
Pediatric Drug Development: Reviewing Challenges and Opportunities by Tracking Innovative Therapies

 ,
, 



Abstract
1. Introduction
2. Study Conception
3. Pediatric Drug Development: The Paradigm Is Shifting
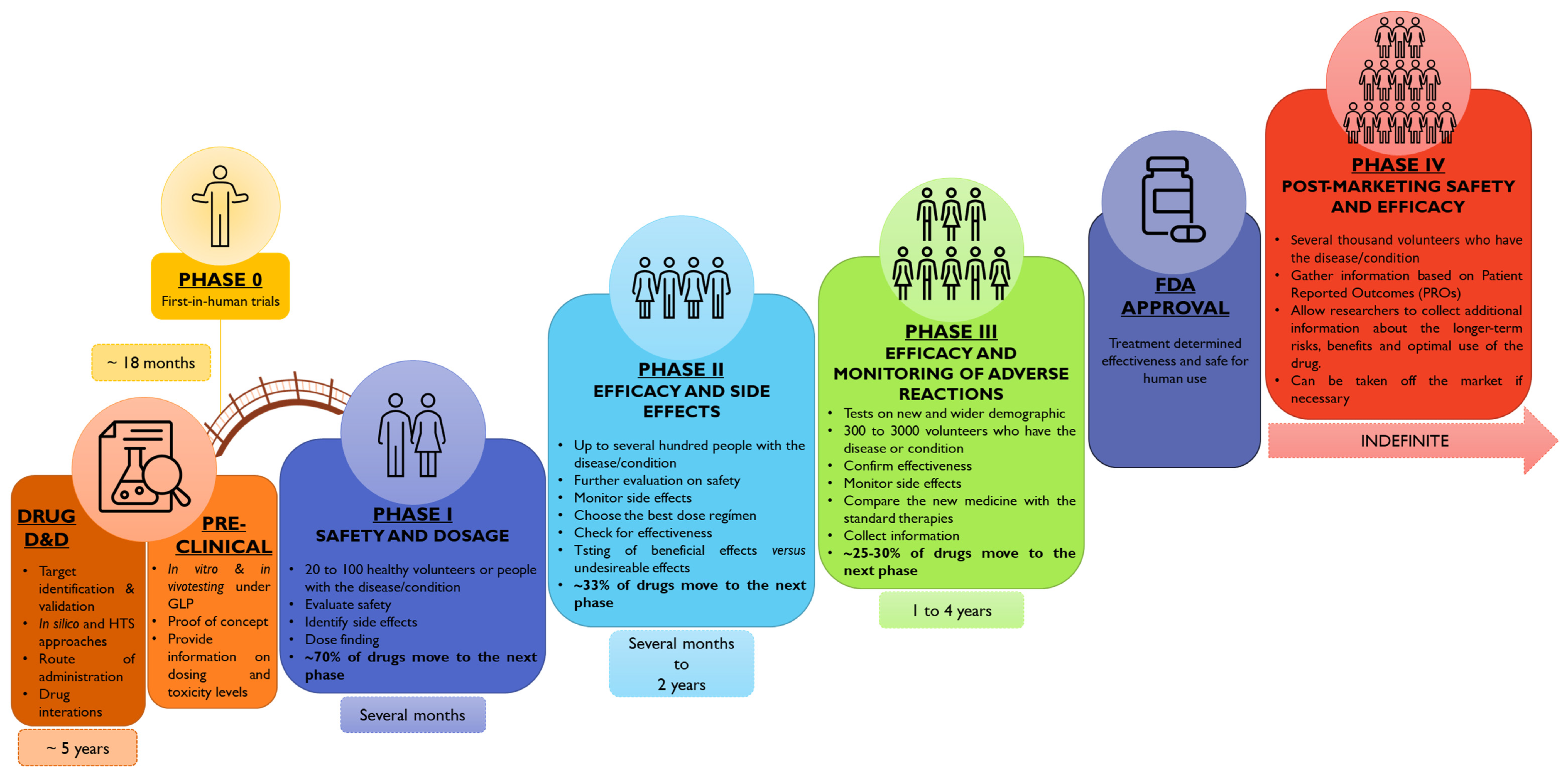
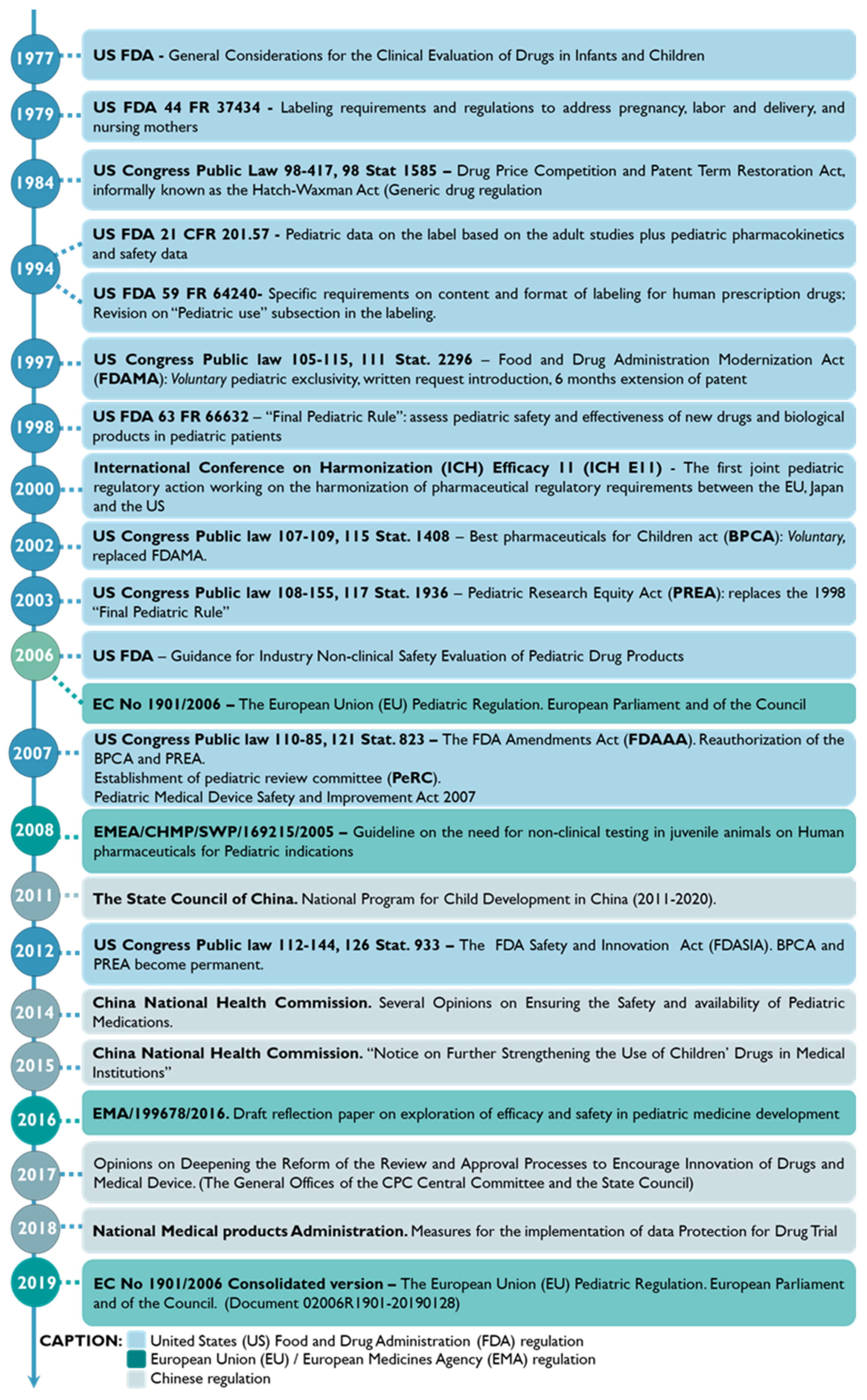
3.1. Snapshot into the Pediatric Drug Development History
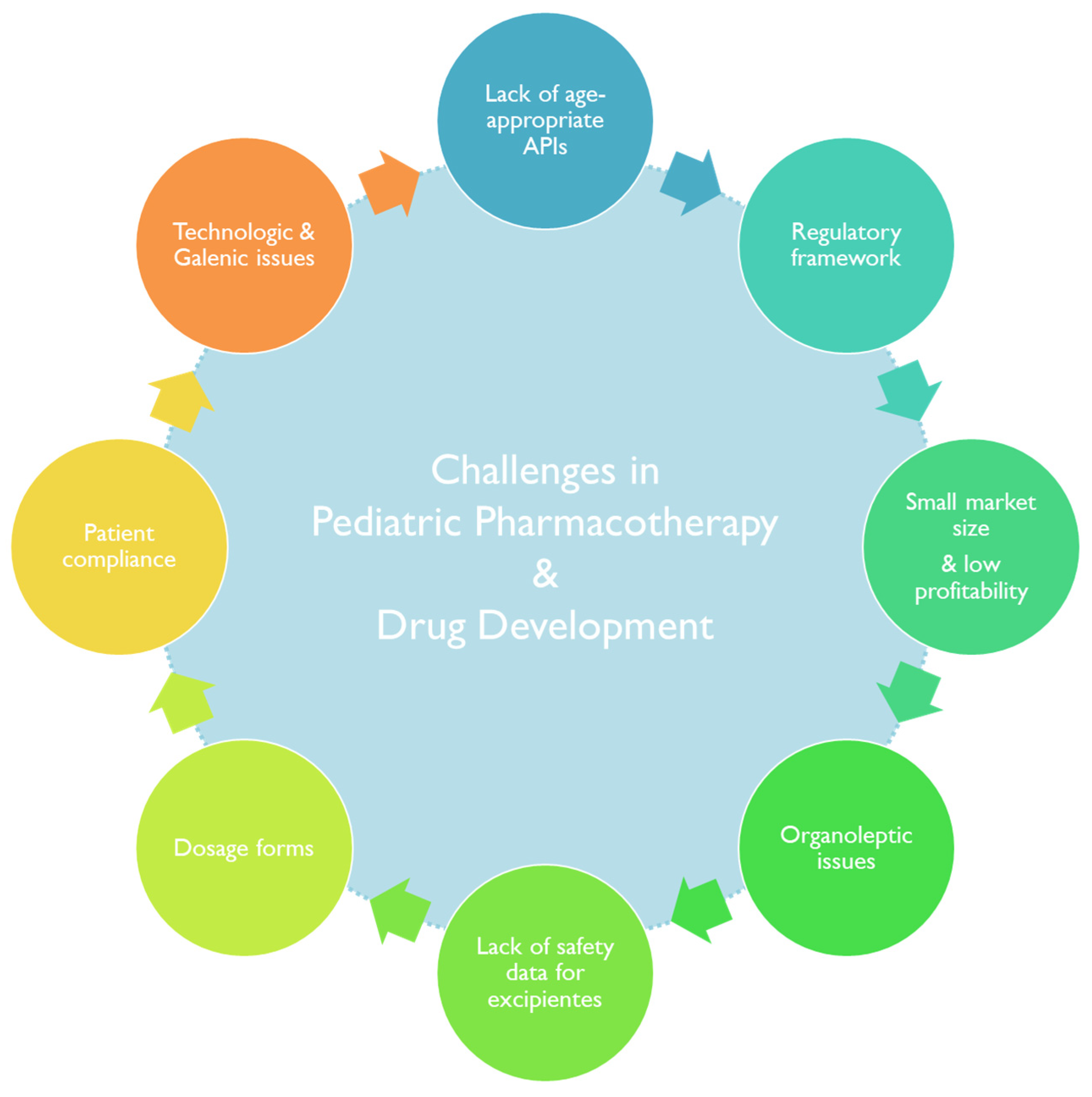
3.2. Constrains in Drug Development for Pediatric Patients
3.2.1. Investments in Pediatric Drug Development and Market Trends
3.2.2. Lack of Approved Active Principal Ingredients for Pediatric-Age Patients
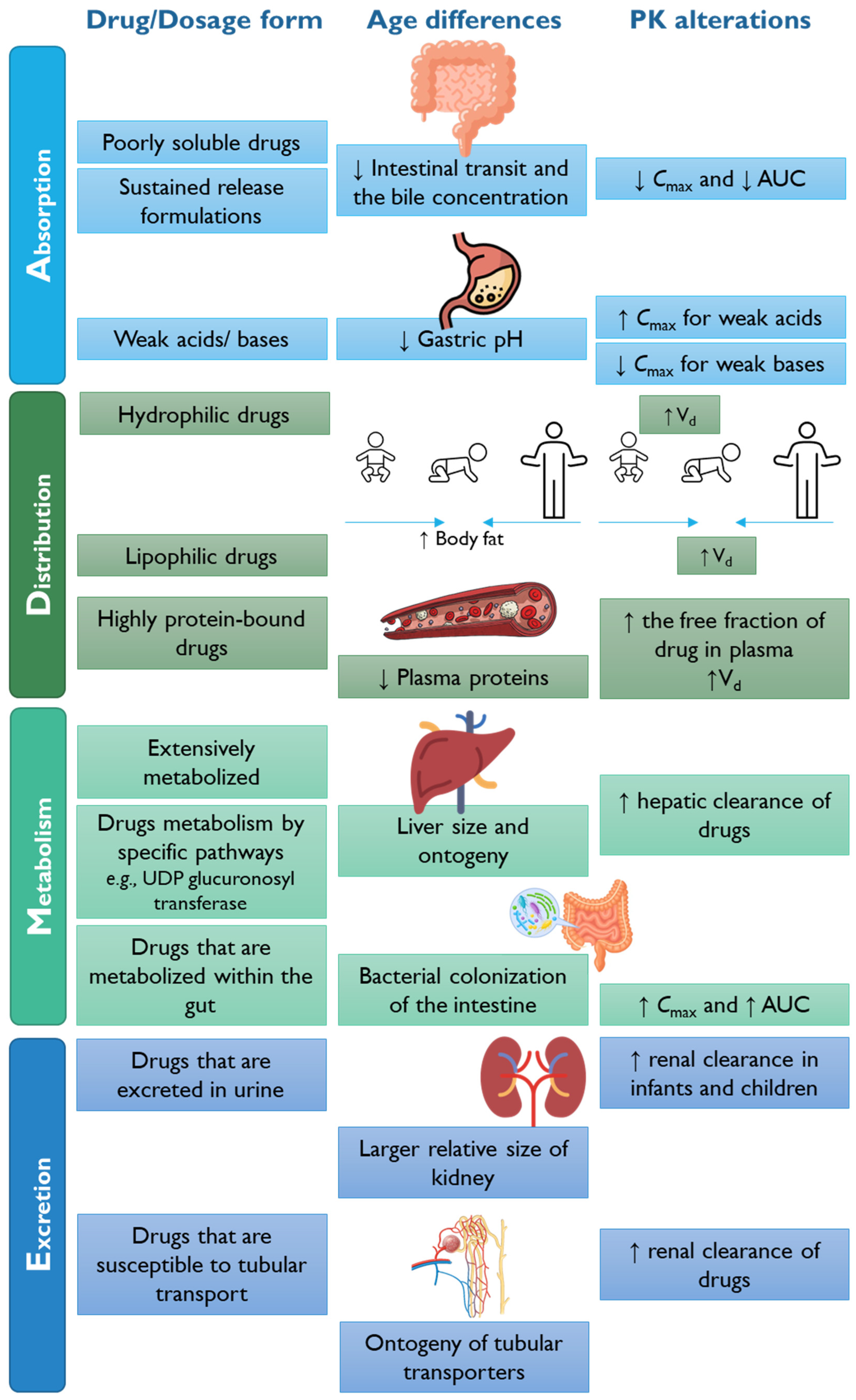
3.2.3. Lack of Pharmacokinetic and Pharmacodynamic Data
3.2.4. Administration Route and Pharmaceutical Dosage Forms in Pediatrics
3.2.5. Excipients
3.2.6. Pediatric Patient Acceptability
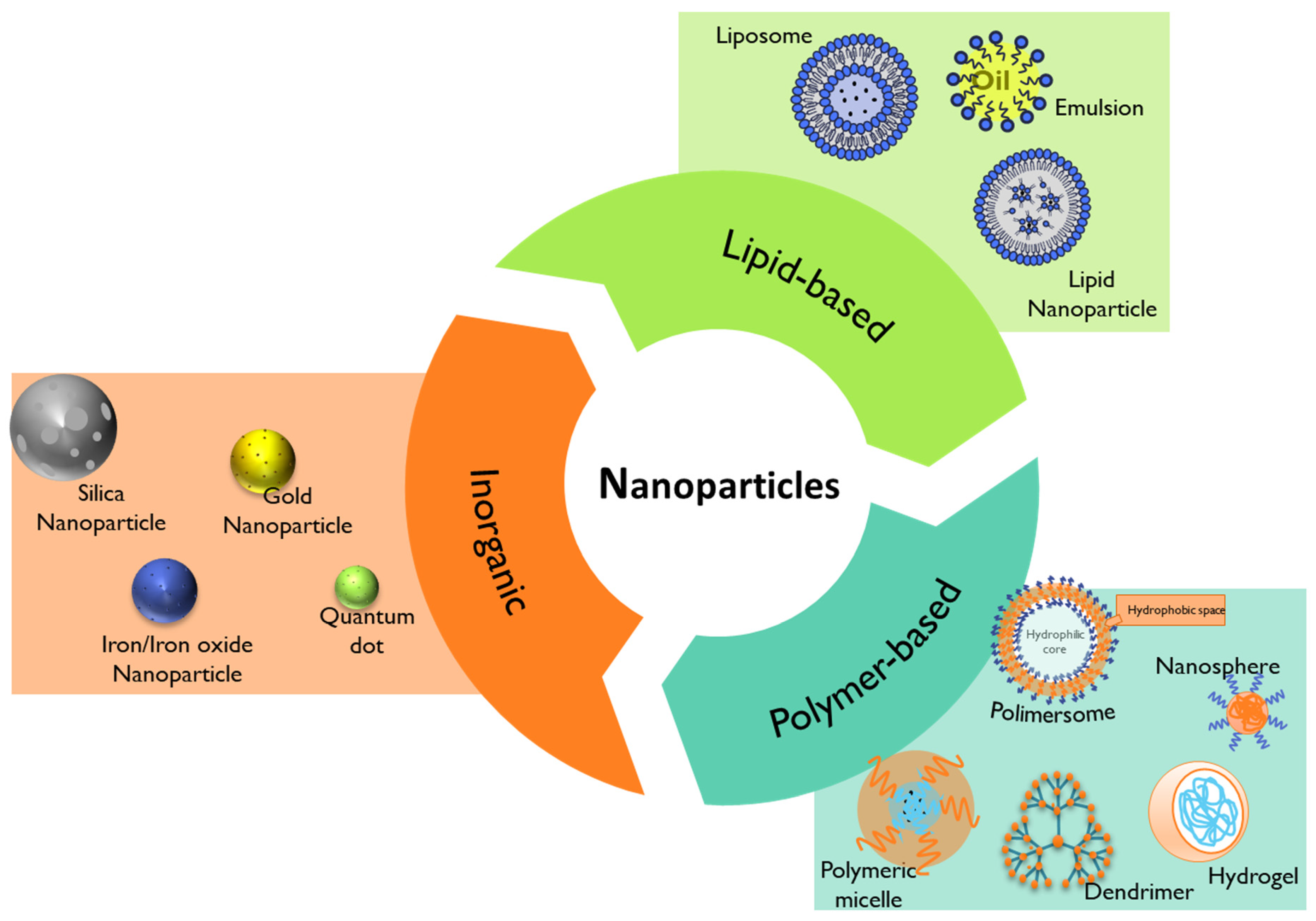
4. Nanomedicine for Pediatric Healthcare
4.1. Lipid-Based Nanoparticles
4.2. Polymer-Based Nanoparticles
4.2.1. Polymeric Micelles
4.2.2. Dendrimers
4.3. Inorganic Nanoparticles
4.4. Challenges in Using Nanotherapy in Pediatrics
5. Advanced Therapy Medicinal Products (ATMPs) for Pediatric Healthcare
5.1. ATMPs—Legal Framework in the European Union
5.2. FDA and EMA-Approved ATMPs in Pediatrics
5.3. Gene Therapy
5.3.1. GTMPs—Guidelines on Quality, Pre-Clinical, and Clinical Aspects
5.4. Cell Therapy
5.4.1. Chimeric Antigen Receptor T Cell Therapy
5.5. Tissue-Engineered Products
5.6. Combined ATMPs
6. Future Perspectives and Final Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Rimsza, M.E.; Hotaling, C.A.J.; Keown, M.E.; Marcin, J.P.; Moskowitz, W.B.; Sigrest, T.D.; Simon, H.K. Definition of a Pediatrician. Pediatrics 2015, 135, 780–781. [Google Scholar] [CrossRef]
- Appropriate ICH Expert Working Group. E11(R1) Addendum: Clinical Investigation of Medicinal Products in the Pediatric Population; Adopted on 18 August 2017. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/e11r1-addendum-clinical-investigation-medicinal-products-pediatric-population (accessed on 12 July 2023).
- Sawyer, S.M.; McNeil, R.; Francis, K.L.; Matskarofski, J.Z.; Patton, G.C.; Bhutta, Z.A.; Esangbedo, D.O.; Klein, J.D. The age of paediatrics. Lancet Child Adolesc. Health 2019, 3, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Hardin, A.P.; Hackell, J.M.; Simon, G.R.; Boudreau, A.D.A.; Baker, C.N.; Barden, G.A.; Meade, K.E.; Moore, S.B.; Richerson, J.; Brown, O.W.; et al. Age limit of pediatrics. Pediatrics 2017, 140, e20172151. [Google Scholar] [CrossRef]
- Maheshwari, M.; Sanwatsarkar, S.; Katakwar, M. Pharmacology related to paediatric anaesthesia. Indian J. Anaesth. 2019, 63, 698. [Google Scholar] [CrossRef]
- Ernest, T.B.; Elder, D.P.; Martini, L.G.; Roberts, M.; Ford, J.L. Developing paediatric medicines: Identifying the needs and recognizing the challenges. J. Pharm. Pharmacol. 2010, 59, 1043–1055. [Google Scholar] [CrossRef]
- Batchelor, H.K.; Marriott, J.F. Formulations for children: Problems and solutions. Br. J. Clin. Pharmacol. 2015, 79, 405–418. [Google Scholar] [CrossRef]
- O’Brien, F.; Clapham, D.; Krysiak, K.; Batchelor, H.; Field, P.; Caivano, G.; Pertile, M.; Nunn, A.; Tuleu, C. Making medicines baby size: The challenges in bridging the formulation gap in neonatal medicine. Int. J. Mol. Sci. 2019, 20, 2688. [Google Scholar] [CrossRef]
- Walsh, J.; Schaufelberger, D.; Iurian, S.; Klein, S.; Batchelor, H.; Turner, R.; Gizurarson, S.; Boltri, L.; Alessandrini, E.; Tuleu, C. Path towards Efficient Paediatric Formulation Development Based on Partnering with Clinical Pharmacologists and Clinicians, a Conect4children Expert Group white Paper; John Wiley and Sons Inc.: Hoboken, NJ, USA, 2021; p. bcp.14989. [Google Scholar]
- Vieira, I.; Sousa, J.J.; Vitorino, C. Paediatric Medicines—Regulatory Drivers, Restraints, Opportunities and Challenges. J. Pharm. Sci. 2021, 110, 1545–1556. [Google Scholar] [CrossRef]
- Ogbonna, J.D.N.; Cunha, E.; Attama, A.A.; Ofokansi, K.C.; Ferreira, H.; Pinto, S.; Gomes, J.; Marx, Í.M.G.; Peres, A.M.; Lobo, J.M.S.; et al. Overcoming Challenges in Pediatric Formulation with a Patient-Centric Design Approach: A Proof-of-Concept Study on the Design of an Oral Solution of a Bitter Drug. Pharmaceuticals 2022, 15, 1331. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Development of Paediatric Medicines: Points to Consider in Formulation. Available online: https://www.who.int/publications/m/item/trs970-annex-5-development-of-paediatric-medicines-points-to-consider-in-formulation (accessed on 12 July 2023).
