
Harnessing Nanomedicine to Potentiate the Chemo-Immunotherapeutic Effects of Doxorubicin and Alendronate Co-Encapsulated in Pegylated Liposomes

 ,
,  ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals Sources
2.2. Formulation of PLAD
2.3. Stability Assays
2.4. In Vitro Uptake and Cytotoxicity Assays
2.5. Animal Studies
2.6. Determination of Dox in Pharmacokinetic and Biodistribution Studies
2.7. Preparation of Radiolabelled PLAD ([111In]In-PLAD) and Formulation
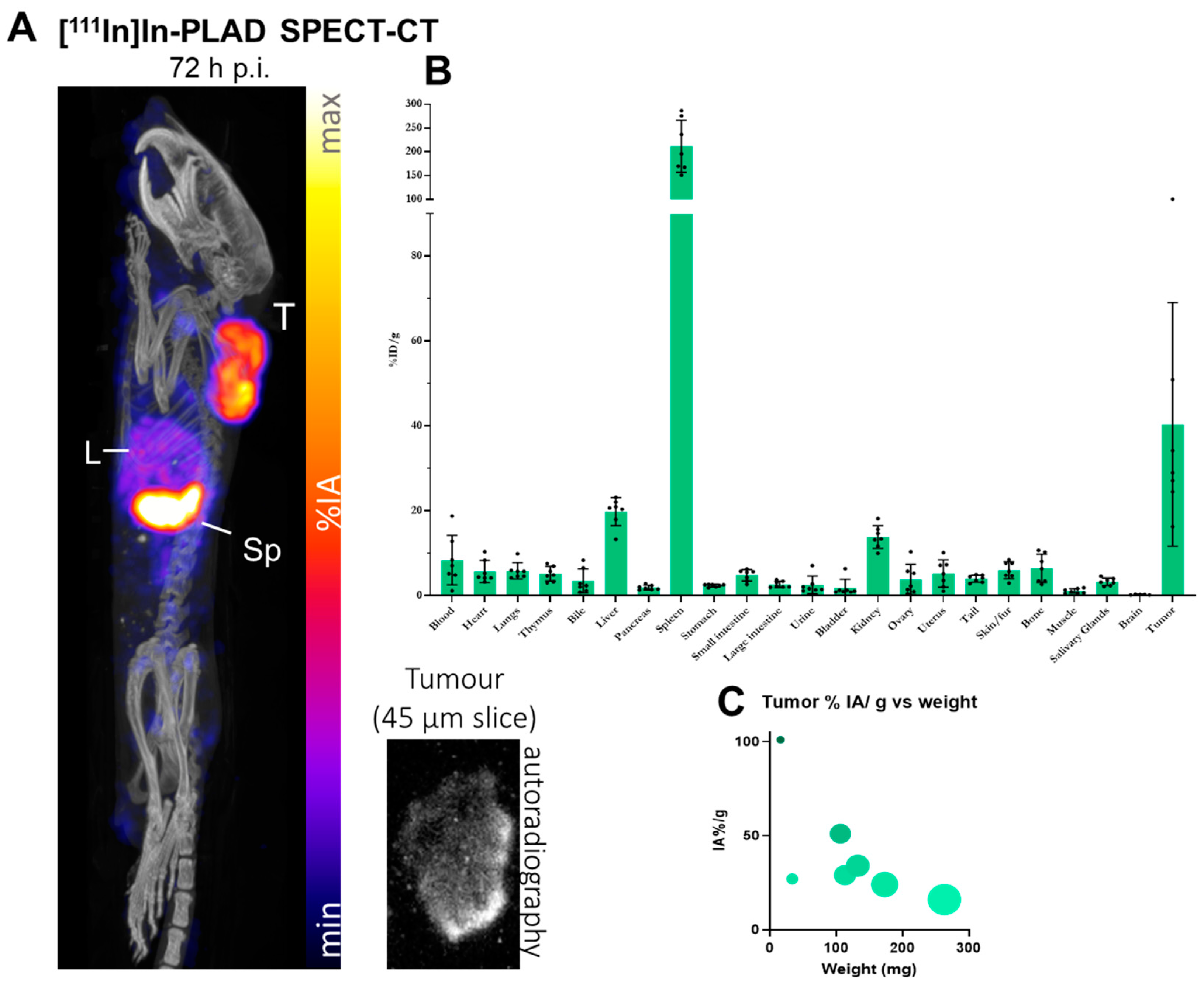
2.8. SPECT/CT Imaging Study with Ex Vivo Biodistribution and Tumor Autoradiography
2.9. Toxicity Studies
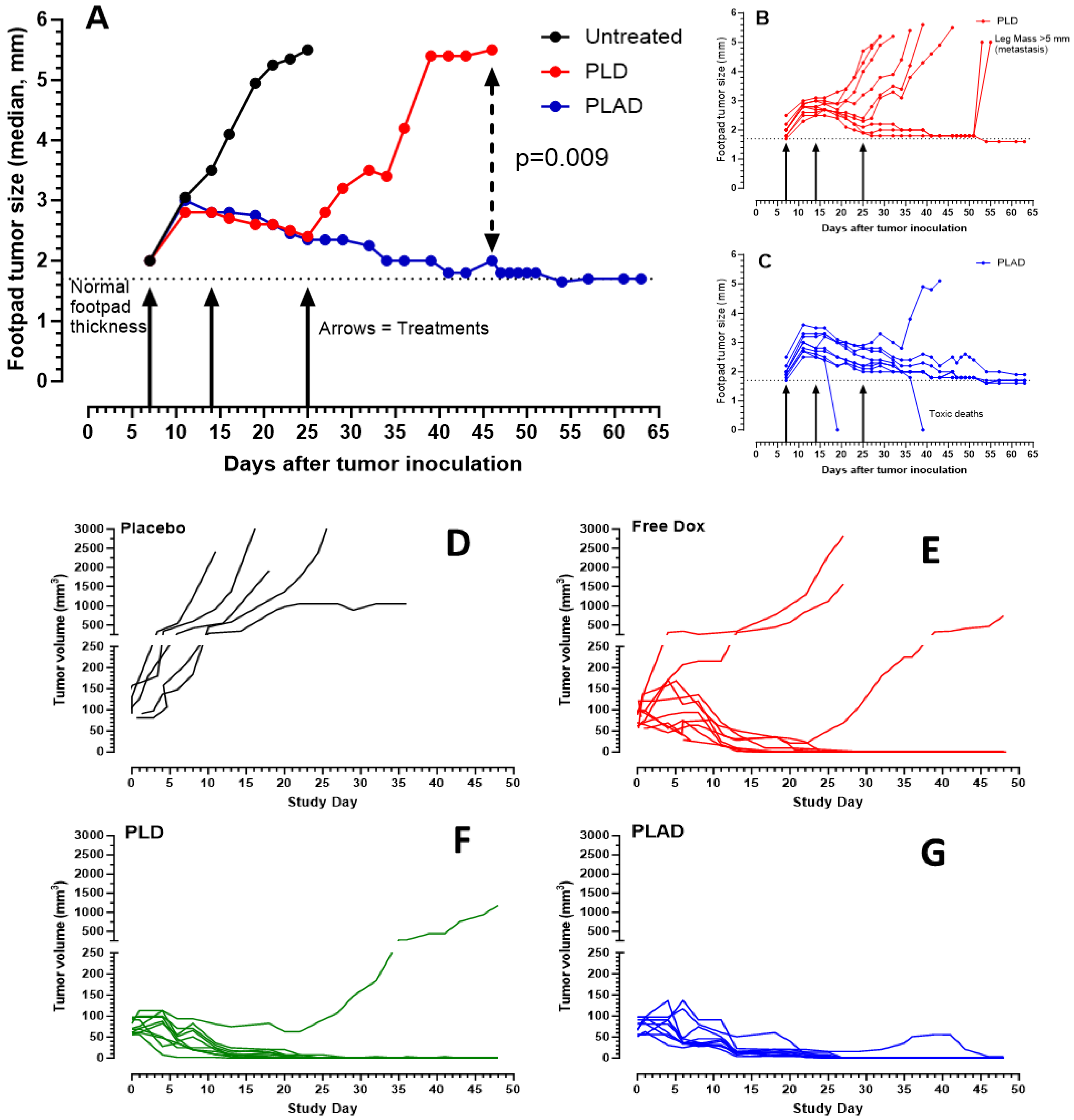
2.10. Antitumor Efficacy
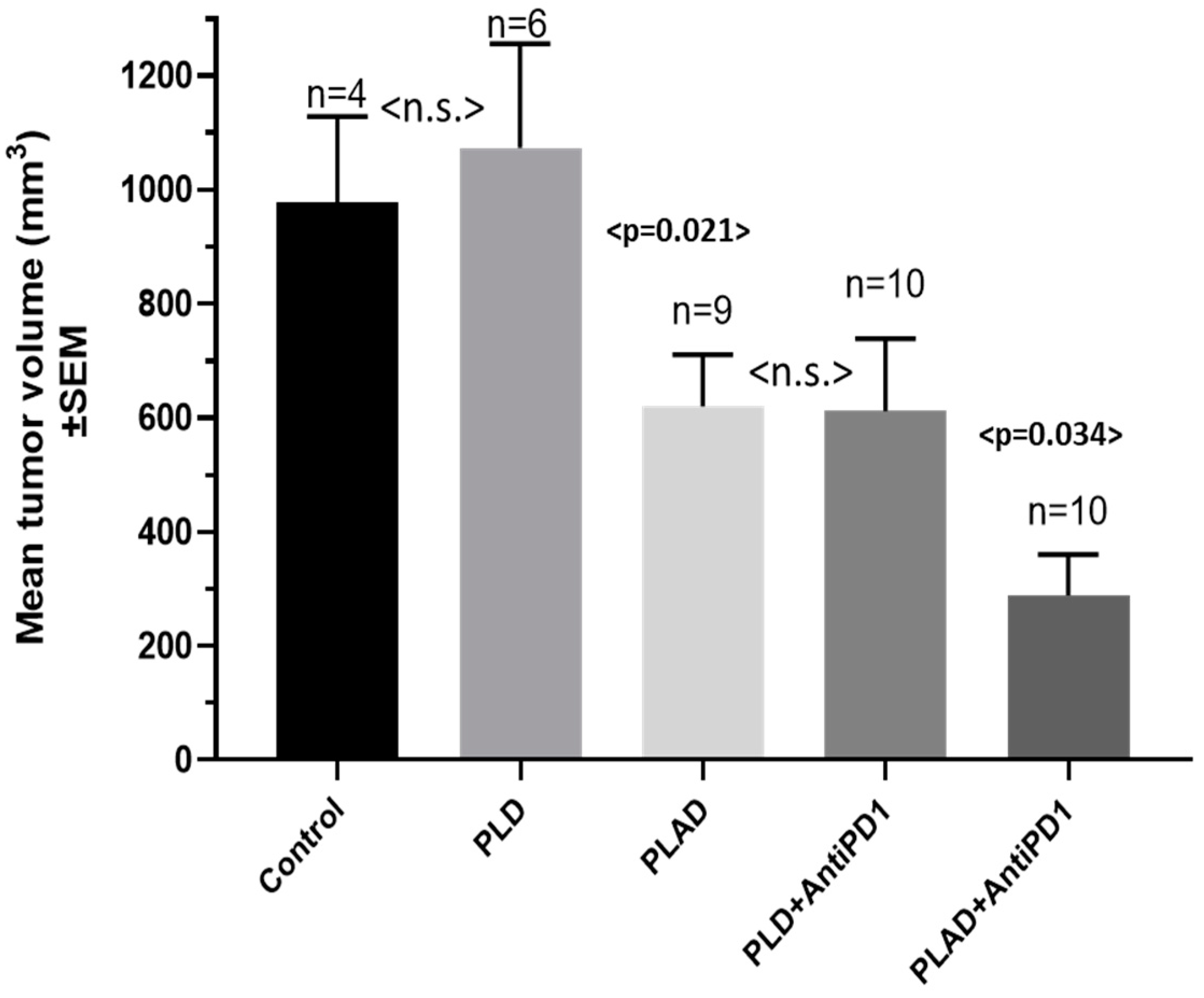
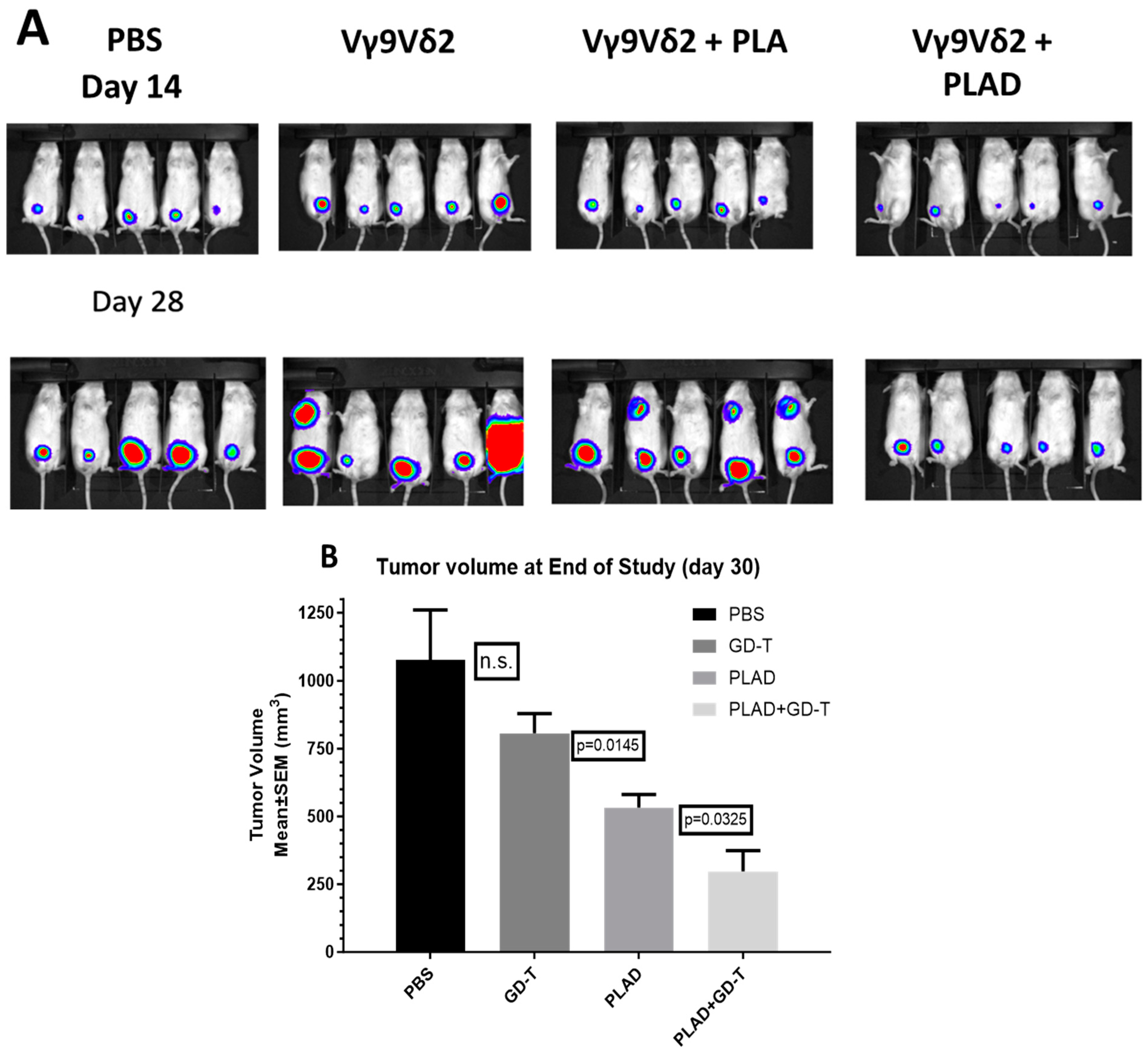
2.11. Therapeutic Studies Combining PLAD with Gamma-Delta T Cell Transfer
3. Results
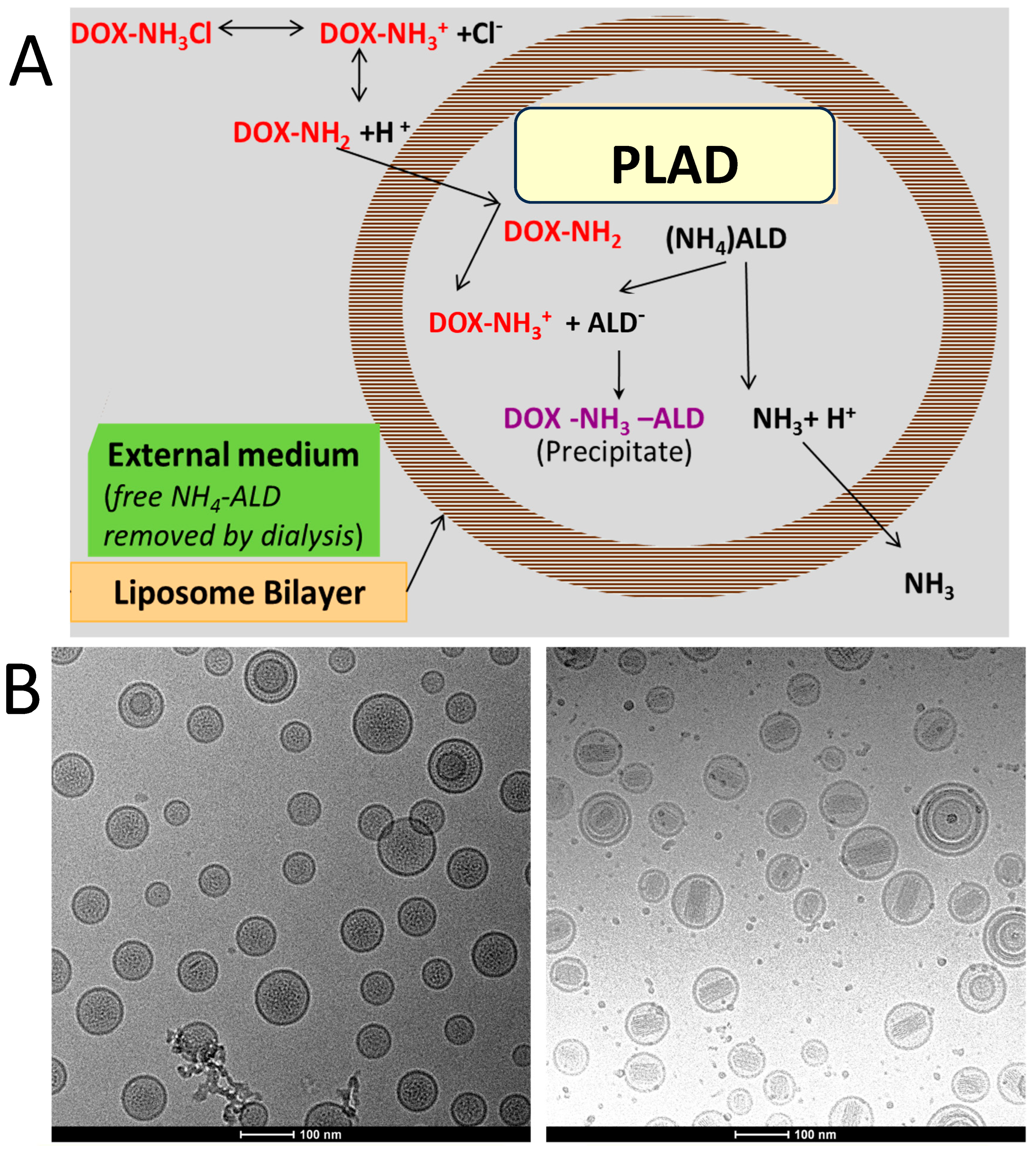
3.1. Formulation and Characterization of PLAD
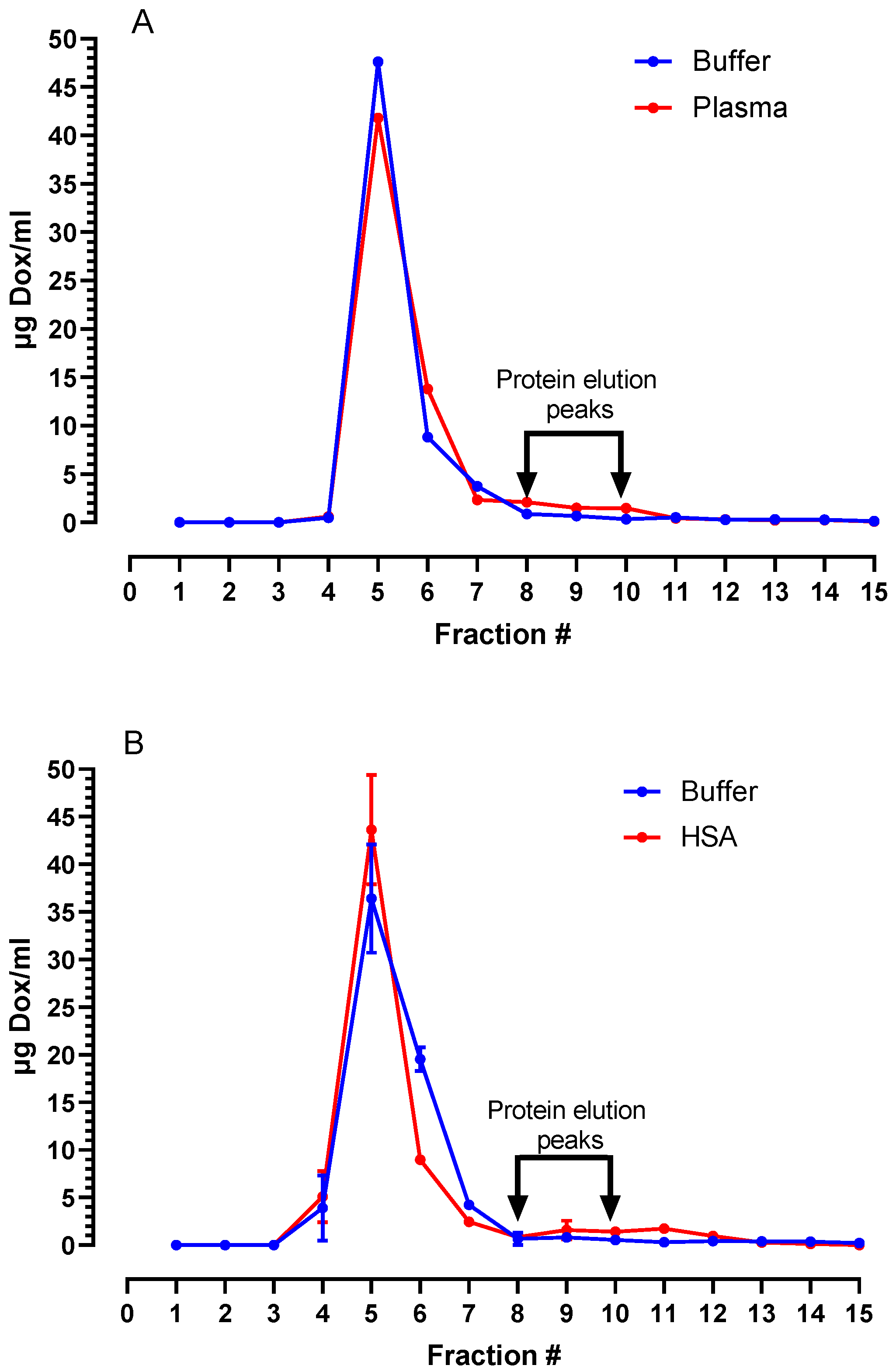
3.2. Stability of PLAD in Biological Fluids
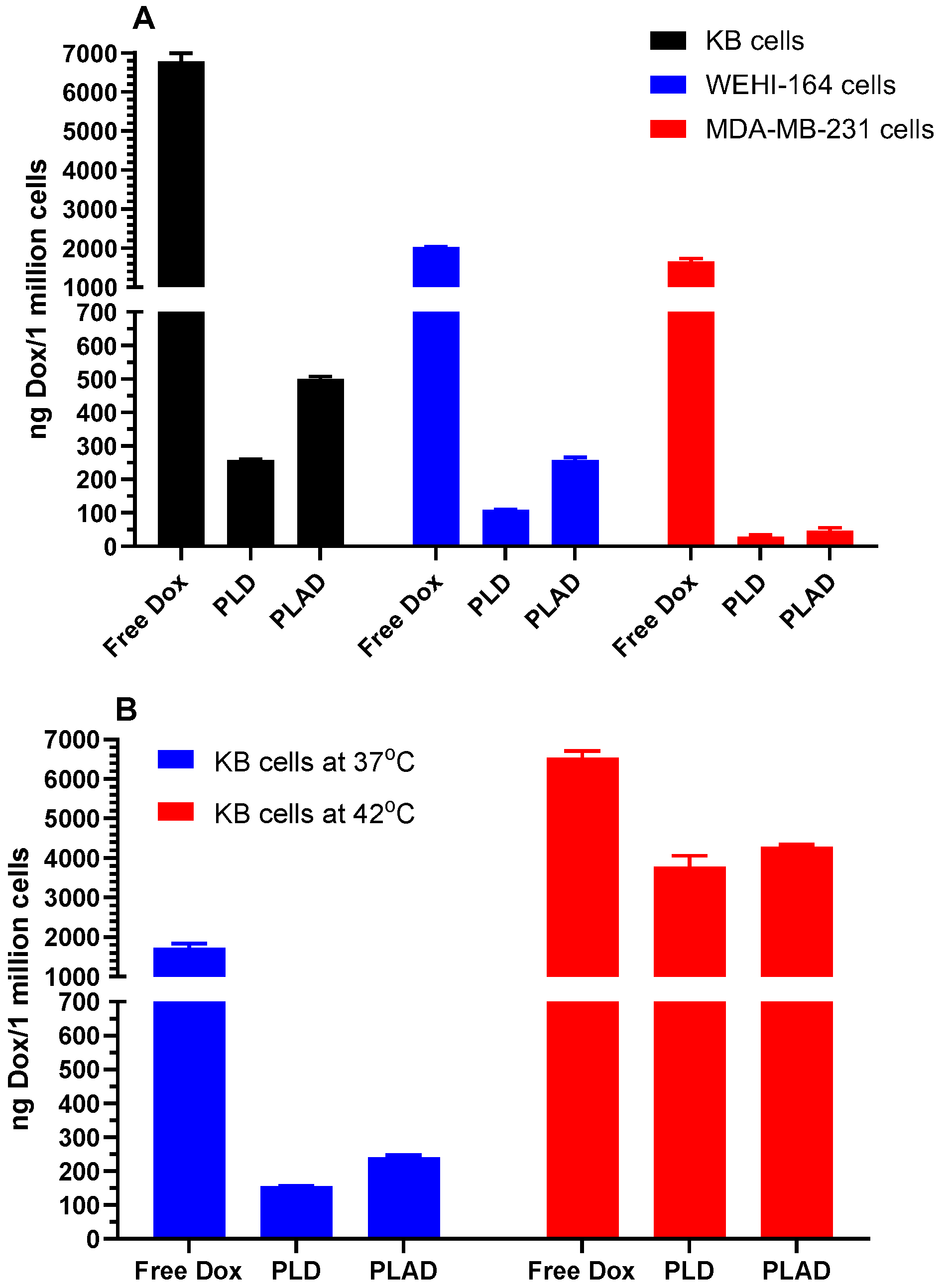
3.3. In Vitro Cell Studies with PLAD: Uptake and Cytotoxicity
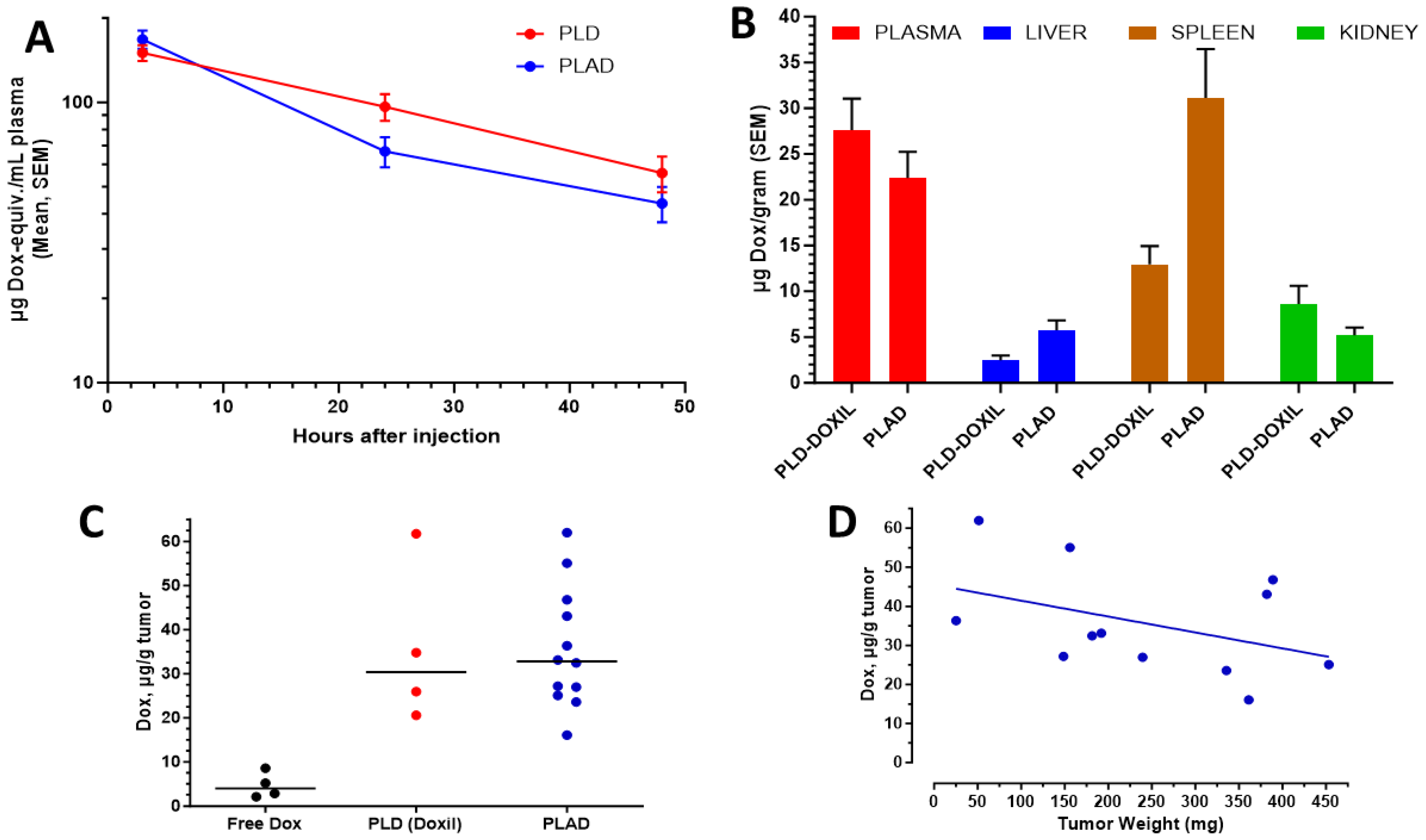
3.4. Pharmacokinetics and Biodistribution
3.5. Imaging Studies of PLAD in Tumor-Bearing Mice
3.6. Toxicity Study
3.7. Therapeutic Activity of PLAD
3.8. PLAD and Gamma-Delta T Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Franco, M.S.; Oliveira, M.C. Liposomes Co- encapsulating Anticancer Drugs in Synergistic Ratios as an Approach to Promote Increased Efficacy and Greater Safety. Anticancer Agents Med. Chem. 2019, 19, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Mayer, L.D. Improving combination cancer therapy: The CombiPlex((R)) development platform. Future Oncol. 2018, 14, 1317–1332. [Google Scholar] [CrossRef] [PubMed]
- Zununi Vahed, S.; Salehi, R.; Davaran, S.; Sharifi, S. Liposome-based drug co-delivery systems in cancer cells. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 71, 1327–1341. [Google Scholar] [CrossRef]
- Dicko, A.; Mayer, L.D.; Tardi, P.G. Use of nanoscale delivery systems to maintain synergistic drug ratios in vivo. Expert. Opin. Drug Deliv. 2010, 7, 1329–1341. [Google Scholar] [CrossRef] [PubMed]
