
Enhancing the Treatment of Uncontrolled Inflammation through the Targeted Delivery of TPCA-1-Loaded Nanoparticles

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
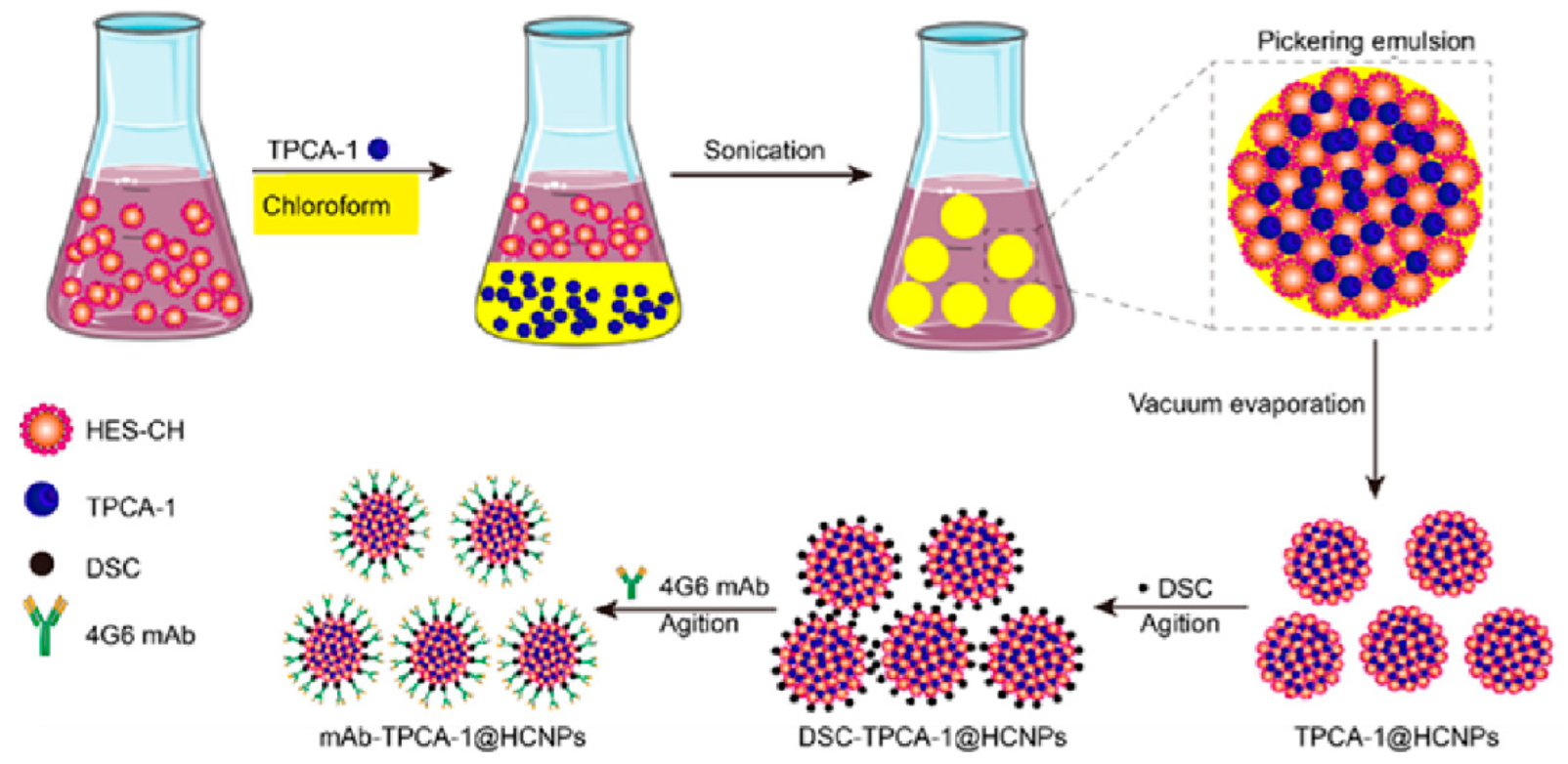
2.2. Preparation of mAb-TPCA-1@HCNPs
2.3. Characterization of mAb-TPCA-1@HCNPs
2.4. Hemolytic Assay
2.5. Cell Culture
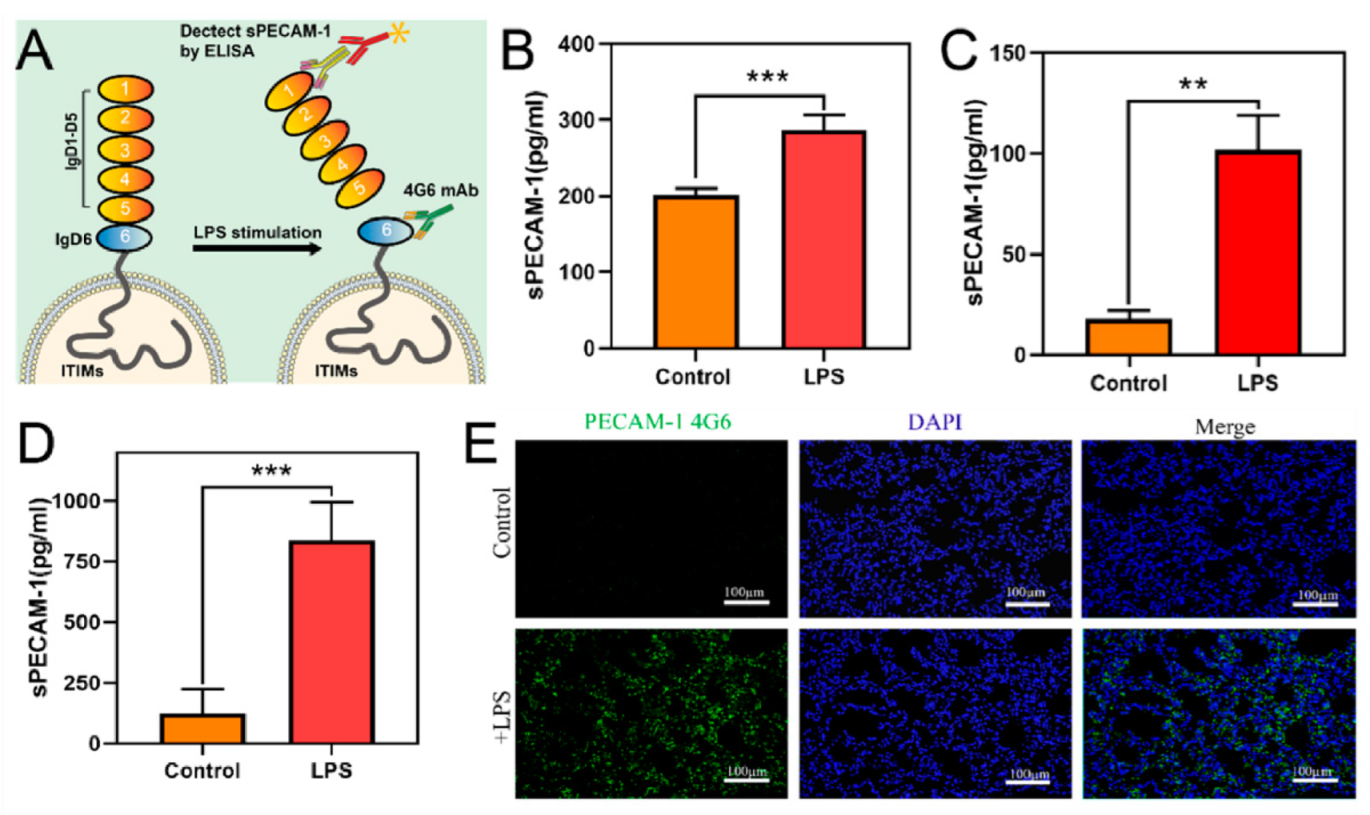
2.6. In Vivo and In Vitro Validation of PECAM-1 Extracellular Segment Cleavage under Inflammatory Stimulation
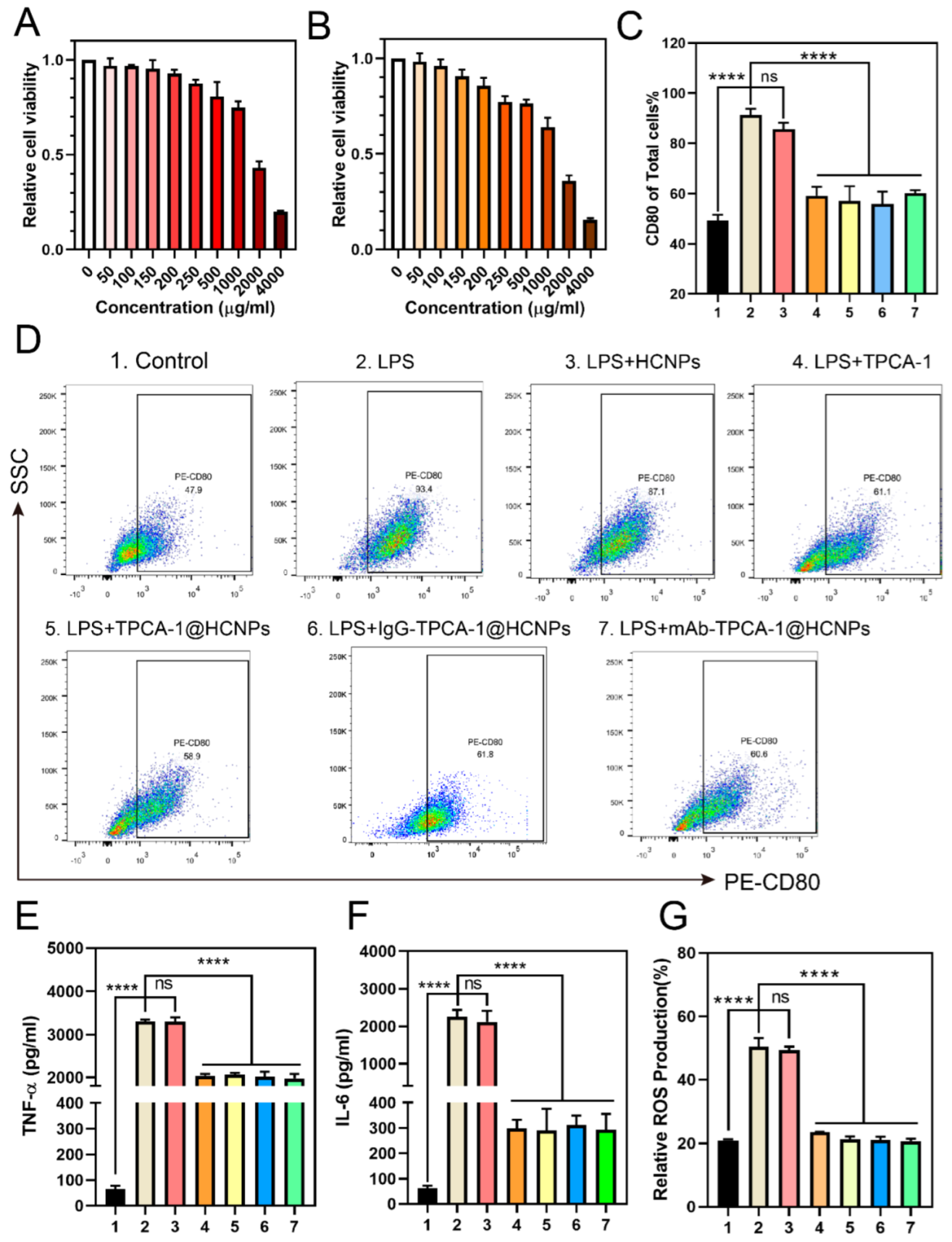
2.7. In Vitro Cell Cytotoxicity Assay
2.8. Cytokine Assay
2.9. Intracellular ROS and NO Measurement
2.10. Macrophage Polarization Analysis
2.11. In Vitro Binding and Cellular Uptake Experiments
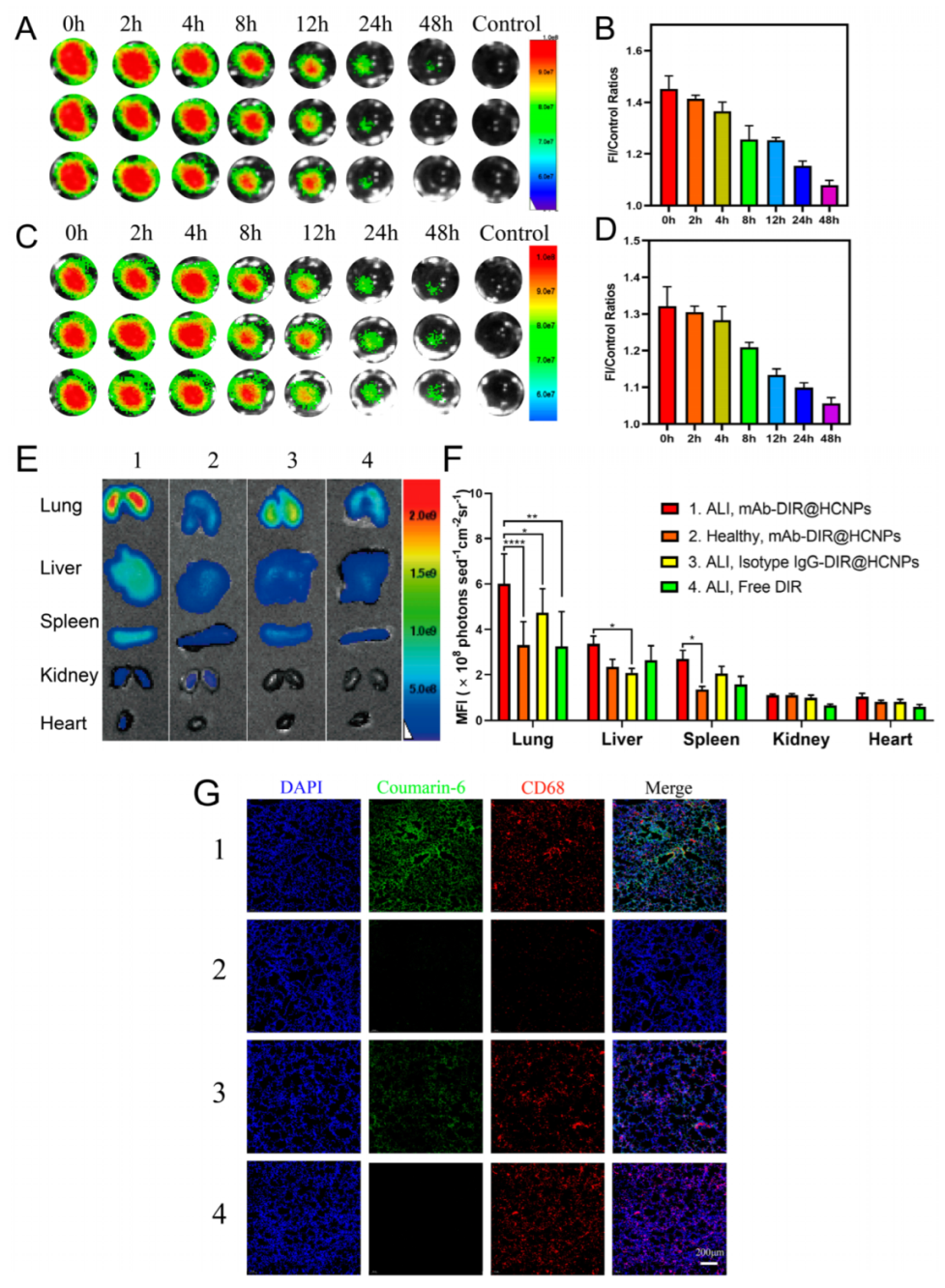
2.12. In Vivo Pharmacokinetics Study of NPs in Mice
2.13. Animal Model Establishment and Treatment
2.14. In Vivo Targeting and Biodistribution of NPs in a Mouse Model
2.15. In Vivo Therapeutic Efficacy Experiment
2.16. Detection of ROS Levels in Mouse Lung Tissue
2.17. Biosafety Evaluation of NPs in Mice
2.18. Statistical Analysis
3. Results
3.1. Preparation and Characterization of mAb-TPCA-1@HCNPs
3.2. In Vivo and In Vitro Validation of PECAM-1 Extracellular Segment Cleavage
3.3. In Vitro Cell Cytotoxicity and Therapeutic Effect
3.4. In Vitro Binding and Cellular Uptake Experiments
3.5. In Vivo Pharmacokinetics and Biodistribution Study
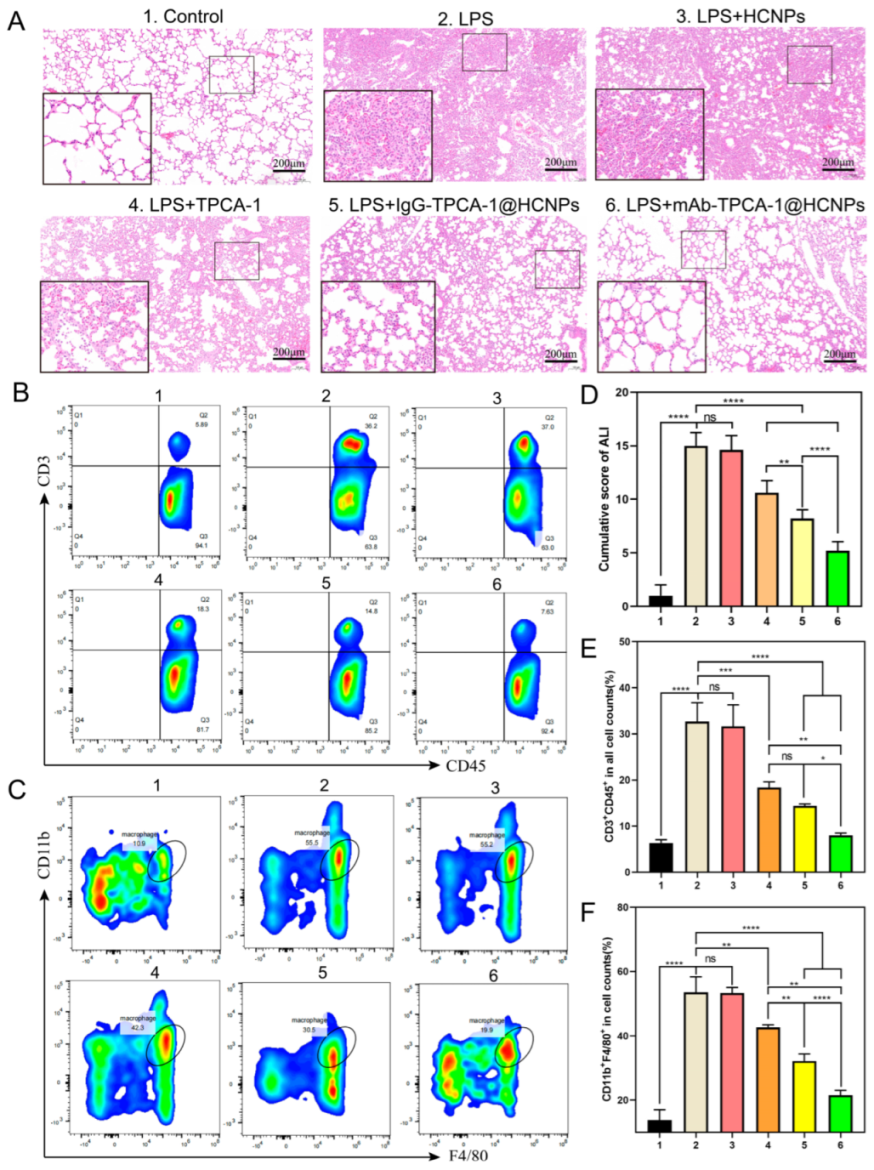
3.6. In Vivo Therapeutic Efficacy Experiment
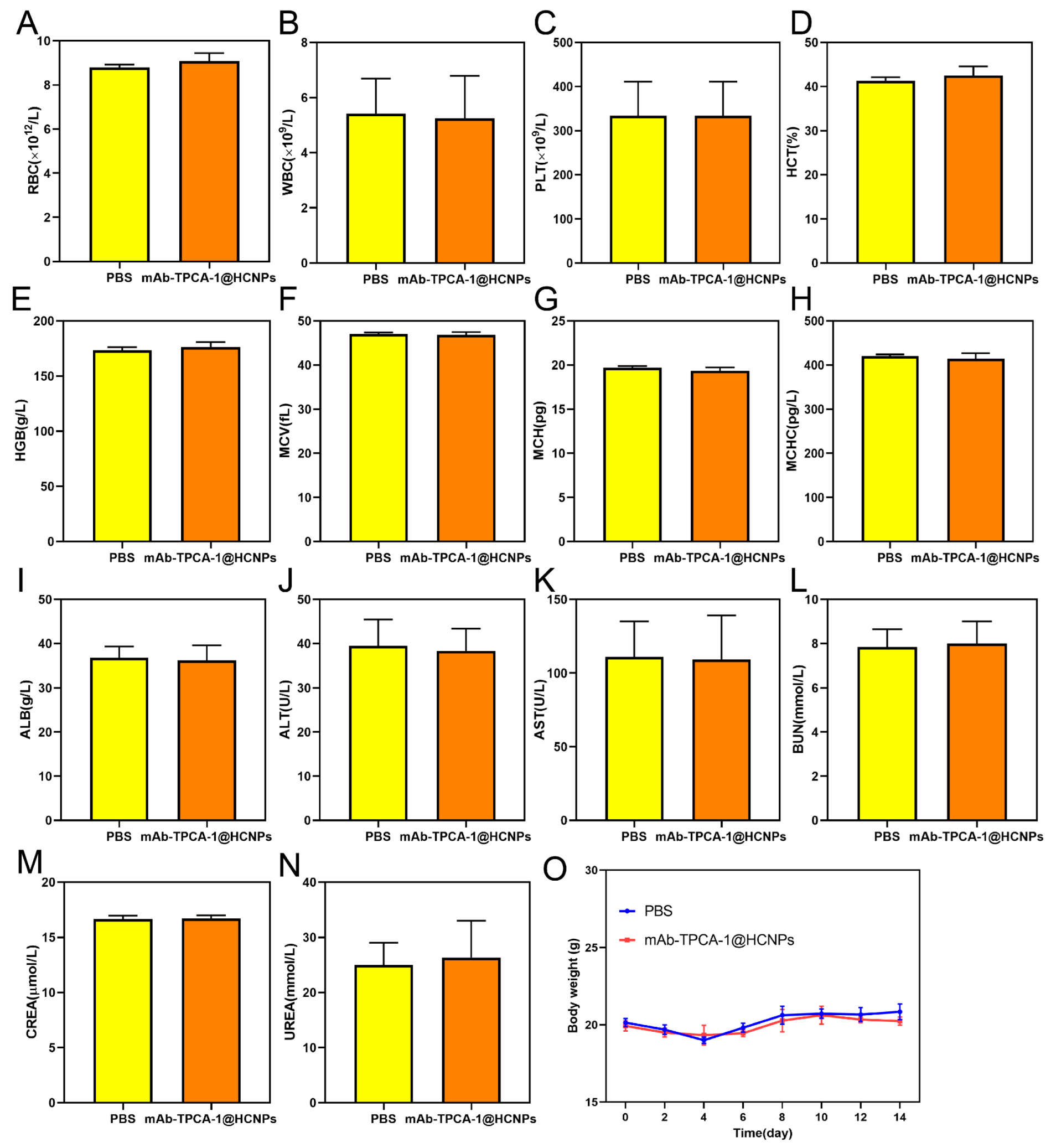
3.7. Biosafety Evaluation in Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gotts, J.E.; Matthay, M.A. Sepsis: Pathophysiology and clinical management. BMJ 2016, 353, i1585. [Google Scholar] [CrossRef]
- Kuypers, F.A. Hyperinflammation, apoptosis, and organ damage. Exp. Biol. Med. 2022, 247, 1112–1123. [Google Scholar] [CrossRef]
- Suberviola, B.; Cuenca Fito, E. Management of hyperinflammation in COVID-19 patients. Rev. Esp. Quimioter. 2022, 35 (Suppl. S3), 6–9. [Google Scholar] [CrossRef]