- Salunke, S.; Giacoia, G.; Tuleu, C. The STEP (Safety and Toxicity of Excipients for Paediatrics) database. Part 1—A need assessment study. Int. J. Pharm. 2012, 435, 101–111. [Google Scholar] [CrossRef]
- Rouaz, K.; Chiclana-Rodríguez, B.; Nardi-Ricart, A.; Suñé-Pou, M.; Mercadé-Frutos, D.; Suñé-Negre, J.M.; Pérez-Lozano, P.; García-Montoya, E. Excipients in the paediatric population: A review. Pharmaceutics 2021, 13, 387. [Google Scholar] [CrossRef] [PubMed]
- Salunke, S.; Brandys, B.; Giacoia, G.; Tuleu, C. The STEP (Safety and Toxicity of Excipients for Paediatrics) database: Part 2—The pilot version. Int. J. Pharm. 2013, 457, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Meyers, R.S.; Thackray, J.; Matson, K.L.; McPherson, C.; Lubsch, L.; Hellinga, R.C.; Hoff, D.S. Key potentially inappropriate drugs in pediatrics: The KIDs list. J. Pediatr. Pharmacol. Ther. 2020, 25, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Rose, K.; Grant-Kels, J.M. The Meanings of “Pediatric Drug Development”. Ther. Innov. Regul. Sci. 2019, 53, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Nishiwaki, S.; Ando, Y. Gap between pediatric and adult approvals of molecular targeted drugs. Sci. Rep. 2020, 10, 17145. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, J.C. The Scarcity of Approved Pediatric High-Risk Medical Devices. JAMA Netw. Open 2021, 4, e2112760. [Google Scholar] [CrossRef]
- Domingues, C.; Santos, A.; Alvarez-Lorenzo, C.; Concheiro, A.; Jarak, I.; Veiga, F.; Barbosa, I.; Dourado, M.; Figueiras, A. Where Is Nano Today and Where Is It Headed? A Review of Nanomedicine and the Dilemma of Nanotoxicology. ACS Nano 2022, 16, 9994–10041. [Google Scholar] [CrossRef]
- Marques, M.S.; Lima, L.A.; Poletto, F.; Contri, R.V.; Kulkamp Guerreiro, I.C. Nanotechnology for the treatment of paediatric diseases: A review. J. Drug Deliv. Sci. Technol. 2022, 75, 103628. [Google Scholar] [CrossRef]
- Pires, L.R.; Vinayakumar, K.B.; Turos, M.; Miguel, V.; Gaspar, J. A Perspective on Microneedle-Based Drug Delivery and Diagnostics in Paediatrics. J. Pers. Med. 2019, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Scientific Foresight (STOA). Therapies for the Future—Advanced Therapies & Nanomedicine. Available online: https://epthinktank.eu/2017/11/22/therapies-for-the-future-advanced-therapies-nanomedicine/ (accessed on 17 July 2023).
- Pizevska, M.; Kaeda, J.; Fritsche, E.; Elazaly, H.; Reinke, P.; Amini, L. Advanced Therapy Medicinal Products’ Translation in Europe: A Developers’ Perspective. Front. Med. 2022, 9, 757647. [Google Scholar] [CrossRef]
- Lederer, C.W.; Koniali, L.; Buerki-Thurnherr, T.; Papasavva, P.L.; La Grutta, S.; Licari, A.; Staud, F.; Bonifazi, D.; Kleanthous, M. Catching Them Early: Framework Parameters and Progress for Prenatal and Childhood Application of Advanced Therapies. Pharmaceutics 2022, 14, 793. [Google Scholar] [CrossRef]
- Kimland, E.; Odlind, V. Off-label drug use in pediatric patients. Clin. Pharmacol. Ther. 2012, 91, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Bucci-Rechtweg, C. Enhancing the Pediatric Drug Development Framework to Deliver Better Pediatric Therapies Tomorrow. Clin. Ther. 2017, 39, 1920–1932. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.A.; Catapano, M.; Hirschfeld, S.; Giaquinto, C. Paediatric drug development: The impact of evolving regulations. Adv. Drug Deliv. Rev. 2014, 73, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Coppes, M.J.; Jackson, C.; Connor, E.M. I-ACT for Children: Helping close the gap in drug approval for adults and children. Pediatr. Res. 2022, 93, 1786–1787. [Google Scholar] [CrossRef] [PubMed]
- Burckart, G.J.; Kim, C. The Revolution in Pediatric Drug Development and Drug Use: Therapeutic Orphans No More. J. Pediatr. Pharmacol. Ther. 2020, 25, 565. [Google Scholar] [CrossRef]
- Greene, J.A.; Podolsky, S.H. Reform, Regulation, and Pharmaceuticals—The Kefauver–Harris Amendments at 50. N. Engl. J. Med. 2012, 367, 1481–1483. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration (FDA). The Drug Development Process. Available online: https://www.fda.gov/patients/learn-about-drug-and-device-approvals/drug-development-process (accessed on 11 January 2022).
