Hybrid Silica-Coated PLGA Nanoparticles for Enhanced Enzyme-Based Therapeutics

 ,
,
Abstract
:1. Introduction
2. Experimental Section
2.1. Preparation of Reagents
2.2. Synthesis of PLGA NPs
2.3. Washing PLGA NPs
2.4. Coating PLGA NPs with Silica
2.5. Washing SiLGA NPs
2.6. Dynamic Light Scattering Measurements of PLGA and SiLGA NPs
2.7. Nanoparticle Tracking Analysis of PLGA and SiLGA NPs
2.8. Transmission Electron Microscopy of PLGA and SiLGA NPs
2.9. Nitrocefin Assays Determine Penicillinase Activity
2.10. Intramuscular Injection of PLGA and SiLGA NPs in Mice
3. Results
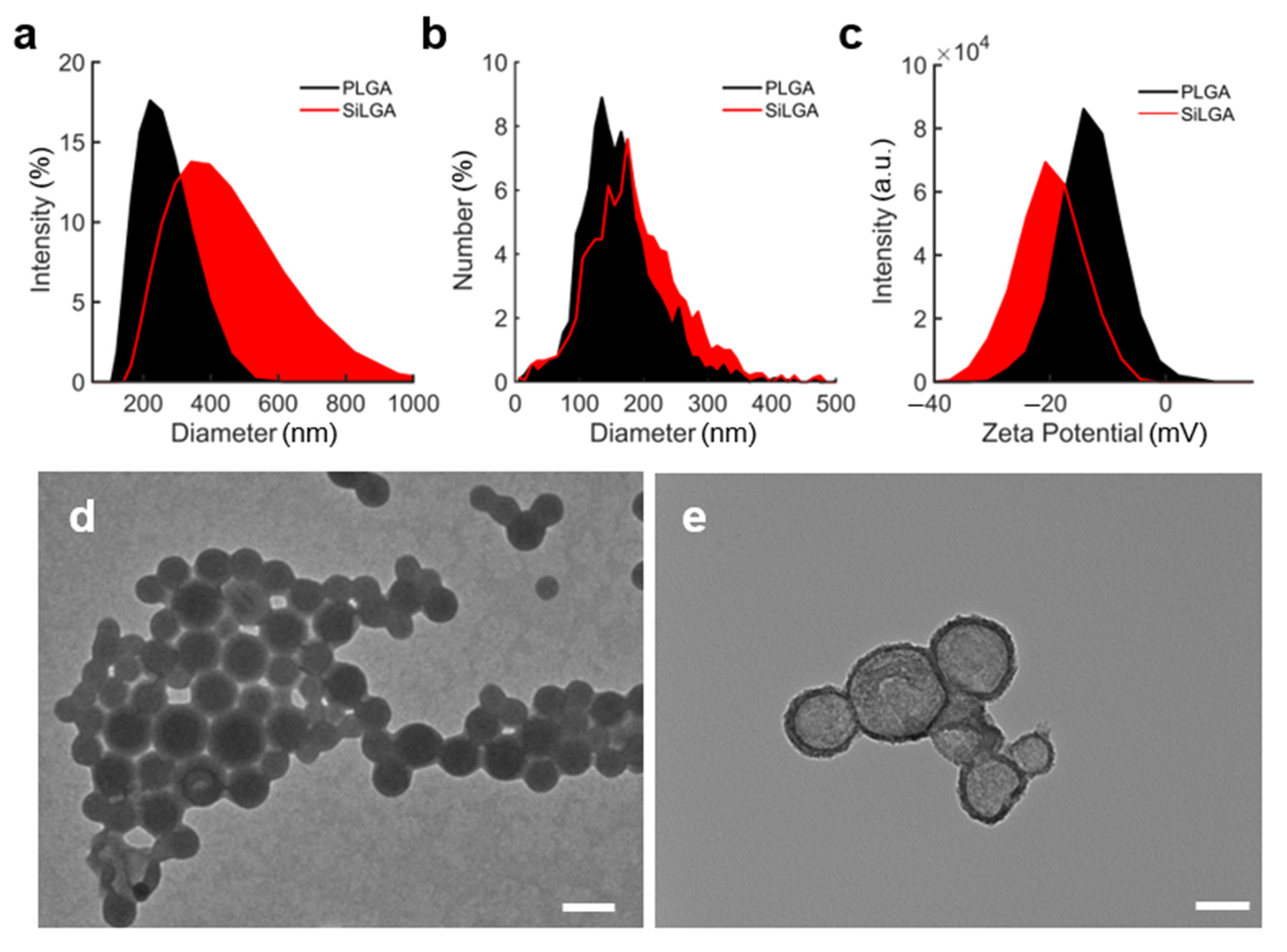
3.1. Structural Characterization of SiLGA NPs
3.2. Enzyme Loading of SiLGA NPs
3.3. SiLGA NPs Protect Enzymes from Proteolysis In Vitro
3.4. SiLGA NPs Slow the Burst Release of Enzymatic Cargo In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Diaz, G.A.; Schulze, A.; McNutt, M.C.; Leão-Teles, E.; Merritt, J.L.; Enns, G.M.; Batzios, S.; Bannick, A.; Zori, R.T.; Sloan, L.S.; et al. Clinical effect and safety profile of pegzilarginase in patients with arginase 1 deficiency. J. Inherit. Metab. Dis. 2021, 44, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Hydery, T.; Coppenrath, V.A. A Comprehensive Review of Pegvaliase, an Enzyme Substitution Therapy for the Treatment of Phenylketonuria. Drug Target Insights 2019, 13, 1177392819857089. [Google Scholar] [CrossRef] [PubMed]
- Longo, N.; Dimmock, D.; Levy, H.; Viau, K.; Bausell, H.; Bilder, D.A.; Burton, B.; Gross, C.; Northrup, H.; Rohr, F.; et al. Evidence- and consensus-based recommendations for the use of pegvaliase in adults with phenylketonuria. Genet. Med. 2019, 21, 1851–1867. [Google Scholar] [CrossRef] [Green Version]
- Longo, N.; Zori, R.; Wasserstein, M.P.; Vockley, J.; Burton, B.K.; Decker, C.; Li, M.; Lau, K.; Jiang, J.; Larimore, K.; et al. Long-term safety and efficacy of pegvaliase for the treatment of phenylketonuria in adults: Combined phase 2 outcomes through PAL-003 extension study. Orphanet J. Rare Dis. 2018, 13, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Xie, Q.; Zhou, H.; Zhang, M.; Shen, J.; Ju, D. Amino Acid Degrading Enzymes and Autophagy in Cancer Therapy. Front. Pharmacol. 2020, 11, 582587. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.A.; Syed, Y.Y.; Keam, S.J. Pegaspargase: A Review in Acute Lymphoblastic Leukaemia. Drugs 2019, 79, 767–777. [Google Scholar] [CrossRef] [Green Version]
- Keating, G.M. Asparaginase Erwinia chrysanthemi (Erwinaze(R)): A guide to its use in acute lymphoblastic leukemia in the USA. BioDrugs 2013, 27, 413–418. [Google Scholar] [CrossRef]
