
Acyldepsipeptide Analogues: A Future Generation Antibiotics for Tuberculosis Treatment

 and
and 
Abstract
:1. Introduction
2. Tuberculosis
2.1. M. tb Epidemiology and Pathophysiology
2.2. Treatment of TB

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TB Dugs | |||
|---|---|---|---|
| Class | Drugs | Mode of Action | References |
| Thioamides | Ethionamide Prothionamide | Inhibit cell wall synthesis | [43,44] |
| Nitroimidazoles | Delamanid | Inhibit mycolic acid synthesis | [45] |
| Ethambutol | Inhibits cell wall synthesis | [46] | |
| Cycloserine | Inhibits cell wall synthesis | [47,48] | |
| Pyrazinamide | Exact target is unclear: Disrupts plasma membrane Disrupts energy metabolism | [49,50] | |
| Diarylquinoline | Bedaquiline | Inhibits ATP synthesis | [51] |
| Aminoglycosides | Kanamycin Streptomycin | Inhibits protein synthesis | [52,53] |
| Cyclic peptides | Capreomycin | Inhibits protein synthesis | [54] |
| Fluoroquinolones | Moxifloxacin Levofloxacin | Inhibits DNA gyrase | [55,56] |
| Para-aminosalicyclic acid | Inhibit folate metabolism | [57,58] | |
| Recent TB Drugs | |||
| Nitroimidazoles | PA-24 | Inhibits mycolic acid | [59] |
| SQ-109 | Inhibits cell wall synthesis | [60] | |
| Meropenem | Inhibits peptidoglycan synthesis | [61] | |
| Benzothiazinones | PBTZ169 PBTZ043 | Inhibits cells wall synthesis | [62,63,64] |
| Imidazopyridine amide | Inhibits cytochrome oxidase | [65,66] | |
| Macrolides | Inhibits protein synthesis | [67,68] | |
| Oxazolidinones | Linezolid Sutezolid | Inhibits protein synthesis | [69,70,71] |
Bacterial Resistance as a Main Limitation for TB Drugs
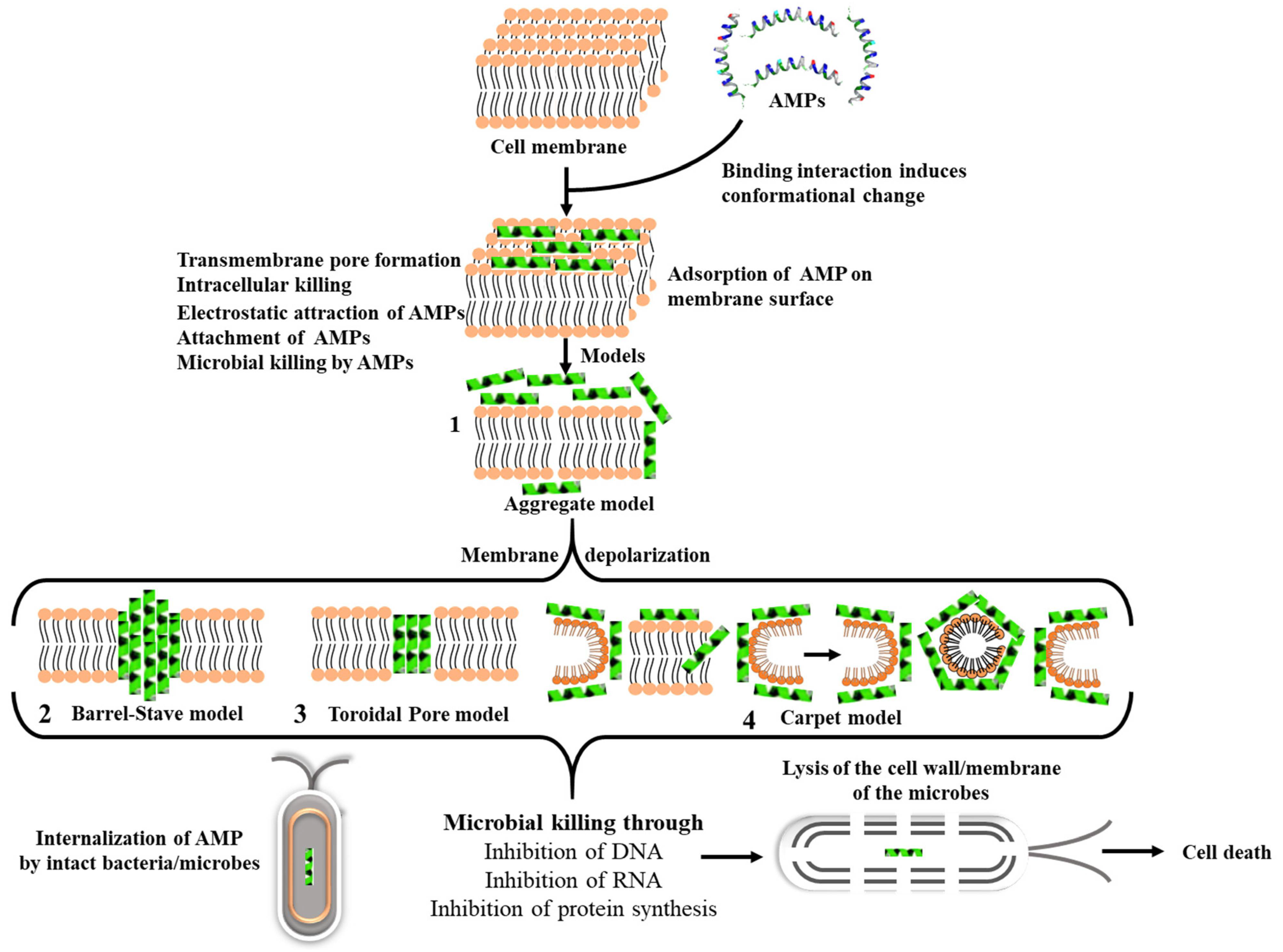
3. AMPs as Future Antibiotics
AMP Mode of Action
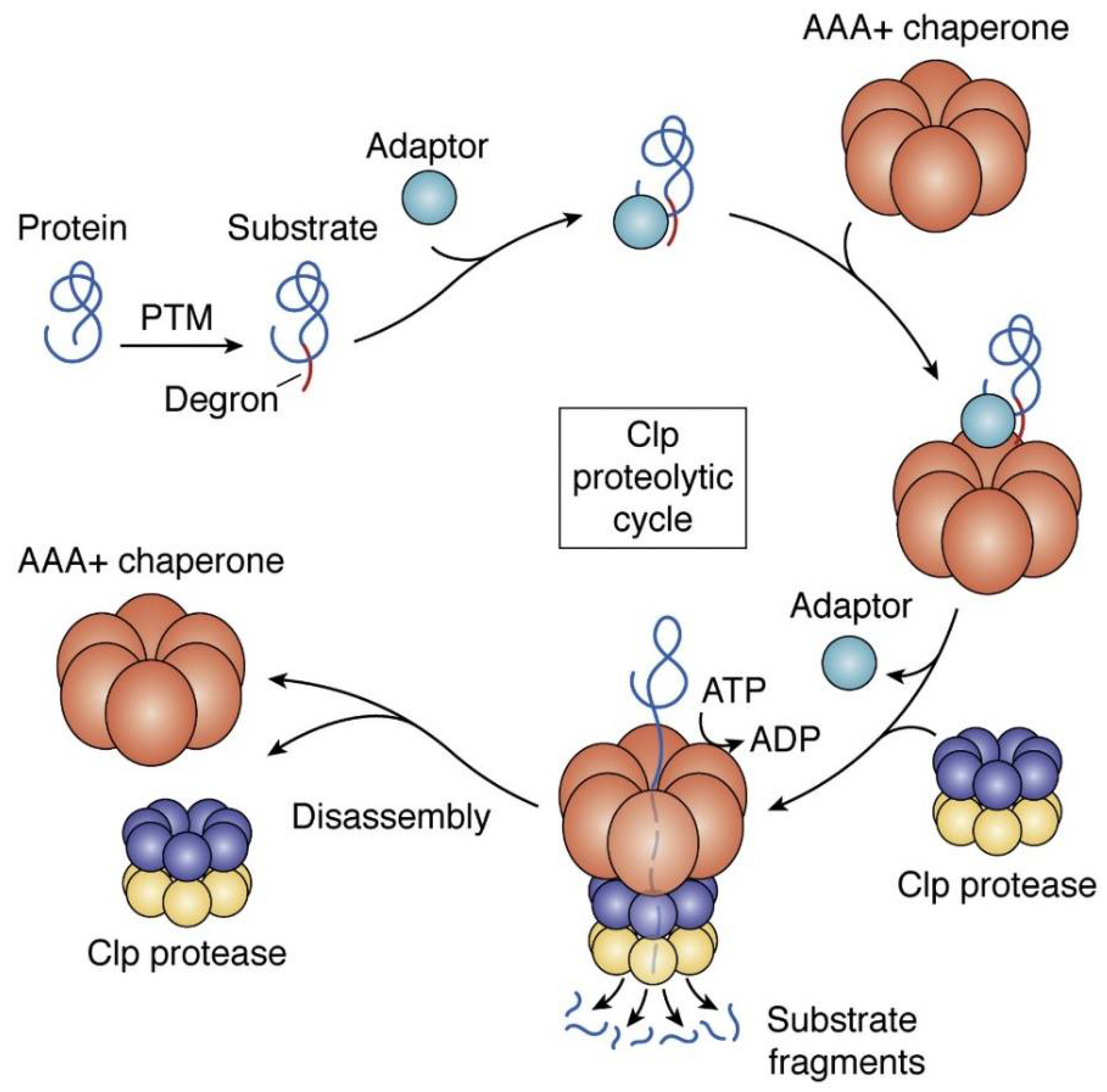
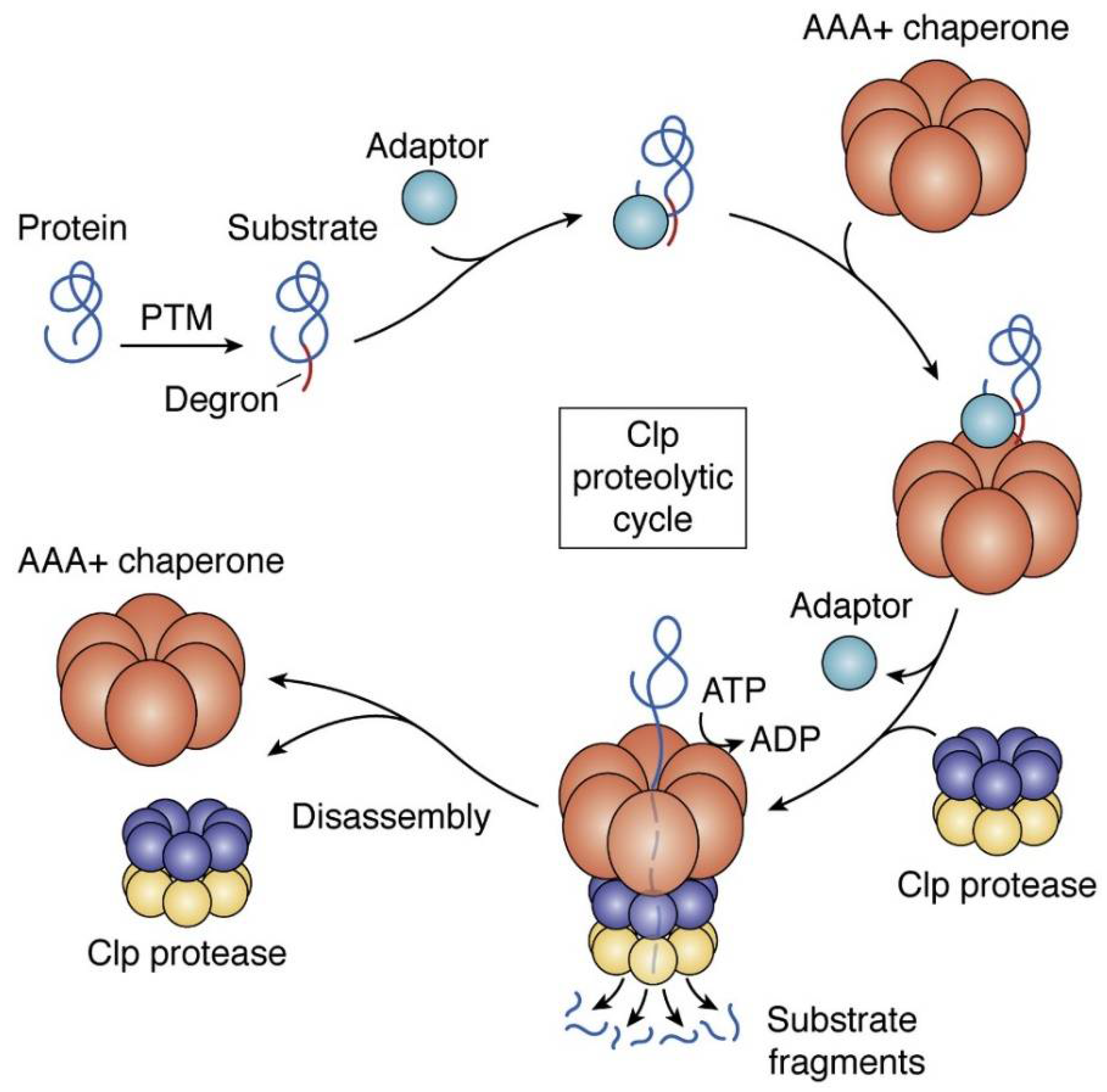
4. ClpP as a Putative Bacterial Therapeutic Target
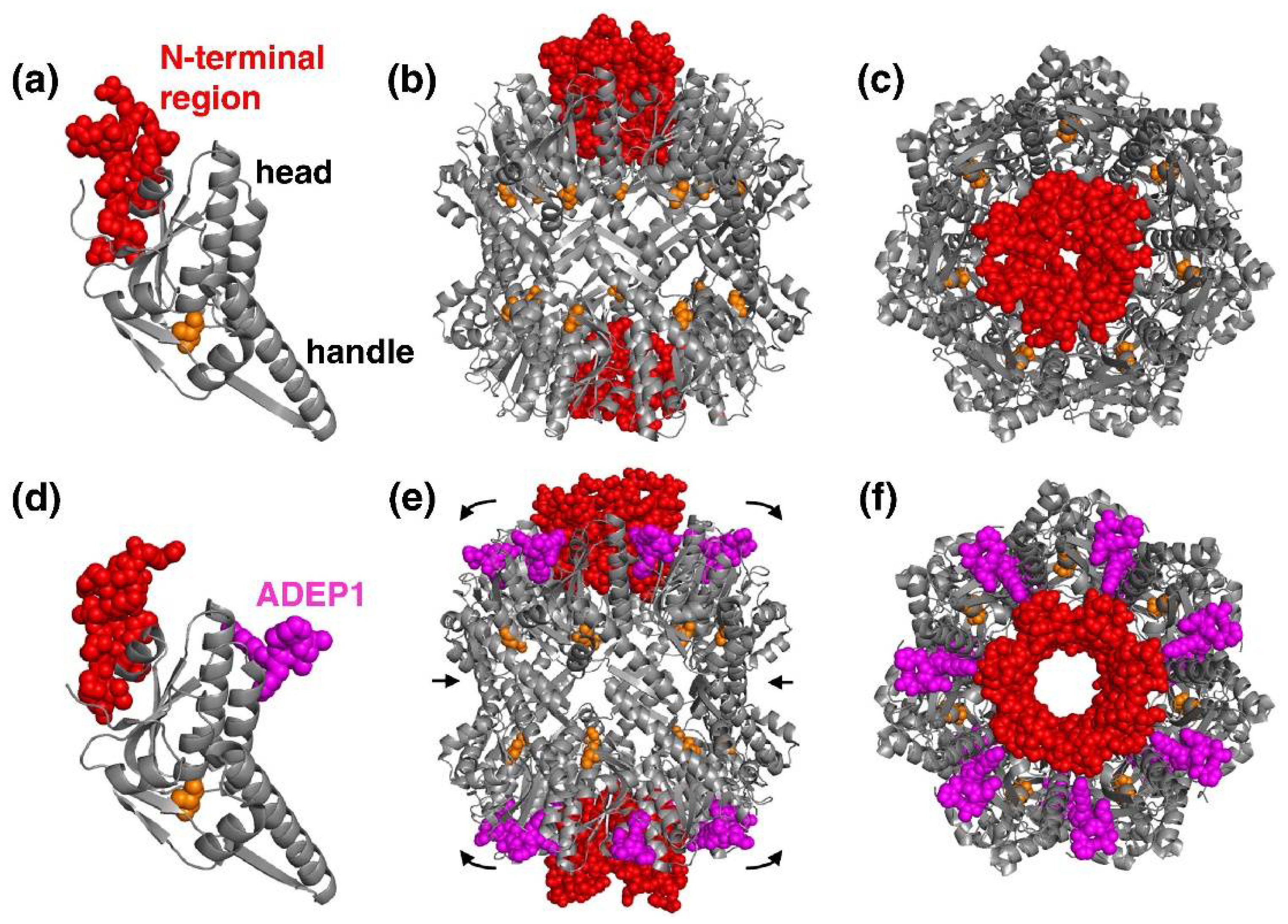
4.1. ADEPs Competes with Clp-ATPases to Deactivate ClpP
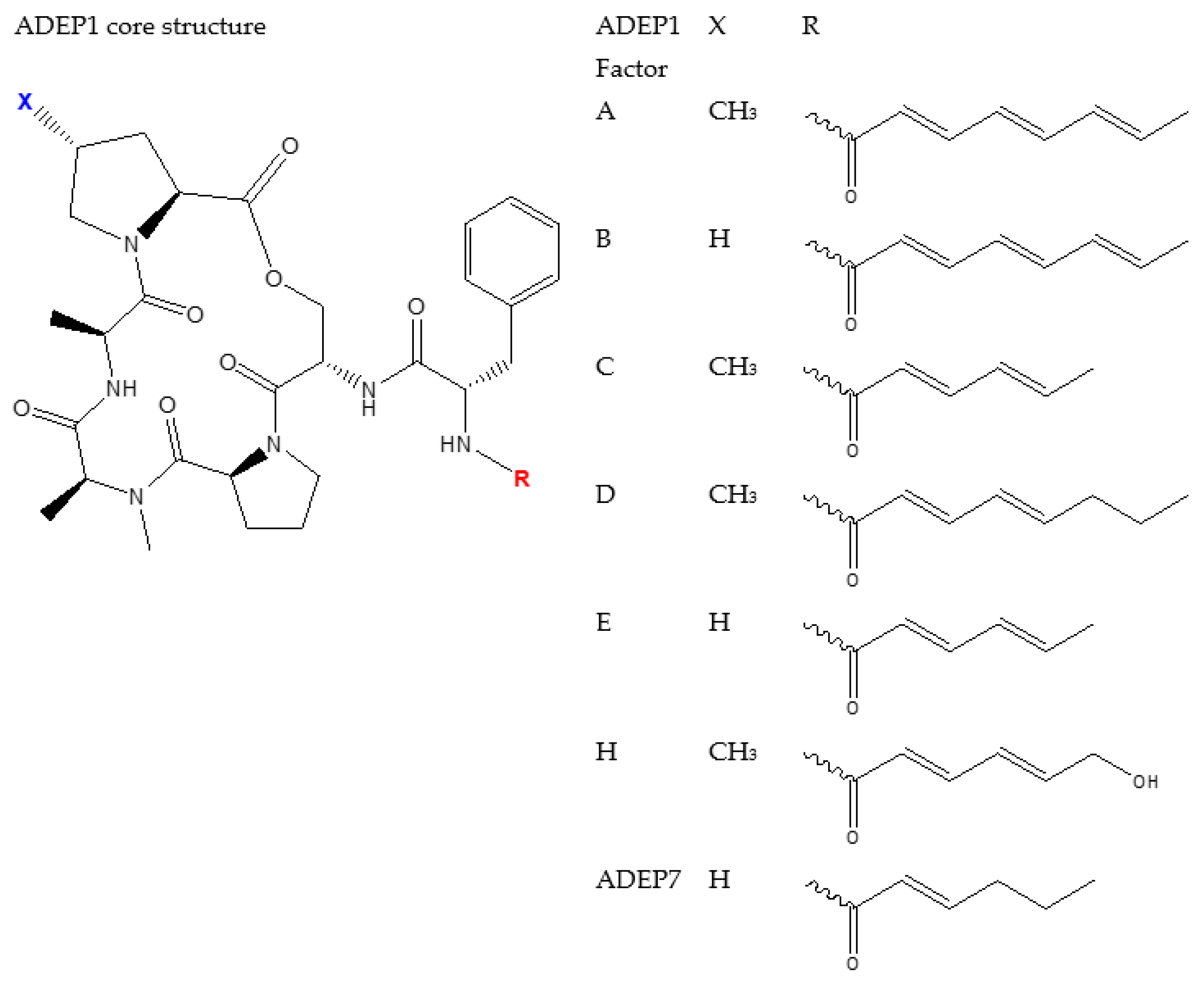
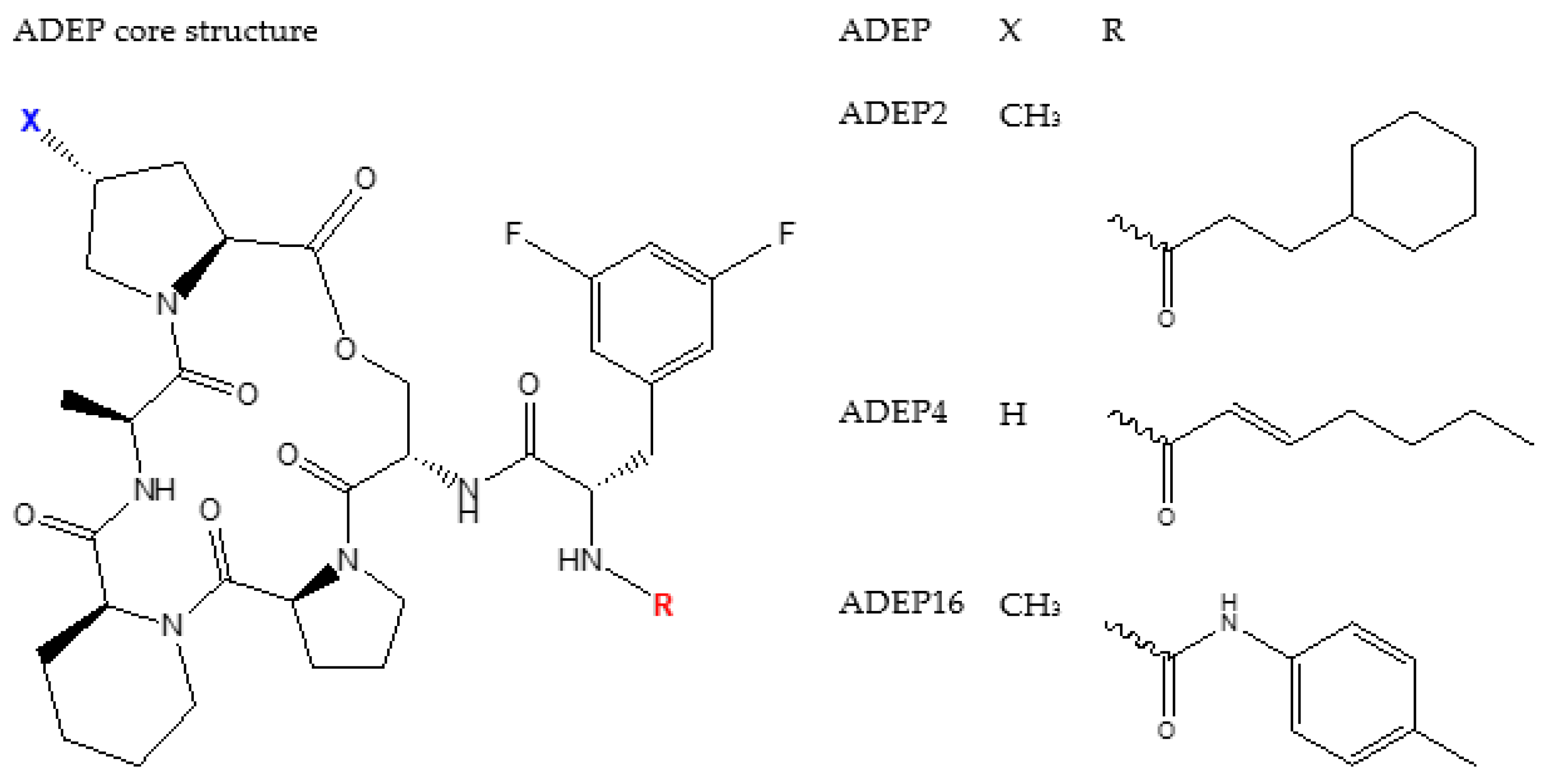
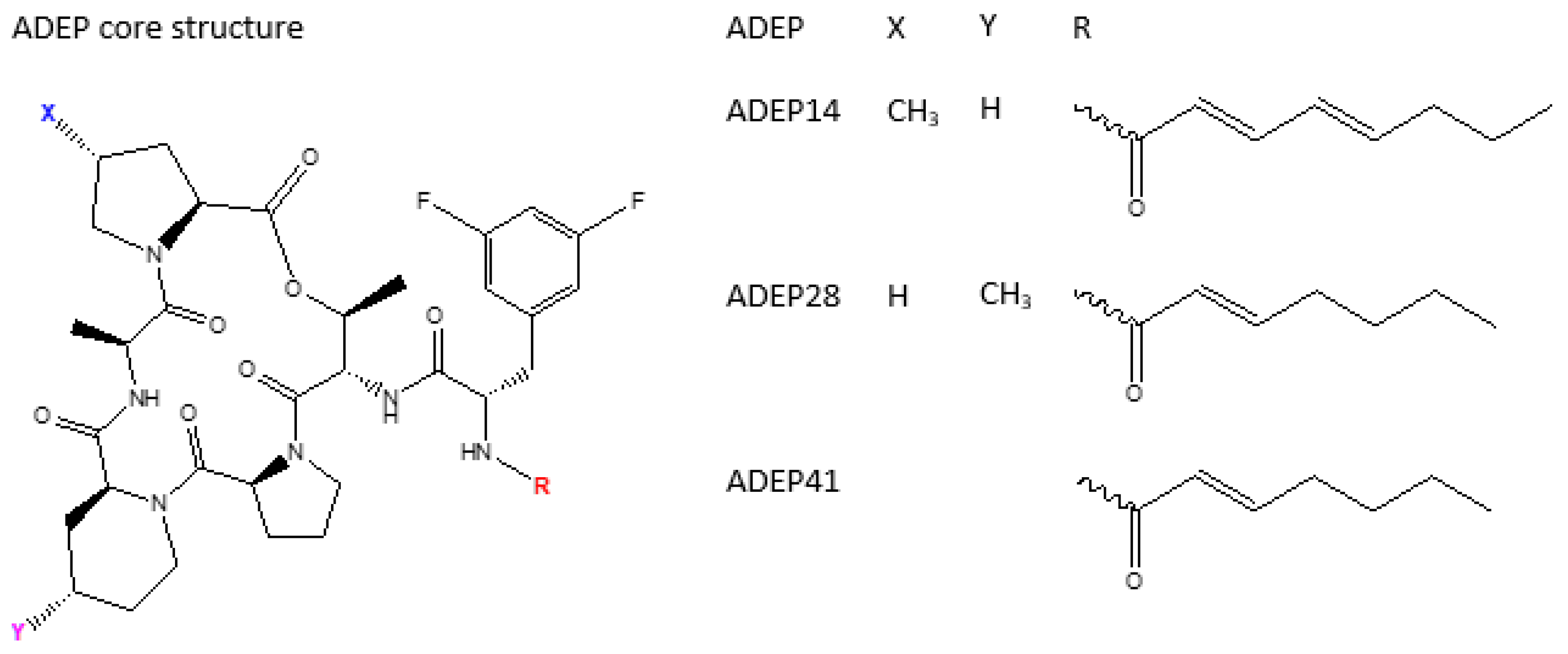
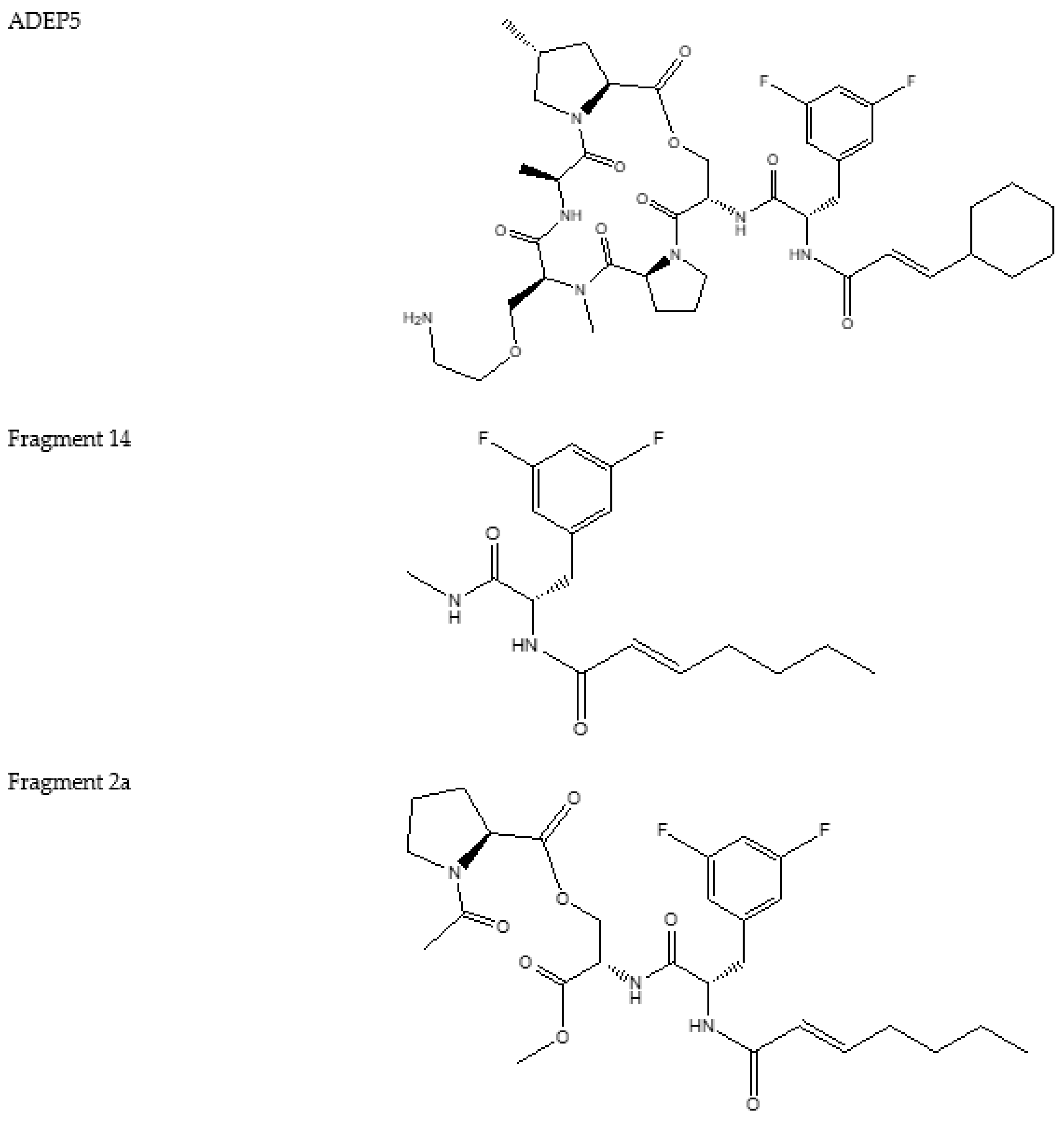
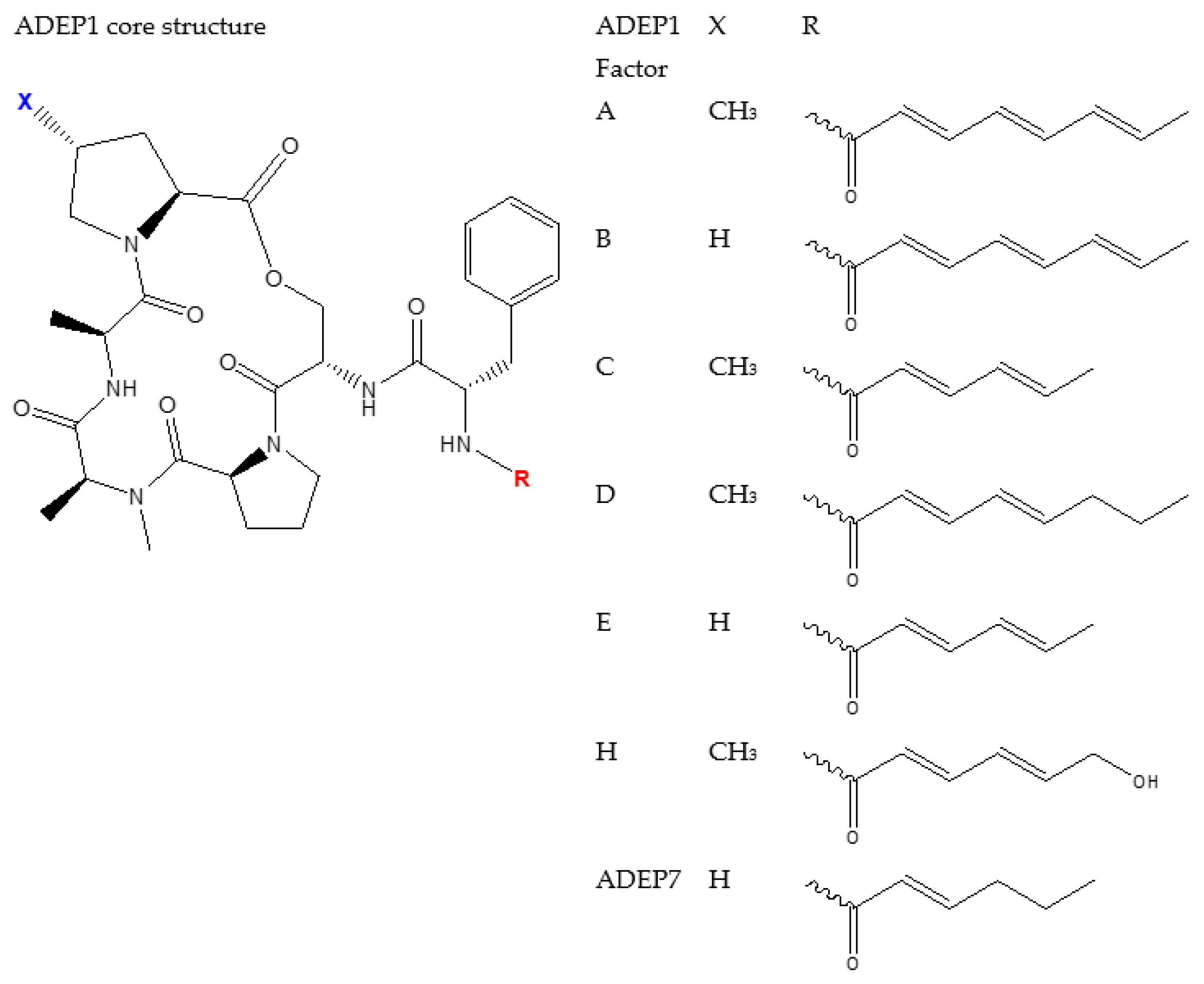
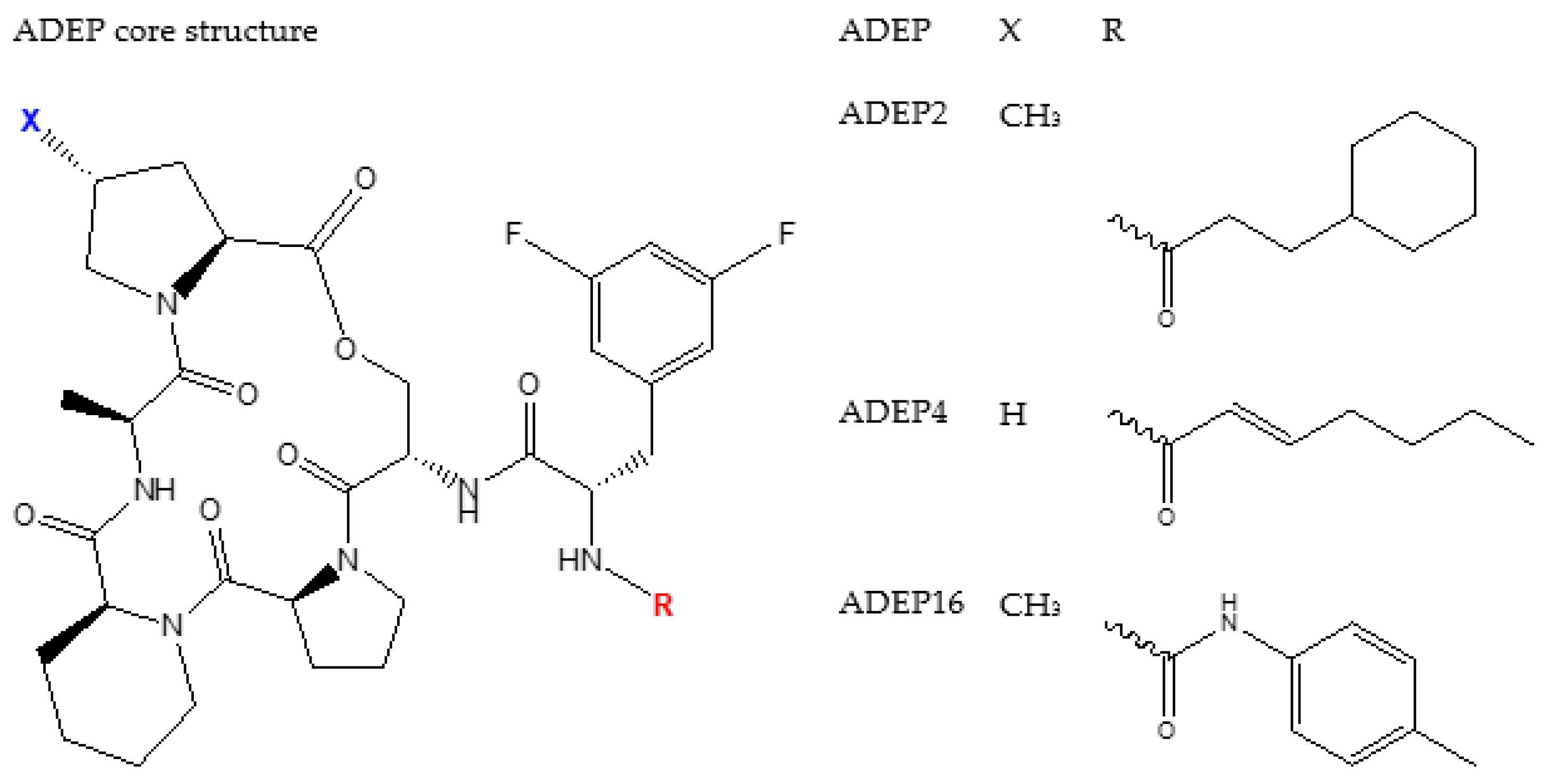
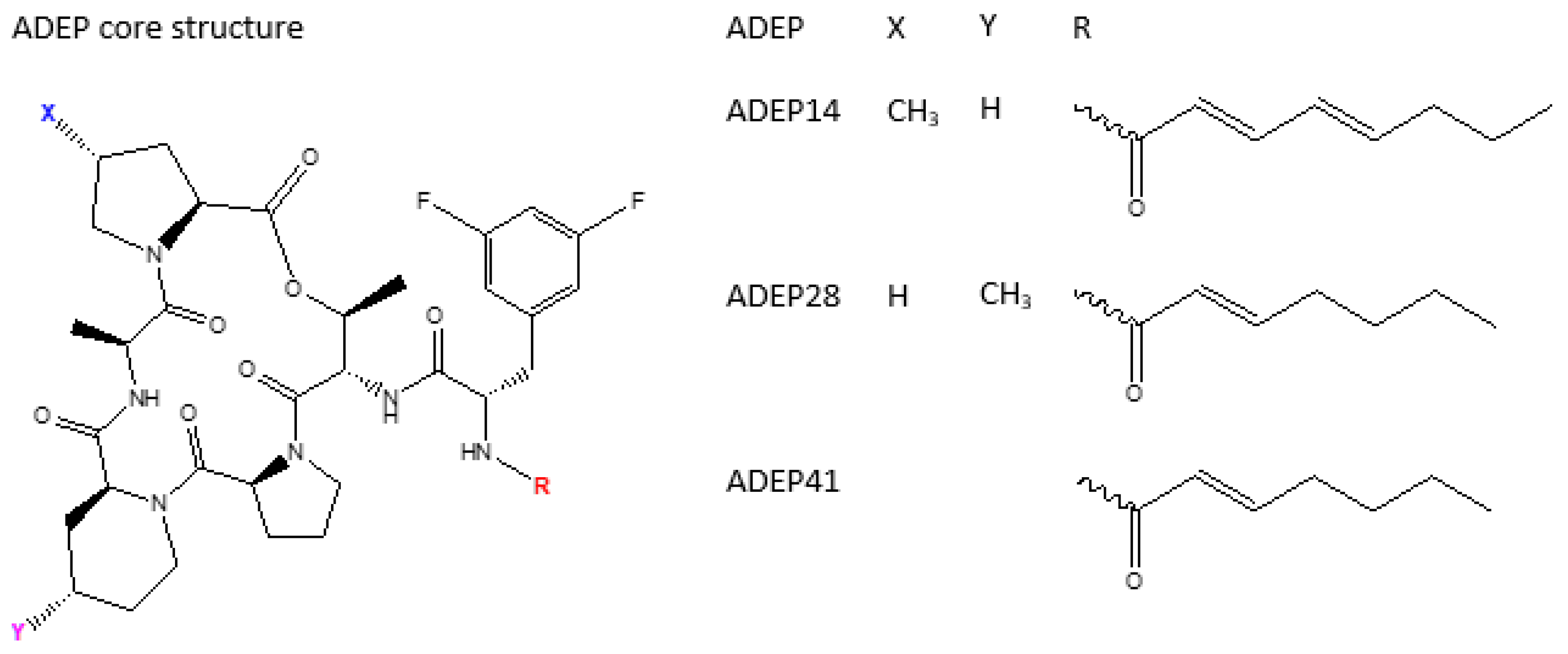