- Réda, C.; Kaufmann, E.; Delahaye-Duriez, A. Machine learning applications in drug development. Comput. Struct. Biotechnol. J. 2020, 18, 241–252. [Google Scholar] [CrossRef]
- Fernandez, E.; Perez, R.; Hernandez, A.; Tejada, P.; Arteta, M.; Ramos, J.T. Factors and Mechanisms for Pharmacokinetic Differences between Pediatric Population and Adults. Pharmaceutics 2011, 3, 53. [Google Scholar] [CrossRef]
- Subramanian, D.; Cruz, C.V.; Garcia-Bournissen, F. Systematic Review of Early Phase Pediatric Clinical Pharmacology Trials. J. Pediatr. Pharmacol. Ther. 2022, 27, 609. [Google Scholar] [CrossRef]
- American Academy of Pediatrics; Committee on Drugs. Guidelines for the ethical conduct of studies to evaluate drugs in pediatric populations. Pediatrics 1977, 60, 91–101. [Google Scholar] [CrossRef]
- Severin, T.; Corriol-Rohou, S.; Bucci-Rechtweg, C.; an Haack, K.; Fuerst-Recktenwald, S.; Lepola, P.; Norjavaara, E.; Dehlinger-Kremer, M.; Haertter, S.; Cheung, S.Y.A. How is the Pharmaceutical Industry Structured to Optimize Pediatric Drug Development? Existing Pediatric Structure Models and Proposed Recommendations for Structural Enhancement. Ther. Innov. Regul. Sci. 2020, 54, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Rose, K. The Challenges of Pediatric Drug Development. Curr. Ther. Res. Clin. Exp. 2019, 90, 128–134. [Google Scholar] [CrossRef]
- Wu, W.; Tang, Z.; Chen, J.; Gao, Y. Pediatric drug development in China: Reforms and challenges. Pharmacol. Res. 2019, 148, 104412. [Google Scholar] [CrossRef]
- Joseph, P.D.; Craig, J.C.; Caldwell, P.H.Y. Clinical trials in children. Br. J. Clin. Pharmacol. 2015, 79, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Shi, F.H.; Huang, S.Y.; Zhang, S.G.; Chen, H.W. The best pharmaceuticals for children—What can we do? Transl. Pediatr. 2020, 9, 86–92. [Google Scholar] [CrossRef]
- The European Parliament and the Council EUR-Lex-02006R1901-20190128-EN. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A02006R1901-20190128 (accessed on 11 January 2022).
- Grand View Research (GVR) Pharmaceutical Manufacturing Market Size Report, 2021–2028. Available online: https://www.grandviewresearch.com/industry-analysis/pharmaceutical-manufacturing-market (accessed on 29 June 2022).
- Milne, C.P. More Efficient Compliance with European Medicines Agency and Food and Drug Administration Regulations for Pediatric Oncology Drug Development: Problems and Solutions. Clin. Ther. 2017, 39, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Zhang, X.; Zhou, L.; Li, L.; Zhang, T. Updated analysis of pediatric clinical studies registered in ClinicalTrials.gov, 2008–2019. BMC Pediatr. 2021, 21, 212. [Google Scholar] [CrossRef]
- EU Clinical Trials Register. Clinical Trials Register, Age Range: “Under 18”. Available online: https://www.clinicaltrialsregister.eu/ctr-search/search (accessed on 14 August 2021).
- Van der Gronde, T.; Uyl-de Groot, C.A.; Pieters, T. Addressing the challenge of high-priced prescription drugs in the era of precision medicine: A systematic review of drug life cycles, therapeutic drug markets and regulatory frameworks. PLoS ONE 2017, 12, e0182613. [Google Scholar] [CrossRef]
- PwC Health Research Institute. Medical Cost Trend: Behind the Numbers 2022: PwC. Available online: https://www.pwc.com/us/en/industries/health-industries/library/behind-the-numbers.html (accessed on 28 January 2022).
- Speer, E.M.; Lee, L.K.; Bourgeois, F.T.; Gitterman, D.; Hay, W.W.; Davis, J.M.; Javier, J.R. The state and future of pediatric research—An introductory overview. Pediatr. Res. 2023, 2023, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Intelligence, M. Pediatric Drugs Market Size & Share Analysis—Industry Research Report—Growth Trends. Available online: https://www.mordorintelligence.com/industry-reports/pediatric-drugs-market (accessed on 3 August 2023).
- Gitterman, D.P.; Langford, W.S.; Hay, W.W. The uncertain fate of the National Institutes of Health (NIH) pediatric research portfolio. Pediatr. Res. 2018, 84, 328–332. [Google Scholar] [CrossRef]
- National Institutes of Health (NIH). RePORT—RePORTER—Search Term “Pedriatic”. Available online: https://reporter.nih.gov/search/Uv2KFJNsBkKhF-a4LG7iOA/projects/charts?shared=true (accessed on 18 January 2022).
- Gitterman, D.P.; Hay, W.W.; Langford, W.S. Making the case for pediatric research: A life-cycle approach and the return on investment. Pediatr. Res. 2022 934 2022, 93, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Pearson, A.D.J.; Weiner, S.L.; Adamson, P.C.; Karres, D.; Reaman, G.; Rousseau, R.; Blanc, P.; Norga, K.; Skolnik, J.; Kearns, P.; et al. ACCELERATE—Five years accelerating cancer drug development for children and adolescents. Eur. J. Cancer 2022, 166, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, A.V.; Modi, N.; de Wildt, S.N.; Aurich, B.; Bakhtadze, S.; Sirvent, F.J.B.; Cabañas, F.; Campbell, L.; Casanova, M.; Charlton, P.; et al. Improving clinical paediatric research and learning from COVID-19: Recommendations by the Conect4Children expert advice group. Pediatr. Res. 2021, 91, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Vinci, R.J. The pediatric workforce: Recent data trends, questions, and challenges for the future. Pediatrics 2021, 147, e2020013292. [Google Scholar] [CrossRef]
- Beleck, A.; Nachman, S. Understanding Pediatric Drug Lag Time: Review of Selected Drug Package Inserts. J. Pediatr. Infect. Dis. Soc. 2021, 10, 509–513. [Google Scholar] [CrossRef]
- Malkawi, W.A.; Alrafayah, E.; Alhazabreh, M.; Abulaila, S.; Al-Ghananeem, A.M. Formulation Challenges and Strategies to Develop Pediatric Dosage Forms. Children 2022, 9, 488. [Google Scholar] [CrossRef]
- Tanaudommongkon, I.; John Miyagi, S.; Green, D.J.; Burnham, J.M.; van den Anker, J.N.; Park, K.; Wu, J.; McCune, S.K.; Yao, L.; Burckart, G.J. Combined Pediatric and Adult Trials Submitted to the US Food and Drug Administration 2012–2018. Clin. Pharmacol. Ther. 2020, 108, 1018. [Google Scholar] [CrossRef]
- Meng, M.; Zhou, Q.; Lei, W.; Tian, M.; Wang, P.; Liu, Y.; Sun, Y.; Chen, Y.; Li, Q. Recommendations on Off-Label Drug Use in Pediatric Guidelines. Front. Pharmacol. 2022, 13, 1. [Google Scholar] [CrossRef]
- Allen, H.C.; Garbe, M.C.; Lees, J.; Aziz, N.; Chaaban, H.; Miller, J.L.; Johnson, P.; DeLeon, S. Off-Label Medication use in Children, More Common than We Think: ASystematic Review of the Literature. J. Okla. State Med. Assoc. 2018, 111, 776. [Google Scholar] [PubMed]
- Noel, G.J.; Nelson, R.M.; Bucci-Rechtweg, C.; Portman, R.; Miller, T.; Green, D.J.; Snyder, D.; Moreno, C.; Hovinga, C.; Connor, E. Inclusion of Adolescents in Adult Clinical Trials: Report of the Institute for Advanced Clinical Trials for Children’s Pediatric Innovation Research Forum. Ther. Innov. Regul. Sci. 2021, 55, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Rosenbaum, S.; Island, R. Developmental Pharmacokinetics in Pediatric Populations. J. Pediatr. Pharmacol. Ther. 2014, 19, 262–276. [Google Scholar] [CrossRef]
- Kelly, L.E.; Sinha, Y.; Barker, C.I.S.; Standing, J.F.; Offringa, M. Useful pharmacodynamic endpoints in children: Selection, measurement, and next steps. Pediatr. Res. 2018, 83, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Reflection Paper: Formulation of Choice for the Paediatric Population (EMEA/CHMP/PEG/194810/2005); European Medicines Agency: Amsterdam, The Netherlands, 2006; pp. 1–45. [Google Scholar]
- Food and Drug Administration (FDA). General Clinical Pharmacology Considerations for Pediatric Studies of Drugs, Including Biological Products. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-clinical-pharmacology-considerations-pediatric-studies-drugs-including-biological-products (accessed on 3 August 2023).
- Conklin, L.S.; Hoffman, E.P.; van den Anker, J. Developmental Pharmacodynamics and Modeling in Pediatric Drug Development. J. Clin. Pharmacol. 2019, 59, S87. [Google Scholar] [CrossRef]
- Sosnik, A.; Carcaboso, A.M. Nanomedicines in the future of pediatric therapy. Adv. Drug Deliv. Rev. 2014, 73, 140–161. [Google Scholar] [CrossRef]
- Batchelor, H.K.; Marriott, J.F. Paediatric pharmacokinetics: Key considerations. Br. J. Clin. Pharmacol. 2015, 79, 395–404. [Google Scholar] [CrossRef]
- Barker, C.I.S.; Standing, J.F.; Kelly, L.E.; Hanly Faught, L.; Needham, A.C.; Rieder, M.J.; de Wildt, S.N.; Offringa, M. Pharmacokinetic studies in children: Recommendations for practice and research. Arch. Dis. Child. 2018, 103, 695. [Google Scholar] [CrossRef]
- Siafaka, P.; Ipekci, E.; Caglar, E.Ş.; Ustundag Okur, N.; Buyukkayhan, D. Current Status of Pediatric Formulations for Chronic and Acute Children’ Diseases: Applications and Future Perspectives. Medeni. Med. J. 2021, 36, 152. [Google Scholar] [CrossRef]
- Leeder, J.S. Translating pharmacogenetics and pharmacogenomics into drug development for clinical pediatrics and beyond. Drug Discov. Today 2004, 9, 567–573. [Google Scholar] [CrossRef]
- Naji-Talakar, S.; Sharma, S.; Martin, L.A.; Barnhart, D.; Prasad, B. Potential implications of DMET ontogeny on the disposition of commonly prescribed drugs in neonatal and pediatric intensive care units. Expert Opin. Drug Metab. Toxicol. 2021, 17, 273. [Google Scholar] [CrossRef] [PubMed]
- Van Groen, B.D.; Pilla Reddy, V.; Badée, J.; Olivares-Morales, A.; Johnson, T.N.; Nicolaï, J.; Annaert, P.; Smits, A.; de Wildt, S.N.; Knibbe, C.A.J.; et al. Pediatric Pharmacokinetics and Dose Predictions: A Report of a Satellite Meeting to the 10th Juvenile Toxicity Symposium. Clin. Transl. Sci. 2021, 14, 29–35. [Google Scholar] [CrossRef]
- Jian, C.; Carpén, N.; Helve, O.; de Vos, W.M.; Korpela, K.; Salonen, A. Early-life gut microbiota and its connection to metabolic health in children: Perspective on ecological drivers and need for quantitative approach. eBioMedicine 2021, 69, 103475. [Google Scholar] [CrossRef] [PubMed]
- Leardini, D.; Venturelli, F.; Baccelli, F.; Cerasi, S.; Muratore, E.; Brigidi, P.; Pession, A.; Prete, A.; Masetti, R. Pharmacomicrobiomics in Pediatric Oncology: The Complex Interplay between Commonly Used Drugs and Gut Microbiome. Int. J. Mol. Sci. 2022, 23, 15387. [Google Scholar] [CrossRef]
- Walsh, J.; Griffin, B.T.; Clarke, G.; Hyland, N.P. Drug–gut microbiota interactions: Implications for neuropharmacology. Br. J. Pharmacol. 2018, 175, 4415. [Google Scholar] [CrossRef] [PubMed]
- Alcorn, J.; McNamara, P.J. Using ontogeny information to build predictive models for drug elimination. Drug Discov. Today 2008, 13, 507–512. [Google Scholar] [CrossRef]
- Anderson, G.D. Children Versus Adults: Pharmacokinetic and Adverse-Effect Differences. Epilepsia 2002, 43, 53–59. [Google Scholar] [CrossRef]
- Johnson, T.N.; Jamei, M.; Rowland-Yeo, K. How Does In Vivo Biliary Elimination of Drugs Change with Age? Evidence from In Vitro and Clinical Data Using a Systems Pharmacology Approach. Drug Metab. Dispos. 2016, 44, 1090–1098. [Google Scholar] [CrossRef]
- Wollmer, E.; Ungell, A.L.; Nicolas, J.M.; Klein, S. Review of paediatric gastrointestinal physiology relevant to the absorption of orally administered medicines. Adv. Drug Deliv. Rev. 2022, 181, 114084. [Google Scholar] [CrossRef]
- Germovsek, E.; Barker, C.I.S.; Sharland, M.; Standing, J.F. Pharmacokinetic–Pharmacodynamic Modeling in Pediatric Drug Development, and the Importance of Standardized Scaling of Clearance. Clin. Pharmacokinet. 2019, 58, 39. [Google Scholar] [CrossRef]
- Barker, C.I.S.; Germovsek, E.; Hoare, R.L.; Lestner, J.M.; Lewis, J.; Standing, J.F. Pharmacokinetic/pharmacodynamic modelling approaches in paediatric infectious diseases and immunology. Adv. Drug Deliv. Rev. 2014, 73, 127–139. [Google Scholar] [CrossRef]
- Zhang, T.; Sun, D.; Shu, Z.; Duan, Z.; Liu, Y.; Du, Q.; Zhang, Y.; Dong, Y.; Wang, T.; Hu, S.; et al. Population Pharmacokinetics and Model-Based Dosing Optimization of Teicoplanin in Pediatric Patients. Front. Pharmacol. 2020, 11, 594562. [Google Scholar] [CrossRef]
- Johnson, T.N.; Small, B.G.; Berglund, E.G.; Rowland Yeo, K. A best practice framework for applying physiologically-based pharmacokinetic modeling to pediatric drug development. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 967–972. [Google Scholar] [CrossRef]
- Khalid, S.; Rasool, M.F.; Masood, I.; Imran, I.; Saeed, H.; Ahmad, T.; Alqahtani, N.S.; Alshammari, F.A.; Alqahtani, F. Application of a physiologically based pharmacokinetic model in predicting captopril disposition in children with chronic kidney disease. Sci. Reports 2023, 13, 2697. [Google Scholar] [CrossRef] [PubMed]
- Ince, I.; Dallmann, A.; Frechen, S.; Coboeken, K.; Niederalt, C.; Wendl, T.; Block, M.; Meyer, M.; Eissing, T.; Burghaus, R.; et al. Predictive Performance of Physiology-Based Pharmacokinetic Dose Estimates for Pediatric Trials: Evaluation with 10 Bayer Small-Molecule Compounds in Children. J. Clin. Pharmacol. 2021, 61, S70–S82. [Google Scholar] [CrossRef]
- Ince, I.; Solodenko, J.; Frechen, S.; Dallmann, A.; Niederalt, C.; Schlender, J.; Burghaus, R.; Lippert, J.; Willmann, S. Predictive Pediatric Modeling and Simulation Using Ontogeny Information. J. Clin. Pharmacol. 2019, 59, S95–S103. [Google Scholar] [CrossRef]
- Liu, X.I.; Dallmann, A.; Wang, Y.M.; Green, D.J.; Burnham, J.M.; Chiang, B.; Wu, P.; Sheng, M.; Lu, K.; van den Anker, J.N.; et al. Monoclonal Antibodies and Fc-Fusion Proteins for Pediatric Use: Dosing, Immunogenicity, and Modeling and Simulation in Data Submitted to the US Food and Drug Administration. J. Clin. Pharmacol. 2019, 59, 1130–1143. [Google Scholar] [CrossRef]
- Lin, W.; Yan, J.H.; Heimbach, T.; He, H. Pediatric Physiologically Based Pharmacokinetic Model Development: Current Status and Challenges. Curr. Pharmacol. Rep. 2018, 4, 491–501. [Google Scholar] [CrossRef]
- Smith, L.; Leggett, C.; Borg, C. Administration of medicines to children: A practical guide. Aust. Prescr. 2022, 45, 188. [Google Scholar] [CrossRef] [PubMed]
- Galande, A.D.; Khurana, N.A.; Mutalik, S. Pediatric dosage forms-challenges and recent developments: A critical review. J. Appl. Pharm. Sci. 2020, 10, 155–166. [Google Scholar] [CrossRef]
- Nadeshkumar, A.; Sathiadas, G.; Sri Ranganathan, S. Administration of oral dosage forms of medicines to children in a resource limited setting. PLoS ONE 2022, 17, e0276379. [Google Scholar] [CrossRef] [PubMed]
- Rautamo, M.; Kvarnström, K.; Sivén, M.; Airaksinen, M.; Lahdenne, P.; Sandler, N. A Focus Group Study about Oral Drug Administration Practices at Hospital Wards—Aspects to Consider in Drug Development of Age-Appropriate Formulations for Children. Pharm. 2020, 12, 109. [Google Scholar] [CrossRef]
- Delgado-Charro, M.B.; Guy, R.H. Effective use of transdermal drug delivery in children. Adv. Drug Deliv. Rev. 2014, 73, 63–82. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.Y.; Kwon, M.; Choi, H.E.; Kim, K.S. Recent advances in transdermal drug delivery systems: A review. Biomater. Res. 2021, 25, 24. [Google Scholar] [CrossRef] [PubMed]
- Hanning, S.M.; Walker, E.; Sutcliffe, E.; Tuleu, C. The rectal route of medicine administration for children: Let’s get to the bottom of it! Eur. J. Pharm. Biopharm. 2020, 157, 25–27. [Google Scholar] [CrossRef]
- Clinical Skills Content Medication Administration: Intramuscular Injections (Pediatric). Available online: https://elsevier.health/en-US/preview/intramuscular-injection-pediatrics (accessed on 4 August 2023).
