
Influence of Intermolecular Interactions on Crystallite Size in Crystalline Solid Dispersions

Abstract
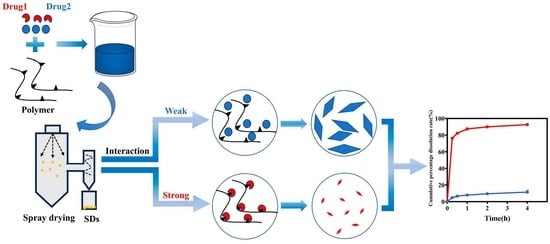
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
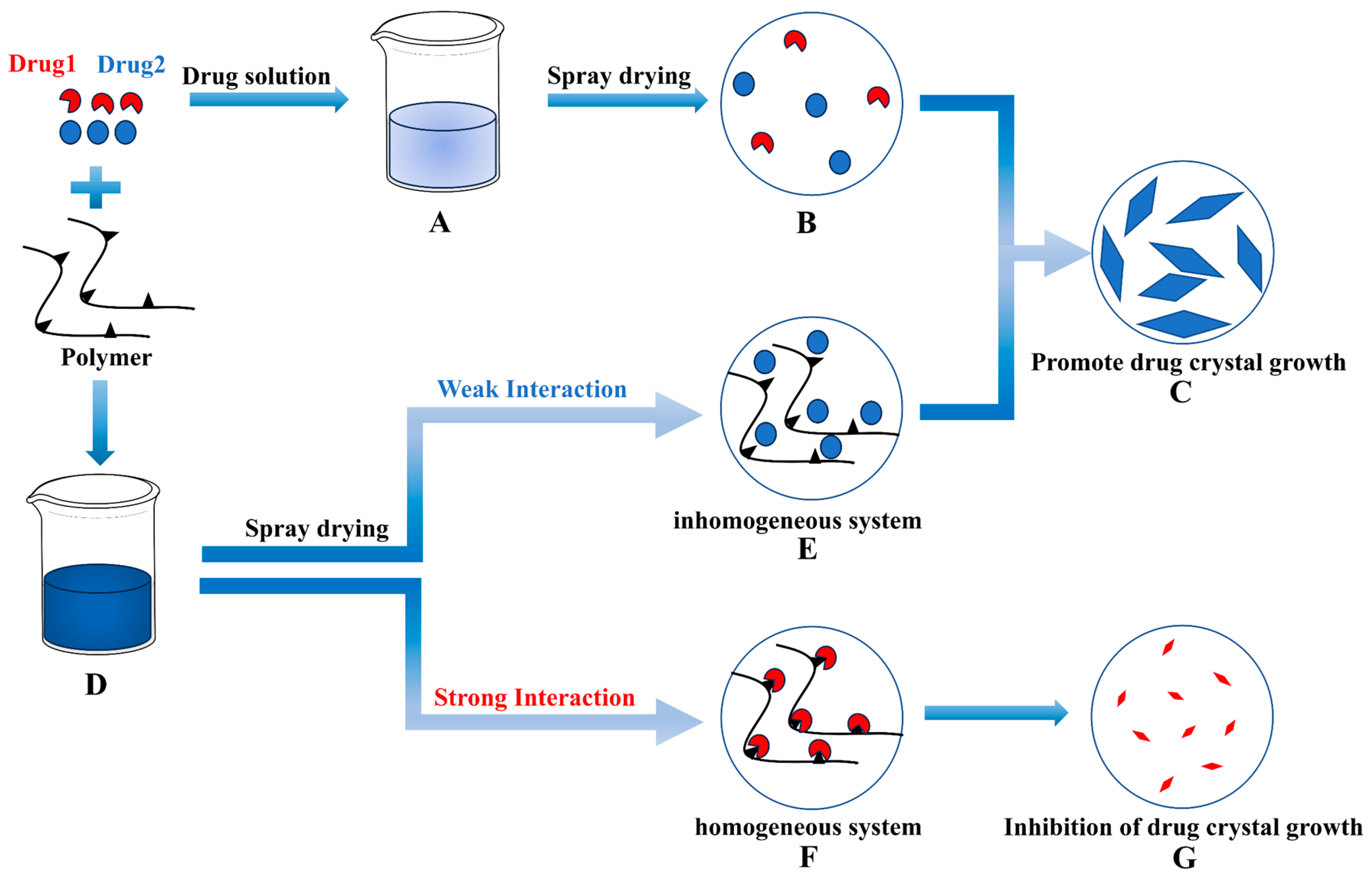{kind=link}
1. Introduction
2. Materials and Methods
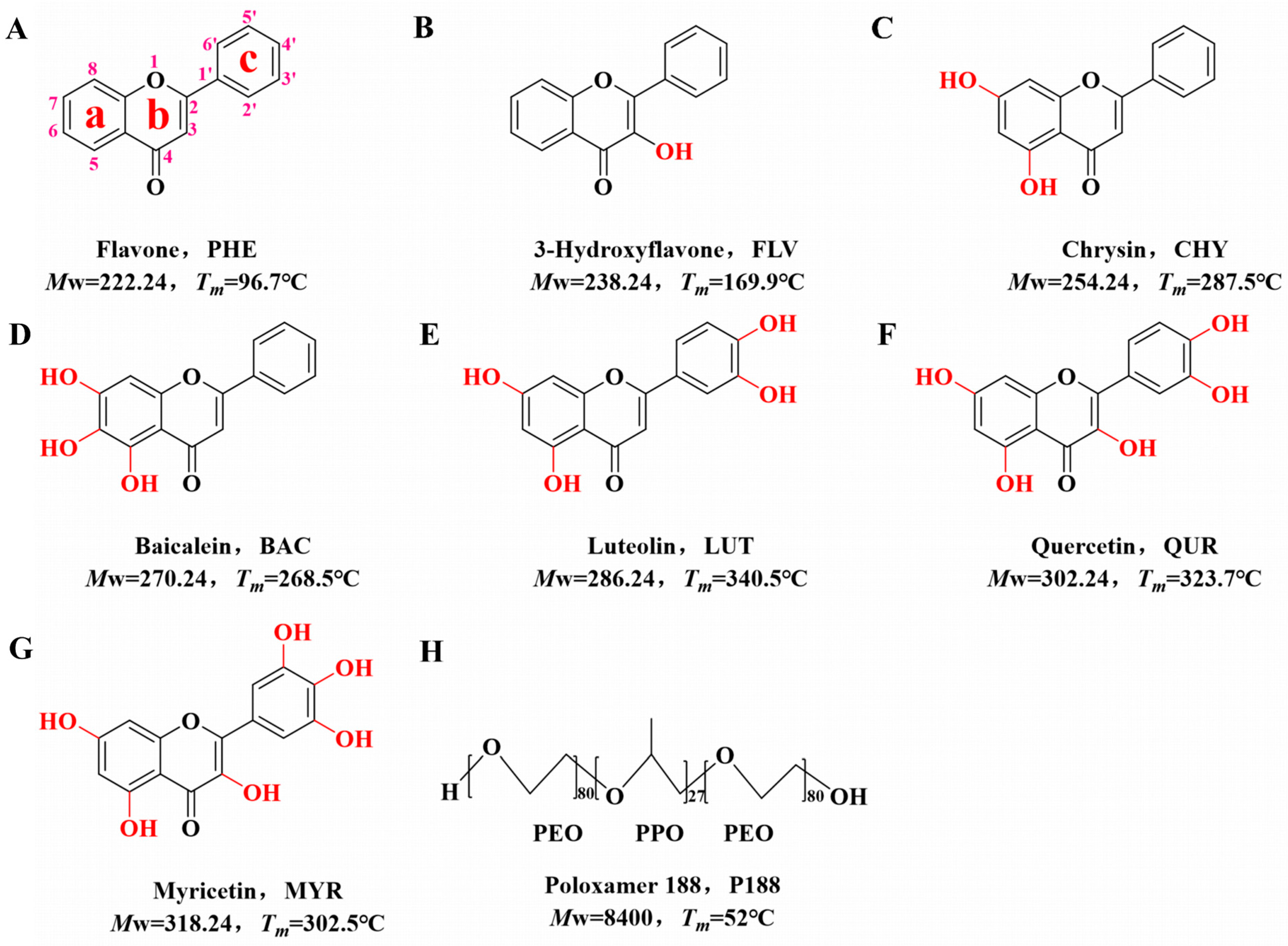
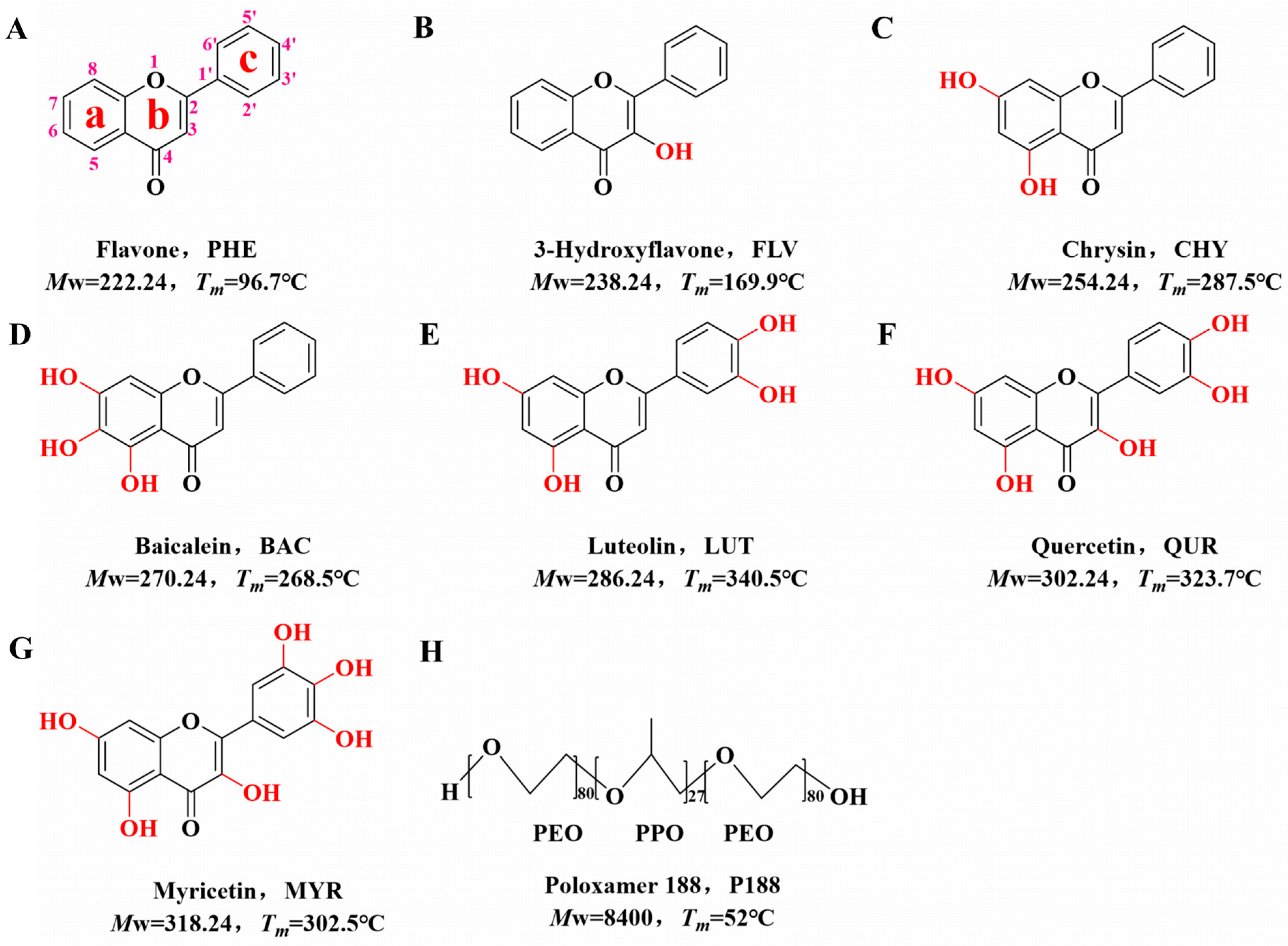
2.1. Materials
2.2. Instruments and Methods
2.2.1. Generation of Flavonoid Drugs and P188 Samples
2.2.2. Theoretical Calculation of Drug–Polymer Solubility Parameters
2.2.3. Differential Scanning Calorimetry (DSC)
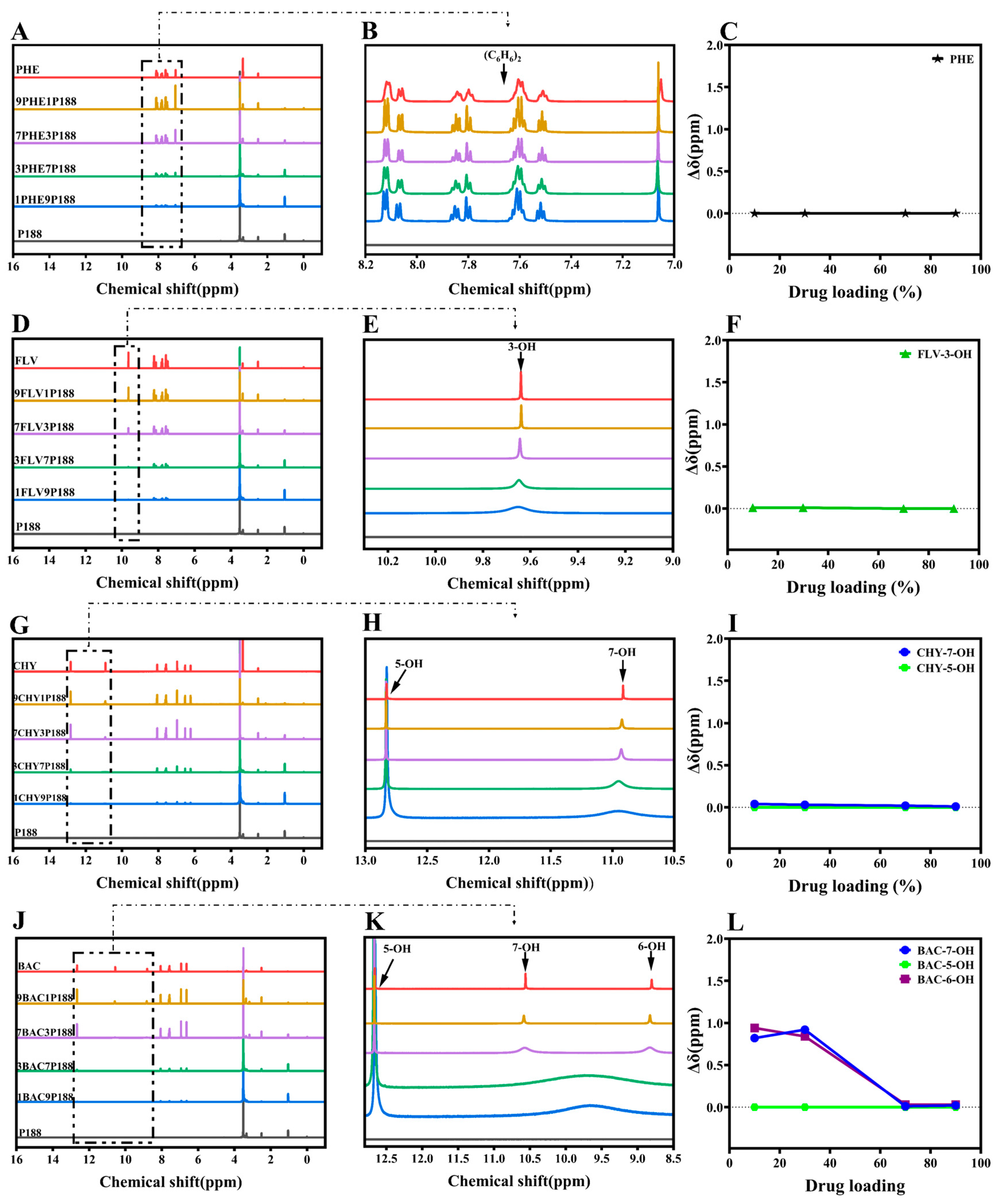
2.2.4. Nuclear Magnetic Resonance (1H-NMR)
2.2.5. Fourier-Transform Infrared Spectroscopy (FT-IR)
2.2.6. Polarized Optical Microscopy (POM)
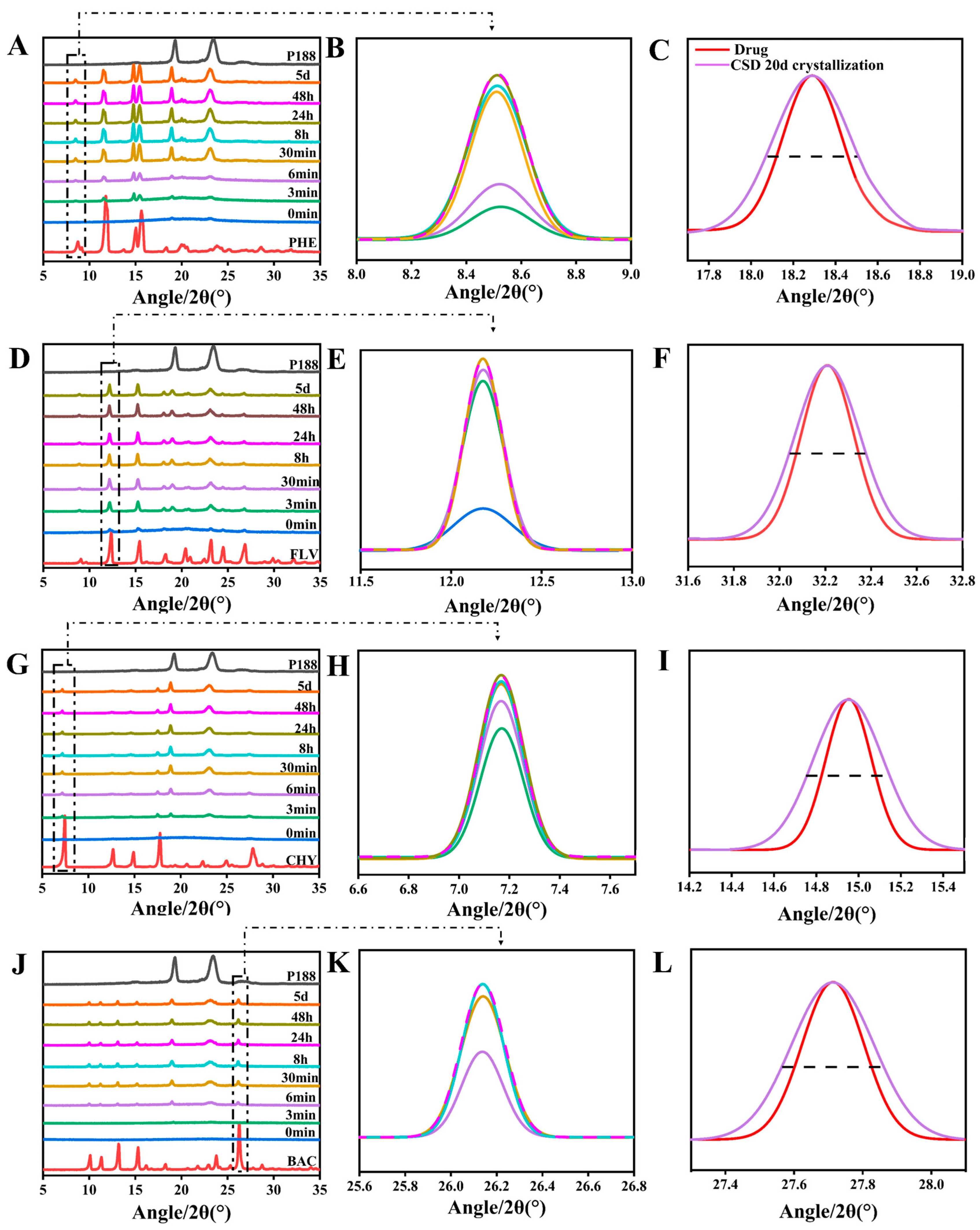
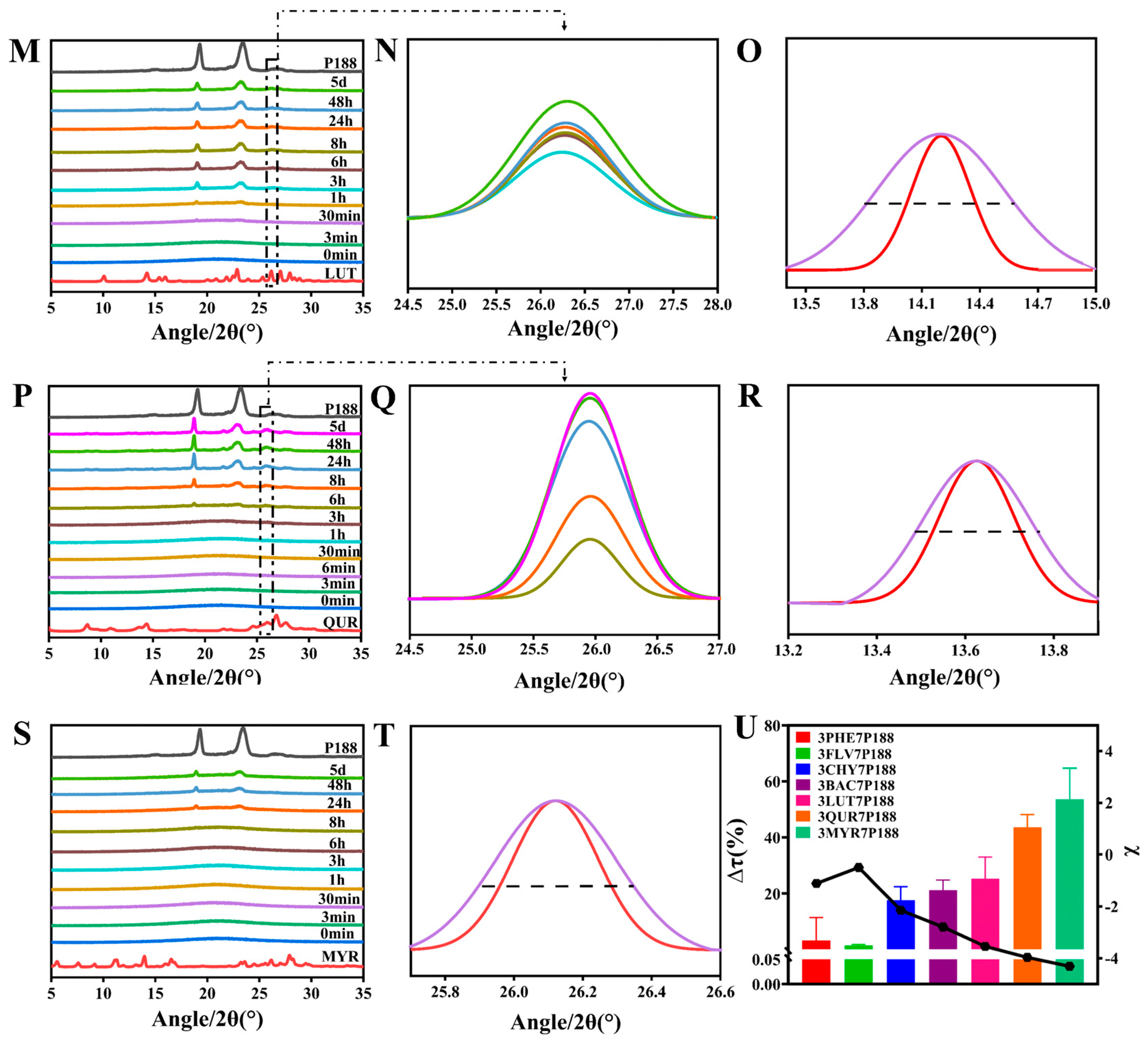
2.2.7. Powder X-ray Diffraction (PXRD)
2.2.8. Particle-Size Analyzer (PSA)
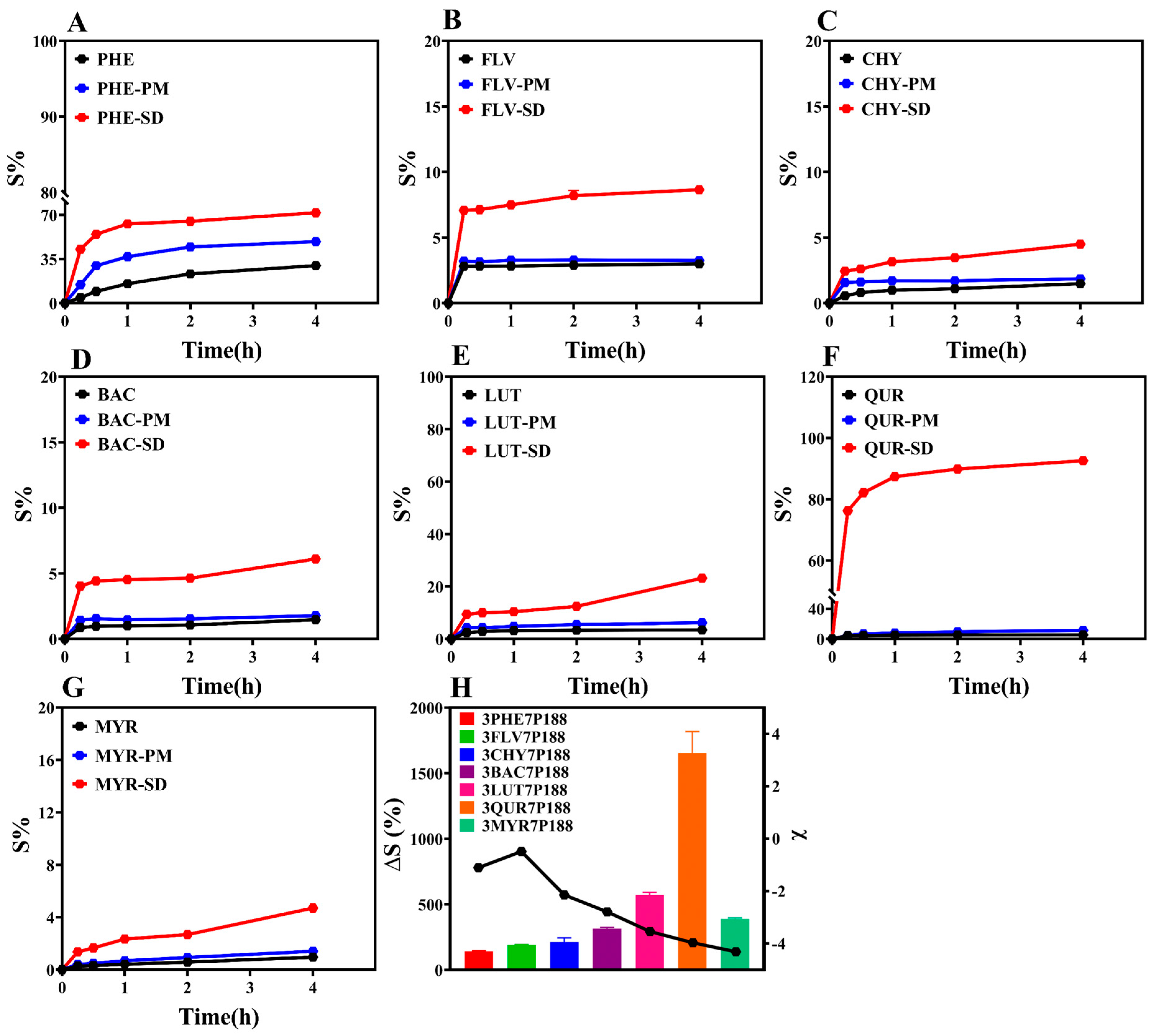
2.2.9. Powder Dissolution
2.2.10. Statistical Analysis
3. Results and Discussion
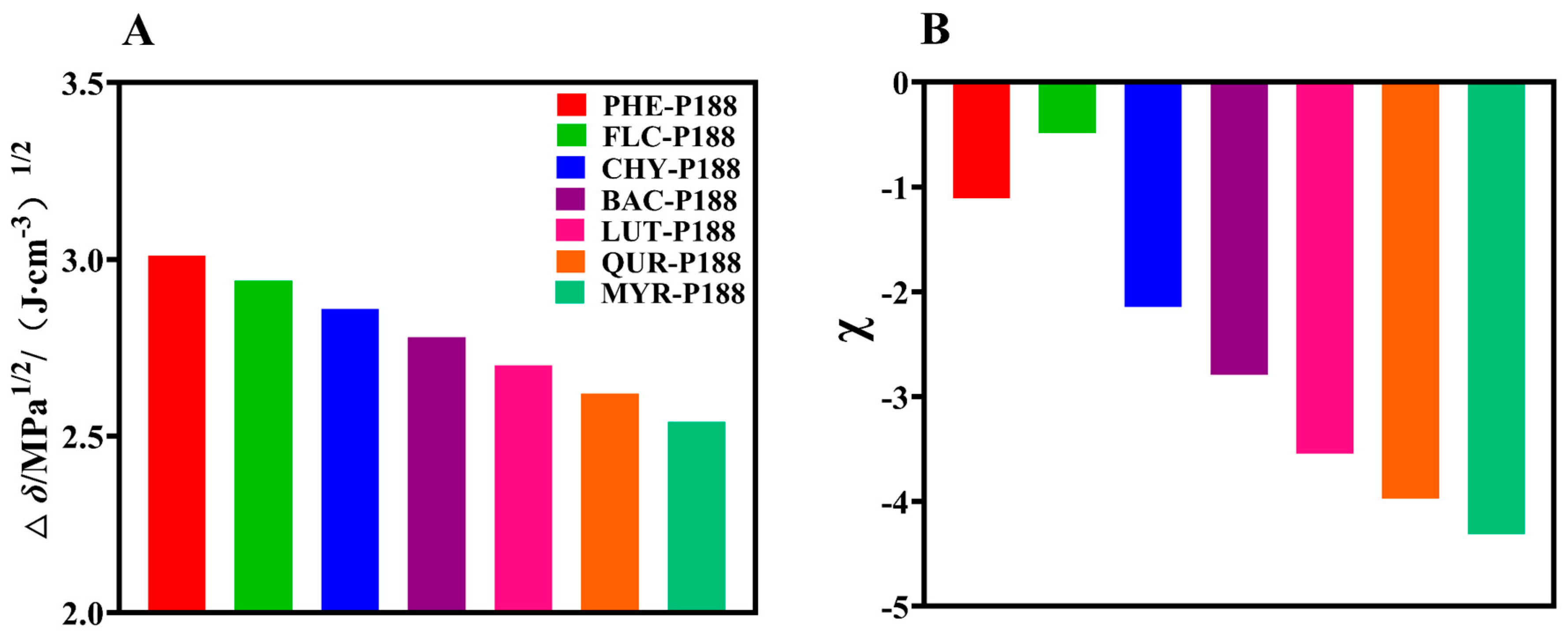
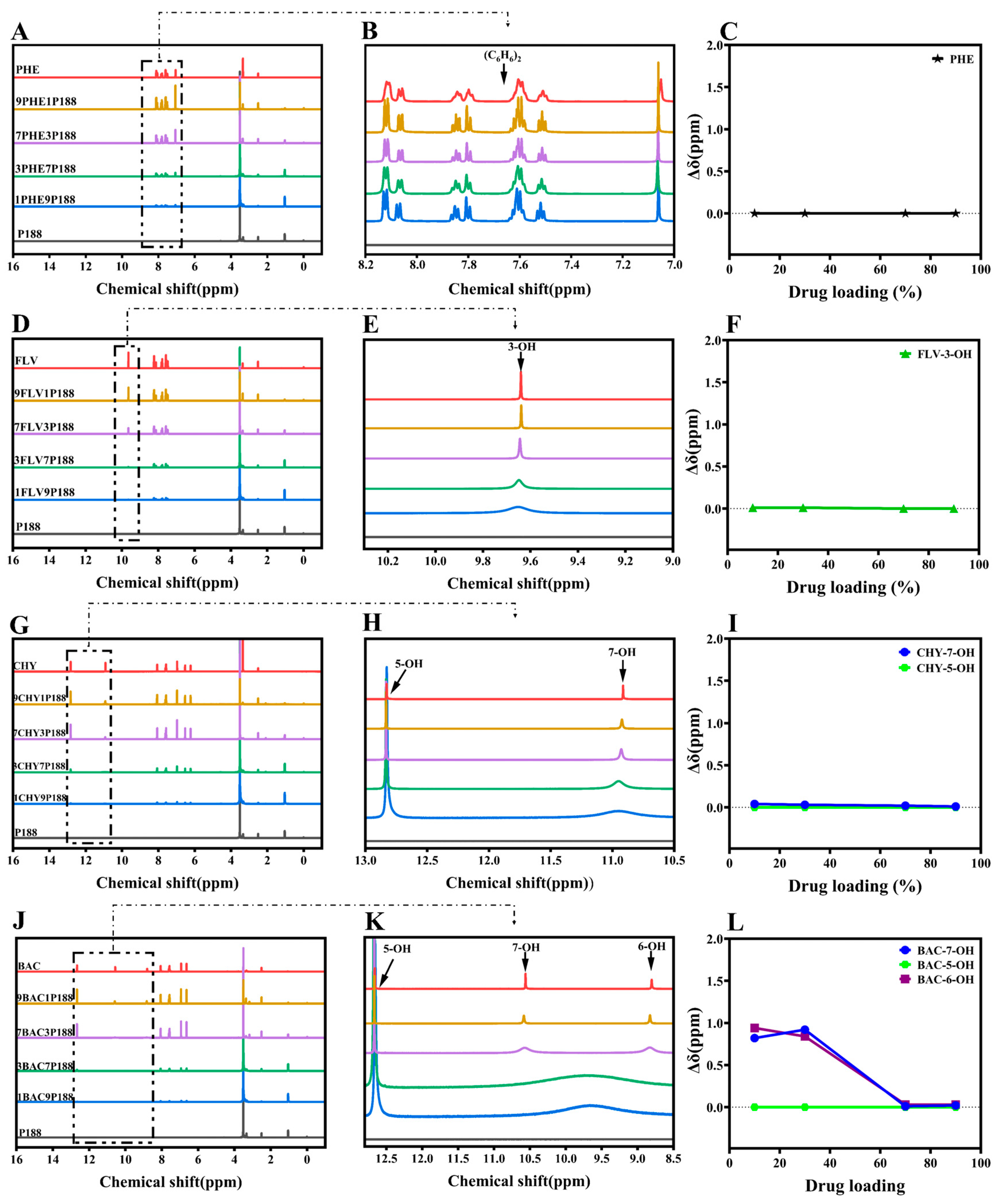
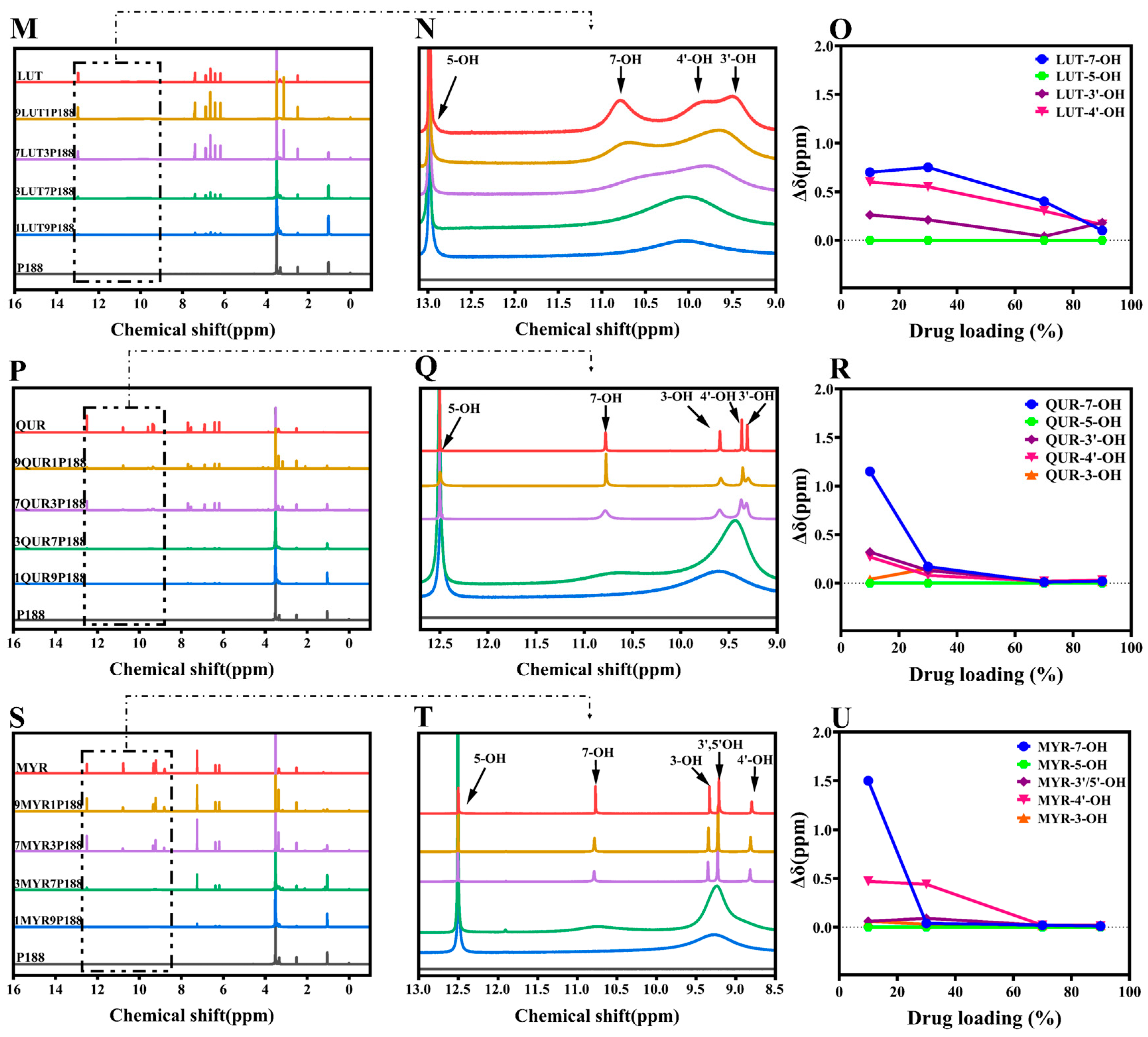
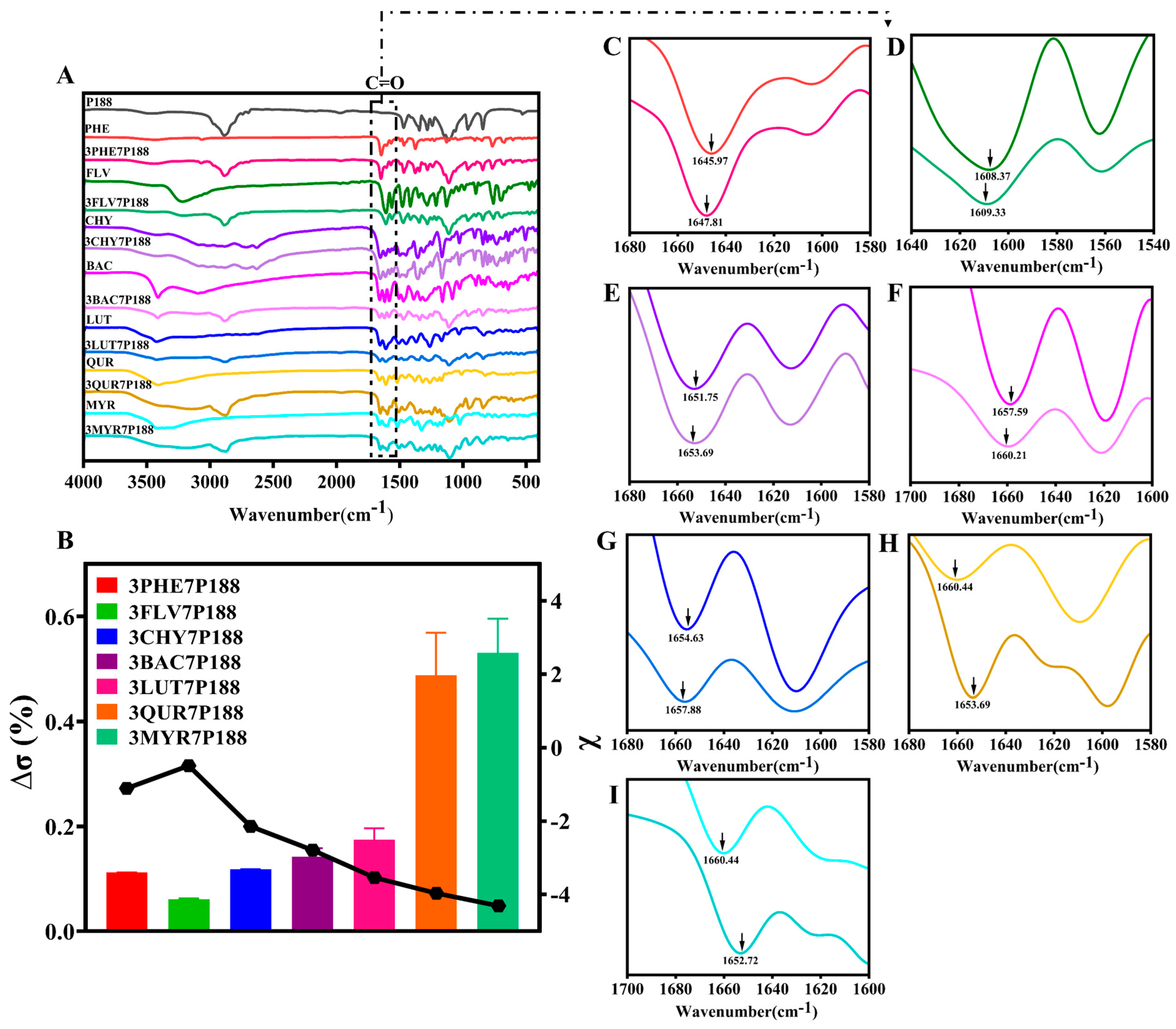
3.1. Evaluation of Intermolecular Interactions between Drugs and P188
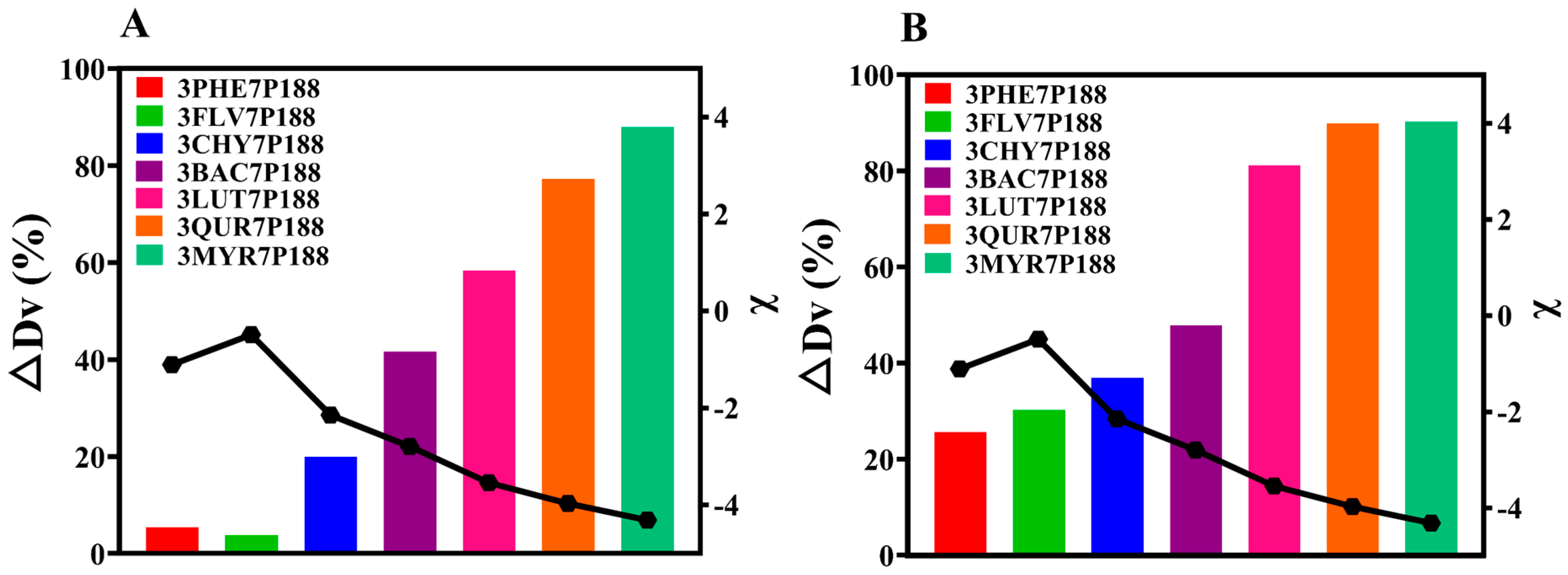
3.2. Investigation of Drug Crystallite Size in CSDs
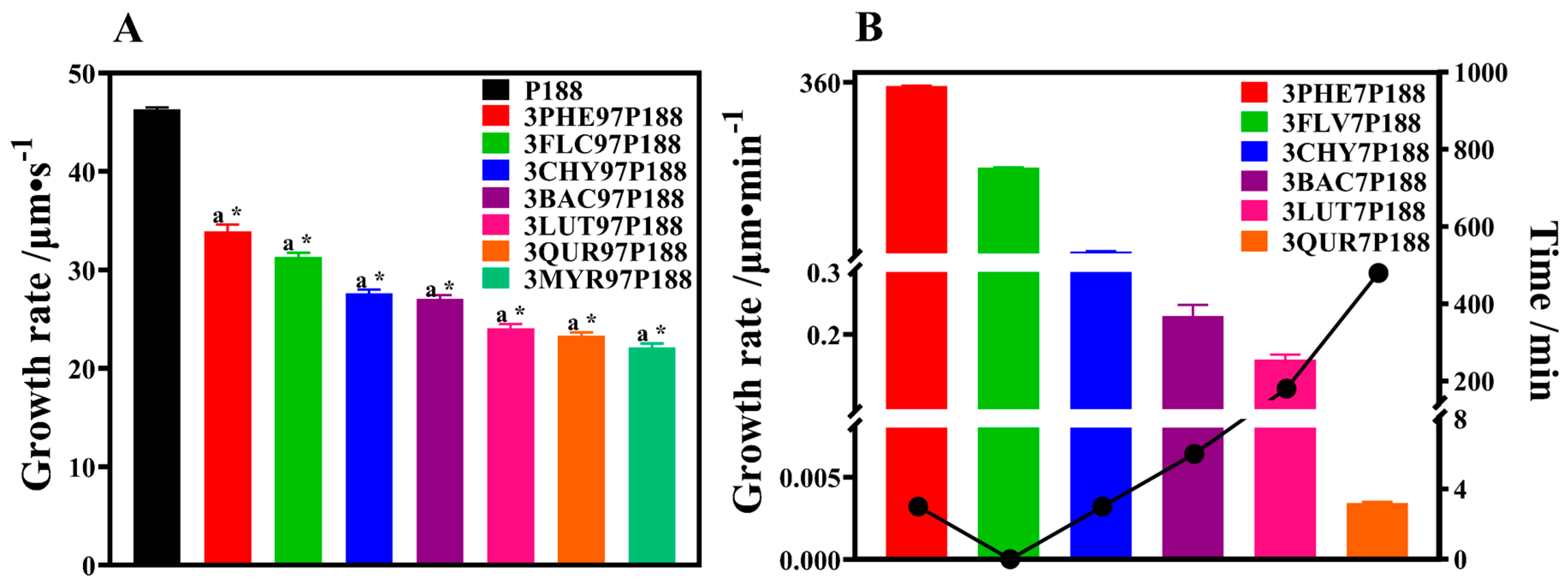
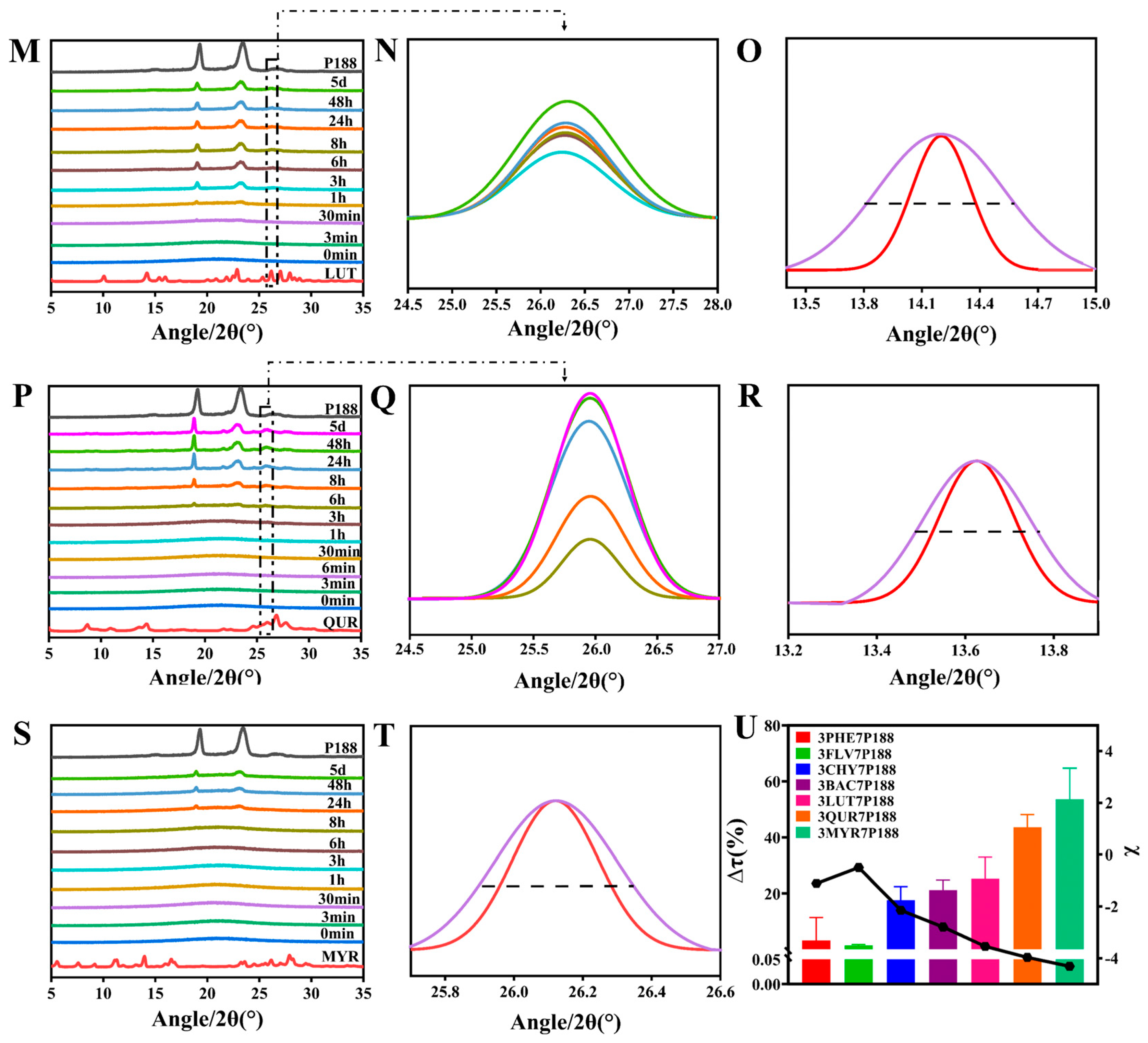
3.2.1. Crystalline Growth Rate
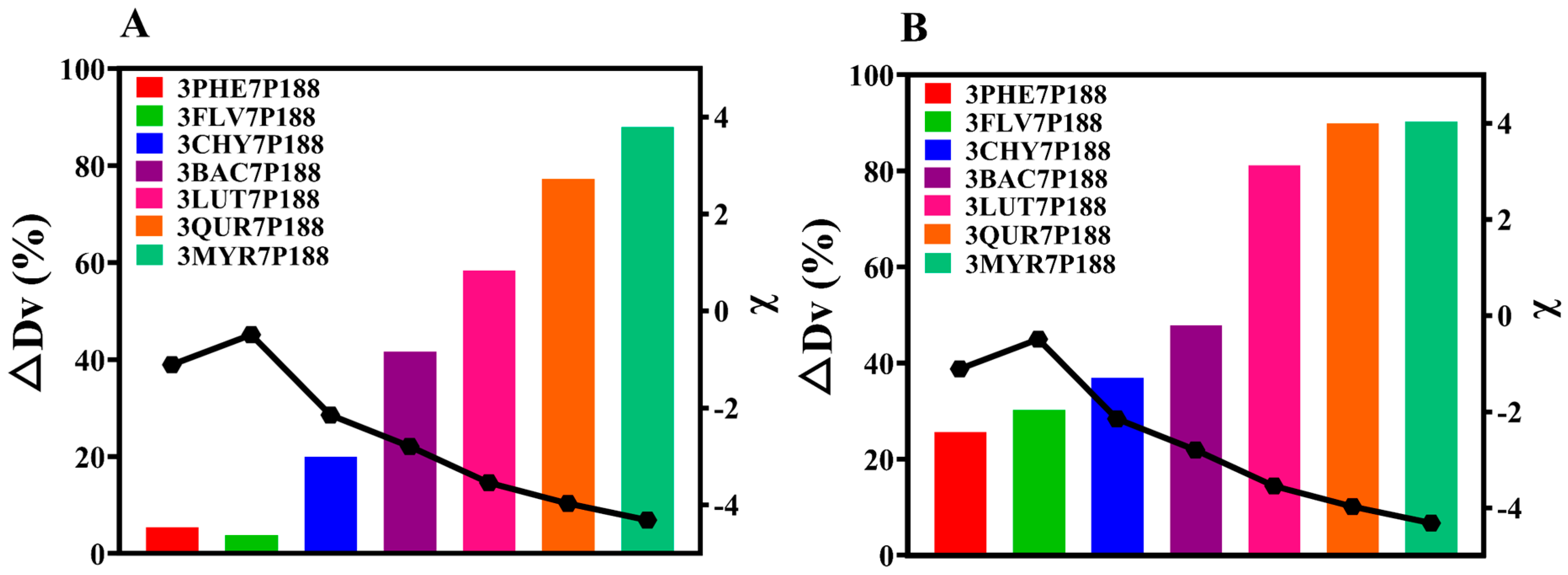
3.2.2. Crystallization Kinetics and Crystalline domain Size
3.2.3. Particle-Size Analysis
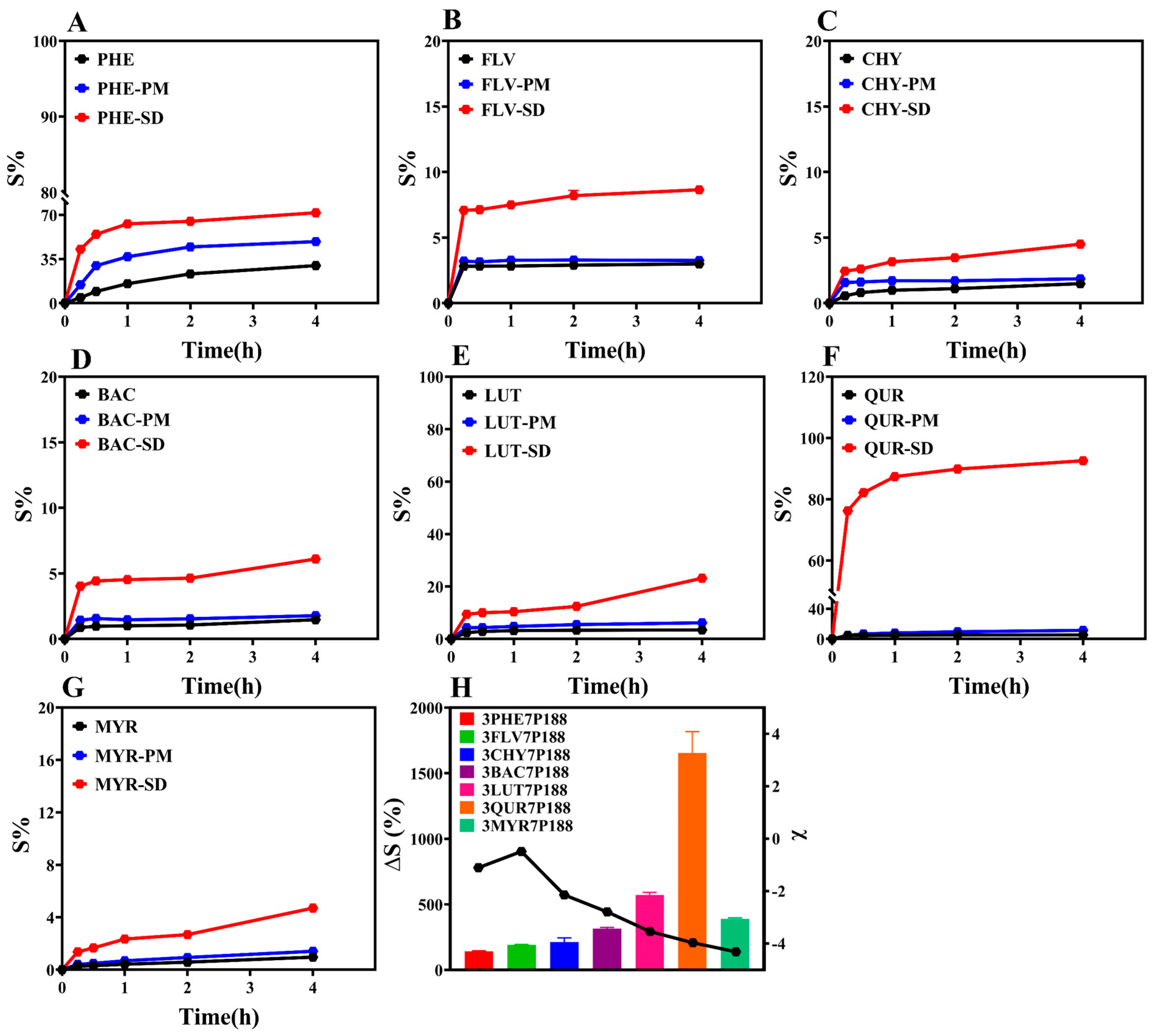
3.3. Powder Dissolution
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Singh, P.A.; Frank, A.; Arora, S.; Sharma, R.; Bajwa, N. Solubility, the Main Concern for Poorly Water-soluble Drugs: Techniques and Alternatives. Lett. Drug Des. Discov. 2023, 20, 163632. [Google Scholar] [CrossRef]
- Paredes, A.J.; McKenna, P.E.; Ramöller, I.K.; Naser, Y.A.; Volpe-Zanutto, F.; Li, M.; Abbate, M.; Zhao, L.; Zhang, C.; Abu-Ershaid, J.M. Microarray patches: Poking a hole in the challenges faced when delivering poorly soluble drugs. Adv. Funct. Mater. 2021, 31, 2005792. [Google Scholar] [CrossRef]
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. Int. J. Pharm. 2011, 420, 1–10. [Google Scholar] [CrossRef]