- Liboiron, B.D.; Mayer, L.D. Nanoscale particulate systems for multidrug delivery: Towards improved combination chemotherapy. Ther. Deliv. 2014, 5, 149–171. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.A.; Gomes, E.R.; De Barros, A.L.B.; Leite, E.A. Co-Encapsulation of Simvastatin and Doxorubicin into pH-Sensitive Liposomes Enhances Antitumoral Activity in Breast Cancer Cell Lines. Pharmaceutics 2023, 15, 369. [Google Scholar] [CrossRef] [PubMed]
- Ghaferi, M.; Raza, A.; Koohi, M.; Zahra, W.; Akbarzadeh, A.; Ebrahimi Shahmabadi, H.; Alavi, S.E. Impact of PEGylated Liposomal Doxorubicin and Carboplatin Combination on Glioblastoma. Pharmaceutics 2022, 14, 2183. [Google Scholar] [CrossRef]
- Prasad, P.; Shuhendler, A.; Cai, P.; Rauth, A.M.; Wu, X.Y. Doxorubicin and mitomycin C co-loaded polymer-lipid hybrid nanoparticles inhibit growth of sensitive and multidrug resistant human mammary tumor xenografts. Cancer Lett. 2013, 334, 263–273. [Google Scholar] [CrossRef]
- Gabizon, A.; Ohana, P.; Amitay, Y.; Gorin, J.; Tzemach, D.; Mak, L.; Shmeeda, H. Liposome co-encapsulation of anti-cancer agents for pharmacological optimization of nanomedicine-based combination chemotherapy. Cancer Drug Resist. 2021, 4, 463–484. [Google Scholar] [CrossRef]
- Mei, K.C.; Liao, Y.P.; Jiang, J.; Chiang, M.; Khazaieli, M.; Liu, X.; Wang, X.; Liu, Q.; Chang, C.H.; Zhang, X.; et al. Liposomal Delivery of Mitoxantrone and a Cholesteryl Indoximod Prodrug Provides Effective Chemo-immunotherapy in Multiple Solid Tumors. ACS Nano 2020, 14, 13343–13366. [Google Scholar] [CrossRef]
- Mayer, L.D.; Tardi, P.; Louie, A.C. CPX-351: A nanoscale liposomal co-formulation of daunorubicin and cytarabine with unique biodistribution and tumor cell uptake properties. Int. J. Nanomed. 2019, 14, 3819–3830. [Google Scholar] [CrossRef] [PubMed]
- Shmeeda, H.; Amitay, Y.; Gorin, J.; Tzemach, D.; Mak, L.; Stern, S.T.; Barenholz, Y.; Gabizon, A. Coencapsulation of alendronate and doxorubicin in pegylated liposomes: A novel formulation for chemoimmunotherapy of cancer. J. Drug Target 2016, 24, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.A.; Patil, Y.; La-Beck, N.M. New insights and evolving role of pegylated liposomal doxorubicin in cancer therapy. Drug Resist. Updates 2016, 29, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Clezardin, P.; Ebetino, F.H.; Fournier, P.G. Bisphosphonates and cancer-induced bone disease: Beyond their antiresorptive activity. Cancer Res. 2005, 65, 4971–4974. [Google Scholar] [CrossRef]
