gu, Y.; Pan, Z.; Li, T.; Hu, M.; Cui, H. MicroRNAs contribute to promyelocyte apoptosis in As2O3-treated APL cells. Cell. Physiol. Biochem. 2013, 32, 1818–1829. [Google Scholar] [CrossRef]
- Fu, Y.; Li, L.; Hou, J.; Li, H.; Lv, C.; Yu, H.; Zhang, X.; Xu, M.; Zhang, M.; Meng, H. miR-139-5p Regulates the Proliferation of Acute Promyelocytic Leukemia Cells by Targeting MNT. J. Oncol. 2021, 2021, 5522051. [Google Scholar] [CrossRef]
- Popov, N.; Wahlström, T.; Hurlin, P.J.; Henriksson, M. Mnt transcriptional repressor is functionally regulated during cell cycle progression. Oncogene 2005, 24, 8326–8337. [Google Scholar] [CrossRef]
- Siu, C.W.; Au, W.Y.; Yung, C.; Kumana, C.R.; Lau, C.P.; Kwong, Y.L.; Tse, H.F. Effects of oral arsenic trioxide therapy on QT intervals in patients with acute promyelocytic leukemia: Implications for long-term cardiac safety. Blood 2006, 108, 103–106. [Google Scholar] [CrossRef]
- Hai, J.J.; Gill, H.; Tse, H.F.; Kumana, C.R.; Kwong, Y.L.; Siu, C.W. Torsade de Pointes during oral arsenic trioxide therapy for acute promyelocytic leukemia in a patient with heart failure. Ann. Hematol. 2015, 94, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Au, W.Y.; Kwong, Y.L. Arsenic trioxide: Safety issues and their management. Acta Pharmacol. Sin. 2008, 29, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Au, W.Y.; Cheung, G.T.; Yuen, T.W.; Kumana, C.R.; Kwong, Y.L. Successful treatment of relapsed acute promyelocytic leukemia in a patient receiving continuous ambulatory peritoneal dialysis with oral arsenic trioxide. Arch. Intern. Med. 2005, 165, 1067–1068. [Google Scholar] [CrossRef]
- Au, W.Y.; Fong, B.M.; Tam, S.; Kwong, Y.L. Feasibility of oral arsenic trioxide treatment for acute promyelocytic leukemia during hemodialysis. Ann. Hematol. 2013, 92, 417–418. [Google Scholar] [CrossRef] [Green Version]
- Au, W.Y.; Tam, S.; Fong, B.M.; Kwong, Y.L. Determinants of cerebrospinal fluid arsenic concentration in patients with acute promyelocytic leukemia on oral arsenic trioxide therapy. Blood 2008, 112, 3587–3590. [Google Scholar] [CrossRef] [PubMed]
- Au, W.Y.; Tam, S.; Kwong, Y.L. Entry of elemental arsenic into the central nervous system in patients with acute promyelocytic leukemia during arsenic trioxide treatment. Leuk. Res. 2008, 32, 357–358. [Google Scholar] [CrossRef]
- Gill, H.; Yim, R.; Lee, H.K.K.; Mak, V.; Lin, S.Y.; Kho, B.; Yip, S.F.; Lau, J.S.M.; Li, W.; Ip, H.W.; et al. Long-term outcome of relapsed acute promyelocytic leukemia treated with oral arsenic trioxide-based reinduction and maintenance regimens: A 15-year prospective study. Cancer 2018, 124, 2316–2326. [Google Scholar] [CrossRef]
- Gill, H.; Kumana, C.R.; Yim, R.; Hwang, Y.Y.; Chan, T.S.Y.; Yip, S.F.; Lee, H.K.K.; Mak, V.; Lau, J.S.M.; Chan, C.C.; et al. Oral arsenic trioxide incorporation into frontline treatment with all-trans retinoic acid and chemotherapy in newly diagnosed acute promyelocytic leukemia: A 5-year prospective study. Cancer 2019, 125, 17. [Google Scholar] [CrossRef]