- Ghosh, S.; Durgvanshi, S.; Han, S.S.; Bhaskar, R.; Sinha, J.K. Therapeutics for the Management of Cytokine Release Syndrome in COVID-19. Curr. Top. Med. Chem. 2023, 23, 128–142. [Google Scholar] [CrossRef]
- Toniati, P.; Piva, S.; Cattalini, M.; Garrafa, E.; Regola, F.; Castelli, F.; Franceschini, F.; Airo, P.; Bazzani, C.; Beindorf, E.A.; et al. Tocilizumab for the treatment of severe COVID-19 pneumonia with hyperinflammatory syndrome and acute respiratory failure: A single center study of 100 patients in Brescia, Italy. Autoimmun. Rev. 2020, 19, 102568. [Google Scholar] [CrossRef]
- Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2021, 397, 1637–1645. [CrossRef]
- Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; Elmahi, E.; et al. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef]
- Chang, C.; Greenspan, A.; Gershwin, M.E. The pathogenesis, diagnosis and clinical manifestations of steroid-induced osteonecrosis. J. Autoimmun. 2020, 110, 102460. [Google Scholar] [CrossRef]
- Li, W.; Huang, Z.; Tan, B.; Chen, G.; Li, X.; Xiong, K.; Zhu, R.; Li, R.; Li, S.; Ye, H.; et al. General recommendation for assessment and management on the risk of glucocorticoid-induced osteonecrosis in patients with COVID-19. J. Orthop. Transl. 2021, 31, 1–9. [Google Scholar] [CrossRef]
- Lambert, N.; Hansen, I.; El Moussaoui, M.; Giot, J.B.; Vercheval, C.; Lommers, É.; Somja, J.; Moutschen, M.; Maquet, P. Lung and liver sarcoidosis-like reaction induced by tocilizumab. Br. J. Clin. Pharmacol. 2021, 87, 4848–4852. [Google Scholar] [CrossRef]
- Chambers, E.; Rounds, S.; Lu, Q. Pulmonary Endothelial Cell Apoptosis in Emphysema and Acute Lung Injury. Adv. Anat. Embryol. Cell Biol. 2018, 228, 63–86. [Google Scholar] [CrossRef] [PubMed]
- Maniatis, N.A.; Kotanidou, A.; Catravas, J.D.; Orfanos, S.E. Endothelial pathomechanisms in acute lung injury. Vasc. Pharmacol. 2008, 49, 119–133. [Google Scholar] [CrossRef]
- Cheng, K.T.; Xiong, S.; Ye, Z.; Hong, Z.; Di, A.; Tsang, K.M.; Gao, X.; An, S.; Mittal, M.; Vogel, S.M.; et al. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J. Clin. Investig. 2017, 127, 4124–4135. [Google Scholar] [CrossRef] [PubMed]