- Teixeira, S.; Branco, L.; Fernandes, M.H.; Costa-Rodrigues, J. Bisphosphonates and Cancer: A Relationship Beyond the Antiresorptive Effects. Mini Rev. Med. Chem. 2019, 19, 988–998. [Google Scholar] [CrossRef]
- La-Beck, N.M.; Liu, X.; Shmeeda, H.; Shudde, C.; Gabizon, A.A. Repurposing amino-bisphosphonates by liposome formulation for a new role in cancer treatment. Semin. Cancer Biol. 2021, 68, 175–185. [Google Scholar] [CrossRef]
- Hodgins, N.O.; Wang, J.T.; Al-Jamal, K.T. Nano-technology based carriers for nitrogen-containing bisphosphonates delivery as sensitisers of γδ T cells for anticancer immunotherapy. Adv. Drug Deliv. Rev. 2017, 114, 143–160. [Google Scholar] [CrossRef]
- Islam, M.R.; Patel, J.; Back, P.I.; Shmeeda, H.; Adamsky, K.; Yang, H.; Alvarez, C.; Gabizon, A.A.; La-Beck, N.M. Comparative effects of free doxorubicin, liposome encapsulated doxorubicin and liposome co-encapsulated alendronate and doxorubicin (PLAD) on the tumor immunologic milieu in a mouse fibrosarcoma model. Nanotheranostics 2022, 6, 451–464. [Google Scholar] [CrossRef]
- Patil, Y.; Shmeeda, H.; Amitay, Y.; Ohana, P.; Kumar, S.; Gabizon, A. Targeting of folate-conjugated liposomes with co-entrapped drugs to prostate cancer cells via prostate-specific membrane antigen (PSMA). Nanomedicine 2018, 14, 1407–1416. [Google Scholar] [CrossRef]
- Gabizon, A.; Horowitz, A.T.; Goren, D.; Tzemach, D.; Shmeeda, H.; Zalipsky, S. In vivo fate of folate-targeted polyethylene-glycol liposomes in tumor-bearing mice. Clin. Cancer Res. 2003, 9, 6551–6559. [Google Scholar]
- Edmonds, S.; Volpe, A.; Shmeeda, H.; Parente-Pereira, A.C.; Radia, R.; Baguna-Torres, J.; Szanda, I.; Severin, G.W.; Livieratos, L.; Blower, P.J.; et al. Exploiting the Metal-Chelating Properties of the Drug Cargo for In Vivo Positron Emission Tomography Imaging of Liposomal Nanomedicines. ACS Nano 2016, 10, 10294–10307. [Google Scholar] [CrossRef] [PubMed]