4.2. ADEP1 Analogues
4.3. ADEPs Activity on M. tb ClpP1P2
4.4. Limitation of ADEPs
4.4.1. Potential Strategies to Improve on ADEP’s Pharmacokinetics and Pharmacodynamics
Chemical Modification
Amino Acid Substitution
Lipophilic Molecules
Nano-Carriers
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Doetsch, R.N. Benjamin Marten and His “New Theory of Consumptions”. Microbiol. Rev. 1978, 42, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Augenstreich, J.; Haanappel, E.; Sayes, F.; Simeone, R.; Guillet, V.; Mazeres, S.; Chalut, C.; Mourey, L.; Brosch, R.; Guilhot, C.; et al. Phthiocerol Dimycocerosates from Mycobacterium Tuberculosis Increase the Membrane Activity of Bacterial Effectors and Host Receptors. Front. Cell Infect. Microbiol. 2020, 10, 420. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural Products in Drug Discovery: Advances and Opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Brötz-Oesterhelt, H.; Beyer, D.; Endermann, R.; Ladel, C.; Schroeder, W.; Hinzen, B.; Raddatz, S.; Paulsen, H.; Henninger, K.; Bandow, J.E.; et al. Dysregulation of Bacterial Proteolytic Machinery by a New Class of Antibiotics. Nat. Med. 2005, 11, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Goodreid, J.D.; Janetzko, J.; Santa Maria, J.P.; Wong, K.S.; Leung, E.; Eger, B.T.; Bryson, S.; Pai, E.F.; Gray-Owen, S.D.; Walker, S.; et al. Development and Characterization of Potent Cyclic Acyldepsipeptide Analogues with Increased Antimicrobial Activity. J. Med. Chem. 2016, 59, 624–646. [Google Scholar] [CrossRef] [PubMed]
- Anunthawan, T.; de la Fuente-Núñez, C.; Hancock, R.E.W.; Klaynongsruang, S. Cationic Amphipathic Peptides KT2 and RT2 Are Taken up into Bacterial Cells and Kill Planktonic and Biofilm Bacteria. Biochim. Biophys. Acta (BBA) Biomembr. 2015, 1848, 1352–1358. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, A.; Martinez, M.; Maturana, P.; Semorile, L.C.; Maffia, P.C. Antimicrobial Peptides: Interaction with Model and Biological Membranes and Synergism with Chemical Antibiotics. Front. Chem. 2018, 6, 204. [Google Scholar] [CrossRef]