- Ainscough, L.P.; Ford, J.L.; Morecroft, C.W.; Peak, M.; Turner, M.A.; Nunn, A.J.; Roberts, M. Accuracy of intravenous and enteral preparations involving small volumes for paediatric use: A review. Eur. J. Hosp. Pharm. 2018, 25, 66–71. [Google Scholar] [CrossRef]
- Linakis, M.W.; Roberts, J.K.; Lala, A.C.; Spigarelli, M.G.; Medlicott, N.J.; Reith, D.M.; Ward, R.M.; Sherwin, C.M.T. Challenges Associated with Route of Administration in Neonatal Drug Delivery. Clin. Pharmacokinet. 2015, 55, 185–196. [Google Scholar] [CrossRef]
- Fonceca, A.M.; Fox Ditcham, W.G.; Everard, M.L.; Devadason, S. Drug Administration by Inhalation in Children. In Kendig’s Disorder of the Respiratory Tract in Children; Elsevier: Amsterdam, The Netherlands, 2019; pp. 257–271.e3. [Google Scholar] [CrossRef]
- Volerman, A.; Kan, K.; Carpenter, D.; Press, V.G. Strategies for Improving Inhalation Technique in Children: A Narrative Review. Patient Prefer. Adherence 2021, 15, 665. [Google Scholar] [CrossRef]
- Khan, D.; Kirby, D.; Bryson, S.; Shah, M.; Rahman Mohammed, A. Paediatric specific dosage forms: Patient and formulation considerations. Int. J. Pharm. 2022, 616, 121501. [Google Scholar] [CrossRef]
- Lajoinie, A.; Henin, E.; Nguyen, K.A.; Malik, S.; Mimouni, Y.; Sapori, J.M.; Bréant, V.; Cochat, P.; Kassai, B. Oral drug dosage forms administered to hospitalized children: Analysis of 117,665 oral administrations in a French paediatric hospital over a 1-year period. Int. J. Pharm. 2016, 500, 336–344. [Google Scholar] [CrossRef]
- Montero-Padilla, S.; Velaga, S.; Morales, J.O. Buccal Dosage Forms: General Considerations for Pediatric Patients. AAPS PharmSciTech 2017, 18, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.K.W.; Xu, Y.; Worsley, A.; Wong, I.C.K. Oral transmucosal drug delivery for pediatric use. Adv. Drug Deliv. Rev. 2014, 73, 50–62. [Google Scholar] [CrossRef]
- Sibum, I.; Hagedoorn, P.; de Boer, A.H.; Frijlink, H.W.; Grasmeijer, F. Challenges for pulmonary delivery of high powder doses. Int. J. Pharm. 2018, 548, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Menditto, E.; Orlando, V.; De Rosa, G.; Minghetti, P.; Musazzi, U.M.; Cahir, C.; Kurczewska-Michalak, M.; Kardas, P.; Costa, E.; Lobo, J.M.S.; et al. Patient Centric Pharmaceutical Drug Product Design—The Impact on Medication Adherence. Pharmaceuticals 2020, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Stegemann, S.; Klingmann, V.; Reidemeister, S.; Breitkreutz, J. Patient-centric drug product development: Acceptability across patient populations—Science and evidence. Eur. J. Pharm. Biopharm. 2023, 188, 1–5. [Google Scholar] [CrossRef]
- Reker, D.; Blum, S.M.; Steiger, C.; Anger, K.E.; Sommer, J.M.; Fanikos, J.; Traverso, G. ‘Inactive’ ingredients in oral medications. Sci. Transl. Med. 2019, 11, eaau6753. [Google Scholar] [CrossRef]
- Adepu, S.; Ramakrishna, S. Controlled Drug Delivery Systems: Current Status and Future Directions. Molecules 2021, 26, 5905. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, G. Safety of Excipients in Pediatric Formulations—A Call for Toxicity Studies in Juvenile Animals? Children 2015, 2, 191. [Google Scholar] [CrossRef]
- Pereira, B.M.P.; Tagkopoulos, I. Benzalkonium chlorides: Uses, regulatory status, and microbial resistance. Appl. Environ. Microbiol. 2019, 85, e00377-19. [Google Scholar] [CrossRef]
- Salunke, S.; Clapham, D.; Agrawal, A.; Hughes, K.; Nunn, T. Best practices for selection of excipients for paediatrics—Workshop reflection. Eur. J. Pharm. Biopharm. 2021, 160, 77–81. [Google Scholar] [CrossRef]
- Salunke, S.; Tuleu, C. The STEP database through the end-users eyes—Usability study. Int. J. Pharm. 2015, 492, 316–331. [Google Scholar] [CrossRef]
- Van Riet-Nales, D.A.; Schobben, A.F.A.M.; Vromans, H.; Egberts, T.C.G.; Rademaker, C.M.A. Safe and effective pharmacotherapy in infants and preschool children: Importance of formulation aspects. Arch. Dis. Child. 2016, 101, 662–669. [Google Scholar] [CrossRef]
- Bianchi, A.; Bottau, P.; Calamelli, E.; Caimmi, S.; Crisafulli, G.; Franceschini, F.; Liotti, L.; Mori, F.; Paglialunga, C.; Saretta, F.; et al. Hypersensitivity to polyethylene glycol in adults and children: An emerging challenge. Acta Bio Medica Atenei Parm. 2021, 92, 2021519. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Sellaturay, P.; Nasser, S.; Islam, S.; Gurugama, P.; Ewan, P.W. Polyethylene glycol (PEG) is a cause of anaphylaxis to the Pfizer/BioNTech mRNA COVID-19 vaccine. Clin. Exp. Allergy 2021, 51, 861. [Google Scholar] [CrossRef]
- Doyle, R.; Melo, M.; Teoh, C.; Wen, S.; Willcocks, S. Safety of mRNA vaccination for COVID-19 in children with polyethylene glycol (PEG)-asparaginase allergy. Pediatr. Allergy Immunol. 2023, 34, e13939. [Google Scholar] [CrossRef] [PubMed]
- Bigini, P.; Gobbi, M.; Bonati, M.; Clavenna, A.; Zucchetti, M.; Garattini, S.; Pasut, G. The role and impact of polyethylene glycol on anaphylactic reactions to COVID-19 nano-vaccines. Nat. Nanotechnol. 2021, 16, 1169–1171. [Google Scholar] [CrossRef]
- Karaaslan, B.G.; Burtecene, N.; Mustu, U.; Ocak, S.; Kasapcopur, O.; Kıykım, A.; Cokugras, H. Evaluation of pediatric patients with suspected polyethylene glycol and polysorbate allergy before mRNA SARS-CoV2 vaccination. Allergol. Immunopathol. 2023, 51, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Kardas, P.; Dabrowa, M.; Witkowski, K. Adherence to treatment in paediatric patients—Results of the nationwide survey in Poland. BMC Pediatr. 2021, 21, 16. [Google Scholar] [CrossRef]
- El-Rachidi, S.; LaRochelle, J.M.; Morgan, J.A. Pharmacists and Pediatric Medication Adherence: Bridging the Gap. Hosp. Pharm. 2017, 52, 124. [Google Scholar] [CrossRef] [PubMed]
- Turner-Bowker, D.M.; An Haack, K.; Krohe, M.; Yaworsky, A.; Vivas, N.; Kelly, M.; Chatterjee, G.; Chaston, E.; Mann, E.; Reaney, M. Development and content validation of the Pediatric Oral Medicines Acceptability Questionnaires (P-OMAQ): Patient-reported and caregiver-reported outcome measures. J. Patient-Rep. Outcomes 2020, 4, 80. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Ranmal, S.; Batchelor, H.K.; Orlu-Gul, M.; Ernest, T.B.; Thomas, I.W.; Flanagan, T.; Tuleu, C. Patient-centred pharmaceutical design to improve acceptability of medicines: Similarities and differences in paediatric and geriatric populations. Drugs 2014, 74, 1871–1889. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.; Cram, A.; Woertz, K.; Breitkreutz, J.; Winzenburg, G.; Turner, R.; Tuleu, C. Playing hide and seek with poorly tasting paediatric medicines: Do not forget the excipients. Adv. Drug Deliv. Rev. 2014, 73, 14–33. [Google Scholar] [CrossRef] [PubMed]
- Plaza-Zamora, J.; Legaz, I.; Osuna, E.; Pérez-Cárceles, M.D. Age and education as factors associated with medication literacy: A community pharmacy perspective. BMC Geriatr. 2020, 20, 501. [Google Scholar] [CrossRef]
- Matson, P.A.; Bakhai, N.; Solomon, B.S.; Flessa, S.; Ramos, J.; Hammond, C.J.; Adger, H. Understanding caregiver acceptance of screening for family substance use in pediatric clinics serving economically disadvantaged children. Subst. Abus. 2022, 43, 282. [Google Scholar] [CrossRef]
- Mfoafo, K.; Tuleu, C.; Hanning, S.; Omidian, H.; Mfoafo, K. Exploring the Potential of Nanotechnology in Pediatric Healthcare: Advances, Challenges, and Future Directions. Pharmaceutics 2023, 15, 1583. [Google Scholar] [CrossRef]
- Fornaguera, C.; García-Celma, M.J. Personalized Nanomedicine: A Revolution at the Nanoscale. J. Pers. Med. 2017, 7, 12. [Google Scholar] [CrossRef]
- Ehmann, F.; Sakai-Kato, K.; Duncan, R.; Pérez De La Ossa, D.H.; Pita, R.; Vidal, J.M.; Kohli, A.; Tothfalusi, L.; Sanh, A.; Tinton, S.; et al. Next-generation nanomedicines and nanosimilars: EU regulators’ initiatives relating to the development and evaluation of nanomedicines. Nanomedicine 2013, 8, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhai, B.; Yang, F.; Chen, Z.; Zhou, Q.; Paiva-Santos, A.C.; Yuan, Z.; Zhou, Y. Nanotechnology-Based Diagnostic and Therapeutic Strategies for Neuroblastoma. Front. Pharmacol. 2022, 13, 908713. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Nogales, C.; González-Fernández, Y.; Aldaz, A.; Couvreur, P.; Blanco-Prieto, M.J. Nanomedicines for pediatric cancers. ACS Nano 2018, 12, 7482–7496. [Google Scholar] [CrossRef]
- Yang, S.; Wallach, M.; Krishna, A.; Kurmasheva, R.; Sridhar, S. Recent Developments in Nanomedicine for Pediatric Cancer. J. Clin. Med. 2021, 10, 1437. [Google Scholar] [CrossRef] [PubMed]
- Rubey, K.M.; Brenner, J.S. Nanomedicine to fight infectious disease. Adv. Drug Deliv. Rev. 2021, 179, 113996. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Godhi, B.S.; Saha, S.; Singh, B.; Dinsa, K.; Bhagchandani, J.; Gautam, A. Use of nanoparticles in pediatric dentistry: A narrative review. J. Int. Oral Health 2022, 14, 357. [Google Scholar]
- Delouise, L.A. Applications of Nanotechnology in Dermatology. J. Investig. Dermatol. 2012, 132, 964–975. [Google Scholar] [CrossRef]
- Trandafir, L.M.; Dodi, G.; Frasinariu, O.; Luca, A.C.; Butnariu, L.I.; Tarca, E.; Moisa, S.M. Tackling Dyslipidemia in Obesity from a Nanotechnology Perspective. Nutrients 2022, 14, 3774. [Google Scholar] [CrossRef] [PubMed]
- Ganta, S.; Talekar, M.; Singh, A.; Coleman, T.P.; Amiji, M.M. Nanoemulsions in Translational Research—Opportunities and Challenges in Targeted Cancer Therapy. AAPS PharmSciTech 2014, 15, 694. [Google Scholar] [CrossRef]
- Sánchez-López, E.; Guerra, M.; Dias-Ferreira, J.; Lopez-Machado, A.; Ettcheto, M.; Cano, A.; Espina, M.; Camins, A.; Garcia, M.L.; Souto, E.B. Current Applications of Nanoemulsions in Cancer Therapeutics. Nanomaterials 2019, 9, 821. [Google Scholar] [CrossRef]
- Basso, J.; Mendes, M.; Cova, T.; Sousa, J.; Pais, A.; Fortuna, A.; Vitorino, R.; Vitorino, C. A Stepwise Framework for the Systematic Development of Lipid Nanoparticles. Biomolecules 2022, 12, 223. [Google Scholar] [CrossRef]
- Grodzinski, P.; Kircher, M.; Goldberg, M.; Gabizon, A. Integrating Nanotechnology into Cancer Care. ACS Nano 2019, 13, 7370–7376. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zannikou, M.; Lofchy, L.; Li, Y.; Gaikwad, H.; Balyasnikova, I.V.; Simberg, D. Liposomal Extravasation and Accumulation in Tumors as Studied by Fluorescence Microscopy and Imaging Depend on the Fluorescent Label. ACS Nano 2021, 15, 11880–11890. [Google Scholar] [CrossRef] [PubMed]
- Tenchov, R.; Bird, R.; Curtze, A.E.; Zhou, Q. Lipid Nanoparticles from Liposomes to mRNA Vaccine Delivery, a Landscape of Research Diversity and Advancement. ACS Nano 2021, 15, 16982–17015. [Google Scholar] [CrossRef]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Praça, F.S.G.; Marinho, H.S.; Martins, M.B.F.; Gaspar, R.; Corvo, M.L.; Medina, W.S.G. Current aspects of breast cancer therapy and diagnosis based on a nanocarrier approach. In Nanostructures for Cancer Therapy; Elsevier: Amsterdam, The Netherlands, 2017; pp. 749–774. [Google Scholar] [CrossRef]
- Forssen, E.A. The design and development of DaunoXome® for solid tumor targeting in vivo. Adv. Drug Deliv. Rev. 1997, 24, 133–150. [Google Scholar] [CrossRef]
- Gu, W.; Andrews, G.P.; Tian, Y. Recent Clinical Successes in Liposomal Nanomedicines. Int. J. Drug Discov. Pharmacol. 2023, 2, 52–59. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Shah, B.D.; Rozario, N.; Turba, E.P.; Bello, C.; Chavez, J.C.; Sokol, L.; Brayer, J.; Lancet, J.E. A Single-Arm, Open-Label Phase 2 Pilot Study of Vyxeos (CPX-351) in Adults with Relapsed or Refractory Acute Lymphoblastic Leukemia. Blood 2021, 138, 4399. [Google Scholar] [CrossRef]
- Li, L.; Xu, Z.P.; Leong, E.W.X.; Ge, R. Lipid Nanoparticles as Delivery Vehicles for Inhaled Therapeutics. Biomedicines 2022, 10, 2179. [Google Scholar] [CrossRef]
- European Medicines Agency (EMA). Arikayce Liposomal. Available online: https://www.ema.europa.eu/en/documents/assessment-report/arikayce-liposomal-epar-public-assessment-report_en.pdf (accessed on 9 August 2023).