- Thakur, M.L.; Welch, M.J.; Joist, J.H.; Coleman, R.E. In-111 Labeled Platelets—Studies on Preparation and Evaluation of in Vitro and in Vivo Functions. Thromb. Res. 1976, 9, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Parente-Pereira, A.C.; Shmeeda, H.; Whilding, L.M.; Zambirinis, C.P.; Foster, J.; van der Stegen, S.J.; Beatson, R.; Zabinski, T.; Brewig, N.; Sosabowski, J.K.; et al. Adoptive immunotherapy of epithelial ovarian cancer with Vgamma9Vdelta2 T cells, potentiated by liposomal alendronic acid. J. Immunol. 2014, 193, 5557–5566. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Gao, H.; Bao, G. Physical Principles of Nanoparticle Cellular Endocytosis. ACS Nano 2015, 9, 8655–8671. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.; Tzemach, D.; Gorin, J.; Mak, L.; Amitay, Y.; Shmeeda, H.; Zalipsky, S. Improved therapeutic activity of folate-targeted liposomal doxorubicin in folate receptor-expressing tumor models. Cancer Chemother. Pharmacol. 2010, 66, 43–52. [Google Scholar] [CrossRef]
- Horowitz, A.T.; Barenholz, Y.; Gabizon, A.A. In vitro cytotoxicity of liposome-encapsulated doxorubicin: Dependence on liposome composition and drug release. Biochim. Biophys. Acta 1992, 1109, 203–209. [Google Scholar] [CrossRef]
- Shmeeda, H.; Amitay, Y.; Gorin, J.; Tzemach, D.; Mak, L.; Ogorka, J.; Kumar, S.; Zhang, J.A.; Gabizon, A. Delivery of zoledronic acid encapsulated in folate-targeted liposome results in potent in vitro cytotoxic activity on tumor cells. J. Control Release 2010, 146, 76–83. [Google Scholar] [CrossRef]
- Keller, R.K.; Fliesler, S.J. Mechanism of aminobisphosphonate action: Characterization of alendronate inhibition of the isoprenoid pathway. Biochem. Biophys. Res. Commun. 1999, 266, 560–563. [Google Scholar] [CrossRef]
- Kopecka, J.; Porto, S.; Lusa, S.; Gazzano, E.; Salzano, G.; Pinzòn-Daza, M.L.; Giordano, A.; Desiderio, V.; Ghigo, D.; De Rosa, G.; et al. Zoledronic acid-encapsulating self-assembling nanoparticles and doxorubicin: A combinatorial approach to overcome simultaneously chemoresistance and immunoresistance in breast tumors. Oncotarget 2016, 7, 20753–20772. [Google Scholar] [CrossRef]
- Gabizon, A.A.; de Rosales, R.T.M.; La-Beck, N.M. Translational considerations in nanomedicine: The oncology perspective. Adv. Drug Deliv. Rev. 2020, 158, 140–157. [Google Scholar] [CrossRef]
- Harrington, K.J.; Rowlinson-Busza, G.; Syrigos, K.N.; Abra, R.M.; Uster, P.S.; Peters, A.M.; Stewart, J.S. Influence of tumour size on uptake of(111)ln-DTPA-labelled pegylated liposomes in a human tumour xenograft model. Br. J. Cancer 2000, 83, 684–688. [Google Scholar] [CrossRef] [PubMed]