- Hansen, A.; Schäfer, I.; Knappe, D.; Seibel, P.; Hoffmann, R. Intracellular Toxicity of Proline-Rich Antimicrobial Peptides Shuttled into Mammalian Cells by the Cell-Penetrating Peptide Penetratin. Antimicrob. Agents Chemother. 2012, 56, 5194–5201. [Google Scholar] [CrossRef]
- Malanovic, N.; Lohner, K. Antimicrobial Peptides Targeting Gram-Positive Bacteria. Pharmaceuticals 2016, 9, 59. [Google Scholar] [CrossRef]
- World Health Organization. The Top 10 Causes of Death; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Gerszten, P.C.; Gerszten, E.; Allison, M.J. Diseases of the Spine in South American Mummies. Neurosurgery 2001, 48, 208–213. [Google Scholar] [CrossRef]
- Riley, R.L.; O’Grady, F. Airborne Infection Transmission and Control. In Molecular Medical Microbiology; Elsevier: Amsterdam, The Netherlands, 1961; pp. 1–180. [Google Scholar]
- Sakula, A. Robert Koch: Centenary of the Discovery of the Tubercle Bacillus, 1882. Thorax 1982, 37, 246–251. [Google Scholar] [CrossRef]
- Rubin, S.A. Tuberculosis. Captain of All These Men of Death. Radiol. Clin. N. Am. 1995, 33, 619–639. [Google Scholar] [CrossRef]
- Wells, W.F. On Air-Borne Infection Study ii. Droplets and Droplet Nuclei. Am. J. Epidemiol. 1934, 20, 611–618. [Google Scholar] [CrossRef]
- Wells, W.F.; Ratcliffe, H.L.; Grumb, C. On the Mechanics of Droplet Nuclei Infection; Quantitative Experimental Air-Borne Tuberculosis in Rabbits. Am. J. Hyg. 1948, 47, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Riley, R.L.; Mills, C.C.; Nyka, W.; Weinstock, N.; Storey, P.B.; Sultan, L.U.; Riley, M.C.; Wells, W.F. Aerial Dissemination of Pulmonary Tuberculosis. A Two-Year Study of Contagion in a Tuberculosis Ward. 1959. Am. J. Epidemiol. 1995, 142, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Fennelly, K.P. Variability of Airborne Transmission of Mycobacterium Tuberculosis: Implications for Control of Tuberculosis in the HIV Era. Clin. Infect. Dis. 2007, 44, 1358–1360. [Google Scholar] [CrossRef] [PubMed]
- Duggal, S.; Chugh, T.D.; Duggal, A.K. HIV and Malnutrition: Effects on Immune System. Clin. Dev. Immunol. 2012, 2012, 784740. [Google Scholar] [CrossRef] [PubMed]
- Trehan, I.; O’Hare, B.A.; Phiri, A.; Heikens, G.T. Challenges in the Management of HIV-Infected Malnourished Children in Sub-Saharan Africa. AIDS Res. Treat. 2012, 2012, 790786. [Google Scholar] [CrossRef]