- Baird, J.A.; Taylor, L.S. Evaluation of amorphous solid dispersion properties using thermal analysis techniques. Adv. Drug Deliv. Rev. 2012, 64, 396–421. [Google Scholar] [CrossRef]
- Van den Mooter, G. The use of amorphous solid dispersions: A formulation strategy to overcome poor solubility and dissolution rate. Drug Discov. Today Technol. 2012, 9, e79–e85. [Google Scholar] [CrossRef]
- Solaiman, A.; Tatari, A.K.; Elkordy, A.A. Naproxen Microparticulate Systems Prepared Using In Situ Crystallisation and Freeze-Drying Techniques. AAPS PharmSciTech 2017, 18, 1438–1446. [Google Scholar] [CrossRef]
- Wang, H.; Li, R.; Rao, Y.; Liu, S.; Hu, C.; Zhang, Y.; Meng, L.; Wu, Q.; Ouyang, Q.; Liang, H. Enhancement of the Bioavailability and Anti-Inflammatory Activity of Glycyrrhetinic Acid via Novel Soluplus®—A Glycyrrhetinic Acid Solid Dispersion. Pharmaceutics 2022, 14, 1797. [Google Scholar] [CrossRef]
- Liu, C.; Liu, Z.; Chen, Y.; Chen, Z.; Chen, H.; Pui, Y.; Qian, F. Oral bioavailability enhancement of β-lapachone, a poorly soluble fast crystallizer, by cocrystal, amorphous solid dispersion, and crystalline solid dispersion. Eur. J. Pharm. Biopharm. 2018, 124, 73–81. [Google Scholar] [CrossRef]
- Hu, C.; Liu, Z.; Liu, C.; Zhang, Y.; Fan, H.; Qian, F. Improvement of Antialveolar Echinococcosis Efficacy of Albendazole by a Novel Nanocrystalline Formulation with Enhanced Oral Bioavailability. ACS Infect. Dis. 2020, 6, 802–810. [Google Scholar] [CrossRef]
- Roduner, E. Size matters: Why nanomaterials are different. Chem. Soc. Rev. 2006, 35, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Mihranyan, A.; Strømme, M. Solubility of fractal nanoparticles. Surf. Sci. 2007, 601, 315–319. [Google Scholar] [CrossRef]
- Galli, C. Experimental determination of the diffusion boundary layer width of micron and submicron particles. Int. J. Pharm. 2006, 313, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Harris, M.T.; Taylor, L.S. Time-resolved SAXS/WAXS study of the phase behavior and microstructural evolution of drug/PEG solid dispersions. Mol. Pharm. 2011, 8, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Harris, M.T.; Taylor, L.S. Modification of crystallization behavior in drug/polyethylene glycol solid dispersions. Mol. Pharm. 