- Goren, D.; Horowitz, A.T.; Tzemach, D.; Tarshish, M.; Zalipsky, S.; Gabizon, A. Nuclear delivery of doxorubicin via folate-targeted liposomes with bypass of multidrug-resistance efflux pump. Clin. Cancer Res. 2000, 6, 1949–1957. [Google Scholar] [PubMed]
- Man, F.; Lim, L.; Volpe, A.; Gabizon, A.; Shmeeda, H.; Draper, B.; Parente-Pereira, A.C.; Maher, J.; Blower, P.J.; Fruhwirth, G.O.; et al. In Vivo PET Tracking of (89)Zr-Labeled Vgamma9Vdelta2 T Cells to Mouse Xenograft Breast Tumors Activated with Liposomal Alendronate. Mol. Ther. 2019, 27, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.A.; Barenholz, Y.; Shmeeda, H.; Maher, J.; Parente-Pereira, A.C. Liposomes Co-Encapsulating a Bisphosphonate and an Amphipathic Agent. US Patent 10085940, 2 October 2018. [Google Scholar]
- Alfayez, M.; Kantarjian, H.; Kadia, T.; Ravandi-Kashani, F.; Daver, N. CPX-351 (vyxeos) in AML. Leuk. Lymphoma 2020, 61, 288–297. [Google Scholar] [CrossRef]
- Feldman, E.J.; Lancet, J.E.; Kolitz, J.E.; Ritchie, E.K.; Roboz, G.J.; List, A.F.; Allen, S.L.; Asatiani, E.; Mayer, L.D.; Swenson, C.; et al. First-in-man study of CPX-351: A liposomal carrier containing cytarabine and daunorubicin in a fixed 5:1 molar ratio for the treatment of relapsed and refractory acute myeloid leukemia. J. Clin. Oncol. 2011, 29, 979–985. [Google Scholar] [CrossRef]
- Batist, G.; Gelmon, K.A.; Chi, K.N.; Miller, W.H.; Jr Chia, S.K.; Mayer, L.D.; Swenson, C.E.; Janoff, A.S.; Louie, A.C. Safety, pharmacokinetics, and efficacy of CPX-1 liposome injection in patients with advanced solid tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 692–700. [Google Scholar] [CrossRef]
- Tardi, P.G.; Dos Santos, N.; Harasym, T.O.; Johnstone, S.A.; Zisman, N.; Tsang, A.W.; Bermudes, D.G.; Mayer, L.D. Drug ratio-dependent antitumor activity of irinotecan and cisplatin combinations in vitro and in vivo. Mol. Cancer Ther. 2009, 8, 2266–2275. [Google Scholar] [CrossRef]
- Zhang, R.X.; Cai, P.; Zhang, T.; Chen, K.; Li, J.; Cheng, J.; Pang, K.S.; Adissu, H.A.; Rauth, A.M.; Wu, X.Y. Polymer-lipid hybrid nanoparticles synchronize pharmacokinetics of co-encapsulated doxorubicin-mitomycin C and enable their spatiotemporal co-delivery and local bioavailability in breast tumor. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 1279–1290. [Google Scholar] [CrossRef]