- Li, R.J.; Jin, R.; Liu, C.; Cao, X.; Manning, M.L.; Di, X.M.; Przepiorka, D.; Namuswe, F.; Deisseroth, A.; Goldberg, K.B.; et al. FDA Approval Summary: Calaspargase Pegol-mknl for Treatment of Acute Lymphoblastic Leukemia in Children and Young Adults. Clin. Cancer Res. 2020, 26, 328–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieters, R.; Hunger, S.P.; Boos, J.; Rizzari, C.; Silverman, L.; Baruchel, A.; Goekbuget, N.; Schrappe, M.; Pui, C.H. L-asparaginase treatment in acute lymphoblastic leukemia: A focus on Erwinia asparaginase. Cancer 2011, 117, 238–249. [Google Scholar] [CrossRef] [Green Version]
- Dinndorf, P.A.; Gootenberg, J.; Cohen, M.H.; Keegan, P.; Pazdur, R. FDA drug approval summary: Pegaspargase (oncaspar) for the first-line treatment of children with acute lymphoblastic leukemia (ALL). Oncologist 2007, 12, 991–998. [Google Scholar] [CrossRef]
- Szlosarek, P.W.; Phillips, M.M.; Pavlyk, I.; Steele, J.; Shamash, J.; Spicer, J.; Kumar, S.; Pacey, S.; Feng, X.; Johnston, A.; et al. Expansion Phase 1 Study of Pegargiminase Plus Pemetrexed and Cisplatin in Patients with Argininosuccinate Synthetase 1–Deficient Mesothelioma: Safety, Efficacy, and Resistance Mechanisms. JTO Clin. Res. Rep. 2020, 1, 100093. [Google Scholar] [CrossRef] [PubMed]
- Szlosarek, P.W.; Steele, J.P.; Nolan, L.; Gilligan, D.; Taylor, P.; Spicer, J.; Lind, M.; Mitra, S.; Shamash, J.; Phillips, M.M.; et al. Arginine Deprivation with Pegylated Arginine Deiminase in Patients with Argininosuccinate Synthetase 1-Deficient Malignant Pleural Mesothelioma: A Randomized Clinical Trial. JAMA Oncol. 2017, 3, 58–66. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Qin, S.; Ryoo, B.Y.; Lu, S.N.; Yen, C.J.; Feng, Y.H.; Lim, H.Y.; Izzo, F.; Colombo, M.; Sarker, D.; et al. Phase III randomized study of second line ADI-PEG 20 plus best supportive care versus placebo plus best supportive care in patients with advanced hepatocellular carcinoma. Ann. Oncol. 2018, 29, 1402–1408. [Google Scholar] [CrossRef]
- Harding, J.J.; Do, R.K.; Dika, I.E.; Hollywood, E.; Uhlitskykh, K.; Valentino, E.; Wan, P.; Hamilton, C.; Feng, X.; Johnston, A.; et al. A phase 1 study of ADI-PEG 20 and modified FOLFOX6 in patients with advanced hepatocellular carcinoma and other gastrointestinal malignancies. Cancer Chemother. Pharmacol. 2018, 82, 429–440. [Google Scholar] [CrossRef]
- Triplett, T.A.; Garrison, K.C.; Marshall, N.; Donkor, M.; Blazeck, J.; Lamb, C.; Qerqez, A.; Dekker, J.D.; Tanno, Y.; Lu, W.C.; et al. Reversal of indoleamine 2,3-dioxygenase-mediated cancer immune suppression by systemic kynurenine depletion with a therapeutic enzyme. Nat. Biotechnol. 2018, 36, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Cramer, S.L.; Saha, A.; Liu, J.; Tadi, S.; Tiziani, S.; Yan, W.; Triplett, K.; Lamb, C.; Alters, S.E.; Rowlinson, S.; et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med. 2017, 23, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.; van der Meer, L.T.; van Leeuwen, F.N. Amino Acid Depletion Therapies: Starving Cancer Cells to Death. Trends Endocrinol. Metab. 2021, 32, 367–381. [Google Scholar] [CrossRef]