2012, 9, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Toth, S.J.; Simpson, G.J.; Hsu, H.-Y.; Taylor, L.S.; Harris, M.T. Crystallization and dissolution behavior of naproxen/polyethylene glycol solid dispersions. J. Phys. Chem. B 2013, 117, 1494–1500. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, L.; Ma, D.; Tang, X.; Zhang, Y.; Yin, T.; Gou, J.; Wang, Y.; He, H. Characterizing and exploring the differences in dissolution and stability between crystalline solid dispersion and amorphous solid dispersion. AAPS PharmSciTech 2020, 21, 262. [Google Scholar] [CrossRef]
- Baird, J.A.; Van Eerdenbrugh, B.; Taylor, L.S. A classification system to assess the crystallization tendency of organic molecules from undercooled melts. J. Pharm. Sci. 2010, 99, 3787–3806. [Google Scholar] [CrossRef]
- Yin, S.X.; Franchini, M.; Chen, J.; Hsieh, A.; Jen, S.; Lee, T.; Hussain, M.; Smith, R. Bioavailability enhancement of a COX-2 inhibitor, BMS-347070, from a nanocrystalline dispersion prepared by spray-drying. J. Pharm. Sci. 2005, 94, 1598–1607. [Google Scholar] [CrossRef]
- Shishulin, A.V.; Fedoseev, V.B.; Shishulina, A.V. Phonon thermal conductivity and phase equilibria of fractal Bi–Sb nanoparticles. Tech. Phys. 2019, 64, 512–517. [Google Scholar] [CrossRef]
- Kim, N.A.; Oh, H.K.; Lee, J.C.; Choi, Y.H.; Jeong, S.H. Comparison of solubility enhancement by solid dispersion and micronized butein and its correlation with in vivo study. J. Pharm. Investig. 2021, 51, 53–60. [Google Scholar] [CrossRef]
- Rask, M.B.; Knopp, M.M.; Olesen, N.E.; Holm, R.; Rades, T. Influence of PVP/VA copolymer composition on drug–polymer solubility. Eur. J. Pharm. Sci. 2016, 85, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Tao, J.; Desikan, S.; Hussain, M.; Smith, R.L. Mechanistic investigation of Pluronic® based nano-crystalline drug-polymer solid dispersions. Pharm. Res. 2007, 24, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yan, Q.; Liu, Y.; Hu, C. Study on the regulation mechanism of effective glass transition temperature on the crystallization of crystalline solid dispersion. Drug Deliv. Transl. Res. 2023, 13, 2677–2689. [Google Scholar] [CrossRef] [PubMed]