- Sprooten, J.; Laureano, R.S.; Vanmeerbeek, I.; Govaerts, J.; Naulaerts, S.; Borras, D.M.; Kinget, L.; Fucikova, J.; Spisek, R.; Jelinkova, L.P.; et al. Trial watch: Chemotherapy-induced immunogenic cell death in oncology. Oncoimmunology 2023, 12, 2219591. [Google Scholar] [CrossRef]
- La-Beck, N.M.; Gabizon, A.A. Nanoparticle Interactions with the Immune System: Clinical Implications for Liposome-Based Cancer Chemotherapy. Front. Immunol. 2017, 8, 416. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.; Cherny, N.; Isacson, R.; AbuRemilah, A.; Shmeeda, H.; Rosengarten, O. A phase 1b study of chemoimmunotherapy with pegylated liposomal Doxorubicin and pembrolizumab in estrogen receptor-positive, endocrine-resistant breast cancer. J. Clin. Oncol. 2021, 39, 1049. [Google Scholar] [CrossRef]
- Lee, E.K.; Xiong, N.; Cheng, S.C.; Barry, W.T.; Penson, R.T.; Konstantinopoulos, P.A.; Hoffman, M.A.; Horowitz, N.; Dizon, D.S.; Stover, E.H.; et al. Combined pembrolizumab and pegylated liposomal doxorubicin in platinum resistant ovarian cancer: A phase 2 clinical trial. Gynecol. Oncol. 2020, 159, 72–78. [Google Scholar] [CrossRef]
- Rossevold, A.H.; Andresen, N.K.; Bjerre, C.A.; Gilje, B.; Jakobsen, E.H.; Raj, S.X.; Falk, R.S.; Russnes, H.G.; Jahr, T.; Mathiesen, R.R.; et al. Atezolizumab plus anthracycline-based chemotherapy in metastatic triple-negative breast cancer: The randomized, double-blind phase 2b ALICE trial. Nat. Med. 2022, 28, 2573–2583. [Google Scholar] [CrossRef]
- Kyte, J.A.; Andresen, N.K.; Russnes, H.G.; Fretland, S.O.; Falk, R.S.; Lingjaerde, O.C.; Naume, B. ICON: A randomized phase IIb study evaluating immunogenic chemotherapy combined with ipilimumab and nivolumab in patients with metastatic hormone receptor positive breast cancer. J. Transl. Med. 2020, 18, 269. [Google Scholar] [CrossRef] [PubMed]
- Man, F.; Lammers, T.; de Rosales, R. Imaging Nanomedicine-Based Drug Delivery: A Review of Clinical Studies. Mol. Imaging Biol. 2018, 20, 683–695. [Google Scholar] [CrossRef]
- Prabhakar, U.; Maeda, H.; Jain, R.K.; Sevick-Muraca, E.M.; Zamboni, W.; Farokhzad, O.C.; Barry, S.T.; Gabizon, A.; Grodzinski, P.; Blakey, D.C. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013, 73, 2412–2417. [Google Scholar] [CrossRef]
