
Composition Dependency of the Flory–Huggins Interaction Parameter in Drug–Polymer Phase Behavior


Abstract
:1. Introduction
2. Materials and Methods
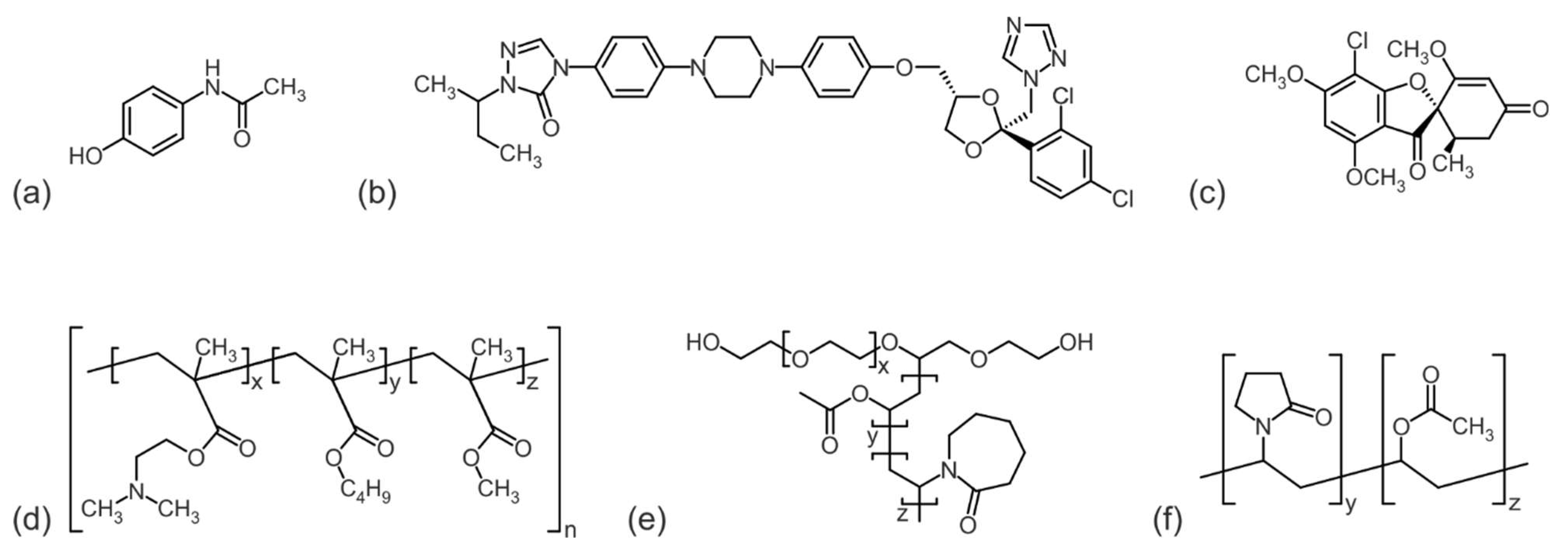
2.1. Materials
2.2. Differential Scanning Calorimetry
2.3. Modeling of Phase Diagrams
3. Results and Discussion
3.1. Composition Dependency of the Flory–Huggins Interaction Parameter
3.2. Improved Modeling of Phase Diagrams Based on the Flory–Huggins Interaction Parameter
3.3. Adoption to ASD Manufacturing Processes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schittny, A.; Huwyler, J.; Puchkov, M. Mechanisms of increased bioavailability through amorphous solid dispersions: A review. Drug Deliv. 2020, 27, 110–127. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Aller, M.; Guillarme, D.; Veuthey, J.-L.; Gurny, R. Strategies for formulating and delivering poorly water-soluble drugs. J. Drug Deliv. Sci. Technol. 2015, 30, 342–351. [Google Scholar] [CrossRef]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Chu, K.R.; Lee, E.; Jeong, S.H.; Park, E.-S. Effect of particle size on the dissolution behaviors of poorly water-soluble drugs. Arch. Pharm. Res. 2012, 35, 1187–1195. [Google Scholar] [CrossRef]
- Bhujbal, S.V.; Mitra, B.; Jain, U.; Gong, Y.; Agrawal, A.; Karki, S.; Taylor, L.; Kumar, S.; Zhou, Q. Pharmaceutical amorphous solid dispersion: A review of manufacturing strategies. Acta Pharm. Sin. B 2021, 11, 2506–2536. [Google Scholar] [CrossRef]
- Hancock, B.C.; Parks, M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric amorphous solid dispersions: A review of amorphization, crystallization, stabilization, solid-state characterization, and aqueous solubilization of biopharmaceutical classification system class II drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef]
- Prudic, A.; Ji, Y.; Sadowski, G. Thermodynamic phase behavior of API/polymer solid dispersions. Mol. Pharm. 2014, 11, 2294–2304. [Google Scholar] [CrossRef]
- Medarević, D.; Djuriš, J.; Barmpalexis, P.; Kachrimanis, K.; Ibrić, S. Analytical and Computational Methods for the Estimation of Drug-Polymer Solubility and Miscibility in Solid Dispersions Development. Pharmaceutics 2019, 11, 372. [Google Scholar] [CrossRef]
- Barmpalexis, P.; Karagianni, A.; Katopodis, K.; Vardaka, E.; Kachrimanis, K. Molecular modelling and simulation of fusion-based amorphous drug dispersions in polymer/plasticizer blends. Eur. J. Pharm. Sci. 2019, 130, 260–268. [Google Scholar] [CrossRef]