- Karavas, E.; Georgarakis, E.; Sigalas, M.P.; Avgoustakis, K.; Bikiaris, D. Investigation of the release mechanism of a sparingly water-soluble drug from solid dispersions in hydrophilic carriers based on physical state of drug, particle size distribution and drug–polymer interactions. Eur. J. Pharm. Biopharm. 2007, 66, 334–347. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, Z.; Qian, F. Crystallization of bifonazole and acetaminophen within the matrix of semicrystalline, PEO–PPO–PEO triblock copolymers. Mol. Pharm. 2015, 12, 590–599. [Google Scholar] [CrossRef]
- dos Santos, K.M.; de Melo Barbosa, R.; Meirelles, L.; Vargas, F.G.A.; da Silva Lins, A.C.; Camara, C.A.; Aragão, C.F.; de Lima Moura, T.F.; Raffin, F.N. Solid dispersion of β-lapachone in PVP K30 and PEG 6000 by spray drying technique. J. Therm. Anal. Calorim. 2021, 146, 2523–2532. [Google Scholar] [CrossRef]
- Hu, C.; Zhang, F.; Fan, H. Evaluation of Drug Dissolution Rate in Co-amorphous and Co-crystal Binary Drug Delivery Systems by Thermodynamic and Kinetic Methods. AAPS PharmSciTech 2021, 22, 1–9. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, X.; Meng, J.; Lu, L.; Du, S.; Xu, H.; Wu, S. Study on the effect of polymer excipients on the dispersibility, interaction, solubility, and scavenging reactive oxygen species of myricetin solid dispersion: Experiment and molecular simulation. ACS Omega 2022, 7, 1514–1526. [Google Scholar] [CrossRef]
- Amponsah-Efah, K.K.; Mistry, P.; Eisenhart, R.; Suryanarayanan, R. The influence of the strength of drug–polymer interactions on the dissolution of amorphous solid dispersions. Mol. Pharm. 2020, 18, 174–186. [Google Scholar] [CrossRef]
- Yao, X.; Kim, S.; Gui, Y.; Chen, Z.; Yu, J.; Jones, K.J.; Yu, L. Amorphous Drug–Polymer Salt with High Stability under Tropical Conditions and Fast Dissolution: The Challenging Case of Lumefantrine-PAA. J. Pharm. Sci. 2021, 110, 3670–3677. [Google Scholar] [CrossRef]
- Chan, S.-Y.; Qi, S.; Craig, D.Q. An investigation into the influence of drug–polymer interactions on the miscibility, processability and structure of polyvinylpyrrolidone-based hot melt extrusion formulations. Int. J. Pharm. 2015, 496, 95–106. [Google Scholar] [CrossRef]