- Yao, S.; Janku, F.; Subbiah, V.; Stewart, J.; Patel, S.P.; Kaseb, A.; Westin, S.N.; Naing, A.; Tsimberidou, A.M.; Hong, D.; et al. Phase 1 trial of ADI-PEG20 plus cisplatin in patients with pretreated metastatic melanoma or other advanced solid malignancies. Br. J. Cancer 2021, 124, 1533–1539. [Google Scholar] [CrossRef]
- Yoshioka, T.; Wada, T.; Uchida, N.; Maki, H.; Yoshida, H.; Ide, N.; Kasai, H.; Hojo, K.; Shono, K.; Maekawa, R.; et al. Anticancer Efficacy in Vivo and in Vitro, Synergy with 5-Fluorouracil, and Safety of Recombinant Methioninase. Cancer Res. 1998, 58, 2583–2587. [Google Scholar]
- Hoffman, R.T.Y.; Li, S.; Han, Q.; Zavala, J., Sr.; Zavala, J., Jr. Methionine Dependence of Cancer and Aging; Hoffman, R., Ed.; Springer Science + Business Media, LLC.: Berlin/Heidelberg, Germany, 1866; pp. 231–242. [Google Scholar]
- Harris, J.M.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003, 2, 214–221. [Google Scholar] [CrossRef]
- Dozier, J.K.; Distefano, M.D. Site-Specific PEGylation of Therapeutic Proteins. Int. J. Mol. Sci. 2015, 16, 25831–25864. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.S.X.; Xu, M.; An, Z.; Tan, X.; Tan, X.; Han, Q.; Miljkovic, D.A.; Yang, M.; Hoffman, R.M. Polyethylene Glycol Conjugation of Recombinant Methioninase for Cancer Therapy. Protein Expr. Purif. 1998, 12, 45–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damodaran, V.F.; Fee, C. Protein PEGylation: An overview of chemistry process considerations. Eur. Pharm. Rev. 2010, 15, 18–26. [Google Scholar]
- Phillips, E.; Penate-Medina, O.; Zanzonico, P.B.; Carvajal, R.D.; Mohan, P.; Ye, Y.; Humm, J.; Gönen, M.; Kalaigian, H.; Schöder, H.; et al. Clinical translation of an ultrasmall inorganic optical-PET imaging nanoparticle probe. Sci. Transl. Med. 2014, 6, 260ra149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang Thi, T.T.; Pilkington, E.H.; Nguyen, D.H.; Lee, J.S.; Park, K.D.; Truong, N.P. The Importance of Poly(ethylene glycol) Alternatives for Overcoming PEG Immunogenicity in Drug Delivery and Bioconjugation. Polymers 2020, 12, 298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Lai, S.K. Anti-PEG immunity: Emergence, characteristics, and unaddressed questions. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2015, 7, 655–677. [Google Scholar] [CrossRef] [Green Version]