- Meyerhoff, A.U.S. Food and Drug Administration approval of AmBisome (liposomal amphotericin B) for treatment of visceral leishmaniasis. Clin. Infect. Dis. 1999, 28, 42–48. [Google Scholar] [CrossRef]
- Ghosh, S.; Carter, K.A.; Lovell, J.F. Liposomal formulations of photosensitizers. Biomaterials 2019, 218, 119341. [Google Scholar] [CrossRef] [PubMed]
- Rouge, J. RNA and nanocarriers: Next generation drug and delivery platform take center stage. Trends Biotechnol. 2023, 41, 281–282. [Google Scholar] [CrossRef]
- Weng, Y.; Xiao, H.; Zhang, J.; Liang, X.J.; Huang, Y. RNAi therapeutic and its innovative biotechnological evolution. Biotechnol. Adv. 2019, 37, 801–825. [Google Scholar] [CrossRef]
- Williams, R.M.; Kapadia, C.; Jaimes, E.A.; Heller, D.A. Nanotargeting to the kidney. In Regenerative Nephrology; Academic Press: Cambridge, MA, USA, 2022; pp. 439–449. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S.; Paulson, J.A. Nanoparticles in the clinic: An update post COVID-19 vaccines. Bioeng. Transl. Med. 2021, 6, e10246. [Google Scholar] [CrossRef]
- Wilson, B.; Geetha, K.M. Lipid nanoparticles in the development of mRNA vaccines for COVID-19. J. Drug Deliv. Sci. Technol. 2022, 74, 103553. [Google Scholar] [CrossRef]
- Yellepeddi, V.K.; Joseph, A.; Nance, E. Pharmacokinetics of nanotechnology-based formulations in pediatric populations. Adv. Drug Deliv. Rev. 2019, 151–152, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Nieto González, N.; Obinu, A.; Rassu, G.; Giunchedi, P.; Gavini, E. Polymeric and Lipid Nanoparticles: Which Applications in Pediatrics? Pharmaceutics 2021, 13, 670. [Google Scholar] [CrossRef]
- Pham, K.; Li, D.; Guo, S.; Penzak, S.; Dong, X. Development and in vivo evaluation of child-friendly lopinavir/ritonavir pediatric granules utilizing novel in situ self-assembly nanoparticles. J. Control Release 2016, 226, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Nogales, C.; Mura, S.; Couvreur, P.; Blanco-Prieto, M.J. Squalenoyl-gemcitabine/edelfosine nanoassemblies: Anticancer activity in pediatric cancer cells and pharmacokinetic profile in mice. Int. J. Pharm. 2020, 582, 119345. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S. Natural Polymer Drug Delivery Systems: Nanoparticles, Plants, and Algae; Springer: Berlin/Heidelberg, Germany, 2016; pp. 1–225. [Google Scholar]
- Prajapati, S.K.; Jain, A.; Jain, A.; Jain, S. Biodegradable polymers and constructs: A novel approach in drug delivery. Eur. Polym. J. 2019, 120, 109191. [Google Scholar] [CrossRef]
- Abourehab, M.A.S.; Pramanik, S.; Abdelgawad, M.A.; Abualsoud, B.M.; Kadi, A.; Ansari, M.J.; Deepak, A. Recent Advances of Chitosan Formulations in Biomedical Applications. Int. J. Mol. Sci. 2022, 23, 10975. [Google Scholar] [CrossRef]
- Kaur, M.; Sharma, A.; Puri, V.; Aggarwal, G.; Maman, P.; Huanbutta, K.; Nagpal, M.; Sangnim, T. Chitosan-Based Polymer Blends for Drug Delivery Systems. Polymers 2023, 15, 2028. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Sharma, G.; Jain, A.; Tiwari, A.; Saraf, S.; Panda, P.K.; Katare, O.P.; Jain, S.K. Systematic optimization of cationic surface engineered mucoadhesive vesicles employing Design of Experiment (DoE): A preclinical investigation. Int. J. Biol. Macromol. 2019, 133, 1142–1155. [Google Scholar] [CrossRef]
- Nerli, G.; Gonçalves, L.M.D.; Cirri, M.; Almeida, A.J.; Maestrelli, F.; Mennini, N.; Mura, P.A. Design, Evaluation and Comparison of Nanostructured Lipid Carriers and Chitosan Nanoparticles as Carriers of Poorly Soluble Drugs to Develop Oral Liquid Formulations Suitable for Pediatric Use. Pharmaceutics 2023, 15, 1305. [Google Scholar] [CrossRef]
- Desai, N.; Rana, D.; Salave, S.; Gupta, R.; Patel, P.; Karunakaran, B.; Sharma, A.; Giri, J.; Benival, D.; Kommineni, N. Chitosan: A Potential Biopolymer in Drug Delivery and Biomedical Applications. Pharmaceutics 2023, 15, 1313. [Google Scholar] [CrossRef] [PubMed]
- Kantak, M.N.; Bharate, S.S. Analysis of clinical trials on biomaterial and therapeutic applications of chitosan: A review. Carbohydr. Polym. 2022, 278, 118999. [Google Scholar] [CrossRef] [PubMed]
- Korelc, K.; Larsen, B.S.; Gašperlin, M.; Tho, I. Water-soluble chitosan eases development of mucoadhesive buccal films and wafers for children. Int. J. Pharm. 2023, 631, 122544. [Google Scholar] [CrossRef]
- Priya Dharshini, K.; Fang, H.; Ramya Devi, D.; Yang, J.X.; Luo, R.H.; Zheng, Y.T.; Brzeziński, M.; Vedha Hari, B.N. pH-sensitive chitosan nanoparticles loaded with dolutegravir as milk and food admixture for paediatric anti-HIV therapy. Carbohydr. Polym. 2021, 256, 117440. [Google Scholar] [CrossRef]
- Dalvi, A.; Ravi, P.R.; Uppuluri, C.T. Rufinamide-Loaded Chitosan Nanoparticles in Xyloglucan-Based Thermoresponsive In Situ Gel for Direct Nose to Brain Delivery. Front. Pharmacol. 2021, 12, 691936. [Google Scholar] [CrossRef] [PubMed]
- Aman, R.; Meshali, M.; Abdelghani, G. Optimization and Formulation of Cinnarizine-Loaded Chitosan Microspheres in Liquid Dosage Form for Pediatric Therapy. Drug Deliv. Lett. 2014, 4, 128–141. [Google Scholar] [CrossRef]
- Severino, P.; Silva, C.; Costa, T.; Silva, H.; Chaud, M.; Santana, M.H.; Souto, E.B. In Vivo Absorption of Didanosine Formulated in Pellets Composed of Chitosan Microspheres. Vivo 2014, 28, 1045–1050. [Google Scholar]
- Mattheolabakis, G.; Milane, L.; Singh, A.; Amiji, M.M. Hyaluronic acid targeting of CD44 for cancer therapy: From receptor biology to nanomedicine. J. Drug Target. 2015, 23, 605–618. [Google Scholar] [CrossRef]
- Pereira, M.; Silva, F.C.; Simões, S.; Ribeiro, H.M.; Almeida, A.J.; Marto, J. Innovative, Sugar-Free Oral Hydrogel as a Co-administrative Vehicle for Pediatrics: A Strategy to Enhance Patient Compliance. AAPS PharmSciTech 2022, 23, 691936. [Google Scholar] [CrossRef] [PubMed]
- Laffleur, F. Novel adhesive hyaluronic acid based solid dosage form for pediatric application. J. Drug Deliv. Sci. Technol. 2018, 44, 213–219. [Google Scholar] [CrossRef]
- Di Cicco, M.; Peroni, D.; Sepich, M.; Tozzi, M.G.; Comberiati, P.; Cutrera, R. Hyaluronic acid for the treatment of airway diseases in children: Little evidence for few indications. Pediatr. Pulmonol. 2020, 55, 2156–2169. [Google Scholar] [CrossRef]
- Sabra, S.; Agwa, M.M. Lactoferrin, a unique molecule with diverse therapeutical and nanotechnological applications. Int. J. Biol. Macromol. 2020, 164, 1046. [Google Scholar] [CrossRef] [PubMed]
- Elzoghby, A.O.; Abdelmoneem, M.A.; Hassanin, I.A.; Abd Elwakil, M.M.; Elnaggar, M.A.; Mokhtar, S.; Fang, J.Y.; Elkhodairy, K.A. Lactoferrin, a multi-functional glycoprotein: Active therapeutic, drug nanocarrier & targeting ligand. Biomaterials 2020, 263, 120355. [Google Scholar] [CrossRef]
- Guzmán-Mejía, F.; Godínez-Victoria, M.; Molotla-Torres, D.E.; Drago-Serrano, M.E. Lactoferrin as a Component of Pharmaceutical Preparations: An Experimental Focus. Pharmaceuticals 2023, 16, 214. [Google Scholar] [CrossRef]
- Janicka, M.; Tomaszewska, M.; Ranoszek-Soliwoda, E.; Celichowski, K.; Grobelny, G.; Szymanski, J.; Conte, M.P.; Rosa, L.; Krzyzowska, M.; Janicka, M.; et al. Lactoferrin-Conjugated Nanoparticles as New Antivirals. Pharmaceutics 2022, 14, 1862. [Google Scholar] [CrossRef]
- Ahmed, F.; Ali, M.J.; Kondapi, A.K. Carboplatin loaded protein nanoparticles exhibit improve anti-proliferative activity in retinoblastoma cells. Int. J. Biol. Macromol. 2014, 70, 572–582. [Google Scholar] [CrossRef]
- Russo, E.; Spallarossa, A.; Tasso, B.; Villa, C.; Brullo, C. Nanotechnology for Pediatric Retinoblastoma Therapy. Pharmaceuticals 2022, 15, 1087. [Google Scholar] [CrossRef] [PubMed]
- Narayana, R.V.L.; Jana, P.; Tomar, N.; Prabhu, V.; Nair, R.M.; Manukonda, R.; Kaliki, S.; Coupland, S.E.; Alexander, J.; Kalirai, H.; et al. Carboplatin- and Etoposide-Loaded Lactoferrin Protein Nanoparticles for Targeting Cancer Stem Cells in Retinoblastoma In Vitro. Investig. Ophthalmol. Vis. Sci. 2021, 62, 13. [Google Scholar] [CrossRef] [PubMed]
- Łukasiewicz, S.; Mikołajczyk, A.; Błasiak, E.; Fic, E.; Dziedzicka-Wasylewska, M. Polycaprolactone Nanoparticles as Promising Candidates for Nanocarriers in Novel Nanomedicines. Pharmaceutics 2021, 13, 191. [Google Scholar] [CrossRef]