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The Role of Cytokines including Interleukin-6 in COVID-19 induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun. Rev. 2020, 19, 102537. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, R.; Seino, K.I. Macrophage activation syndrome and COVID-19. Inflamm. Regen. 2020, 40, 19. [Google Scholar] [CrossRef]
- Newman, P.J. The role of PECAM-1 in vascular cell biology. Ann. N. Y. Acad. Sci. 1994, 714, 165–174. [Google Scholar] [CrossRef]
- Lertkiatmongkol, P.; Liao, D.; Mei, H.; Hu, Y.; Newman, P.J. Endothelial functions of platelet/endothelial cell adhesion molecule-1 (CD31). Curr. Opin. Hematol. 2016, 23, 253–259. [Google Scholar] [CrossRef]
- Newman, P.J. The biology of PECAM-1. J. Clin. Investig. 1997, 99, 3–8. [Google Scholar] [CrossRef]
- Paddock, C.; Zhou, D.; Lertkiatmongkol, P.; Newman, P.J.; Zhu, J. Structural basis for PECAM-1 homophilic binding. Blood 2016, 127, 1052–1061. [Google Scholar] [CrossRef]
- Kjaergaard, A.G.; Dige, A.; Krog, J.; Tønnesen, E.; Wogensen, L. Soluble adhesion molecules correlate with surface expression in an in vitro model of endothelial activation. Basic Clin. Pharmacol. Toxicol. 2013, 113, 273–279. [Google Scholar] [CrossRef]
- Eugenin, E.A.; Gamss, R.; Buckner, C.; Buono, D.; Klein, R.S.; Schoenbaum, E.E.; Calderon, T.M.; Berman, J.W. Shedding of PECAM-1 during HIV infection: A potential role for soluble PECAM-1 in the pathogenesis of NeuroAIDS. J. Leukoc. Biol. 2006, 79, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Fornasa, G.; Groyer, E.; Clement, M.; Dimitrov, J.; Compain, C.; Gaston, A.T.; Varthaman, A.; Khallou-Laschet, J.; Newman, D.K.; Graff-Dubois, S.; et al. TCR stimulation drives cleavage and shedding of the ITIM receptor CD31. J. Immunol. 2010, 184, 5485–5492. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tang, J.; Shuai, W.; Meng, J.; Feng, J.; Han, Z. Macrophage polarization and its role in the pathogenesis of acute lung injury/acute respiratory distress syndrome. Inflamm. Res. 2020, 69, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H. Vascular permeability in cancer and infection as related to macromolecular drug delivery, with emphasis on the EPR effect for tumor-selective drug targeting. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Azzopardi, E.A.; Ferguson, E.L.; Thomas, D.W. The enhanced permeability retention effect: A new paradigm for drug targeting in infection. J. Antimicrob. Chemother. 2013, 68, 257–274. [Google Scholar] [CrossRef]
- Durymanov, M.; Kamaletdinova, T.; Lehmann, S.E.; Reineke, J. Exploiting passive nanomedicine accumulation at sites of enhanced vascular permeability for non-cancerous applications. J. Control. Release 2017, 261, 10–22. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, J.; Yang, Y.; Pu, X.; Zhao, J.; Zhang, N. Targeted and Combined TPCA-1-Gold Nanocage Therapy for In Vivo Treatment of Inflammatory Arthritis. AAPS PharmSciTech 2020, 21, 298. [Google Scholar] [CrossRef]
- Qiao, Q.; Liu, X.; Cui, K.; Li, X.; Tian, T.; Yu, Y.; Niu, B.; Kong, L.; Yang, C.; Zhang, Z. Hybrid Biomimetic Nanovesicles to Drive High Lung Biodistribution and Prevent Cytokine Storm for ARDS Treatment. ACS Nano 2022, 16, 15124–15140. [Google Scholar] [CrossRef]
- Ma, Q.; Fan, Q.; Xu, J.; Bai, J.; Han, X.; Dong, Z.; Zhou, X.; Liu, Z.; Gu, Z.; Wang, C. Calming Cytokine Storm in Pneumonia by Targeted Delivery of TPCA-1 Using Platelet-Derived Extracellular Vesicles. Matter 2020, 3, 287–301. [Google Scholar] [CrossRef]
- Gad, S.C.; Sharp, K.L.; Montgomery, C.; Payne, J.D.; Goodrich, G.P. Evaluation of the toxicity of intravenous delivery of auroshell particles (gold-silica nanoshells). Int. J. Toxicol. 2012, 31, 584–594. [Google Scholar] [CrossRef]
- Kolosnjaj-Tabi, J.; Javed, Y.; Lartigue, L.; Volatron, J.; Elgrabli, D.; Marangon, I.; Pugliese, G.; Caron, B.; Figuerola, A.; Luciani, N.; et al. The One Year Fate of Iron Oxide Coated Gold Nanoparticles in Mice. ACS Nano 2015, 9, 7925–7939. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, D.K.; Zhang, Q.; Franklin, J.L.; Coffey, R.J. Extracellular vesicles and nanoparticles: Emerging complexities. Trends Cell Biol. 2023, 33, 667–681. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, S.; He, L.; Yu, H.; Yu, H. Hydroxyethyl starch 130/0.4 for volume replacement therapy in surgical patients: A systematic review and meta-analysis of randomized controlled trials. Perioper. Med. 2021, 10, 16. [Google Scholar] [CrossRef]