- McSweeney, M.D.; Versfeld, Z.C.; Carpenter, D.M.; Lai, S.K. Physician Awareness of Immune Responses to Polyethylene Glycol-Drug Conjugates. Clin. Transl. Sci. 2018, 11, 162–165. [Google Scholar] [CrossRef]
- Kozma, G.T.; Shimizu, T.; Ishida, T.; Szebeni, J. Anti-PEG antibodies: Properties, formation, testing and role in adverse immune reactions to PEGylated nano-biopharmaceuticals. Adv. Drug Deliv. Rev. 2020, 154, 163–175. [Google Scholar] [CrossRef]
- Huckaby, J.T.; Jacobs, T.M.; Li, Z.; Perna, R.J.; Wang, A.; Nicely, N.I.; Lai, S.K. Structure of an anti-PEG antibody reveals an open ring that captures highly flexible PEG polymers. Commun. Chem. 2020, 3, 124. [Google Scholar] [CrossRef]
- Ju, Y.; Lee, W.S.; Pilkington, E.H.; Kelly, H.G.; Li, S.; Selva, K.J.; Wragg, K.M.; Subbarao, K.; Nguyen, T.H.; Rowntree, L.C.; et al. Anti-PEG Antibodies Boosted in Humans by SARS-CoV-2 Lipid Nanoparticle mRNA Vaccine. ACS Nano 2022, 16, 11769–11780. [Google Scholar] [CrossRef]
- Verma, A.; Chen, K.; Bender, C.; Gorney, N.; Leonard, W.; Barnette, P. PPEGylated E. coli asparaginase desensitization: An effective and feasible option for pediatric patients with acute lymphoblastic leukemia who have developed hypersensitivity to pegaspargase in the absence of asparaginase Erwinia chrysanthemi availability. Pediatr. Hematol. Oncol. 2019, 36, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Veronese, F.M. Peptide and protein PEGylation-a review of problems and solutions. Biomaterials 2001, 22, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y.; McCormack, P.L. Exenatide Extended-Release: An Updated Review of Its Use in Type 2 Diabetes Mellitus. Drugs 2015, 75, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Operti, M.C.; Bernhardt, A.; Grimm, S.; Engel, A.; Figdor, C.G.; Tagit, O. PLGA-based nanomedicines manufacturing: Technologies overview and challenges in industrial scale-up. Int. J. Pharm. 2021, 605, 120807. [Google Scholar] [CrossRef] [PubMed]
- Donnez, J.; Dewart, P.J.; Hedon, B.; Perino, A.; Schindler, A.E.; Blumberg, J.; Querleu, D. Equivalence of the 3-month and 28-day formulations of triptorelin with regard to achievement and maintenance of medical castration in women with endometriosis. Fertil. Steril. 2004, 81, 297–304. [Google Scholar] [CrossRef]
- Mundargi, R.C.; Babu, V.R.; Rangaswamy, V.; Patel, P.; Aminabhavi, T.M. Nano/micro technologies for delivering macromolecular therapeutics using poly(D,L-lactide-co-glycolide) and its derivatives. J. Control Release 2008, 125, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Chan, G.; Hu, Y.; Hu, H.; Ouyang, D. A Comprehensive Map of FDA-Approved Pharmaceutical Products. Pharmaceutics 2018, 10, 263. [Google Scholar] [CrossRef] [Green Version]