- Grossen, P.; Witzigmann, D.; Sieber, S.; Huwyler, J. PEG-PCL-based nanomedicines: A biodegradable drug delivery system and its application. J. Control Release 2017, 260, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Xu, X.; Barwe, S.P.; Yang, X.; Czymmek, K.; Waldman, S.A.; Mason, R.W.; Jia, X.; Rajasekaran, A.K. Dexamethasone-loaded Block Copolymer Nanoparticles Induce Leukemia Cell Deathand Enhances Therapeutic Efficacy: A Novel Application in PediatricNanomedicine. Mol. Pharm. 2013, 10, 2199. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377. [Google Scholar] [CrossRef]
- Ni, M.Z.; Xiong, M.; Zhang, X.C.; Cai, G.P.; Chen, H.W.; Zeng, Q.M.; Yu, Z.C. Poly(lactic-co-glycolic acid) nanoparticles conjugated with CD133 aptamers for targeted salinomycin delivery to CD133+ osteosarcoma cancer stem cells. Int. J. Nanomed. 2015, 10, 2537–2554. [Google Scholar] [CrossRef]
- Jarak, I.; Varela, C.L.; Tavares da Silva, E.; Roleira, F.F.M.; Veiga, F.; Figueiras, A. Pluronic-based nanovehicles: Recent advances in anticancer therapeutic applications. Eur. J. Med. Chem. 2020, 206, 112526. [Google Scholar] [CrossRef]
- De Castro, K.C.; Coco, J.C.; dos Santos, É.M.; Ataide, J.A.; Martinez, R.M.; do Nascimento, M.H.M.; Prata, J.; da Fonte, P.R.M.L.; Severino, P.; Mazzola, P.G.; et al. Pluronic® triblock copolymer-based nanoformulations for cancer therapy: A 10-year overview. J. Control Release 2023, 353, 802–822. [Google Scholar] [CrossRef]
- Figueiras, A.; Domingues, C.; Jarak, I.; Santos, A.I.; Parra, A.; Pais, A.; Alvarez-Lorenzo, C.; Concheiro, A.; Kabanov, A.; Cabral, H.; et al. New Advances in Biomedical Application of Polymeric Micelles. Pharmaceutics 2022, 14, 1700. [Google Scholar] [CrossRef] [PubMed]
- Domingues, C.; Alvarez-Lorenzo, C.; Concheiro, A.; Veiga, F.; Figueiras, A. Nanotheranostic Pluronic-Like Polymeric Micelles: Shedding Light into the Dark Shadows of Tumors. Mol. Pharm. 2019, 16, 4757–4774. [Google Scholar] [CrossRef]
- Khodaei, A.; Jahanmard, F.; Madaah Hosseini, H.R.; Bagheri, R.; Dabbagh, A.; Weinans, H.; Amin Yavari, S. Controlled temperature-mediated curcumin release from magneto-thermal nanocarriers to kill bone tumors. Bioact. Mater. 2022, 11, 107–117. [Google Scholar] [CrossRef]
- Jia, Y.; Jiang, Y.; He, Y.; Zhang, W.; Zou, J.; Magar, K.T.; Boucetta, H.; Teng, C.; He, W. Approved Nanomedicine against Diseases. Pharmaceutics 2023, 15, 774. [Google Scholar] [CrossRef] [PubMed]
- Bernabeu, E.; Cagel, M.; Lagomarsino, E.; Moretton, M.; Chiappetta, D.A. Paclitaxel: What has been done and the challenges remain ahead. Int. J. Pharm. 2017, 526, 474–495. [Google Scholar] [CrossRef]
- Werner, M.E.; Cummings, N.D.; Sethi, M.; Wang, E.C.; Sukumar, R.; Moore, D.T.; Wang, A.Z. Preclinical Evaluation of Genexol-PM, a Nanoparticle Formulation of Paclitaxel, as a Novel Radiosensitizer for the Treatment of Non-Small Cell Lung Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 463. [Google Scholar] [CrossRef]
- Ranade, A.A.; Joshi, D.A.; Phadke, G.K.; Patil, P.P.; Kasbekar, R.B.; Apte, T.G.; Dasare, R.R.; Mengde, S.D.; Parikh, P.M.; Bhattacharyya, G.S.; et al. Clinical and economic implications of the use of nanoparticle paclitaxel (Nanoxel) in India. Ann. Oncol. 2013, 24, v6–v12. [Google Scholar] [CrossRef]
- Huang, D.; Wu, D. Biodegradable dendrimers for drug delivery. Mater. Sci. Eng. C. Mater. Biol. Appl. 2018, 90, 713–727. [Google Scholar] [CrossRef]
- Michlewska, S.; Ionov, M.; Szwed, A.; Rogalska, A.; Del Olmo, N.S.; Ortega, P.; Denel, M.; Jacenik, D.; Shcharbin, D.; de la Mata, F.J.; et al. Ruthenium Dendrimers against Human Lymphoblastic Leukemia 1301 Cells. Int. J. Mol. Sci. 2020, 21, 4119. [Google Scholar] [CrossRef]
- Chittasupho, C.; Aonsri, C.; Imaram, W. Targeted dendrimers for antagonizing the migration and viability of NALM-6 lymphoblastic leukemia cells. Bioorg. Chem. 2021, 107, 104601. [Google Scholar] [CrossRef] [PubMed]
- Guido, C.; Baldari, C.; Maiorano, G.; Mastronuzzi, A.; Carai, A.; Quintarelli, C.; De Angelis, B.; Cortese, B.; Gigli, G.; Palamà, I.E. Nanoparticles for Diagnosis and Target Therapy in Pediatric Brain Cancers. Diagnostics 2022, 12, 173. [Google Scholar] [CrossRef] [PubMed]
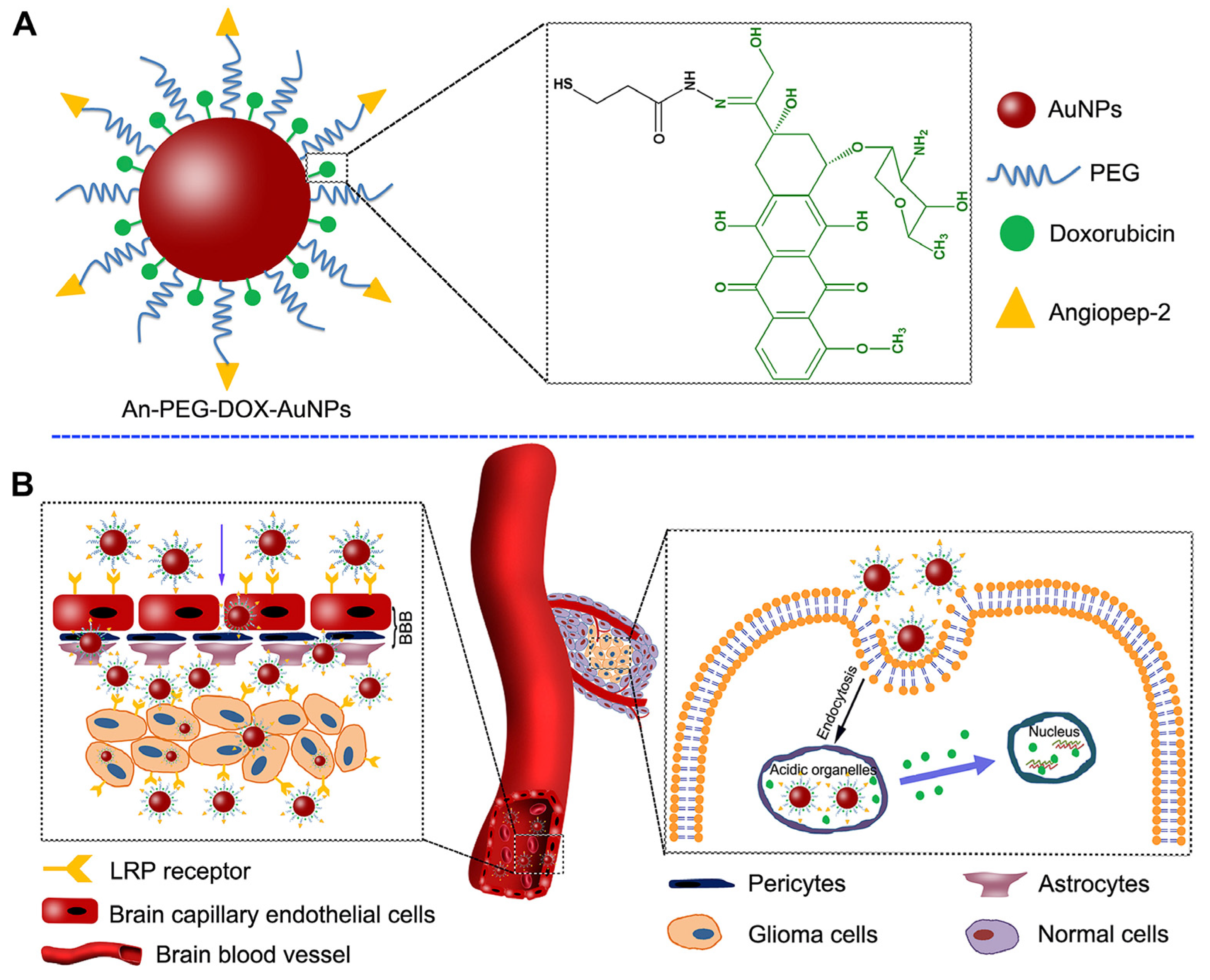
- Ruan, S.; Yuan, M.; Zhang, L.; Hu, G.; Chen, J.; Cun, X.; Zhang, Q.; Yang, Y.; He, Q.; Gao, H. Tumor microenvironment sensitive doxorubicin delivery and release to glioma using angiopep-2 decorated gold nanoparticles. Biomaterials 2015, 37, 425–435. [Google Scholar] [CrossRef]
- Morford, L.L.; Bowman, C.J.; Blanset, D.L.; Bøgh, I.B.; Chellman, G.J.; Halpern, W.G.; Weinbauer, G.F.; Coogan, T.P. Preclinical safety evaluations supporting pediatric drug development with biopharmaceuticals: Strategy, challenges, current practices. Birth Defects Res. Part B Dev. Reprod. Toxicol. 2011, 92, 359–380. [Google Scholar] [CrossRef] [PubMed]
- Rasool, M.; Malik, A.; Waquar, S.; Arooj, M.; Zahid, S.; Asif, M.; Shaheen, S.; Hussain, A.; Ullah, H.; Gan, S.H. New challenges in the use of nanomedicine in cancer therapy. Bioengineered 2022, 13, 759. [Google Scholar] [CrossRef] [PubMed]
- Dugershaw, B.B.; Aengenheister, L.; Hansen, S.S.K.; Hougaard, K.S.; Buerki-Thurnherr, T. Recent insights on indirect mechanisms in developmental toxicity of nanomaterials. Part. Fibre Toxicol. 2020, 17, 31. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A. Safety and Toxicity Implications of Multifunctional Drug Delivery Nanocarriers on Reproductive Systems In Vitro and In Vivo. Front. Toxicol. 2022, 4, 895667. [Google Scholar] [CrossRef] [PubMed]
- López-Paniagua, M.; de la Mata, A.; Galindo, S.; Blázquez, F.; Calonge, M.; Nieto-Miguel, T. Advanced Therapy Medicinal Products for the Eye: Definitions and Regulatory Framework. Pharmaceutics 2021, 13, 347. [Google Scholar] [CrossRef]
- European Union Regulation (EC) No 1394/2007 of the European Parliament and of the Council of 13 November 2007 on Advanced Therapy Medicinal Products and Amending Directive 2001/83/EC and Regulation (EC) No 726/2004 (Text with EEA relevance). Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2007:324:0121:0137:en:PDF (accessed on 17 July 2023).
- European Medicines Agency (EMA). Support for Advanced-Therapy Developers | GLP Requirements. Available online: https://www.ema.europa.eu/en/human-regulatory/research-development/advanced-therapies/support-advanced-therapy-developers (accessed on 10 September 2023).
- Detela, G.; Lodge, A. EU Regulatory Pathways for ATMPs: Standard, Accelerated and Adaptive Pathways to Marketing Authorisation. Mol. Ther. Methods Clin. Dev. 2019, 13, 205–232. [Google Scholar] [CrossRef]
- Iglesias-Lopez, C.; Agustí, A.; Vallano, A.; Obach, M. Current landscape of clinical development and approval of advanced therapies. Mol. Ther. Methods Clin. Dev. 2021, 23, 606–618. [Google Scholar] [CrossRef]
- Iglesias-Lopez, C.; Agustí, A.; Obach, M.; Vallano, A. Regulatory Framework for Advanced Therapy Medicinal Products in Europe and United States. Front. Pharmacol. 2019, 10, 921. [Google Scholar] [CrossRef]
- Smith, M.D.; Brune, J.C.; Wildemann, B.; Pruss, A. Whither Advanced Therapy Medicinal Products? Transfus. Med. Hemotherapy 2013, 40, 449. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration (FDA). Approved Cellular and Gene Therapy Products. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products (accessed on 9 August 2023).
- El-Kadiry, A.E.H.; Rafei, M.; Shammaa, R. Cell Therapy: Types, Regulation, and Clinical Benefits. Front. Med. 2021, 8, 756029. [Google Scholar] [CrossRef]
- Bashor, C.J.; Hilton, I.B.; Bandukwala, H.; Smith, D.M.; Veiseh, O. Engineering the next generation of cell-based therapeutics. Nat. Rev. Drug Discov. 2022, 21, 655–675. [Google Scholar] [CrossRef]
- Buckland, K.F.; Bobby Gaspar, H. Gene and cell therapy for children—New medicines, new challenges? Adv. Drug Deliv. Rev. 2014, 73, 162–169. [Google Scholar] [CrossRef]
- Ligon, J.A.; Wessel, K.M.; Shah, N.N.; Glod, J. Adoptive Cell Therapy in Pediatric and Young Adult Solid Tumors: Current Status and Future Directions. Front. Immunol. 2022, 13, 846346. [Google Scholar] [CrossRef]
- European Medicines Agency (EMA). Advanced Therapy Medicinal Products: Overview. Available online: https://www.ema.europa.eu/en/human-regulatory/overview/advanced-therapy-medicinal-products-overview (accessed on 18 July 2023).