- Au, W.Y.; Kumana, C.R.; Lee, H.K.; Lin, S.Y.; Liu, H.; Yeung, D.Y.; Lau, J.S.; Kwong, Y.L. Oral arsenic trioxide-based maintenance regimens for first complete remission of acute promyelocytic leukemia: A 10-year follow-up study. Blood 2011, 118, 6535–6543. [Google Scholar] [CrossRef]
- Dai, J.; Weinberg, R.S.; Waxman, S.; Jing, Y. Malignant cells can be sensitized to undergo growth inhibition and apoptosis by arsenic trioxide through modulation of the glutathione redox system. Blood 1999, 93, 268–277. [Google Scholar] [CrossRef]
- Grad, J.M.; Bahlis, N.J.; Reis, I.; Oshiro, M.M.; Dalton, W.S.; Boise, L.H. Ascorbic acid enhances arsenic trioxide-induced cytotoxicity in multiple myeloma cells. Blood 2001, 98, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Au, W.Y.; Kwong, Y.L. Frequent varicella zoster reactivation associated with therapeutic use of arsenic trioxide: Portents of an old scourge. J. Am. Acad. Dermatol. 2005, 53, 890–892. [Google Scholar] [CrossRef] [PubMed]
- Lo-Coco, F.; Avvisati, G.; Vignetti, M.; Thiede, C.; Orlando, S.M.; Iacobelli, S.; Ferrara, F.; Fazi, P.; Cicconi, L.; Di Bona, E.; et al. Retinoic Acid and Arsenic Trioxide for Acute Promyelocytic Leukemia. N. Engl. J. Med. 2013, 369, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Platzbecker, U.; Avvisati, G.; Cicconi, L.; Thiede, C.; Paoloni, F.; Vignetti, M.; Ferrara, F.; Divona, M.; Albano, F.; Efficace, F.; et al. Improved Outcomes with Retinoic Acid and Arsenic Trioxide Compared with Retinoic Acid and Chemotherapy in Non–High-Risk Acute Promyelocytic Leukemia: Final Results of the Randomized Italian-German APL0406 Trial. J. Clin. Oncol. 2017, 35, 605–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravandi, F.; Koumenis, I.; Johri, A.; Tallman, M.; Roboz, G.J.; Strickland, S.; Garcia-Manero, G.; Borthakur, G.; Naqvi, K.; Meyer, M.; et al. Oral arsenic trioxide ORH-2014 pharmacokinetic and safety profile in patients with advanced hematologic disorders. Haematologica 2020, 105, 1567–1574. [Google Scholar] [CrossRef] [PubMed]
- Grant, S. ATRA and ATO team up against NPM1. Blood 2015, 125, 3369–3371. [Google Scholar] [CrossRef]
- El Hajj, H.; Dassouki, Z.; Berthier, C.; Raffoux, E.; Ades, L.; Legrand, O.; Hleihel, R.; Sahin, U.; Tawil, N.; Salameh, A.; et al. Retinoic acid and arsenic trioxide trigger degradation of mutated NPM1, resulting in apoptosis of AML cells. Blood 2015, 125, 3447–3454. [Google Scholar] [CrossRef]
- Martelli, M.P.; Gionfriddo, I.; Mezzasoma, F.; Milano, F.; Pierangeli, S.; Mulas, F.; Pacini, R.; Tabarrini, A.; Pettirossi, V.; Rossi, R.; et al. Arsenic trioxide and all-trans retinoic acid target NPM1 mutant oncoprotein levels and induce apoptosis in NPM1-mutated AML cells. Blood 2015, 125, 3455–3465. [Google Scholar] [CrossRef]
- Cho, H.; Jang, J.E.; Eom, J.-I.; Jeung, H.-K.; Chung, H.; Kim, J.S.; Cheong, J.-W.; Min, Y.H. Arsenic trioxide synergistically promotes the antileukaemic activity of venetoclax by downregulating Mcl-1 in acute myeloid leukaemia cells. Exp. Hematol. Oncol. 2021, 10, 28. [Google Scholar] [CrossRef]