- Golombek, S.K.; May, J.N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumor targeting via EPR: Strategies to enhance patient responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef]
- Kim, J.; Cho, H.; Lim, D.K.; Joo, M.K.; Kim, K. Perspectives for Improving the Tumor Targeting of Nanomedicine via the EPR Effect in Clinical Tumors. Int. J. Mol. Sci. 2023, 24, 10082. [Google Scholar] [CrossRef]
- Rajan, R.; Sabnani, M.K.; Mavinkurve, V.; Shmeeda, H.; Mansouri, H.; Bonkoungou, S.; Le, A.D.; Wood, L.M.; Gabizon, A.A.; La-Beck, N.M. Liposome-induced immunosuppression and tumor growth is mediated by macrophages and mitigated by liposome-encapsulated alendronate. J. Control Release 2018, 271, 139–148. [Google Scholar] [CrossRef]
- La-Beck, N.M.; Nguyen, D.T.; Le, A.D.; Alzghari, S.K.; Trinh, S.T. Optimizing Patient Outcomes with PD-1/PD-L1 Immune Checkpoint Inhibitors for the First-Line Treatment of Advanced Non-Small Cell Lung Cancer. Pharmacotherapy 2020, 40, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef] [PubMed]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guerin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- La-Beck, N.M.; Jean, G.W.; Huynh, C.; Alzghari, S.K.; Lowe, D.B. Immune Checkpoint Inhibitors: New Insights and Current Place in Cancer Therapy. Pharmacotherapy 2015, 35, 963–976. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Rébé, C. Using immunogenic cell death to improve anticancer efficacy of immune checkpoint inhibitors: From basic science to clinical application. Immunol. Rev. 2023. [Google Scholar] [CrossRef]
- Trinh, S.; Le, A.; Gowani, S.; La-Beck, N.M. Management of Immune-Related Adverse Events Associated with Immune Checkpoint Inhibitor Therapy: A Minireview of Current Clinical Guidelines. Asia Pac. J. Oncol. Nurs. 2019, 6, 154–160. [Google Scholar] [CrossRef]
- Shi, F.; Huang, X.; Hong, Z.; Lu, N.; Huang, X.; Liu, L.; Liang, T.; Bai, X. Improvement strategy for immune checkpoint blockade: A focus on the combination with immunogenic cell death inducers. Cancer Lett. 2023, 562, 216167. [Google Scholar] [CrossRef]