- Papanikolaou, E.; Bosio, A. The Promise and the Hope of Gene Therapy. Front. Genome Ed. 2021, 3, 618346. [Google Scholar] [CrossRef] [PubMed]
- Gowing, G.; Svendsen, S.; Svendsen, C.N. Ex vivo gene therapy for the treatment of neurological disorders. Prog. Brain Res. 2017, 230, 99–132. [Google Scholar] [CrossRef] [PubMed]
- Soofiyani, S.R.; Baradaran, B.; Lotfipour, F.; Kazemi, T.; Mohammadnejad, L. Gene Therapy, Early Promises, Subsequent Problems, and RecentBreakthroughs. Adv. Pharm. Bull. 2013, 3, 249. [Google Scholar] [CrossRef]
- Naldini, L. Genetic engineering of hematopoiesis: Current stage of clinical translation and future perspectives. EMBO Mol. Med. 2019, 11, e9958. [Google Scholar] [CrossRef]
- Aiuti, A.; Roncarolo, M.G.; Naldini, L. Gene therapy for ADA-SCID, the first marketing approval of an ex vivo gene therapy in Europe: Paving the road for the next generation of advanced therapy medicinal products. EMBO Mol. Med. 2017, 9, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral hematopoietic stem cell gene therapy in patients with wiskott-aldrich syndrome. Science 2013, 341, 1233151. [Google Scholar] [CrossRef]
- Hacein-Bey Abina, S.; Gaspar, H.B.; Blondeau, J.; Caccavelli, L.; Charrier, S.; Buckland, K.; Picard, C.; Six, E.; Himoudi, N.; Gilmour, K.; et al. Outcomes following gene therapy in patients with severe Wiskott-Aldrich syndrome. JAMA 2015, 313, 1550–1563. [Google Scholar] [CrossRef]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Eichler, F.; Duncan, C.; Musolino, P.L.; Orchard, P.J.; De Oliveira, S.; Thrasher, A.J.; Armant, M.; Dansereau, C.; Lund, T.C.; Miller, W.P.; et al. Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N. Engl. J. Med. 2017, 377, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef]
- Sessa, M.; Lorioli, L.; Fumagalli, F.; Acquati, S.; Redaelli, D.; Baldoli, C.; Canale, S.; Lopez, I.D.; Morena, F.; Calabria, A.; et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: An ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 2016, 388, 476–487. [Google Scholar] [CrossRef]
- Marktel, S.; Scaramuzza, S.; Cicalese, M.P.; Giglio, F.; Galimberti, S.; Lidonnici, M.R.; Calbi, V.; Assanelli, A.; Bernardo, M.E.; Rossi, C.; et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent ß-thalassemia. Nat. Med. 2019, 25, 234–241. [Google Scholar] [CrossRef]
- De Ravin, S.S.; Wu, X.; Moir, S.; Anaya-O’Brien, S.; Kwatemaa, N.; Littel, P.; Theobald, N.; Choi, U.; Su, L.; Marquesen, M.; et al. Lentiviral hematopoietic stem cell gene therapy for X-linked severe combined immunodeficiency. Sci. Transl. Med. 2016, 8, 335ra57. [Google Scholar] [CrossRef]
- Wang, J.; Meng, F.; Kim, B.K.; Ke, X.; Yeo, Y. In-vitro and in-vivo difference in gene delivery by lithocholic acid-polyethyleneimine conjugate. Biomaterials 2019, 217, 119296. [Google Scholar] [CrossRef]
- Li, L.; He, Z.Y.; Wei, X.W.; Gao, G.P.; Wei, Y.Q. Challenges in CRISPR/CAS9 Delivery: Potential Roles of Nonviral Vectors. Hum. Gene Ther. 2015, 26, 452–462. [Google Scholar] [CrossRef]
- Godbey, W.T.; Wu, K.K.; Mikos, A.G. Size matters: Molecular weight affects the efficiency of poly(ethylenimine) as a gene delivery vehicle. J. Biomed. Mater. Res. 1999, 45, 268–275. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Zhao, P.; Chen, Z.; Lin, Q. Hyperbranched-star PEI-g-PEG as a nonviral vector with efficient uptake and hypotoxicity for retinoblastoma gene therapy application. Colloid Interface Sci. Commun. 2022, 50, 100647. [Google Scholar] [CrossRef]
- Domingues, C.S.D.C.; Serambeque, B.P.; Laranjo Cândido, M.S.; Marto, C.M.M.; Veiga, F.J.B.; Sarmento Antunes Cruz Ribeiro, A.B.; Figueiras, A.R.R.; Botelho, M.F.R.; Dourado, M.A.R.F. Epithelial-mesenchymal transition and microRNAs: Challenges and future perspectives in oral cancer. Head Neck 2018, 40, 2304–2313. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.; Domingues, C.; Jarak, I.; Veiga, F.; Figueiras, A. Osteosarcoma from the unknown to the use of exosomes as a versatile and dynamic therapeutic approach. Eur. J. Pharm. Biopharm. 2022, 170, 91–111. [Google Scholar] [CrossRef] [PubMed]
- Zu, H.; Gao, D. Non-viral Vectors in Gene Therapy: Recent Development, Challenges, and Prospects. AAPS J. 2021, 23, 78. [Google Scholar] [CrossRef] [PubMed]
- Bahadir, E.B.; Sezgintürk, M.K. Poly(amidoamine) (PAMAM): An emerging material for electrochemical bio(sensing) applications. Talanta 2016, 148, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, L.; Shao, N.; Hu, Z.; Chen, H.; Xu, L.; Wang, C.; Cheng, Y.; Xiao, J. Triazine-modified dendrimer for efficient TRAIL gene therapy in osteosarcoma. Acta Biomater. 2015, 17, 115–124. [Google Scholar] [CrossRef]
- Flores-Mejía, R.; Fragoso-Vázquez, M.J.; Pérez-Blas, L.G.; Parra-Barrera, A.; Hernández-Castro, S.S.; Estrada-Pérez, A.R.; Rodrígues, J.; Lara-Padilla, E.; Ortiz-Morales, A.; Correa-Basurto, J. Chemical characterization (LC–MS–ESI), cytotoxic activity and intracellular localization of PAMAM G4 in leukemia cells. Sci. Rep. 2021, 11, 8210. [Google Scholar] [CrossRef]
- Kandasamy, G.; Danilovtseva, E.N.; Annenkov, V.V.; Krishnan, U.M. Poly(1-vinylimidazole) polyplexes as novel therapeutic gene carriers for lung cancer therapy. Beilstein J. Nanotechnol. 2020, 11, 354. [Google Scholar] [CrossRef]
- Pack, D.W.; Putnam, D.; Langer, R. Design of Imidazole-Containing Endosomolytic Biopolymers for Gene Delivery. Biotechnol. Bioeng. 2000, 67, 217–223. [Google Scholar] [CrossRef]
- Jeon, W.Y.; Choi, Y.B.; Kim, H.H. Ultrasonic synthesis and characterization of poly(acrylamide)-co-poly(vinylimidazole)@MWCNTs composite for use as an electrochemical material. Ultrason. Sonochem. 2018, 43, 73–79. [Google Scholar] [CrossRef]
- Abdellatif Soliman, S.M.; Sanad, M.F.; Shalan, A.E. Synthesis, characterization and antimicrobial activity applications of grafted copolymer alginate-g-poly(N-vinyl imidazole). RSC Adv. 2021, 11, 11541. [Google Scholar] [CrossRef]
- Massoudi, S.; Bagheri, M.; Beygi Khosrowshahi, Y.; Hosseini, M. Antibacterial and cytotoxicity assessment of poly (N-vinyl imidazole)/nitrogen-doped graphene quantum dot nanocomposite hydrogels. Polym. Bull. 2023, 80, 6471–6494. [Google Scholar] [CrossRef]
- MacLaughlin, F.C.; Mumper, R.J.; Wang, J.; Tagliaferri, J.M.; Gill, I.; Hinchcliffe, M.; Rolland, A.P. Chitosan and depolymerized chitosan oligomers as condensing carriers for in vivo plasmid delivery. J. Control Release 1998, 56, 259–272. [Google Scholar] [CrossRef]
- Nurunnabi, M.; Revuri, V.; Huh, K.M.; Lee, Y.K. Polysaccharide based nano/microformulation: An effective and versatile oral drug delivery system. In Nanostructures for Oral Medicine; Elsevier: Amsterdam, The Netherlands, 2017; pp. 409–433. [Google Scholar] [CrossRef]
- Ritthidej, G.C. Nasal Delivery of Peptides and Proteins with Chitosan and Related Mucoadhesive Polymers. In Peptide and Protein Delivery; Academic Press: Cambridge, MA, USA, 2011; pp. 47–68. [Google Scholar] [CrossRef]
- Ta, H.T.; Dass, C.R.; Larson, I.; Choong, P.F.M.; Dunstan, D.E. A chitosan hydrogel delivery system for osteosarcoma gene therapy with pigment epithelium-derived factor combined with chemotherapy. Biomaterials 2009, 30, 4815–4823. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, Y.; Qi, H.; Cui, W.; Zhang, L.; Fu, X.; He, X.; Liu, M.; Li, P.F.; Yu, T. CRISPR/Cas9 therapeutics: Progress and prospects. Signal Transduct. Target. Ther. 2023, 8, 36. [Google Scholar] [CrossRef]
- Vigliotti, V.S.; Martinez, I. Public health applications of CRISPR: How children’s health can benefit. Semin. Perinatol. 2018, 42, 531. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Simões, M.L.; Marois, E.; Dimopoulos, G. CRISPR/Cas9 -mediated gene knockout of Anopheles gambiae FREP1 suppresses malaria parasite infection. PLOS Pathog. 2018, 14, e1006898. [Google Scholar] [CrossRef] [PubMed]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.-S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef]
- Webber, B.R.; Osborn, M.J.; McElroy, A.N.; Twaroski, K.; Lonetree, C.; DeFeo, A.P.; Xia, L.; Eide, C.; Lees, C.J.; McElmurry, R.T.; et al. CRISPR/Cas9-based genetic correction for recessive dystrophic epidermolysis bullosa. NPJ Regen. Med. 2016, 1, 16014. [Google Scholar] [CrossRef] [PubMed]
- Dhandapani, J.P. Butterfly Children/Epidermolysis Bullosa. Pondicherry J. Nurs. 2021, 14, 66–68. [Google Scholar] [CrossRef]
- Grand View Research (GVR). Cell Therapy Market Size, Share And Growth Report, 2030. Available online: https://www.grandviewresearch.com/industry-analysis/cell-therapy-market (accessed on 9 August 2023).
- Mordor Intelligence Cell Therapy Market—Size, Trends, Industry Analysis & Growth. Available online: https://www.mordorintelligence.com/industry-reports/cell-therapy-market (accessed on 9 August 2023).
- Azar, J.; Bahmad, H.F.; Daher, D.; Moubarak, M.M.; Hadadeh, O.; Monzer, A.; Bitar, S.A.; Jamal, M.; Al-Sayegh, M.; Abou-Kheir, W. The Use of Stem Cell-Derived Organoids in Disease Modeling: An Update. Int. J. Mol. Sci. 2021, 22, 7667. [Google Scholar] [CrossRef]
- Clarke, M.F. Clinical and Therapeutic Implications of Cancer Stem Cells. N. Engl. J. Med. 2019, 380, 2237–2245. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Hao, J.; Wang, L.; Tan, Y.; Tian, Y.; Li, S.; Ma, A.; Fu, B.; Dai, J.; Zhai, P.; et al. Developing Standards to Support the Clinical Translation of Stem Cells. Stem Cells Transl. Med. 2021, 10, S85–S95. [Google Scholar] [CrossRef]
- Zhang, K.; Cheng, K. Stem cell-derived exosome versus stem cell therapy. Nat. Rev. Bioeng. 2023, 1, 608–609. [Google Scholar] [CrossRef]
- Ji, Y.; Hu, C.; Chen, Z.; Li, Y.; Dai, J.; Zhang, J.; Shu, Q. Clinical trials of stem cell-based therapies for pediatric diseases: A comprehensive analysis of trials registered on ClinicalTrials.gov and the ICTRP portal site. Stem Cell Res. Ther. 2022, 13, 307. [Google Scholar] [CrossRef]
- Yáñez-Muñoz, R.J.; Grupp, S.A. CAR-T in the clinic: Drive with care. Gene Ther. 2018, 25, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, M.; Joechner, A.; Li, Z.; Yang, S.F.; Schlegel, P. Development of CAR T Cell Therapy in Children—A Comprehensive Overview. J. Clin. Med. 2022, 11, 2158. [Google Scholar] [CrossRef] [PubMed]
- Blache, U.; Popp, G.; Dünkel, A.; Koehl, U.; Fricke, S. Potential solutions for manufacture of CAR T cells in cancer immunotherapy. Nat. Commun. 2022, 13, 5225. [Google Scholar] [CrossRef]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Hucks, G.; Rheingold, S.R. The journey to CAR T cell therapy: The pediatric and young adult experience with relapsed or refractory B-ALL. Blood Cancer J. 2019, 9, 10. [Google Scholar] [CrossRef]
- Moretti, A.; Ponzo, M.; Nicolette, C.A.; Tcherepanova, I.Y.; Biondi, A.; Magnani, C.F. The Past, Present, and Future of Non-Viral CAR T Cells. Front. Immunol. 2022, 13, 867013. [Google Scholar] [CrossRef]
- Zhao, N.; Song, Y.; Xie, X.; Zhu, Z.; Duan, C.; Nong, C.; Wang, H.; Bao, R. Synthetic biology-inspired cell engineering in diagnosis, treatment, and drug development. Signal Transduct. Target. Ther. 2023, 8, 112. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, K.; Zambaiti, E.; De Coppi, P. Regenerative medicine: Current research and perspective in pediatric surgery. Pediatr. Surg. Int. 2023, 39, 167. [Google Scholar] [CrossRef] [PubMed]
- Lally, C.; Joyce, K.; Pandit, A. Biomaterials enhancing performance of cell and nucleic-acid therapies: An opportunity in the brain. Biomater. Biosyst. 2022, 5, 100036. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, G.C.; Lanyi, K.; Inskip, A.; Ogunbayo, O.J.; Brhlikova, P.; Craig, D. A pipeline analysis of advanced therapy medicinal products. Drug Discov. Today 2023, 28, 103549. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Commercial Name | Active Agent | Composition (Molar Ratio) | Indication | Approval Year | Ref. |
|---|---|---|---|---|---|
| Cancer | |||||
| Doxil® | Doxorubicin | HSPC:Cholesterol:PEG 2000-DSPE (56:39:5) | Kaposi’s sarcoma, ovarian cancer, multiple myeloma | 1995 | [146] |
| DaunoXome® | Daunorubicin | DSPC:Cholesterol (10:5) | Kaposi’s sarcoma | 1996 | [147,148] |
| DepoCyt® | Cytarabin and AraC | Cholesterol:Triolein:DOPC:DPPG (11:1:7:1) | Complication of lymphoma lymphomatous meningitis | 1999 | [149] |
| Myocet® | Doxorubicin | EPC:Cholesterol (55:45 molar ratio) | Metastatic breast cancer | 2000 | [150] |
| Mepact® | Mifamurtide | DOPS:POPC (3:7) | Osteosarcoma | 2004 | [150] |
| Marqibo® | Vincristin | Sphingomyelin:Cholesterol (60:40) | Acute lymphoblastic leukemia | 2012 | [149] |
| OnivydeTM | Irinotecan | Metastatic pancreatic adenocarcinoma | 2015 | [150] | |
| Vyxeos® | 1:5 molar ratio of daunorubicin:cytarabine | DSPC:DSPG:Cholesterol (7:2:1) | Acute myeloid leukemia | 2017 | [151] |
| Infection | |||||
| Arikayce® | Amikacin | DPPC:Cholesterol (2:1 weight ratio) | Pulmonary infection caused by Mycobacterium avium | 2018 | [152,153] |
| Abelcet® | Amphotericin B | DMPC:DMPG (7:3) | Fungal infections | 1995 | [150] |
| Amphotec® | Amphotericin B | Cholesteryl sulphate:Amphotericin B (1:1 molar ratio) | Fungal infections | 1996 | [150] |
| AmBisome® | Amphotericin B | HSPC:DSPG:Cholesterol:Amphotericin B (2:0.8:1:0.4) | Fungal/protozoal infections | 1997 | [154] |
| Pain management | |||||
| DepoDurTM | Morphine sulfate | DOPC, DPPG, Cholesterol, Triolein | Pain management | 2004 | [150] |
| Exparel® | Bupivacaine | DEPC, DPPG, Cholesterol and Tricaprylin | Pain management | 2011 | [150] |
| Photodynamic therapy (Ophtalmic) | |||||
| Visudyne® | Verteporfin (Photosensitizer) | Verteporphin:EPG:DMPC (1:3:5) | Wet age-related macular degeneration, myopia, ocular histoplasmosis | 2000 | [149,155] |
| Nucleic acid therapy | |||||
| OnpattroTM (Patisiran) | siRNA lipid formulation designed to target transthyretin (TTR) mRNA in the liver cells | Hereditary transthyretin amyloidosis (hATTR) | 2018 | [156,157,158] | |
| Vaccines | |||||
| Epaxal® | Inactivated hepatitis A virus (strain RGSB) | DOPC:DOPE (75:25) | Hepatitis A | 1993 | [150] |