- Tan, R.; Wan, Y.; Yang, X. Hydroxyethyl starch and its derivatives as nanocarriers for delivery of diagnostic and therapeutic agents towards cancers. Biomater. Transl. 2020, 1, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Yang, D.; Long, T.; Yuan, L.; Qiu, S.; Li, D.; Mu, C.; Ge, L. pH-Sensitive nanoparticles based on amphiphilic imidazole/cholesterol modified hydroxyethyl starch for tumor chemotherapy. Carbohydr. Polym. 2022, 277, 118827. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhu, H.; Hu, J.; Zhao, Y.; Wang, Q.; Wan, J.; Yang, Y.; Xu, H.; Yang, X. Highly compressed assembly of deformable nanogels into nanoscale suprastructures and their application in nanomedicine. ACS Nano 2011, 5, 2671–2680. [Google Scholar] [CrossRef]
- Davarpanah, L.; Vahabzadeh, F. Formation of oil-in-water (O/W) pickering emulsions via complexation between β-cyclodextrin and selected organic solvents. Starch-Stärke 2012, 64, 898–913. [Google Scholar] [CrossRef]
- Hu, H.; Xiao, C.; Wu, H.; Li, Y.; Zhou, Q.; Tang, Y.; Yu, C.; Yang, X.; Li, Z. Nanocolloidosomes with Selective Drug Release for Active Tumor-Targeted Imaging-Guided Photothermal/Chemo Combination Therapy. ACS Appl. Mater. Interfaces 2017, 9, 42225–42238. [Google Scholar] [CrossRef]
- Dormont, F.; Brusini, R.; Cailleau, C.; Reynaud, F.; Peramo, A.; Gendron, A.; Mougin, J.; Gaudin, F.; Varna, M.; Couvreur, P. Squalene-based multidrug nanoparticles for improved mitigation of uncontrolled inflammation in rodents. Sci. Adv. 2020, 6, eaaz5466. [Google Scholar] [CrossRef]
- Tanaka, A.; Minoguchi, K.; Chen, X.; Oda, N.; Yokoe, T.; Yamamoto, Y.; Yamamoto, M.; Watanabe, Y.; Ohta, S.; Xu, X.; et al. Activated protein C attenuates leukocyte elastase-induced lung injury in mice. Shock 2008, 30, 153–158. [Google Scholar] [CrossRef]