- Islam, M.R.; Patel, J.; Back, P.I.; Shmeeda, H.; Kallem, R.R.; Shudde, C.; Markiewski, M.; Putnam, W.C.; Gabizon, A.A.; La-Beck, N.M. Pegylated Liposomal Alendronate Biodistribution, Immune Modulation, and Tumor Growth Inhibition in a Murine Melanoma Model. Biomolecules 2023, 13, 1309. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLAD Batch (Batch Size) | Vesicle Size nm | PDI | Zeta Potential mV | Osmolality mOsm/kg | pH | ALD mg/g | Cholesterol mg/g | mPEG2000-DSPE mg/g | HSPC mg/g | DOX-HCl mg/g 1 |
|---|---|---|---|---|---|---|---|---|---|---|
| Batch 1 (0.5 L) | 110.3 | 0.058 | −12.13 | 317 | 7.1 | 0.5 | 1.62 | 1.36 | 4.6 | 0.9 |
| Batch 2 (1.5 L) | 99.8 | 0.028 | −13.41 | 291 | 6.7 | 0.6 | 1.62 | 1.21 | 4.3 | 0.9 |
| Cell Line | KB | MDA-MB-231 | Wehi-164 | 4T1 | M109 |
|---|---|---|---|---|---|
| Free Dox | 0.07 | 0.6 | 0.5 | 2.2 | 0.1 |
| PLD | 4.8 | 15.9 | 18.75 | >50 | 8.0 |
| PLAD-1 | 0.6 | 5.1 | 4.6 | 1.0 | |
| PLAD-2 | 2.0 | 11.7 | 6.6 | 24.6 | |
| Free Ald | >50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gabizon, A.; Shmeeda, H.; Draper, B.; Parente-Pereira, A.; Maher, J.; Carrascal-Miniño, A.; de Rosales, R.T.M.; La-Beck, N.M. Harnessing Nanomedicine to Potentiate the Chemo-Immunotherapeutic Effects of Doxorubicin and Alendronate Co-Encapsulated in Pegylated Liposomes. Pharmaceutics 2023, 15, 2606. https://doi.org/10.3390/pharmaceutics15112606
Gabizon A, Shmeeda H, Draper B, Parente-Pereira A, Maher J, Carrascal-Miniño A, de Rosales RTM, La-Beck NM. Harnessing Nanomedicine to Potentiate the Chemo-Immunotherapeutic Effects of Doxorubicin and Alendronate Co-Encapsulated in Pegylated Liposomes. Pharmaceutics. 2023; 15(11):2606. https://doi.org/10.3390/pharmaceutics15112606
Chicago/Turabian StyleGabizon, Alberto, Hilary Shmeeda, Benjamin Draper, Ana Parente-Pereira, John Maher, Amaia Carrascal-Miniño, Rafael T. M. de Rosales, and Ninh M. La-Beck. 2023. "Harnessing Nanomedicine to Potentiate the Chemo-Immunotherapeutic Effects of Doxorubicin and Alendronate Co-Encapsulated in Pegylated Liposomes" Pharmaceutics 15, no. 11: 2606. https://doi.org/10.3390/pharmaceutics15112606
APA StyleGabizon, A., Shmeeda, H., Draper, B., Parente-Pereira, A., Maher, J., Carrascal-Miniño, A., de Rosales, R. T. M., & La-Beck, N. M. (2023). Harnessing Nanomedicine to Potentiate the Chemo-Immunotherapeutic Effects of Doxorubicin and Alendronate Co-Encapsulated in Pegylated Liposomes. Pharmaceutics, 15(11), 2606. https://doi.org/10.3390/pharmaceutics15112606