| Inflexal®V | Inactivated hemaglutinine of Influenza virus strains A and B | DOPC:DOPE (75:25) | Influenza | 1997 | [150] |
| mRNA-1273 | mRNA | Positively charged lipid:PEGylated lipid:Cholesterol:DSPC (50:1.5:38.5:10) | COVID-19 | 2020, Emergency Use Authorization | [159,160] |
| BNT162b2 | mRNA | Positively charged lipid:PEGylated lipid:Cholesterol:DSPC (46.3:1.6:42.7:9.4) | COVID-19 | 2020, Emergency Use Authorization | [159,160] |
| Name | Lipids Used for Liposomes | Adult PK Parameters | Pediatric PK Parameters | Ratios of Pediatric Versus Adult PK Parameters | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dose | AUC0–∞ (ng/mL·h) | Cmax (ng/mL) | Tmax (h) | T1/2 (h) | CL (mL/min) | Dose | AUC0–∞ (ng/ml.hr) | Cmax (ng/mL) | Tmax (h) | T1/2 (h) | CL (mL/min) | AUC | Cmax | Tmax | T1/2 | CL | ||
| Marqibo® (Vincristine sulfate) | Sphingomyelin and cholesterol | 2.25 mg/m2, i.v | 14,566 | 1220 | 3.7 | 7.66 | 5.75 | 2.25 mg/m2, i.v | 31,043 | 2150 | 1.12 | 10.7 | 1.2 | 2.13 | 1.76 | 0.3 | 1.39 | 0.2 |
| SPI-77 (Cisplatin) | Soy PC, cholesterol and MPEG-DSPE. | 200 mg/m2, i.v | 13,850,680 | 82,538 | N/A | 103 | 0.29 | 200 mg/m2, i.v | 24,004,000 | 2,414,000 | N/A | 78 | 0.15 | 1.73 | 29.24 | N/A | 0.75 | 0.51 |
| DepoCyt® (Cytarabine) | DOPC, DPPG, cholesterol | 25 mg I/T | 355,000 | 25,000 | N/A | 229 | 0.09 | 25 mg I/T | 363,700 | 21,300 | N/A | 59.3 | 0.24 | 1.02 | 0.85 | N/A | 0.25 | 2.66 |
| DaunoXome® (Daunorubicin) | DSPC, cholesterol | 80 mg/m2, i.v | 10,330 | 400 | N/A | 0.77 | 233 | 80 mg/m2, i.v | 108,206 | 900 | N/A | 12.63 | 7.6 | 10.47 | 2.25 | N/A | 16.4 | 0.03 |
| AmBisome® (Amphotericin B) | Soy PC, DSPG, alpha tocopherol, cholesterol | 2 mg/kg i.v | 288,000 | 22,900 | N/A | 6 | 0.16 | 5 mg/kg i.v | 442,000 | 46,200 | N/A | 12.6 | 0.75 | 0.61 | 0.8 | N/A | 2.1 | 4.68 |
| NCT Number | Phase | Study Status | Conditions | Interventions |
|---|---|---|---|---|
| Cancer | ||||
| NCT05739630 | II and III | Recruiting | Acute Leukemia | Mitoxantrone liposome anti-thymocyte globulin |
| NCT04293562 | III | Recruiting | Acute Myeloid Leukemia | Liposome-encapsulated daunorubicin-cytarabine, among others |
| NCT04606108 | II | Recruiting | Soft Tissue Sarcoma | Camrelizumab in combination with liposome doxorubicin and ifosfamide |
| NCT05656248 | II | Recruiting | Myeloid Neoplasm | Dual-drug liposomal encapsulation of cytarabine and daunorubicin (CPX-351) |
| NCT05620862 | I | Recruiting | Lymphoma, Solid Tumors | Mitoxantrone hydrochloride liposome |
| NCT05457829 | II | Not Yet Recruiting | Rhabdomyosarcoma, Child | Doxorubicin Hydrochloride Liposome+IrinotecanTemozolomide+Irinotecan+Vincristine |
| NCT04915612 | I | Recruiting | Acute Myeloid Leukemia Arising from previous Myelodysplastic Syndrome | Gemtuzumab ozogamicin liposome-encapsulated daunorubicin-cytarabine |
| NCT05926492 | II | Not Yet Recruiting | Osteosarcoma | Surufatinib plus chemotherapy, liposomal doxorubicin |
| NCT04996160 | I | Recruiting | Acute Lymphoblastic Leukemia | Palbociclib, Dexamethasone, Bortezomib, Liposomal Doxorubicin |
| NCT04546620 | II | Recruiting | Diffuse Large B Cell Lymphoma | R-CHOP, R-CHOP + acalabrutinib, Liposomal doxorubicin |
| NCT04199026 | Early Phase I | Recruiting | Metastatic Sarcoma|Recurrent Sarcoma|Resectable Sarcoma | Liposomal doxorubicin, among others |
| NCT05518383 | IV | Recruiting | Luymphoma | Liposomal doxorubicin, among others |
| NCT05315336 | III | Not Yet Recruiting | Hemophagocytic Lymphohistiocytosis | Liposomal doxorubicin, etoposide, and methylprednisolone (L-DEP) and PD-1 antibody |
| NCT05561036 | III | Recruiting | Desmoid Tumor | Liposome doxorubicin |
| NCT05675410 | III | Recruiting | Lugano Classification Limited Stage Hodgkin Lymphoma AJCC v8 | Liposomal doxorubicin, among others |
| NCT04791228 | II | Recruiting | Solid tumors | Lyso-thermosensitive Liposomal Doxorubicin |
| NCT04984174 | Recruiting | Pancreatic Cancer | Liposomal Irinotecan | |
| NCT05576532 | II | Recruiting | T-lymphoblastic Lymphoma | BCL2 Inhibitor plus IM2 regimen, Liposome mitoxantrone |
| NCT05711628 | III | Not Yet Recruiting | Lymphoma | Pegylated Liposomal Doxorubicin Hydrochloride among others |
| NCT04589741 | II | Recruiting | Soft Tissue Sarcoma | Toripalimab, Liposome adriamycin |
| NCT05210374 | I | Recruiting | Relapsed Sarcomas | Liposomal doxorubicin, among others |
| Other pathologies | ||||
| NCT05730920 | IV | Recruiting | Adolescent/Juvenile Idiopathic Scoliosis | Liposomal bupivacaine |
| NCT05714176 | IV | Not Yet Recruiting | Chronic Kidney Disease (CKD) | Ferric Pyrophosphate Liposomal |
| NCT05468372 | II | Recruiting | Mucormycosis; Pulmonary (Etiology) | Liposomal Amphotericin B |
| NCT04799236 | III | Recruiting | Mucosal Leishmaniasis | Liposomal Amphotericin B, among others |
| Chitosan | Model Drug | Aim | Refs. |
|---|---|---|---|
| Chitosan with different molecular weights, degrees of deacetylation (DDA), and patterns of deacetylation | Prednisolone | To develop child-friendly solid dosage forms, e.g., oromucosal films and wafers | [173] |
| Low-molecular-weight chitosan (CS, 50–190 kDa, 75–85% deacetylation degree) | Cephalosporin | To formulate effective oral solutions of poorly soluble drugs suitable | [170] |
| Chitosan from Portunus Sanguinolentus | Dolutegravir | To adjust the dose | [174] |
| Medium molecular weight chitosan (190–310 KDa; 75–85% deacetylated) | Rufinamide | To reduce dose and dose frequency of Rufinamide by formulating Rufinamide-loaded chitosan nanoparticles suspended in a solution of a thermo-responsive polymer–tamarind seed xyloglucan for in situ gelling | [175] |
| Chitosan (90–95% deacetylation degree) | Cinnarizine | To develop chitosan microspheres for oral pediatric formulation with improved stability, organoleptic properties, and easier administration | [176] |
| Chitosan (approx. MW 296.6 kDa and deacetylation 82.83 ± 3.63%) | Didanosine | To make chitosan granules containing didanosine incorporated in chitosan microspheres, to facilitate handling and deglutition | [177] |
| NCT Number | Brief Summary | Conditions | Completion Date | Study Results |
|---|---|---|---|---|
| NCT00707486 | The purpose of this study is to determine whether the HemCon Dental Dressing is effective in stopping bleeding during dental surgeries. | Tooth Extractions | 1 July 2009 | YES |
| NCT01597817 | To evaluate the effect of a textile coated with chitosan in atopic dermatitis (AD) treatment as well as its impact on systemic inflammation and skin microbiome. | Atopic Dermatitis | 1 December 2012 | NO |
| NCT01950546 | To evaluate the effectiveness of nanosilver fluoride for controlling the growth of S. mutans present in the dental plaque of children. | Dental Caries | 1 January 2015 | NO |
| NCT02789033 | To assess the efficacy of the combination of isosorbide dinitrate spray and chitosan in diabetic foot ulcers. | Diabetic Foot Ulcers | 1 August 2015 | YES |
| NCT02668055 | To evaluate the slow-release Tb4 collagen and chitosan porous sponge scaffolds skin substitutes and the effectiveness of clinical trials for the treatment of difficult-to-heal wounds and security. | Wounds | 1 December 2015 | NO |
| NCT05475444 | PLGA nanoparticles coated with chitosan polymer were prepared and then incorporated in in situ gel to be injected into root canals of patients suffering from bacterial infection of their endodontics. | Bacterial Infections Oral | 15 March 2020 | NO |
| NCT04365270 | To assess the antibacterial effect on carious dentine of glass ionomers when modified with chitosan and/or titanium dioxide nanoparticles versus the control group of modification with chlorhexidine when used in primary molars. | Caries | 5 January 2021 | NO |
| NCT03421717 | Peri-implantitis is an inflammation in the mucosa surrounding an oral implant with loss of the supporting bone. The goals of peri-implantitis treatment are to resolve inflammation and arrest disease progression. | Periimplantitis|Peri-implant Mucositis | 8 April 2021 | NO |
| NCT04906291 | To verify the caries-preventive efficacy of toothpaste containing biomimetic hydroxyapatite (H.A.) complex in children compared to traditional fluoridated toothpaste. | Caries | 31 October 2021 | NO |
| NCT04481945 | To assess antimicrobial activity of nanosilver- and chitosan-inserted C sealer | Endodontic Disease | 1 January 2022 | NO |
| NCT04005872 | The management of deep carious lesions. | Deep Caries | 30 November 2022 | NO |
| Tradename | Formulation | Intervention | Approval Year |
|---|---|---|---|
| INFeD® | Iron Dextran Injection USP | Iron-deficient anemia | 1992 |
| DexFerrum® | Iron Dextran Injection USP | Iron-deficient anemia | 1996 |
| Ferrlecit® | Ferric gluconate (Rx) | Iron deficiency in chronic kidney disease | 1999 |
| Venofer® | Iron sucrose injection | Iron deficiency in chronic kidney disease | 2000 |
| Feraheme® | Ferumoxytol injection | Iron deficiency in chronic kidney disease | 2009 |
| Injectafer® | Ferric carboxymaltose injection | Iron-deficient anemia | 2013 |
| Name | Type of ATMP | Indication | Approval |
|---|---|---|---|
| Cell therapy medicinal product. | |||
| Allocord Clevecord Ducord Hemacord | Cord blood HPC | Indicated for use in unrelated-donor hematopoietic progenitor stem cell transplantation procedures. | FDA |
| Ebvallo | Allogeneic T-cell immunotherapy | To treat adults and children from 2 years of age who, after receiving an organ or a bone marrow-transplantation, develop a blood cancer called Epstein–Barr virus positive post-transplant lymphoproliferative disease (EBV + PTLD). | EMA |
| Omisirge | Cord blood nicotinamide modified allogeneic hematopoietic progenitor cells | Used in adults and pediatric patients 12 years and older with hematologic malignancies who have planned umbilical cord blood transplantation following myeloablative conditioning to reduce the time to neutrophil recovery and the incidence of infection. | FDA |
| Gene therapy medicinal product. | |||
| Elevidys | Non-replicating, recombinant adeno-associated virus for delivery of Micro-dystrophine gene | To treat ambulatory pediatric patients aged 4–5 years with Duchenne muscular dystrophy (DMD) with a confirmed mutation in the micro-dystrophine gene. | FDA |
| Luxturna | Adeno-associated viral vector serotype 2 for delivery of RPE65 gene | To treat adult and pediatric patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy. | FDA EMA |
| Skysona | Autologous CD34+ enriched HSCs transduced ex vivo with lentiviral vector encoding ABCD1 complementary deoxyribonucleic acid (cDNA) for human adrenoleukodystrophy protein | To slow the progression of neurologic dysfunction in male patients 4–17 years of age with early, active cerebral adrenoleukodystrophy with no available sibling hematopoietic stem cell donor. | FDA No longer authorized by EMA |
| Vyjuvek | Herpes-simplex virus type 1 vector for delivery of COL7A1 gene | To treat wounds in patients with 6 months of age and older with dystrophic epidermolysis bullosa with mutation(s) in the collagen type VII alpha 1 chain (COL7A1) gene. | FDA |
| Zynteglo | Autologous CD34+ enriched hematopoietic stem cells transduced ex vivo with lentiglobin BB305 lentiviral vector encoding beta-A-T87Q-globin gene | Treatment of adult and pediatric patients with ß-thalassemia who require regular red blood cell (RBC) transfusions. | FDA No longer authorized by EMA |
| Zolgensma | Adeno-associated viral vector serotype 9 encoding the Survival Motor Neuron 1 gene | Treatment of Spinal Muscular Atrophy (Type I) for pediatric patients under 2 years of age. | FDA EMA |
| Strimvelis | Autologous CD34+ enriched HSC transduced with retroviral vector that encodes for the human ADA cDNA sequence | To treat patients with severe combined immunodeficiency due to adenosine deaminase deficiency (ADA-SCID). | EMA |
| Kymriah | Genetically modified (chimeric antigen receptor) autologous T cell immunotherapy | To treat children or young adults (up to 25 years old) with Acute Lymphoblastic Leukemia. | EMA |
| Libmeldy | Autologous hematopoietic stem and progenitor cell transfected ex vivo with lentiviral vector encoding the human Arylsulfatase A gene | To treat children with metachromatic leukodystrophy. | EMA |
| Upstaza | Adeno-associated viral vector encoding L-amino Acid Decarboxylase gene | To treat adults and children aged 18 months and older with severe aromatic L-amino acid decarboxylase deficiency with a genetically confirmed diagnosis. | EMA |
| Tissue engineered product | |||
| Rethymic | Allogeneic processed thymus tissue | Immune reconstitution in pediatric patients with congenital athymia. | FDA |
| Spherox | Chondrocyte spheroids | To repair defects to the cartilage in the knee in patients who are experiencing symptoms (such as pain and problems moving the knee) in adults and adolescents. | EMA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domingues, C.; Jarak, I.; Veiga, F.; Dourado, M.; Figueiras, A. Pediatric Drug Development: Reviewing Challenges and Opportunities by Tracking Innovative Therapies. Pharmaceutics 2023, 15, 2431. https://doi.org/10.3390/pharmaceutics15102431
Domingues C, Jarak I, Veiga F, Dourado M, Figueiras A. Pediatric Drug Development: Reviewing Challenges and Opportunities by Tracking Innovative Therapies. Pharmaceutics. 2023; 15(10):2431. https://doi.org/10.3390/pharmaceutics15102431
Chicago/Turabian StyleDomingues, Cátia, Ivana Jarak, Francisco Veiga, Marília Dourado, and Ana Figueiras. 2023. "Pediatric Drug Development: Reviewing Challenges and Opportunities by Tracking Innovative Therapies" Pharmaceutics 15, no. 10: 2431. https://doi.org/10.3390/pharmaceutics15102431
APA StyleDomingues, C., Jarak, I., Veiga, F., Dourado, M., & Figueiras, A. (2023). Pediatric Drug Development: Reviewing Challenges and Opportunities by Tracking Innovative Therapies. Pharmaceutics, 15(10), 2431. https://doi.org/10.3390/pharmaceutics15102431