- Belperio, J.A.; Keane, M.P.; Burdick, M.D.; Londhe, V.; Xue, Y.Y.; Li, K.; Phillips, R.J.; Strieter, R.M. Critical role for CXCR2 and CXCR2 ligands during the pathogenesis of ventilator-induced lung injury. J. Clin. Investig. 2002, 110, 1703–1716. [Google Scholar] [CrossRef]
- Xu, S.; Cui, F.; Huang, D.; Zhang, D.; Zhu, A.; Sun, X.; Cao, Y.; Ding, S.; Wang, Y.; Gao, E.; et al. PD-L1 monoclonal antibody-conjugated nanoparticles enhance drug delivery level and chemotherapy efficacy in gastric cancer cells. Int. J. Nanomed. 2019, 14, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Arabi, L.; Badiee, A.; Mosaffa, F.; Jaafari, M.R. Targeting CD44 expressing cancer cells with anti-CD44 monoclonal antibody improves cellular uptake and antitumor efficacy of liposomal doxorubicin. J. Control. Release 2015, 220, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Wang, Q.; Shi, Y.; Huang, Y.; Zhang, J.; Zhang, X.; Lin, G. Nanotechnology-mediated immunochemotherapy combined with docetaxel and PD-L1 antibody increase therapeutic effects and decrease systemic toxicity. J. Control. Release 2018, 286, 369–380. [Google Scholar] [CrossRef]
- Yu, Q.; Qiu, Y.; Chen, X.; Wang, X.; Mei, L.; Wu, H.; Liu, K.; Liu, Y.; Li, M.; Zhang, Z.; et al. Chemotherapy priming of the Pancreatic Tumor Microenvironment Promotes Delivery and Anti-Metastasis Efficacy of Intravenous Low-Molecular-Weight Heparin-Coated Lipid-siRNA Complex. Theranostics 2019, 9, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Chen, S.; Xu, C.Y.; Ma, Y.; Qian, H.; Xu, Y.; Chen, H.; Wang, X.; He, G.; Lu, Y.; et al. PEGylated rhenium nanoclusters: A degradable metal photothermal nanoagent for cancer therapy. Chem. Sci. 2019, 10, 5435–5443. [Google Scholar] [CrossRef] [PubMed]
- Naganuma, Y.; Satoh, K.; Yi, Q.; Asazuma, N.; Yatomi, Y.; Ozaki, Y. Cleavage of platelet endothelial cell adhesion molecule-1 (PECAM-1) in platelets exposed to high shear stress. J. Thromb. Haemost. 2004, 2, 1998–2008. [Google Scholar] [CrossRef]
- Villar, J.; Muros, M.; Cabrera-Benítez, N.E.; Valladares, F.; López-Hernández, M.; Flores, C.; Martín-Barrasa, J.L.; Blanco, J.; Liu, M.; Kacmarek, R.M. Soluble platelet-endothelial cell adhesion molecule-1, a biomarker of ventilator-induced lung injury. Crit. Care 2014, 18, R41. [Google Scholar] [CrossRef]