- Jeong, Y.H.; Hur, Y.-G.; Lee, H.; Kim, S.; Cho, J.-E.; Chang, J.; Shin, S.J.; Lee, H.; Kang, Y.A.; Cho, S.-N.; et al. Discrimination between Active and Latent Tuberculosis Based on Ratio of Antigen-Specific to Mitogen-Induced IP-10 Production. J. Clin. Microbiol. 2015, 53, 504–510. [Google Scholar] [CrossRef]
- Zumla, A.; Raviglione, M.; Hafner, R.; Fordham von Reyn, C. Tuberculosis. N. Engl. J. Med. 2013, 368, 745–755. [Google Scholar] [CrossRef]
- Bermudez, L.E.; Sangari, F.J. Mycobacterial Invasion of Epithelial Cells. In Bacterial Invasion into Eukaryotic Cells: Subcellular Biochemistry; Oelschlaeger, T.A., Hacker, J., Eds.; Subcellular Biochemistry; Springer: Boston, MA, USA, 2000; pp. 231–249. ISBN 978-1-4757-4580-1. [Google Scholar]
- Schlesinger, L.S. Macrophage Phagocytosis of Virulent but Not Attenuated Strains of Mycobacterium Tuberculosis Is Mediated by Mannose Receptors in Addition to Complement Receptors. J. Immunol. 1993, 150, 2920–2930. [Google Scholar] [PubMed]
- Gaynor, C.D.; McCormack, F.X.; Voelker, D.R.; McGowan, S.E.; Schlesinger, L.S. Pulmonary Surfactant Protein a Mediates Enhanced Phagocytosis of Mycobacterium Tuberculosis by a Direct Interaction with Human Macrophages. J. Immunol. 1995, 155, 5343–5351. [Google Scholar] [PubMed]
- Nathan, C.F.; Hibbs, J.B. Role of Nitric Oxide Synthesis in Macrophage Antimicrobial Activity. Curr. Opin. Immunol. 1991, 3, 65–70. [Google Scholar] [CrossRef]
- Silva Miranda, M.; Breiman, A.; Allain, S.; Deknuydt, F.; Altare, F. The Tuberculous Granuloma: An Unsuccessful Host Defence Mechanism Providing a Safety Shelter for the Bacteria? Clin. Dev. Immunol. 2012, 2012, 139127. [Google Scholar] [CrossRef] [PubMed]
- Peddireddy, V.; Doddam, S.N.; Ahmed, N. Mycobacterial Dormancy Systems and Host Responses in Tuberculosis. Front. Immunol. 2017, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Grange, J.M. Is the Eradication of Tuberculosis ‘yesterday’s Ambition’ or ‘tomorrow’s Triumph’? Clin. Med. 2010, 10, 450–453. [Google Scholar] [CrossRef]
- WHO. Global Tuberculosis Report 2021; WHO: Geneva, Switzerland, 2021; Available online: https://www.who.int/publications/i/item/9789240037021, (accessed on 1 June 2022).