- Hååg, P.; Olsson, M.; Forsberg, J.; Lindberg, M.L.; Stenerlöw, B.; Zong, D.; Kanter, L.; Lewensohn, R.; Viktorsson, K.; Zhivotovsky, B.; et al. Caspase-2 is a mediator of apoptotic signaling in response to gemtuzumab ozogamicin in acute myeloid leukemia. Cell Death Discov. 2022, 8, 284. [Google Scholar] [CrossRef]
- Mesbahi, Y.; Zekri, A.; Ghaffari, S.H.; Tabatabaie, P.S.; Ahmadian, S.; Ghavamzadeh, A. Blockade of JAK2/STAT3 intensifies the anti-tumor activity of arsenic trioxide in acute myeloid leukemia cells: Novel synergistic mechanism via the mediation of reactive oxygen species. Eur. J. Pharmacol. 2018, 834, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C.-J.; Voorhees, P.; Kolesar, J.; Ahuja, H.G.; Sánchez, F.C.; Rodríguez, G.; Kim, K.S.; Werndli, J.E.; Bailey, H.; Kahl, B. Phase II study of arsenic trioxide and ascorbic acid for relapsed or refractory lymphoid malignancies: A Wisconsin Oncology Network study. Hematol. Oncol. 2009, 2009, 27. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Chen, Z.; McCarty, N. Synergistic anticancer effects of arsenic trioxide with bortezomib in mantle cell lymphoma. Am. J. Hematol. 2012, 87, 1057–1064. [Google Scholar] [CrossRef]
- Wang, M.L.; Rule, S.; Martin, P.; Goy, A.; Auer, R.; Kahl, B.S.; Jurczak, W.; Advani, R.H.; Romaguera, J.E.; Williams, M.E.; et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2013, 369, 507–516. [Google Scholar] [CrossRef] [Green Version]
- Moodad, S.; El Hajj, R.; Hleihel, R.; Hajjar, L.; Tawil, N.; Karam, M.; Hamie, M.; Abou Merhi, R.; El Sabban, M.; El Hajj, H. Lenalidomide in Combination with Arsenic Trioxide: An Effective Therapy for Primary Effusion Lymphoma. Cancers 2020, 12, 2483. [Google Scholar] [CrossRef]
- Yan, L.; Majerciak, V.; Zheng, Z.-M.; Lan, K. Towards better understanding of KSHV life cycle: From transcription and posttranscriptional regulations to pathogenesis. Virol. Sin. 2019, 34, 135–161. [Google Scholar] [CrossRef] [PubMed]
- DeCotiis, J.L.; Lukac, D.M. KSHV and the Role of Notch Receptor Dysregulation in Disease Progression. Pathogens 2017, 6, 34. [Google Scholar] [CrossRef]
- Xia, L.; Tan, S.; Zhou, Y.; Lin, J.; Wang, H.; Oyang, L.; Tian, Y.; Liu, L.; Su, M.; Wang, H.; et al. Role of the NFκB-signaling pathway in cancer. Onco Targets Ther. 2018, 11, 2063–2073. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, J.; Lou, B.; Wu, R.; Wang, G.; Lu, C.; Wang, H.; Pi, J.; Xu, Y. The role of reactive oxygen species in arsenic toxicity. Biomolecules 2020, 10, 240. [Google Scholar] [CrossRef]
- Huang, W.; Zeng, Y.C. A candidate for lung cancer treatment: Arsenic trioxide. Clin. Transl. Oncol. 2019, 21, 1115–1126. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Zhao, S.; Li, Q.; Li, Y.; Kawamoto, K. Arsenic trioxide-mediated Notch pathway inhibition depletes the cancer stem-like cell population in gliomas. Cancer Lett. 2010, 292, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.J.; Yang, M.H.; Zheng, J.C.; Li, B.; Nie, W. Arsenic trioxide inhibits cancer stem-like cells via down-regulation of Gli1 in lung cancer. Am. J. Transl. Res. 2016, 8, 1133–1143. [Google Scholar] [PubMed]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.Y.; Lam, S.K.; Li, Y.Y.; Ho, J.C. Arsenic trioxide-induced cytotoxicity in small cell lung cancer via altered redox homeostasis and mitochondrial integrity. Int. J. Oncol. 