- Long, M.E.; Mallampalli, R.K.; Horowitz, J.C. Pathogenesis of pneumonia and acute lung injury. Clin. Sci. 2022, 136, 747–769. [Google Scholar] [CrossRef]
- Öcal, S. SARS-CoV-2 and lung injury: Dysregulation of immune response but not hyperimmune response as in “cytokine storm syndrome”. Clin. Respir. J. 2022, 16, 13–16. [Google Scholar] [CrossRef]
- Otifi, H.M.; Adiga, B.K. Endothelial Dysfunction in Covid-19 Infection. Am. J. Med. Sci. 2022, 363, 281–287. [Google Scholar] [CrossRef]
- Martin, F.P.; Jacqueline, C.; Poschmann, J.; Roquilly, A. Alveolar Macrophages: Adaptation to Their Anatomic Niche during and after Inflammation. Cells 2021, 10, 2720. [Google Scholar] [CrossRef]
- Lee, J.W.; Chun, W.; Lee, H.J.; Min, J.H.; Kim, S.M.; Seo, J.Y.; Ahn, K.S.; Oh, S.R. The Role of Macrophages in the Development of Acute and Chronic Inflammatory Lung Diseases. Cells 2021, 10, 897. [Google Scholar] [CrossRef] [PubMed]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Joffre, J.; Hellman, J.; Ince, C.; Ait-Oufella, H. Endothelial Responses in Sepsis. Am. J. Respir. Crit. Care Med. 2020, 202, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Hakanpaa, L.; Kiss, E.A.; Jacquemet, G.; Miinalainen, I.; Lerche, M.; Guzmán, C.; Mervaala, E.; Eklund, L.; Ivaska, J.; Saharinen, P. Targeting β1-integrin inhibits vascular leakage in endotoxemia. Proc. Natl. Acad. Sci. USA 2018, 115, E6467–E6476. [Google Scholar] [CrossRef] [PubMed]
- Kellner, M.; Noonepalle, S.; Lu, Q.; Srivastava, A.; Zemskov, E.; Black, S.M. ROS Signaling in the Pathogenesis of Acute Lung Injury (ALI) and Acute Respiratory Distress Syndrome (ARDS). Adv. Exp. Med. Biol. 2017, 967, 105–137. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, S.; Xiang, D.; Ju, L.; Shen, D.; Wang, X.; Wang, Y. Friend or Foe? The Roles of Antioxidants in Acute Lung Injury. Antioxidants 2021, 10, 1956. [Google Scholar] [CrossRef] [PubMed]