- Genovese, S.; Mannucci, E.; Ceriello, A. A Review of the Long-Term Efficacy, Tolerability, and Safety of Exenatide Once Weekly for Type 2 Diabetes. Adv. Ther. 2017, 34, 1791–1814. [Google Scholar] [CrossRef] [Green Version]
- Singhal, A.; Morris, V.B.; Labhasetwar, V.; Ghorpade, A. Nanoparticle-mediated catalase delivery protects human neurons from oxidative stress. Cell Death Dis. 2013, 4, e903. [Google Scholar] [CrossRef] [Green Version]
- Hua, Y.; Su, Y.; Zhang, H.; Liu, N.; Wang, Z.; Gao, X.; Gao, J.; Zheng, A. Poly(lactic-co-glycolic acid) microsphere production based on quality by design: A review. Drug Deliv. 2021, 28, 1342–1355. [Google Scholar] [CrossRef]
- Lu, B.; Lv, X.; Le, Y. Chitosan-Modified PLGA Nanoparticles for Control-Released Drug Delivery. Polymers 2019, 11, 304. [Google Scholar] [CrossRef]
- Chen, H.; Xie, L.Q.; Qin, J.; Jia, Y.; Cai, X.; Nan, W.; Yang, W.; Lv, F.; Zhang, Q.Q. Surface modification of PLGA nanoparticles with biotinylated chitosan for the sustained in vitro release and the enhanced cytotoxicity of epirubicin. Colloids Surf. B Biointerfaces 2016, 138, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lima, I.A.; Khalil, N.M.; Tominaga, T.T.; Lechanteur, A.; Sarmento, B.; Mainardes, R.M. Mucoadhesive chitosan-coated PLGA nanoparticles for oral delivery of ferulic acid. Artif. Cell. Nanomed. Biotechnol. 2018, 46, 993–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annenkov Vadim, V.; Danilovtseva, E.N.; Pal’shin, V.A.; Verkhozina, O.g.N.; Zelinskiy, S.N.; Krishnan, U.M. Silicic acid condensation under the influence of water-soluble polymers: From biology to new materials. RSC Adv. 2017, 7, 20995–21027. [Google Scholar] [CrossRef] [Green Version]
- Ortac, I.; Simberg, D.; Yeh, Y.S.; Yang, J.; Messmer, B.; Trogler, W.C.; Tsien, R.Y.; Esener, S. Dual-porosity hollow nanoparticles for the immunoprotection and delivery of nonhuman enzymes. Nano Lett. 2014, 14, 3023–3032. [Google Scholar] [CrossRef]
- Sapre, A.A.; Yong, G.; Ruff, L.E.; Plaut, J.S.; Sayar, Z.; Agarwal, A.; Martinez, J.; Nguyen, T.N.; Liu, Y.T.; Messmer, B.T.; et al. Silica cloaking of adenovirus enhances gene delivery while reducing immunogenicity. J. Control. Release 2019, 297, 48–59. [Google Scholar] [CrossRef]
- Yang, J.; Lind, J.U.; Trogler, W.C. Synthesis of Hollow Silica and Titania Nanospheres. Chem. Mater. 2008, 20, 2875–2877. [Google Scholar] [CrossRef]