- Sarode, A.L.; Sandhu, H.; Shah, N.; Malick, W.; Zia, H. Hot melt extrusion (HME) for amorphous solid dispersions: Predictive tools for processing and impact of drug–polymer interactions on supersaturation. Eur. J. Pharm. Sci. 2013, 48, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Shen, X.; Tong, C.; Zhang, S.; Chen, Q.; Li, Y.; Li, S. Gossypin: A flavonoid with diverse pharmacological effects. Chem. Biol. Drug Des. 2023, 101, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yang, J.; Xie, Y. Improvement strategies for the oral bioavailability of poorly water-soluble flavonoids: An overview. Int. J. Pharm. 2019, 570, 118642. [Google Scholar] [CrossRef]
- Pápay, Z.E.; Kállai-Szabó, N.; Ludányi, K.; Klebovich, I.; Antal, I. Development of oral site-specific pellets containing flavonoid extract with antioxidant activity. Eur. J. Pharm. Sci. 2016, 95, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Petrussa, E.; Braidot, E.; Zancani, M.; Peresson, C.; Bertolini, A.; Patui, S.; Vianello, A. Plant flavonoids—Biosynthesis, transport and involvement in stress responses. Int. J. Mol. Sci. 2013, 14, 14950–14973. [Google Scholar] [CrossRef] [PubMed]
- Pool, H.; Quintanar, D.; de Dios Figueroa, J.; Mano, C.M.; Bechara, J.E.H.; Godínez, L.A.; Mendoza, S. Antioxidant effects of quercetin and catechin encapsulated into PLGA nanoparticles. J. Nanomater. 2012, 2012, 86. [Google Scholar] [CrossRef]
- Zheng, Y.-Z.; Zhou, Y.; Liang, Q.; Chen, D.-F.; Guo, R. A theoretical study on the hydrogen-bonding interactions between flavonoids and ethanol/water. J. Mol. Model. 2016, 22, 1–10. [Google Scholar] [CrossRef]
- Hildebrand, J.H. Solubility of Non-Electrolytes, 2nd ed.; Reinhold Publishing Corp.: New York, NY, USA; Chapman & Hall, Ltd.: London, UK, 1936; p. 203. [Google Scholar]
- Mohammad, M.A.; Alhalaweh, A.; Velaga, S.P. Hansen solubility parameter as a tool to predict cocrystal formation. Int. J. Pharm. 2011, 407, 63–71. [Google Scholar] [CrossRef]
- Turpin, E.R.; Taresco, V.; Al-Hachami, W.A.; Booth, J.; Treacher, K.; Tomasi, S.; Alexander, C.; Burley, J.; Laughton, C.A.; Garnett, M.C. In silico screening for solid dispersions: The trouble with solubility parameters and χFH. Mol. Pharm. 2018, 15, 4654–4667. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Tao, J.; Zhang, G.G.; Yu, L. Solubilities of crystalline drugs in polymers: An improved analytical method and comparison of solubilities of indomethacin and nifedipine in PVP, PVP/VA, and PVAc. J. Pharm. Sci. 2010, 99, 4023–4031. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Trivino, A.; Prasad, D.; Chauhan, H. Investigation and correlation of drug polymer miscibility and molecular interactions by various approaches for the preparation of amorphous solid dispersions. Eur. J. Pharm. Sci. 2015, 71, 12–24. [Google Scholar] [CrossRef]