- Jankute, M.; Cox, J.A.G.; Harrison, J.; Besra, G.S. Assembly of the Mycobacterial Cell Wall. Annu. Rev. Microbiol. 2015, 69, 405–423. [Google Scholar] [CrossRef]
- Rolain, J.-M.; Abat, C.; Jimeno, M.-T.; Fournier, P.-E.; Raoult, D. Do We Need New Antibiotics? Clin. Microbiol. Infect. 2016, 22, 408–415. [Google Scholar] [CrossRef]
- Yocum, R.R.; Rasmussen, J.R.; Strominger, J.L. The Mechanism of Action of Penicillin. Penicillin Acylates the Active Site of Bacillus Stearothermophilus D-Alanine Carboxypeptidase. J. Biol. Chem. 1980, 255, 3977–3986. [Google Scholar] [CrossRef]
- Fleming, A. On the Antibacterial Action of Cultures of a Penicillium, with Special Reference to Their Use in the Isolation of B. Influenzæ. Br. J. Exp. Pathol. 1929, 10, 226–236. [Google Scholar] [CrossRef]
- Tan, S.Y.; Tatsumura, Y. Alexander Fleming (1881–1955): Discoverer of Penicillin. Singap. Med. J. 2015, 56, 366–367. [Google Scholar] [CrossRef] [PubMed]
- Caltrider, P.G. Pyocyanine. In Antibiotics: Volume I Mechanism of Action; Gottlieb, D., Shaw, P.D., Eds.; Springer: Berlin/Heidelberg, Germany, 1967; pp. 117–121. ISBN 978-3-662-38439-8. [Google Scholar]
- Aminov, R.I. A Brief History of the Antibiotic Era: Lessons Learned and Challenges for the Future. Front. Microbiol. 2010, 1, 1–7. [Google Scholar] [CrossRef] [PubMed]
- History of TB Drugs—Which Was the First Drug? TBFacts. Available online: https://tbfacts.org/history-of-tb-drugs/#:~:text=The%20first%20clinical%20treatments%20of,for%20the%20treatment%20of%20tuberculosis (accessed on 1 June 2022).
- Takayama, K.; Wang, C.; Besra, G.S. Pathway to Synthesis and Processing of Mycolic Acids in Mycobacterium Tuberculosis. Clin. Microbiol. Rev. 2005, 18, 81–101. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.T.; Kim, T.-O.; Shin, H.-J.; Ko, Y.C.; Hun Choe, Y.; Kim, H.-R.; Kwon, Y.-S. Bedaquiline and Delamanid for the Treatment of Multidrug-Resistant Tuberculosis: A Multicentre Cohort Study in Korea. Eur. Respir. J. 2018, 51, 1702467. [Google Scholar] [CrossRef]
- Seung, K.J.; Keshavjee, S.; Rich, M.L. Multidrug-Resistant Tuberculosis and Extensively Drug-Resistant Tuberculosis. Cold Spring Harb. Perspect. Med. 2015, 5, a017863. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.R.; Nunn, P.; Dheda, K.; Schaaf, H.S.; Zignol, M.; van Soolingen, D.; Jensen, P.; Bayona, J. Multidrug-Resistant and Extensively Drug-Resistant Tuberculosis: A Threat to Global Control of Tuberculosis. Lancet 2010, 375, 1830–1843. [Google Scholar] [CrossRef]
- Nishida, C.R.; Ortiz de Montellano, P.R. Bioactivation of Anti-Tuberculosis Thioamide and Thiourea Prodrugs by Bacterial and Mammalian Flavin Monooxygenases. Chem. Biol. Interact. 2011, 192, 21–25. [Google Scholar] [CrossRef]
- Shetty, A.; Dick, T. Mycobacterial Cell Wall Synthesis Inhibitors Cause Lethal ATP Burst. Front. Microbiol. 2018, 9, 1898. [Google Scholar] [CrossRef]
- Thakare, R.; Soni, I.; Dasgupta, A.; Chopra, S. Delamanid for the Treatment of Pulmonary Multidrug-Resistant Tuberculosis. Drugs Today 2015, 51, 117–123. [Google Scholar] [CrossRef]
- Schubert, K.; Sieger, B.; Meyer, F.; Giacomelli, G.; Böhm, K.; Rieblinger, A.; Lindenthal, L.; Sachs, N.; Wanner, G.; Bramkamp, M. The Antituberculosis Drug Ethambutol Selectively Blocks Apical Growth in CMN Group Bacteria. mBio 2017, 8, e02213-16. [Google Scholar] [CrossRef] [Green Version]
- Batson, S.; de Chiara, C.; Majce, V.; Lloyd, A.J.; Gobec, S.; Rea, D.; Fülöp, V.; Thoroughgood, C.W.; Simmons, K.J.; Dowson, C.G.; et al. Inhibition of D-Ala:D-Ala Ligase through a Phosphorylated Form of the Antibiotic D-Cycloserine. Nat. Commun. 2017, 8, 1939. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, W.A.; Alsultan, A.; Al-Shaer, M.H.; An, G.; Ahmed, S.; Alkabab, Y.; Banu, S.; Barbakadze, K.; Houpt, E.; Kipiani, M.; et al. Cycloserine Population Pharmacokinetics and Pharmacodynamics in Patients with Tuberculosis. Antimicrob. Agents Chemother. 2019, 63, e00055-19. [Google Scholar] [CrossRef]
- Zhang, Y.; Wade, M.M.; Scorpio, A.; Zhang, H.; Sun, Z. Mode of Action of Pyrazinamide: Disruption of Mycobacterium Tuberculosis Membrane Transport and Energetics by Pyrazinoic Acid. J. Antimicrob. Chemother. 2003, 52, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Black, P.A.; Warren, R.M.; Louw, G.E.; van Helden, P.D.; Victor, T.C.; Kana, B.D. Energy Metabolism and Drug Efflux in Mycobacterium Tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 2491–2503. [Google Scholar] [CrossRef] [PubMed]
- Sarathy, J.P.; Gruber, G.; Dick, T. Re-Understanding the Mechanisms of Action of the Anti-Mycobacterial Drug Bedaquiline. Antibiotics 2019, 8, 261. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, P.; Rodríguez-Cano, F.; Zerolo, F.J.; Casal, M. Investigation of the in Vitro Activity of Streptomycin against Mycobacterium Tuberculosis. Microb. Drug Resist. 2002, 8, 147–149. [Google Scholar] [CrossRef]
- Chulluncuy, R.; Espiche, C.; Nakamoto, J.A.; Fabbretti, A.; Milón, P. Conformational Response of 30S-Bound IF3 to A-Site Binders Streptomycin and Kanamycin. Antibiotics 2016, 5, 38. [Google Scholar] [CrossRef]
- Lin, Y.; Li, Y.; Zhu, N.; Han, Y.; Jiang, W.; Wang, Y.; Si, S.; Jiang, J. The Antituberculosis Antibiotic Capreomycin Inhibits Protein Synthesis by Disrupting Interaction between Ribosomal Proteins L12 and L10. Antimicrob. Agents Chemother. 2014, 58, 2038–2044. [Google Scholar] [CrossRef]
- Tunitskaya, V.L.; Khomutov, A.R.; Kochetkov, S.N.; Kotovskaya, S.K.; Charushin, V.N. Inhibition of DNA Gyrase by Levofloxacin and Related Fluorine-Containing Heterocyclic Compounds. Acta Nat. 2011, 3, 94–99. [Google Scholar] [CrossRef]
- Chopra, S.; Matsuyama, K.; Tran, T.; Malerich, J.P.; Wan, B.; Franzblau, S.G.; Lun, S.; Guo, H.; Maiga, M.C.; Bishai, W.R.; et al. Evaluation of Gyrase B as a Drug Target in Mycobacterium Tuberculosis. J. Antimicrob. Chemother. 2012, 67, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Gruber, T.; Barry, C.E.; Boshoff, H.I.; Rhee, K.Y. Para-Aminosalicylic Acid Acts as an Alternative Substrate of Folate Metabolism in Mycobacterium Tuberculosis. Science 2013, 339, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Howe, M.D.; Kordus, S.L.; Cole, M.S.; Bauman, A.A.; Aldrich, C.C.; Baughn, A.D.; Minato, Y. Methionine Antagonizes Para-Aminosalicylic Acid Activity via Affecting Folate Precursor Biosynthesis in Mycobacterium Tuberculosis. Front. Cell. Infect. Microbiol. 2018, 8, 399. [Google Scholar] [CrossRef]
- Manjunatha, U.; Boshoff, H.I.; Barry, C.E. The Mechanism of Action of PA-824. Commun. Integr. Biol. 2009, 2, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Tahlan, K.; Wilson, R.; Kastrinsky, D.B.; Arora, K.; Nair, V.; Fischer, E.; Barnes, S.W.; Walker, J.R.; Alland, D.; Barry, C.E.; et al. SQ109 Targets MmpL3, a Membrane Transporter of Trehalose Monomycolate Involved in Mycolic Acid Donation to the Cell Wall Core of Mycobacterium Tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 1797–1809. [Google Scholar] [CrossRef] [PubMed]
- Catalão, M.J.; Filipe, S.R.; Pimentel, M. Revisiting Anti-Tuberculosis Therapeutic Strategies That Target the Peptidoglycan Structure and Synthesis. Front. Microbiol. 2019, 10, 190. [Google Scholar] [CrossRef]
- Vilchèze, C. Mycobacterial Cell Wall: A Source of Successful Targets for Old and New Drugs. Appl. Sci. 2020, 10, 2278. [Google Scholar] [CrossRef]
- Makarov, V.; Lechartier, B.; Zhang, M.; Neres, J.; Sar, A.M.V.D.; Raadsen, S.A.; Hartkoorn, R.C.; Ryabova, O.B.; Vocat, A.; Decosterd, L.A.; et al. Towards a New Combination Therapy for Tuberculosis with next Generation Benzothiazinones. EMBO Mol. Med. 2014, 6, 372–383. [Google Scholar] [CrossRef]
- Makarov, V.; Manina, G.; Mikusova, K.; Möllmann, U.; Ryabova, O.; Saint-Joanis, B.; Dhar, N.; Pasca, M.R.; Buroni, S.; Lucarelli, A.P.; et al. Benzothiazinones Kill Mycobacterium Tuberculosis by Blocking Arabinan Synthesis. Science 2009, 324, 801–804. [Google Scholar] [CrossRef]
- O’Malley, T.; Alling, T.; Early, J.V.; Wescott, H.A.; Kumar, A.; Moraski, G.C.; Miller, M.J.; Masquelin, T.; Hipskind, P.A.; Parish, T. Imidazopyridine Compounds Inhibit Mycobacterial Growth by Depleting ATP Levels. Antimicrob. Agents Chemother. 2018, 62, e02439-17. [Google Scholar] [CrossRef]
- Lu, P.; Asseri, A.H.; Kremer, M.; Maaskant, J.; Ummels, R.; Lill, H.; Bald, D. The Anti-Mycobacterial Activity of the Cytochrome Bcc Inhibitor Q203 Can Be Enhanced by Small-Molecule Inhibition of Cytochrome Bd. Sci. Rep. 2018, 8, 2625. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.J.; Krasnykh, O.; Pan, D.; Petukhova, V.; Yu, G.; Liu, Y.; Liu, H.; Hong, S.; Wang, Y.; Wan, B.; et al. Structure-Activity Relationships of Macrolides against Mycobacterium Tuberculosis. Tuberculosis 2008, 88, S49–S63. [Google Scholar] [CrossRef]
- Van der Paardt, A.-F.; Wilffert, B.; Akkerman, O.W.; de Lange, W.C.M.; van Soolingen, D.; Sinha, B.; van der Werf, T.S.; Kosterink, J.G.W.; Alffenaar, J.-W.C. Evaluation of Macrolides for Possible Use against Multidrug-Resistant Mycobacterium Tuberculosis. Eur. Respir. J. 2015, 46, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sala, C.; Dhar, N.; Vocat, A.; Sambandamurthy, V.K.; Sharma, S.; Marriner, G.; Balasubramanian, V.; Cole, S.T. In Vitro and In Vivo Activities of Three Oxazolidinones against Nonreplicating Mycobacterium Tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 3217–3223. [Google Scholar] [CrossRef] [PubMed]
- Alcalá, L.; Ruiz-Serrano, M.J.; Pérez-Fernández Turégano, C.; García de Viedma, D.; Díaz-Infantes, M.; Marín-Arriaza, M.; Bouza, E. In Vitro Activities of Linezolid against Clinical Isolates of Mycobacterium Tuberculosis That Are Susceptible or Resistant to First-Line Antituberculous Drugs. Antimicrob. Agents Chemother. 2003, 47, 416–417. [Google Scholar] [CrossRef] [PubMed]
- Wallis, R.S.; Dawson, R.; Friedrich, S.O.; Venter, A.; Paige, D.; Zhu, T.; Silvia, A.; Gobey, J.; Ellery, C.; Zhang, Y.; et al. Mycobactericidal Activity of Sutezolid (PNU-100480) in Sputum (EBA) and Blood (WBA) of Patients with Pulmonary Tuberculosis. PLoS ONE 2014, 9, e94462. [Google Scholar] [CrossRef]
- Gaynes, R.; Monnet, D. The Contribution of Antibiotic Use on the Frequency of Antibiotic Resistance in Hospitals. In Ciba Foundation Symposium 207—Antibiotic Resistance: Origins, Evolution, Selection and Spread: Antibiotic Resistance: Origins, Evolution, Selection and Spread: Ciba Foundation Symposium 207; John Wiley & Sons, Ltd.: Chichester, UK, 2007; pp. 47–56. [Google Scholar]
- Coates, A.; Hu, Y.; Bax, R.; Page, C. The Future Challenges Facing the Development of New Antimicrobial Drugs. Nat. Rev. Drug Discov. 2002, 1, 895–910. [Google Scholar] [CrossRef]
- Tang, Y.-W.; Li, H.; Griffin, J.P.; Haas, D.W.; D’Agata, E.M.C. Rapidly Increasing Prevalence of Penicillin-Resistant Streptococcus Pneumoniae in Middle Tennessee: A 10-Year Clinical and Molecular Analysis. J. Clin. Microbiol. 2002, 40, 395–399. [Google Scholar] [CrossRef]
- Feikin, D.R.; Schuchat, A.; Kolczak, M.; Barrett, N.L.; Harrison, L.H.; Lefkowitz, L.; McGeer, A.; Farley, M.M.; Vugia, D.J.; Lexau, C.; et al. Mortality from Invasive Pneumococcal Pneumonia in the Era of Antibiotic Resistance, 1995–1997. Am. J. Public Health 2000, 90, 223–229. [Google Scholar]
- Mamishi, S.; Moradkhani, S.; Mahmoudi, S.; Hosseinpour-Sadeghi, R.; Pourakbari, B. Penicillin-Resistant Trend of Streptococcus Pneumoniae in Asia: A Systematic Review. Iran. J. Microbiol. 2014, 6, 198–210. [Google Scholar]
- Brennan, P.J. Structure, Function, and Biogenesis of the Cell Wall of Mycobacterium Tuberculosis. Tuberculosis 2003, 83, 91–97. [Google Scholar] [CrossRef]
- Chao, M.C.; Rubin, E.J. Letting Sleeping Dos Lie: Does Dormancy Play a Role in Tuberculosis? Annu. Rev. Microbiol. 2010, 64, 293–311. [Google Scholar] [CrossRef] [PubMed]
- Danilchanka, O.; Pavlenok, M.; Niederweis, M. Role of Porins for Uptake of Antibiotics by Mycobacterium Smegmatis. Antimicrob. Agents Chemother. 2008, 52, 3127–3134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephan, J.; Mailaender, C.; Etienne, G.; Daffé, M.; Niederweis, M. Multidrug Resistance of a Porin Deletion Mutant of Mycobacterium Smegmatis. Antimicrob. Agents Chemother. 2004, 48, 4163–4170. [Google Scholar] [CrossRef] [PubMed]
- Kardan Yamchi, J.; Haeili, M.; Gizaw Feyisa, S.; Kazemian, H.; Hashemi Shahraki, A.; Zahednamazi, F.; Imani Fooladi, A.A.; Feizabadi, M.M. Evaluation of Efflux Pump Gene Expression among Drug Susceptible and Drug Resistant Strains of Mycobacterium Tuberculosis from Iran. Infect. Genet. Evol. 2015, 36, 23–26. [Google Scholar] [CrossRef]
- Wivagg, C.N.; Bhattacharyya, R.P.; Hung, D.T. Mechanisms of β-Lactam Killing and Resistance in the Context of Mycobacterium Tuberculosis. J. Antibiot. 2014, 67, 645–654. [Google Scholar] [CrossRef]
- Almeida Da Silva, P.E.A.; Palomino, J.C. Molecular Basis and Mechanisms of Drug Resistance in Mycobacterium Tuberculosis: Classical and New Drugs. J. Antimicrob. Chemother. 2011, 66, 1417–1430. [Google Scholar] [CrossRef]
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural Mechanism for Rifampicin Inhibition of Bacterial RNA Polymerase. Cell 2001, 104, 901–912. [Google Scholar] [CrossRef]
- Centers for Disease Control (CDC) Nosocomial Transmission of Multidrug-Resistant Tuberculosis among HIV-Infected Persons—Florida and New York, 1988–1991. MMWR Morb. Mortal. Wkly. Rep. 1991, 40, 585–591.