2015, 46, 1067–1078. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Peng, X.; Yang, D.; Guo, M.; Xu, X.; Yin, F.; Wang, Y.; Huang, J.; Zhan, L.; Qi, Z. Bcl-2 hijacks the arsenic trioxide resistance in SH-SY5Y cells. J. Cell Mol. Med. 2022, 26, 563–569. [Google Scholar] [CrossRef]
- Haybar, H.; Shahrabi, S.; Rezaeeyan, H.; Jodat, H.; Saki, N. Strategies to inhibit arsenic trioxide-induced cardiotoxicity in acute promyelocytic leukemia. J. Cell Physiol. 2019, in press. [Google Scholar] [CrossRef]
- Meier, J.A.; Larner, A.C. Toward a new STATe: The role of STATs in mitochondrial function. Semin. Immunol. 2014, 26, 20–28. [Google Scholar] [CrossRef]
- Leung, L.L.; Lam, S.K.; Li, Y.Y.; Ho, J.C.M. Tumour growth-suppressive effect of arsenic trioxide in squamous cell lung carcinoma. Oncol. Lett. 2017, 14, 3748–3754. [Google Scholar] [CrossRef]
- Lam, S.K.; Li, Y.Y.; Zheng, C.Y.; Leung, L.L.; Ho, J.C. E2F1 downregulation by arsenic trioxide in lung adenocarcinoma. Int. J. Oncol. 2014, 45, 2033–2043. [Google Scholar] [CrossRef]
- Bobé, P.; Bonardelle, D.; Benihoud, K.; Opolon, P.; Chelbi-Alix, M.K. Arsenic trioxide: A promising novel therapeutic agent for lymphoproliferative and autoimmune syndromes in MRL/lpr mice. Blood 2006, 108, 3967–3975. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, J.; Chen, Q.; Zhou, L.; Li, M.; Qiu, H.; Sun, A.; Wu, D. Efficacy of venetoclax in combination with azacitidine followed by haploidentical transplantation in refractory acute myeloid leukaemia and mixed phenotype acute leukaemia. Br. J. Haematol. 2020, 189, e200–e204. [Google Scholar] [CrossRef] [PubMed]
- Waxman, S.; Anderson, K.C. History of the Development of Arsenic Derivatives in Cancer Therapy. Oncologist 2001, 6, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Cajas, L.J.; Casallas, A.; Medina, Y.F.; Quintana, G.; Rondón, F. Pannus y artritis reumatoide: Evolución histórica y fisiopatológica. Rev. Colomb. Reumatol. 2019, 26, 118–128. [Google Scholar] [CrossRef]
- Luo, F.; Zhuang, Y.; Sides, M.D.; Sanchez, C.G.; Shan, B.; White, E.S.; Lasky, J.A. Arsenic trioxide inhibits transforming growth factor-β1-induced fibroblast to myofibroblast differentiation in vitro and bleomycin induced lung fibrosis in vivo. Respir. Res. 2014, 15, 51. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Xu, M.; Zhang, X.; Niu, Q.; Hu, Y.; Li, Y.; Li, S. Bi-directional regulation of TGF-β/Smad pathway by arsenic: A systemic review and meta-analysis of in vivo and in vitro studies. Life Sci. 2019, 220, 92–105. [Google Scholar] [CrossRef]
- Li, C.; Zhang, J.; Wang, W.; Wang, H.; Zhang, Y.; Zhang, Z. Data on arsenic trioxide modulates Treg/Th17/Th1/Th2 cells in treatment-naïve rheumatoid arthritis patients and collagen-induced arthritis model mice. Data Brief. 2019, 27, 104615. [Google Scholar] [CrossRef]