- Baranović, G.; Šegota, S. Infrared spectroscopy of flavones and flavonols. Reexamination of the hydroxyl and carbonyl vibrations in relation to the interactions of flavonoids with membrane lipids. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 192, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, L.; Cheng, G.; Li, L.; Liu, X.; Huang, Q. Preparation and recognition properties of molecularly imprinted nanofiber membrane of chrysin. Polymers 2022, 14, 2398. [Google Scholar] [CrossRef]
- Tong, M.; Wu, X.; Zhang, S.; Hua, D.; Li, S.; Yu, X.; Wang, J.; Zhang, Z. Application of TPGS as an efflux inhibitor and a plasticizer in baicalein solid dispersion. Eur. J. Pharm. Sci. 2022, 168, 106071. [Google Scholar] [CrossRef] [PubMed]
- Alshehri, S.; Imam, S.S.; Altamimi, M.A.; Hussain, A.; Shakeel, F.; Elzayat, E.; Mohsin, K.; Ibrahim, M.; Alanazi, F. Enhanced dissolution of luteolin by solid dispersion prepared by different methods: Physicochemical characterization and antioxidant activity. ACS Omega 2020, 5, 6461–6471. [Google Scholar] [CrossRef]
- Van Hecke, E.; Benali, M. Solid dispersions of quercetin-PEG matrices: Miscibility prediction, preparation and characterization. Food Biosci. 2022, 49, 101868. [Google Scholar] [CrossRef]
- Mukesh, S.; Joshi, P.; Bansal, A.K.; Kashyap, M.C.; Mandal, S.K.; Sathe, V.; Sangamwar, A.T. Amorphous salts solid dispersions of celecoxib: Enhanced biopharmaceutical performance and physical stability. Mol. Pharm. 2021, 18, 2334–2348. [Google Scholar] [CrossRef]
- Gupta, J.; Nunes, C.; Vyas, S.; Jonnalagadda, S. Prediction of solubility parameters and miscibility of pharmaceutical compounds by molecular dynamics simulations. J. Phys. Chem. B 2011, 115, 2014–2023. [Google Scholar] [CrossRef]
- Liu, L.; Chen, L.; Müllers, W.; Serno, P.; Qian, F. Water-Resistant Drug–Polymer Interaction Contributes to the Formation of Nano-Species during the Dissolution of Felodipine Amorphous Solid Dispersions. Mol. Pharm. 2022, 19, 2888–2899. [Google Scholar] [CrossRef] [PubMed]