- Shen, S.; Wu, Y.; Liu, Y.; Wu, D. High drug-loading nanomedicines: Progress, current status, and prospects. Int. J. Nanomed. 2017, 12, 4085–4109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alibolandi, M.; Farzad, S.A.; Mohammadi, M.; Abnous, K.; Taghdisi, S.M.; Kalalinia, F.; Ramezani, M. Tetrac-decorated chitosan-coated PLGA nanoparticles as a new platform for targeted delivery of SN38. Artif. Cell. Nanomed. Biotechnol. 2018, 46, 1003–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maggi, M.; Mittelman, S.D.; Parmentier, J.H.; Colombo, G.; Meli, M.; Whitmire, J.M.; Merrell, D.S.; Whitelegge, J.; Scotti, C. A protease-resistant Escherichia coli asparaginase with outstanding stability and enhanced anti-leukaemic activity in vitro. Sci. Rep. 2017, 7, 14479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zolnik, B.S.; Leary, P.E.; Burgess, D.J. Elevated temperature accelerated release testing of PLGA microspheres. J. Control. Release 2006, 112, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Faisant, N.; Siepmann, J.; Benoit, J. PLGA-based microparticles: Elucidation of mechanisms and a new, simple mathematical model quantifying drug release. Eur. J. Pharm. Sci. 2002, 115, 355–366. [Google Scholar]
- Keles, H.; Naylor, A.; Clegg, F.; Sammon, C. Investigation of factors influencing the hydrolytic degradation of single PLGA microparticles. Polym. Degrad. Stab. 2015, 119, 228–241. [Google Scholar] [CrossRef] [Green Version]
- Tracy, M.A.; Ward, K.L.; Firouzabadian, L.; Wang, Y.; Dong, N.; Qian, R.; Zhang, Y. Factors affecting the degradation rate of poly(lactide-co-glycolide) microspheres in vivo and in vitro. Biomaterials 1999, 20, 1057–1062. [Google Scholar] [CrossRef]
- Chiu, M.; Taurino, G.; Bianchi, M.G.; Kilberg, M.S.; Bussolati, O. Asparagine Synthetase in Cancer: Beyond Acute Lymphoblastic Leukemia. Front. Oncol. 2019, 9, 1480. [Google Scholar] [CrossRef] [PubMed]
- Brito, A.E.M.; Pessoa, A., Jr.; Converti, A.; Rangel-Yagui, C.O.; Silva, J.A.D.; Apolinario, A.C. Poly (lactic-co-glycolic acid) nanospheres allow for high l-asparaginase encapsulation yield and activity. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 98, 524–534. [Google Scholar] [CrossRef]
- Singh, M.; Hassan, N.; Verma, D.; Thakur, P.; Panda, B.P.; Panda, A.K.; Sharma, R.K.; Mirza, A.; Mansoor, S.; Alrokayan, S.H.; et al. Design of expert guided investigation of native L-asparaginase encapsulated long-acting cross-linker-free poly (lactic-co-glycolic) acid nanoformulation in an Ehrlich ascites tumor model. Saudi Pharm. J. 2020, 28, 719–728. [Google Scholar] [CrossRef]
- Quesada, M.; Muniesa, C.; Botella, P. Hybrid PLGA-Organosilica Nanoparticles with Redox-Sensitive Molecular Gates. Chem. Mater. 2013, 25, 2597–2602. [Google Scholar] [CrossRef]