- Frieden, T.R.; Sterling, T.; Pablos-Mendez, A.; Kilburn, J.O.; Cauthen, G.M.; Dooley, S.W. The Emergence of Drug-Resistant Tuberculosis in New York City. N. Engl. J. Med. 1993, 328, 521–526. [Google Scholar] [CrossRef]
- Rullán, J.V.; Herrera, D.; Cano, R.; Moreno, V.; Godoy, P.; Peiró, E.F.; Castell, J.; Ibañez, C.; Ortega, A.; Agudo, L.S.; et al. Nosocomial Transmission of Multidrug-Resistant Mycobacterium Tuberculosis in Spain. Emerg. Infect. Dis. 1996, 2, 125–129. [Google Scholar] [CrossRef]
- Shah, N.S.; Wright, A.; Bai, G.-H.; Barrera, L.; Boulahbal, F.; Martín-Casabona, N.; Drobniewski, F.; Gilpin, C.; Havelková, M.; Lepe, R.; et al. Worldwide Emergence of Extensively Drug-Resistant Tuberculosis. Emerg. Infect. Dis. 2007, 13, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Migliori, G.B.; Ortmann, J.; Girardi, E.; Besozzi, G.; Lange, C.; Cirillo, D.M.; Ferrarese, M.; De Iaco, G.; Gori, A.; Raviglione, M.C. Extensively Drug-Resistant Tuberculosis, Italy and Germany. Emerg. Infect. Dis. 2007, 13, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Velayati, A.A.; Masjedi, M.R.; Farnia, P.; Tabarsi, P.; Ghanavi, J.; ZiaZarifi, A.H.; Hoffner, S.E. Emergence of New Forms of Totally Drug-Resistant Tuberculosis Bacilli: Super Extensively Drug-Resistant Tuberculosis or Totally Drug-Resistant Strains in Iran. Chest 2009, 136, 420–425. [Google Scholar] [CrossRef]
- Shah, N.S.; Richardson, J.; Moodley, P.; Moodley, S.; Babaria, P.; Ramtahal, M.; Heysell, S.K.; Li, X.; Moll, A.P.; Friedland, G.; et al. Increasing Drug Resistance in Extensively Drug-Resistant Tuberculosis, South Africa. Emerg. Infect. Dis. 2011, 17, 510–513. [Google Scholar] [CrossRef]
- Powers, J.H. Antimicrobial Drug Development—The Past, the Present, and the Future. Clin. Microbiol. Infect. 2004, 10 (Suppl. S4), 23–31. [Google Scholar] [CrossRef]
- Butler, M.S.; Buss, A.D. Natural Products—the Future Scaffolds for Novel Antibiotics? Biochem. Pharmacol. 2006, 71, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Hair, P.I.; Keam, S.J. Daptomycin: A Review of Its Use in the Management of Complicated Skin and Soft-Tissue Infections and Staphylococcus Aureus Bacteraemia. Drugs 2007, 67, 1483–1512. [Google Scholar] [CrossRef]
- Zappia, G.; Menendez, P.; Monache, G.D.; Misiti, D.; Nevola, L.; Botta, B. The Contribution of Oxazolidinone Frame to the Biological Activity of Pharmaceutical Drugs and Natural Products. Mini Rev. Med. Chem. 2007, 7, 389–409. [Google Scholar] [CrossRef]
- Xie, M.; Liu, D.; Yang, Y. Anti-Cancer Peptides: Classification, Mechanism of Action, Reconstruction and Modification. Open Biol. 2020, 10, 200004. [Google Scholar] [CrossRef]
- Rubattu, S.; Volpe, M. Natriuretic Peptides in the Cardiovascular System: Multifaceted Roles in Physiology, Pathology and Therapeutics. Int. J. Mol. Sci. 2019, 20, 3991. [Google Scholar] [CrossRef]
- Wang, R.; Zhao, H.; Pan, X.; Orfila, C.; Lu, W.; Ma, Y. Preparation of Bioactive Peptides with Antidiabetic, Antihypertensive, and Antioxidant Activities and Identification of A-glucosidase Inhibitory Peptides from Soy Protein. Food Sci. Nutr. 2019, 7, 1848–1856. [Google Scholar] [CrossRef] [PubMed]
- Gordon, Y.J.; Romanowski, E.G.; McDermott, A.M. A Review of Antimicrobial Peptides and Their Therapeutic Potential as Anti-Infective Drugs. Curr. Eye Res. 2005, 30, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zeng, X.; Yang, Q.; Qiao, S. Antimicrobial Peptides as Potential Alternatives to Antibiotics in Food Animal Industry. Int. J. Mol. Sci. 2016, 17, 603. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The Expanding Scope of Antimicrobial Peptide Structures and Their Modes of Action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef]
- Ho, Y.-H.; Shah, P.; Chen, Y.-W.; Chen, C.-S. Systematic Analysis of Intracellular-Targeting Antimicrobial Peptides, Bactenecin 7, Hybrid of Pleurocidin and Dermaseptin, Proline–Arginine-Rich Peptide, and Lactoferricin B, by Using Escherichia Coli Proteome Microarrays. Mol. Cell Proteom. 2016, 15, 1837–1847. [Google Scholar] [CrossRef]
- Yount, N.Y.; Yeaman, M.R. Structural Congruence among Membrane-Active Host Defense Polypeptides of Diverse Phylogeny. Biochim. Biophys. Acta (BBA) Biomembr. 2006, 1758, 1373–1386. [Google Scholar] [CrossRef]
- Gorr, S.-U.; Abdolhosseini, M. Antimicrobial Peptides and Periodontal Disease. J. Clin. Periodontol. 2011, 38 (Suppl. S11), 126–141. [Google Scholar] [CrossRef]
- Wu, X.; Wang, Z.; Li, X.; Fan, Y.; He, G.; Wan, Y.; Yu, C.; Tang, J.; Li, M.; Zhang, X.; et al. In Vitro and In Vivo Activities of Antimicrobial Peptides Developed Using an Amino Acid-Based Activity Prediction Method. Antimicrob. Agents Chemother. 2014, 58, 5342–5349. [Google Scholar] [CrossRef]
- Forde, É.; Schütte, A.; Reeves, E.; Greene, C.; Humphreys, H.; Mall, M.; Fitzgerald-Hughes, D.; Devocelle, M. Differential In Vitro and In Vivo Toxicities of Antimicrobial Peptide Prodrugs for Potential Use in Cystic Fibrosis. Antimicrob. Agents Chemother. 2016, 60, 2813–2821. [Google Scholar] [CrossRef]
- Travis, S.M.; Anderson, N.N.; Forsyth, W.R.; Espiritu, C.; Conway, B.D.; Greenberg, E.P.; McCray, P.B.; Lehrer, R.I.; Welsh, M.J.; Tack, B.F. Bactericidal Activity of Mammalian Cathelicidin-Derived Peptides. Infect. Immun. 2000, 68, 2748–2755. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Falla, T.J. Antimicrobial Peptides: Broad-Spectrum Antibiotics from Nature. Clin. Microbiol. Infect. 1996, 1, 226–229. [Google Scholar] [CrossRef]
- Rathinakumar, R.; Walkenhorst, W.F.; Wimley, W.C. Broad-Spectrum Antimicrobial Peptides by Rational Combinatorial Design and High-Throughput Screening: The Importance of Interfacial Activity. J. Am. Chem. Soc. 2009, 131, 7609–7617. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hu, Y.; Yang, Y.; Lu, Z.; Wang, Y. Antimicrobial Resistance in Livestock: Antimicrobial Peptides Provide a New Solution for a Growing Challenge. Anim. Front. 2018, 8, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef]
- Omardien, S.; Brul, S.; Zaat, S.A.J. Antimicrobial Activity of Cationic Antimicrobial Peptides against Gram-Positives: Current Progress Made in Understanding the Mode of Action and the Response of Bacteria. Front. Cell Dev. Biol. 2016, 4, 111. [Google Scholar] [CrossRef]
- Volzing, K.; Borrero, J.; Sadowsky, M.J.; Kaznessis, Y.N. Antimicrobial Peptides Targeting Gram-Negative Pathogens, Produced and Delivered by Lactic Acid Bacteria. ACS Synth. Biol. 2013, 2, 643–650. [Google Scholar] [CrossRef]
- Gruenheid, S.; Moual, H. Resistance to Antimicrobial Peptides in Gram-Negative Bacteria. FEMS Microbiol. Lett. 2012, 330, 81–89. [Google Scholar] [CrossRef]
- Bondaryk, M.; Staniszewska, M.; Zielińska, P.; Urbańczyk-Lipkowska, Z. Natural Antimicrobial Peptides as Inspiration for Design of a New Generation Antifungal Compounds. J. Fungi 2017, 3, 46. [Google Scholar] [CrossRef]
- Sun, Y.; Shang, D. Inhibitory Effects of Antimicrobial Peptides on Lipopolysaccharide-Induced Inflammation. Mediat. Inflamm. 2015, 2015, 167572. [Google Scholar] [CrossRef]
- Mangoni, M.L.; McDermott, A.M.; Zasloff, M. Antimicrobial Peptides and Wound Healing: Biological and Therapeutic Considerations. Exp. Dermatol. 2016, 25, 167–173. [Google Scholar] [CrossRef]
- Yu, G.; Baeder, D.Y.; Regoes, R.R.; Rolff, J. Combination Effects of Antimicrobial Peptides. Antimicrob. Agents Chemother. 2016, 60, 1717–1724. [Google Scholar] [CrossRef] [PubMed]
- Dubos, R.J. Studies on a bactericidal agent extracted from a soil bacillus. J. Exp. Med. 1939, 70, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuji, T.; Gallo, R.L. Antimicrobial Peptides: Old Molecules with New Ideas. J. Invest Dermatol. 2012, 132, 887–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zasloff, M. Magainins, a Class of Antimicrobial Peptides from Xenopus Skin: Isolation, Characterization of Two Active Forms, and Partial CDNA Sequence of a Precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453. [Google Scholar] [CrossRef]
- Balls, A.K.; Hale, W.S.; Harris, T.H. A Crystalline Protein Obtained from a Lipoprotein of Wheat Flour. Cereal Chem. 1942, 19, 279–288. [Google Scholar]
- Coll, P.; Garrigó, M.; Moreno, C.; Martí, N. Routine Use of Gen-Probe Amplified Mycobacterium Tuberculosis Direct (MTD) Test for Detection of Mycobacterium Tuberculosis with Smear-Positive and Smear-Negative Specimens. Int. J. Tuberc. Lung Dis. 2003, 7, 886–891. [Google Scholar]
- De Caleya, R.F.; Gonzalez-Pascual, B.; García-Olmedo, F.; Carbonero, P. Susceptibility of Phytopathogenic Bacteria to Wheat Purothionins in Vitro. Appl. Microbiol. 1972, 23, 998–1000. [Google Scholar] [CrossRef]
- Wilson, D.N.; Guichard, G.; Innis, C.A. Antimicrobial Peptides Target Ribosomes. Oncotarget 2015, 6, 16826–16827. [Google Scholar] [CrossRef]
- Bechinger, B.; Gorr, S.-U. Antimicrobial Peptides. J. Dent. Res. 2017, 96, 254–260. [Google Scholar] [CrossRef]
- Fadaka, A.O.; Sibuyi, N.R.S.; Madiehe, A.M.; Meyer, M. Nanotechnology-Based Delivery Systems for Antimicrobial Peptides. Pharmaceutics 2021, 13, 1795. [Google Scholar] [CrossRef]
- Raheem, N.; Straus, S.K. Mechanisms of Action for Antimicrobial Peptides with Antibacterial and Antibiofilm Functions. Front. Microbiol. 2019, 10, 2866. [Google Scholar] [CrossRef] [PubMed]