- Metre, S.; Mukesh, S.; Samal, S.K.; Chand, M.; Sangamwar, A.T. Enhanced biopharmaceutical performance of rivaroxaban through polymeric amorphous solid dispersion. Mol. Pharm. 2018, 15, 652–668. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Understanding the generation and maintenance of supersaturation during the dissolution of amorphous solid dispersions using modulated DSC and 1H NMR. Int. J. Pharm. 2018, 536, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.-Y.; Chen, L.; Liu, F.; Chen, L.-J.; Meng, M.; Sun, H.-Q.; Zhang, Y.-M. Preparation and activity evaluation of chrysin-β-d-galactopyranoside. Arch. Pharm. Res. 2016, 39, 1433–1440. [Google Scholar] [CrossRef]
- Jiang, J.; Dong, H. Preparation and characterization of high-purity baicalein. J. Beijing Univ. Chem. Technol. 2008, 35, 31. [Google Scholar]
- Liu, S.; Zhang, S.; Su, Y.; Liu, Q.; Liao, X. A Theoretical Study on Nuclear Magnetic Resonance Spectra of Three Flavonol Derivatives. Chin. J. Magn. Reson. 2007, 24, 175. [Google Scholar]
- Biela, M.; Rimarčík, J.; Senajová, E.; Kleinová, A.; Klein, E. Antioxidant action of deprotonated flavonoids: Thermodynamics of sequential proton-loss electron-transfer. Phytochemistry 2020, 180, 112528. [Google Scholar] [CrossRef]
- Janeiro, P.; Corduneanu, O.; Oliveira Brett, A.M. Chrysin and (±)-taxifolin electrochemical oxidation mechanisms. Electroanal. Int. J. Devoted Fundam. Pract. Asp. Electroanal. 2005, 17, 1059–1064. [Google Scholar] [CrossRef]
- Taft, R.W.; Kamlet, M.J. The solvatochromic comparison method. 2. The.alpha.-scale of solvent hydrogen-bond donor (HBD) acidities. J. Am. Chem. Soc. 1976, 98, 2886–2894. [Google Scholar] [CrossRef]
- Seitsonen, A.P.; Idrissi, A.; Protti, S.; Mezzetti, A. Solvent effects on the vibrational spectrum of 3-hydroxyflavone. J. Mol. Liquids 2019, 275, 723–728. [Google Scholar] [CrossRef]
- Ali, H.M.; Ali, I.H. Structure-antioxidant activity relationships, QSAR, DFT calculation, and mechanisms of flavones and flavonols. Med. Chem. Res. 2019, 28, 2262–2269. [Google Scholar] [CrossRef]
- Barzegar, A. The role of intramolecular H-bonds predominant effects in myricetin higher antioxidant activity. Comput. Theor. Chem. 2017, 1115, 239–247. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, C.; Chen, Z.; Su, C.; Hageman, M.; Hussain, M.; Haskell, R.; Stefanski, K.; Qian, F. Drug–polymer–water interaction and its implication for the dissolution performance of amorphous solid dispersions. Mol. Pharm. 2015, 12, 576–589. [Google Scholar] [CrossRef]