- Georg, P.; Astaburuaga-García, R.; Bonaguro, L.; Brumhard, S.; Michalick, L.; Lippert, L.J.; Kostevc, T.; Gäbel, C.; Schneider, M.; Streitz, M.; et al. Complement activation induces excessive T cell cytotoxicity in severe COVID-19. Cell 2022, 185, 493–512.e425. [Google Scholar] [CrossRef]
- Muhammad, W.; Zhai, Z.; Wang, S.; Gao, C. Inflammation-modulating nanoparticles for pneumonia therapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2022, 14, e1763. [Google Scholar] [CrossRef]
- Li, X.; Gong, N.; Tian, F.; Zhang, S.; Zhang, Y.; Wang, Y.; Qing, G.; Wang, Y.; Li, F.; Xu, Y.; et al. Suppression of cytokine release syndrome during CAR-T-cell therapy via a subcutaneously injected interleukin-6-adsorbing hydrogel. Nat. Biomed. Eng. 2023, 7, 1129–1141. [Google Scholar] [CrossRef]
- Huang, C.; Dong, L.; Zhao, B.; Lu, Y.; Huang, S.; Yuan, Z.; Luo, G.; Xu, Y.; Qian, W. Anti-inflammatory hydrogel dressings and skin wound healing. Clin. Transl. Med. 2022, 12, e1094. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Z.; Tang, L.; Luo, L.; Luo, W.; Li, Y.; Wang, X.; Huang, L.; Hu, Y.; Mei, H. Enhancing the Treatment of Uncontrolled Inflammation through the Targeted Delivery of TPCA-1-Loaded Nanoparticles. Pharmaceutics 2023, 15, 2435. https://doi.org/10.3390/pharmaceutics15102435
Chen Z, Tang L, Luo L, Luo W, Li Y, Wang X, Huang L, Hu Y, Mei H. Enhancing the Treatment of Uncontrolled Inflammation through the Targeted Delivery of TPCA-1-Loaded Nanoparticles. Pharmaceutics. 2023; 15(10):2435. https://doi.org/10.3390/pharmaceutics15102435
Chicago/Turabian StyleChen, Zhaozhao, Lu Tang, Lili Luo, Wenjing Luo, Yingying Li, Xindi Wang, Linlin Huang, Yu Hu, and Heng Mei. 2023. "Enhancing the Treatment of Uncontrolled Inflammation through the Targeted Delivery of TPCA-1-Loaded Nanoparticles" Pharmaceutics 15, no. 10: 2435. https://doi.org/10.3390/pharmaceutics15102435
APA StyleChen, Z., Tang, L., Luo, L., Luo, W., Li, Y., Wang, X., Huang, L., Hu, Y., & Mei, H. (2023). Enhancing the Treatment of Uncontrolled Inflammation through the Targeted Delivery of TPCA-1-Loaded Nanoparticles. Pharmaceutics, 15(10), 2435. https://doi.org/10.3390/pharmaceutics15102435