- Heggannavar, G.B.; Vijeth, S.; Kariduraganavar, M.Y. Development of dual drug loaded PLGA based mesoporous silica nanoparticles and their conjugation with Angiopep-2 to treat glioma. J. Drug Deliv. Sci. Technol. 2019, 53, 101157. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Active Enzyme | Enzyme Formulation(s) | FDA Status | Disease(s) | Reference(s) |
|---|---|---|---|---|
| asparaginase | colaspase; crisantaspase; pegaspargase; calaspargase pegol-mknl | approved | acute lymphoblastic leukemia; lymphoblastic lymphoma | [5,6,7,8,9,10,17,21,22,24] |
| phenylalanine ammonia lyase | pegvaliase | approved | phenylketonuria | [2,3,4,5] |
| arginine deiminase | pegargiminase; ADI-PEG 20 | phase 2 | metastatic melanoma; hepatocellular carcinoma; mesothelioma | [5,11,12,13,14,17,18] |
| arginase | pegzilarginase | phase 3; phase 1 | Arginase 1 deficiency; melanoma, leukemia, lymphoma | [1,5,17] |
| methioninase | phase 1 | lung carcinoma, breast cancer, renal cancer, lymphoma | [5,19,20,23] | |
| kynureninase | PEG-KYNase | preclinical research | melanoma, breast carcinoma, colon carcinoma | [15] |
| cysteinase | preclinical research | prostate cancer, breast cancer | [16] |
| Commercial Name(s) | Active Ingredient | Disease(s) | Reference(s) |
|---|---|---|---|
| Decapeptyl®; Decapeptyl™ SR; Trelstar™ Depot | triptorelin | prostate cancer, endometriosis | [36,37,38,39] |
| Bydureon®; Bydureon Bcise® | exenatide | type 2 diabetes | [35,39,40] |
| Sandostatin LAR®; Sandostatin LAR® Depot | octreotide | acromegaly | [36,38,39] |
| Suprecur® MP | buserelin | prostate cancer, endometriosis | [36,38] |
| Somatuline® LA | lanreotide | acromegaly | [36,38,39] |
| Lupron Depot® | leuprolide | prostate cancer | [36,38,39] |
| Nutropin Depot® | somatropin | pediatric growth hormone deficiency | [36,38] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gustafson, K.T.; Mokhtari, N.; Manalo, E.C.; Montoya Mira, J.; Gower, A.; Yeh, Y.-S.; Vaidyanathan, M.; Esener, S.C.; Fischer, J.M. Hybrid Silica-Coated PLGA Nanoparticles for Enhanced Enzyme-Based Therapeutics. Pharmaceutics 2023, 15, 143. https://doi.org/10.3390/pharmaceutics15010143
Gustafson KT, Mokhtari N, Manalo EC, Montoya Mira J, Gower A, Yeh Y-S, Vaidyanathan M, Esener SC, Fischer JM. Hybrid Silica-Coated PLGA Nanoparticles for Enhanced Enzyme-Based Therapeutics. Pharmaceutics. 2023; 15(1):143. https://doi.org/10.3390/pharmaceutics15010143
Chicago/Turabian StyleGustafson, Kyle T., Negin Mokhtari, Elise C. Manalo, Jose Montoya Mira, Austin Gower, Ya-San Yeh, Mukanth Vaidyanathan, Sadik C. Esener, and Jared M. Fischer. 2023. "Hybrid Silica-Coated PLGA Nanoparticles for Enhanced Enzyme-Based Therapeutics" Pharmaceutics 15, no. 1: 143. https://doi.org/10.3390/pharmaceutics15010143