- Malanovic, N.; Lohner, K. Gram-Positive Bacterial Cell Envelopes: The Impact on the Activity of Antimicrobial Peptides. Biochim. Biophys. Acta (BBA) Biomembr. 2016, 1858, 936–946. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-Y.; Yan, Z.-B.; Meng, Y.-M.; Hong, X.-Y.; Shao, G.; Ma, J.-J.; Cheng, X.-R.; Liu, J.; Kang, J.; Fu, C.-Y. Antimicrobial Peptides: Mechanism of Action, Activity and Clinical Potential. Mil. Med. Res. 2021, 8, 48. [Google Scholar] [CrossRef] [PubMed]
- Benfield, A.H.; Henriques, S.T. Mode-of-Action of Antimicrobial Peptides: Membrane Disruption vs. Intracellular Mechanisms. Front. Med. Technol. 2020, 2, 610997. [Google Scholar] [CrossRef]
- Hsu, C.-H. Structural and DNA-Binding Studies on the Bovine Antimicrobial Peptide, Indolicidin: Evidence for Multiple Conformations Involved in Binding to Membranes and DNA. Nucleic Acids Res. 2005, 33, 4053–4064. [Google Scholar] [CrossRef] [Green Version]
- Subbalakshmi, C.; Sitaram, N. Mechanism of Antimicrobial Action of Indolicidin. FEMS Microbiol. Lett. 1998, 160, 91–96. [Google Scholar] [CrossRef]
- Park, C.B.; Kim, H.S.; Kim, S.C. Mechanism of Action of the Antimicrobial Peptide Buforin II: Buforin II Kills Microorganisms by Penetrating the Cell Membrane and Inhibiting Cellular Functions. Biochem. Biophys. Res. Commun. 1998, 244, 253–257. [Google Scholar] [CrossRef]
- Linde, C.M.A.; Hoffner, S.E.; Refai, E.; Andersson, M. In Vitro Activity of PR-39, a Proline-Arginine-Rich Peptide, against Susceptible and Multi-Drug-Resistant Mycobacterium Tuberculosis. J. Antimicrob. Chemother. 2001, 47, 575–580. [Google Scholar] [CrossRef]
- Boman, H.G.; Agerberth, B.; Boman, A. Mechanisms of Action on Escherichia Coli of Cecropin P1 and PR-39, Two Antibacterial Peptides from Pig Intestine. Infect. Immun. 1993, 61, 2978–2984. [Google Scholar] [CrossRef]
- Culp, E.; Wright, G.D. Bacterial Proteases, Untapped Antimicrobial Drug Targets. J. Antibiot. 2017, 70, 366–377. [Google Scholar] [CrossRef]
- Yu, A.Y.H.; Houry, W.A. ClpP: A Distinctive Family of Cylindrical Energy-dependent Serine Proteases. FEBS Lett. 2007, 581, 3749–3757. [Google Scholar] [CrossRef] [PubMed]
- Brötz-Oesterhelt, H.; Sass, P. Bacterial Caseinolytic Proteases as Novel Targets for Antibacterial Treatment. Int. J. Med. Microbiol. 2014, 304, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Elsholz, A.K.W.; Birk, M.S.; Charpentier, E.; Turgay, K. Functional Diversity of AAA+ Protease Complexes in Bacillus Subtilis. Front. Mol. Biosci. 2017, 4, 44. [Google Scholar] [CrossRef] [PubMed]
- Malik, I.T.; Brötz-Oesterhelt, H. Conformational Control of the Bacterial Clp Protease by Natural Product Antibiotics. Nat. Prod. Rep. 2017, 34, 815–831. [Google Scholar] [CrossRef] [PubMed]
- Frees, D.; Gerth, U.; Ingmer, H. Clp Chaperones and Proteases Are Central in Stress Survival, Virulence and Antibiotic Resistance of Staphylococcus Aureus. Int. J. Med. Microbiol. 2014, 304, 142–149. [Google Scholar] [CrossRef]
- Frees, D.; Savijoki, K.; Varmanen, P.; Ingmer, H. Clp ATPases and ClpP Proteolytic Complexes Regulate Vital Biological Processes in Low GC, Gram-positive Bacteria. Mol. Microbiol. 2007, 63, 1285–1295. [Google Scholar] [CrossRef]
- Dohmen, R.J.; Wu, P.; Varshavsky, A. Heat-Inducible Degron: A Method for Constructing Temperature-Sensitive Mutants. Science 1994, 263, 1273–1276. [Google Scholar] [CrossRef]
- Montandon, C.; Friso, G.; Liao, J.-Y.R.; Choi, J.; van Wijk, K.J. In Vivo Trapping of Proteins Interacting with the Chloroplast CLPC1 Chaperone: Potential Substrates and Adaptors. J. Proteome Res. 2019, 18, 2585–2600. [Google Scholar] [CrossRef]
- Lee, B.-G.; Kim, M.K.; Song, H.K. Structural Insights into the Conformational Diversity of ClpP from Bacillus Subtilis. Mol. Cells 2011, 32, 589–595. [Google Scholar] [CrossRef]
- Bouchnak, I.; Wijk, K.J. van Structure, Function, and Substrates of Clp AAA+ Protease Systems in Cyanobacteria, Plastids, and Apicoplasts: A Comparative Analysis. J. Biol. Chem. 2021, 296. [Google Scholar] [CrossRef]
- Wolf, N.M.; Lee, H.; Zagal, D.; Nam, J.; Oh, D.; Lee, H.; Suh, J.; Pauli, G.F.; Cho, S.; Abad-Zapatero, C. Structure of the N-terminal Domain of ClpC1 in Complex with the Antituberculosis Natural Product Ecumicin Reveals Unique Binding Interactions. Acta. Cryst. 2020, 76, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Mei, Z.; Qi, Y.; Yan, C.; Hu, Q.; Wang, J.; Shi, Y. Structure and Mechanism of the Hexameric MecA–ClpC Molecular Machine. Nature 2011, 471, 7338. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Kim, J.-Y.; Anderson, J.R.; Akopian, T.; Hong, S.; Jin, Y.-Y.; Kandror, O.; Kim, J.-W.; Lee, I.-A.; Lee, S.-Y.; et al. The Cyclic Peptide Ecumicin Targeting ClpC1 Is Active against Mycobacterium Tuberculosis In Vivo. Antimicrob. Agents Chemother. 2015, 59, 880–889. [Google Scholar] [CrossRef] [PubMed]
- Gavrish, E.; Sit, C.S.; Cao, S.; Kandror, O.; Spoering, A.; Peoples, A.; Ling, L.; Fetterman, A.; Hughes, D.; Bissell, A.; et al. Lassomycin, a Ribosomally Synthesized Cyclic Peptide, Kills Mycobacterium Tuberculosis by Targeting the ATP-Dependent Protease ClpC1P1P2. Chem. Biol. 2014, 21, 509–518. [Google Scholar] [CrossRef]
- Akopian, T.; Kandror, O.; Raju, R.M.; UnniKrishnan, M.; Rubin, E.J.; Goldberg, A.L. The Active ClpP Protease from M. Tuberculosis Is a Complex Composed of a Heptameric ClpP1 and a ClpP2 Ring. EMBO J. 2012, 31, 1529–1541. [Google Scholar] [CrossRef] [Green Version]
- Vasudevan, D.; Rao, S.P.S.; Noble, C.G. Structural Basis of Mycobacterial Inhibition by Cyclomarin A. J. Biol. Chem. 2013, 288, 30883–30891. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Jung, I.P.; Ha, N.R.; Kim, A.R.; Kim, S.H.; Yoon, M.Y. Mutation Analysis of the Interactions between Mycobacterium Tuberculosis Caseinolytic Protease C1 (ClpC1) and Ecumicin. Int. J. Biol. Macromol. 2017, 101, 348–357. [Google Scholar] [CrossRef]
- Böttcher, T.; Sieber Stephan, A. β-Lactones as Privileged Structures for the Active-Site Labeling of Versatile Bacterial Enzyme Classes. Angew. Chem. Int. Ed. 2008, 47, 4600–4603. [Google Scholar] [CrossRef]
- Michel, K.H.; Kastner, R.E. A54556 Antibiotics and Process for Production Thereof. Antibiotic A54556, a Complex of 8 Individual Factors, Produced by Submerged, Aerobic Fermentation of New Streptomyces hawaiiensis NRRL 15010. The Complex and the Individual, Separated Factors are Active against Staphylococcus and Streptococcus which are Penicillin Resistant. U.S. Patent US-4492650-A, 8 January 1985. [Google Scholar]
- Brötz-Oesterhelt, H.; Vorbach, A. Reprogramming of the Caseinolytic Protease by ADEP Antibiotics: Molecular Mechanism, Cellular Consequences, Therapeutic Potential. Front. Mol. Biosci. 2021, 8, 690902. [Google Scholar] [CrossRef]
- Lee, B.-G.; Park, E.Y.; Lee, K.-E.; Jeon, H.; Sung, K.H.; Paulsen, H.; Rübsamen-Schaeff, H.; Brötz-Oesterhelt, H.; Song, H.K. Structures of ClpP in Complex with Acyldepsipeptide Antibiotics Reveal Its Activation Mechanism. Nat. Struct. Mol. Biol. 2010, 17, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Li, D.H.S.; Chung, Y.S.; Gloyd, M.; Joseph, E.; Ghirlando, R.; Wright, G.D.; Cheng, Y.-Q.; Maurizi, M.R.; Guarné, A.; Ortega, J. Acyldepsipeptide Antibiotics Induce the Formation of a Structured Axial Channel in ClpP: A Model for the ClpX/ClpA-Bound State of ClpP. Chem. Biol. 2010, 17, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-I.; Levchenko, I.; Fraczkowska, K.; Fraczkowska, R.V.; Sauer, R.T.; Baker, T.A. Molecular Determinants of Complex Formation between Clp/Hsp100 ATPases and the ClpP Peptidase. Nat. Struct. Biol. 2001, 8, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Sowole, M.A.; Alexopoulos, J.A.; Cheng, Y.-Q.; Ortega, J.; Konermann, L. Activation of ClpP Protease by ADEP Antibiotics: Insights from Hydrogen Exchange Mass Spectrometry. J. Mol. Biol. 2013, 425, 4508–4519. [Google Scholar] [CrossRef]
- Gersch, M.; List, A.; Groll, M.; Sieber, S.A. Insights into Structural Network Responsible for Oligomerization and Activity of Bacterial Virulence Regulator Caseinolytic Protease P (ClpP) Protein. J. Biol. Chem. 2012, 287, 9484–9494. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Kandror, O.; Akopian, T.; Dharkar, P.; Wlodawer, A.; Maurizi, M.R.; Goldberg, A.L. Structure and Functional Properties of the Active Form of the Proteolytic Complex, ClpP1P2, from Mycobacterium Tuberculosis. J. Biol. Chem. 2016, 291, 7465–7476. [Google Scholar] [CrossRef]
- Goodreid, J.D.; Wong, K.; Leung, E.; McCaw, S.E.; Gray-Owen, S.D.; Lough, A.; Houry, W.A.; Batey, R.A. Total Synthesis and Antibacterial Testing of the A54556 Cyclic Acyldepsipeptides Isolated from Streptomyces Hawaiiensis. J. Nat. Prod. 2014, 77, 2170–2181. [Google Scholar] [CrossRef]
- Hinzen, B.; Raddatz, S.; Paulsen, H.; Lampe, T.; Schumacher, A.; Häbich, D.; Hellwig, V.; Benet-Buchholz, J.; Endermann, R.; Labischinski, H.; et al. Medicinal Chemistry Optimization of Acyldepsipeptides of the Enopeptin Class Antibiotics. Chem. Med. Chem. 2006, 1, 689–693. [Google Scholar] [CrossRef]
- Conlon, B.P.; Nakayasu, E.S.; Fleck, L.E.; LaFleur, M.D.; Isabella, V.M.; Coleman, K.; Leonard, S.N.; Smith, R.D.; Adkins, J.N.; Lewis, K. Activated ClpP Kills Persisters and Eradicates a Chronic Biofilm Infection. Nature 2013, 503, 365–370. [Google Scholar] [CrossRef]