- Pezzoli, R.; Lyons, J.G.; Gately, N.; Higginbotham, C.L. Investigation of miscibility estimation methods between indomethacin and poly (vinylpyrrolidone-co-vinyl acetate). Int. J. Pharm. 2018, 549, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, M.; Liu, T.; Gao, Z.; Ye, Q.; Tan, X.; Hou, Y.; Sun, J.; Wang, D.; He, Z. Co-amorphous solid dispersion systems of lacidipine-spironolactone with improved dissolution rate and enhanced physical stability. Asian J. Pharm. Sci. 2019, 14, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Gedat, E.; Schreiber, A.; Findenegg, G.H.; Shenderovich, I.; Limbach, H.H.; Buntkowsky, G. Stray field gradient NMR reveals effects of hydrogen bonding on diffusion coefficients of pyridine in mesoporous silica. Magn. Reson. Chem. 2010, 39, 104739. [Google Scholar] [CrossRef]
- Taylor, L.S.; Zografi, G. Spectroscopic characterization of interactions between PVP and indomethacin in amorphous molecular dispersions. Pharm. Res. 1997, 14, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Mureşan-Pop, M.; Pop, M.; Borodi, G.; Todea, M.; Nagy-Simon, T.; Simon, S. Solid dispersions of Myricetin with enhanced solubility: Formulation, characterization and crystal structure of stability-impeding Myricetin monohydrate crystals. J. Mol. Struct. 2017, 1141, 607–614. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, H.; Zhang, Y.; Liu, Y.; Guo, Y.; Hu, C. Influence of Intermolecular Interactions on Crystallite Size in Crystalline Solid Dispersions. Pharmaceutics 2023, 15, 2493. https://doi.org/10.3390/pharmaceutics15102493
Huang H, Zhang Y, Liu Y, Guo Y, Hu C. Influence of Intermolecular Interactions on Crystallite Size in Crystalline Solid Dispersions. Pharmaceutics. 2023; 15(10):2493. https://doi.org/10.3390/pharmaceutics15102493
Chicago/Turabian StyleHuang, Hua, Yong Zhang, Yao Liu, Yufei Guo, and Chunhui Hu. 2023. "Influence of Intermolecular Interactions on Crystallite Size in Crystalline Solid Dispersions" Pharmaceutics 15, no. 10: 2493. https://doi.org/10.3390/pharmaceutics15102493
APA StyleHuang, H., Zhang, Y., Liu, Y., Guo, Y., & Hu, C. (2023). Influence of Intermolecular Interactions on Crystallite Size in Crystalline Solid Dispersions. Pharmaceutics, 15(10), 2493. https://doi.org/10.3390/pharmaceutics15102493