- Guerrero, P.A.; McCarty, J.H. TGF-β Activation and Signaling in Angiogenesis. In Physiologic and Pathologic Angiogenesis; IntechOpen: London, UK, 2017. [Google Scholar]
- Fang, Y.; Zhang, Z. Arsenic trioxide as a novel anti-glioma drug: A review. Cell Mol. Biol. Lett. 2020, 25, 44. [Google Scholar] [CrossRef]
- Amodeo, V.; Deli, A.; Betts, J.; Bartesaghi, S.; Zhang, Y.; Richard-Londt, A.; Ellis, M.; Roshani, R.; Vouri, M.; Galavotti, S.; et al. A PML/Slit Axis Controls Physiological Cell Migration and Cancer Invasion in the CNS. Cell Rep. 2017, 20, 411–426. [Google Scholar] [CrossRef]
- Weinhäuser, I.; Pereira-Martins, D.A.; Ortiz, C.; Silveira, D.R.; Simões, L.A.A.; Bianco, T.M.; Araujo, C.L.; Koury, L.C.; Melo, R.A.M.; Bittencourt, R.I.; et al. Reduced SLIT2 is Associated with Increased Cell Proliferation and Arsenic Trioxide Resistance in Acute Promyelocytic Leukemia. Cancers 2020, 12, 11. [Google Scholar] [CrossRef]
- Lin, L.T.; Liu, S.Y.; Leu, J.D.; Chang, C.Y.; Chiou, S.H.; Lee, T.C.; Lee, Y.J. Arsenic trioxide-mediated suppression of miR-182-5p is associated with potent anti-oxidant effects through up-regulation of SESN2. Oncotarget 2018, 9, 16028–16042. [Google Scholar] [CrossRef]
- Pasha, M.; Eid, A.H.; Eid, A.A.; Gorin, Y.; Munusamy, S. Sestrin2 as a Novel Biomarker and Therapeutic Target for Various Diseases. Oxidative Med. Cell. Longev. 2017, 2017, 3296294. [Google Scholar] [CrossRef] [PubMed]
- Macip, S.; Igarashi, M.; Fang, L.; Chen, A.; Pan, Z.Q.; Lee, S.W.; Aaronson, S.A. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. Embo J. 2002, 21, 2180–2188. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Manzano, C.; Fueyo, J.; Kyritsis, A.P.; McDonnell, T.J.; Steck, P.A.; Levin, V.A.; Yung, W.K. Characterization of p53 and p21 functional interactions in glioma cells en route to apoptosis. J. Natl. Cancer Inst. 1997, 89, 1036–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chin, L.; Kumana, C.R.; Kwong, Y.-L.; Gill, H. The Development and Clinical Applications of Oral Arsenic Trioxide for Acute Promyelocytic Leukaemia and Other Diseases. Pharmaceutics 2022, 14, 1945. https://doi.org/10.3390/pharmaceutics14091945
Chin L, Kumana CR, Kwong Y-L, Gill H. The Development and Clinical Applications of Oral Arsenic Trioxide for Acute Promyelocytic Leukaemia and Other Diseases. Pharmaceutics. 2022; 14(9):1945. https://doi.org/10.3390/pharmaceutics14091945
Chicago/Turabian StyleChin, Lynn, Cyrus R. Kumana, Yok-Lam Kwong, and Harinder Gill. 2022. "The Development and Clinical Applications of Oral Arsenic Trioxide for Acute Promyelocytic Leukaemia and Other Diseases" Pharmaceutics 14, no. 9: 1945. https://doi.org/10.3390/pharmaceutics14091945
APA StyleChin, L., Kumana, C. R., Kwong, Y. -L., & Gill, H. (2022). The Development and Clinical Applications of Oral Arsenic Trioxide for Acute Promyelocytic Leukaemia and Other Diseases. Pharmaceutics, 14(9), 1945. https://doi.org/10.3390/pharmaceutics14091945