- Griffith, E.C.; Zhao, Y.; Singh, A.P.; Conlon, B.P.; Tangallapally, R.; Shadrick, W.R.; Liu, J.; Wallace, M.J.; Yang, L.; Elmore, J.M.; et al. Ureadepsipeptides as ClpP Activators. ACS Infect. Dis. 2019, 5, 1915–1925. [Google Scholar] [CrossRef]
- Carney, D.W.; Schmitz, K.R.; Truong, J.V.; Sauer, R.T.; Sello, J.K. Restriction of the Conformational Dynamics of the Cyclic Acyldepsipeptide Antibiotics Improves Their Antibacterial Activity. J. Am. Chem. Soc. 2014, 136, 1922–1929. [Google Scholar] [CrossRef]
- Carney, D.W.; Compton, C.L.; Schmitz, K.R.; Stevens, J.P.; Sauer, R.T.; Sello, J.K. A Simple Fragment of the Cyclic Acyldepsipeptides Is Necessary and Sufficient for ClpP Activation and Antibacterial Activity. ChemBioChem 2014, 15, 2216–2220. [Google Scholar] [CrossRef] [PubMed]
- Raju, R.M.; Unnikrishnan, M.; Rubin, D.H.F.; Krishnamoorthy, V.; Kandror, O.; Akopian, T.N.; Goldberg, A.L.; Rubin, E.J. Mycobacterium Tuberculosis ClpP1 and ClpP2 Function Together in Protein Degradation and Are Required for Viability in Vitro and During Infection. PLoS Pathog. 2012, 8, e1002511. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.R.; Sauer, R.T. Substrate Delivery by the AAA+ ClpX and ClpC1 Unfoldases Activates the Mycobacterial ClpP1P2 Peptidase. Mol. Microbiol. 2014, 93, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Alhuwaider, A.A.H.; Dougan, D.A. AAA+ Machines of Protein Destruction in Mycobacteria. Front. Mol. Biosci. 2017, 4, 49. [Google Scholar] [CrossRef] [Green Version]
- Personne, Y.; Brown, A.C.; Schuessler, D.L.; Parish, T. Mycobacterium Tuberculosis ClpP Proteases Are Co-Transcribed but Exhibit Different Substrate Specificities. PLoS ONE 2013, 8, e60228. [Google Scholar] [CrossRef]
- Schmitz, K.R.; Carney, D.W.; Sello, J.K.; Sauer, R.T. Crystal Structure of Mycobacterium Tuberculosis ClpP1P2 Suggests a Model for Peptidase Activation by AAA+ Partner Binding and Substrate Delivery. Proc. Natl. Acad. Sci. USA 2014, 111, E4587–E4595. [Google Scholar] [CrossRef]
- Famulla, K.; Sass, P.; Malik, I.; Akopian, T.; Kandror, O.; Alber, M.; Hinzen, B.; Ruebsamen-Schaeff, H.; Kalscheuer, R.; Goldberg, A.L.; et al. Acyldepsipeptide Antibiotics Kill Mycobacteria by Preventing the Physiological Functions of the ClpP1P2 Protease: ADEPs Target ClpP1P2 of M. Tuberculosis. Mol. Microbiol. 2016, 101, 194–209. [Google Scholar] [CrossRef]
- Joshi, S.A.; Hersch, G.L.; Baker, T.A.; Sauer, R.T. Communication between ClpX and ClpP during Substrate Processing and Degradation. Nat. Struct. Mol. Biol. 2004, 11, 404–411. [Google Scholar] [CrossRef]
- Leodolter, J.; Warweg, J.; Weber-Ban, E. The Mycobacterium Tuberculosis ClpP1P2 Protease Interacts Asymmetrically with Its ATPase Partners ClpX and ClpC1. PLoS ONE 2015, 10, e0125345. [Google Scholar] [CrossRef]
- Ollinger, J.; O’Malley, T.; Kesicki, E.A.; Odingo, J.; Parish, T. Validation of the Essential ClpP Protease in Mycobacterium Tuberculosis as a Novel Drug Target. J. Bacteriol. 2012, 194, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.L.; Richard, J. Challenges in the Delivery of Peptide Drugs: An Industry Perspective. Ther. Deliv. 2015, 6, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, J.; Falciani, C.; Roscia, G.; Pollini, S.; Bindi, S.; Scali, S.; Arrieta, U.C.; Gómez-Vallejo, V.; Quercini, L.; Ibba, E.; et al. In Vitro and in Vivo Efficacy, Toxicity, Bio-Distribution and Resistance Selection of a Novel Antibacterial Drug Candidate. Sci. Rep. 2016, 6, 26077. [Google Scholar] [CrossRef]
- Huggins, D.J.; Sherman, W.; Tidor, B. Rational Approaches to Improving Selectivity in Drug Design. J. Med. Chem. 2012, 55, 1424–1444. [Google Scholar] [CrossRef] [PubMed]
- Joo, S.H. Cyclic Peptides as Therapeutic Agents and Biochemical Tools. Biomol. Ther. 2012, 20, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Doherty, T.; Waring, A.J.; Ruchala, P.; Hong, M. Roles of Arginine and Lysine Residues in the Translocation of a Cell-Penetrating Peptide from 13C, 31P and 19F Solid-State NMR. Biochemistry 2009, 48, 4587–4595. [Google Scholar] [CrossRef]
- Chatterjee, J.; Gilon, C.; Hoffman, A.; Kessler, H. N-Methylation of Peptides: A New Perspective in Medicinal Chemistry. Acc. Chem. Res. 2008, 41, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Buccini, D.F.; Cardoso, M.H.; Franco, O.L. Antimicrobial Peptides and Cell-Penetrating Peptides for Treating Intracellular Bacterial Infections. Front. Cell Infect. Microbiol. 2020, 10, 612931. [Google Scholar] [CrossRef]
- Lee, H.; Lim, S.I.; Shin, S.-H.; Lim, Y.; Koh, J.W.; Yang, S. Conjugation of Cell-Penetrating Peptides to Antimicrobial Peptides Enhances Antibacterial Activity. ACS Omega 2019, 4, 15694–15701. [Google Scholar] [CrossRef]
- Vale, N.; Ferreira, A.; Matos, J.; Fresco, P.; Gouveia, M.J. Amino Acids in the Development of Prodrugs. Molecules 2018, 23, 2318. [Google Scholar] [CrossRef]
- Renukuntla, J.; Vadlapudi, A.D.; Patel, A.; Boddu, S.H.S.; Mitra, A.K. Approaches for Enhancing Oral Bioavailability of Peptides and Proteins. Int. J. Pharm. 2013, 447, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, J.; Rechenmacher, F.; Kessler, H. N-Methylation of Peptides and Proteins: An Important Element for Modulating Biological Functions. Angew. Chem. Int. Ed. Engl. 2013, 52, 254–269. [Google Scholar] [CrossRef] [PubMed]
- Doedens, L.; Opperer, F.; Cai, M.; Beck, J.G.; Dedek, M.; Palmer, E.; Hruby, V.J.; Kessler, H. Multiple N-Methylation of MT-II Backbone Amide Bonds Leads to Melanocortin Receptor Subtype HMC1R Selectivity: Pharmacological and Conformational Studies. J. Am. Chem. Soc. 2010, 132, 8115–8128. [Google Scholar] [CrossRef]
- Neil, E.; Marsh, G. Towards the Nonstick Egg: Designing Fluorous Proteins. Chem. Biol. 2000, 7, R153–R157. [Google Scholar] [CrossRef]
- Čižinauskas, V.; Elie, N.; Brunelle, A.; Briedis, V. Fatty Acids Penetration into Human Skin Ex Vivo: A TOF-SIMS Analysis Approach. Biointerphases 2017, 12, 011003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irby, D.; Du, C.; Li, F. Lipid–Drug Conjugate for Enhancing Drug Delivery. Mol. Pharm. 2017, 14, 1325–1338. [Google Scholar] [CrossRef]
- De Jong, W.H.; Borm, P.J. Drug Delivery and Nanoparticles: Applications and Hazards. Int. J. Nanomed. 2008, 3, 133–149. [Google Scholar] [CrossRef]
- Guisbiers, G.; Mejía-Rosales, S.; Leonard Deepak, F. Nanomaterial Properties: Size and Shape Dependencies. Available online: https://www.hindawi.com/journals/jnm/2012/180976/ (accessed on 31 August 2018).
- Mandal, A. Copper Nanomaterials as Drug Delivery System against Infectious Agents and Cancerous Cells. J. Appl. Life Sci. Int. 2017, 15, 1–8. [Google Scholar] [CrossRef]
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation Design for Poorly Water-Soluble Drugs Based on Biopharmaceutics Classification System: Basic Approaches and Practical Applications. Int. J. Pharm 2011, 420, 1–10. [Google Scholar] [CrossRef]
- Kou, L.; Bhutia, Y.D.; Yao, Q.; He, Z.; Sun, J.; Ganapathy, V. Transporter-Guided Delivery of Nanoparticles to Improve Drug Permeation across Cellular Barriers and Drug Exposure to Selective Cell Types. Front. Pharm. 2018, 9. [Google Scholar] [CrossRef]
- Martínez-Carmona, M.; Lozano, D.; Colilla, M.; Vallet-Regí, M. Selective Topotecan Delivery to Cancer Cells by Targeted PH-Sensitive Mesoporous Silica Nanoparticles. RSC Adv. 2016, 6, 50923–50932. [Google Scholar] [CrossRef]
- Ren, G.; Hu, D.; Cheng, E.W.C.; Vargas-Reus, M.A.; Reip, P.; Allaker, R.P. Characterisation of Copper Oxide Nanoparticles for Antimicrobial Applications. Int. J. Antimicrob. Agents 2009, 33, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Redding, J.E.; Wiley, P.A.; Wen, L.; McConnell, J.S.; Zhang, B. Mutagenicity Evaluation of Metal Oxide Nanoparticles by the Bacterial Reverse Mutation Assay. Chemosphere 2010, 79, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Quan, Q.; Xie, J.; Gao, H.; Yang, M.; Zhang, F.; Liu, G.; Lin, X.; Wang, A.; Eden, H.S.; Lee, S.; et al. HSA Coated Iron Oxide Nanoparticles as Drug Delivery Vehicles for Cancer Therapy. Mol. Pharm. 2011, 8, 1669–1676. [Google Scholar] [CrossRef] [PubMed]
- Jafari, A.R.; Mosavi, T.; Mosavari, N.; Majid, A.; Movahedzade, F.; Tebyaniyan, M.; Kamalzadeh, M.; Dehgan, M.; Jafari, S.; Arastoo, S. Mixed Metal Oxide Nanoparticles Inhibit Growth of Mycobacterium Tuberculosis into THP-1 Cells. Int. J. Mycobacteriol. 2016, 5, S181–S183. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cobongela, S.Z.Z.; Makatini, M.M.; Mdluli, P.S.; Sibuyi, N.R.S. Acyldepsipeptide Analogues: A Future Generation Antibiotics for Tuberculosis Treatment. Pharmaceutics 2022, 14, 1956. https://doi.org/10.3390/pharmaceutics14091956
Cobongela SZZ, Makatini MM, Mdluli PS, Sibuyi NRS. Acyldepsipeptide Analogues: A Future Generation Antibiotics for Tuberculosis Treatment. Pharmaceutics. 2022; 14(9):1956. https://doi.org/10.3390/pharmaceutics14091956
Chicago/Turabian StyleCobongela, Sinazo Z. Z., Maya M. Makatini, Phumlane S. Mdluli, and Nicole R. S. Sibuyi. 2022. "Acyldepsipeptide Analogues: A Future Generation Antibiotics for Tuberculosis Treatment" Pharmaceutics 14, no. 9: 1956. https://doi.org/10.3390/pharmaceutics14091956