- Walden, D.M.; Bundey, Y.; Jagarapu, A.; Antontsev, V.; Chakravarty, K.; Varshney, J. Molecular Simulation and Statistical Learning Methods toward Predicting Drug-Polymer Amorphous Solid Dispersion Miscibility, Stability, and Formulation Design. Molecules 2021, 26, 182. [Google Scholar] [CrossRef] [PubMed]
- Kyeremateng, S.O.; Pudlas, M.; Woehrle, G.H. A fast and reliable empirical approach for estimating solubility of crystalline drugs in polymers for hot melt extrusion formulations. J. Pharm. Sci. 2014, 103, 2847–2858. [Google Scholar] [CrossRef] [PubMed]
- Lehmkemper, K.; Kyeremateng, S.O.; Heinzerling, O.; Degenhardt, M.; Sadowski, G. Long-Term Physical Stability of PVP- and PVPVA-Amorphous Solid Dispersions. Mol. Pharm. 2017, 14, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Thakore, S.D.; Akhtar, J.; Jain, R.; Paudel, A.; Bansal, A.K. Analytical and Computational Methods for the Determination of Drug-Polymer Solubility and Miscibility. Mol. Pharm. 2021, 18, 2835–2866. [Google Scholar] [CrossRef] [PubMed]
- Prudic, A.; Kleetz, T.; Korf, M.; Ji, Y.; Sadowski, G. Influence of copolymer composition on the phase behavior of solid dispersions. Mol. Pharm. 2014, 11, 4189–4198. [Google Scholar] [CrossRef]
- Prudic, A.; Ji, Y.; Luebbert, C.; Sadowski, G. Influence of humidity on the phase behavior of API/polymer formulations. Eur. J. Pharm. Biopharm. 2015, 94, 352–362. [Google Scholar] [CrossRef]
- Flory, P.J. Thermodynamics of High Polymer Solutions. J. Chem. Phys. 1941, 9, 660. [Google Scholar] [CrossRef]
- Huggins, M.L. Thermodynamic properties of solutions of long-chain compounds. Ann. N. Y. Acad. Sci. 1942, 43, 1–32. [Google Scholar] [CrossRef]
- Marsac, P.J.; Shamblin, S.L.; Taylor, L.S. Theoretical and practical approaches for prediction of drug-polymer miscibility and solubility. Pharm. Res. 2006, 23, 2417–2426. [Google Scholar] [CrossRef]
- Paudel, A.; van Humbeeck, J.; van den Mooter, G. Theoretical and experimental investigation on the solid solubility and miscibility of naproxen in poly(vinylpyrrolidone). Mol. Pharm. 2010, 7, 1133–1148. [Google Scholar] [CrossRef]
- Wlodarski, K.; Sawicki, W.; Kozyra, A.; Tajber, L. Physical stability of solid dispersions with respect to thermodynamic solubility of tadalafil in PVP-VA. Eur. J. Pharm. Biopharm. 2015, 96, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Tao, J.; Zhang, G.G.Z.; Yu, L. Solubilities of crystalline drugs in polymers: An improved analytical method and comparison of solubilities of indomethacin and nifedipine in PVP, PVP/VA, and PVAc. J. Pharm. Sci. 2010, 99, 4023–4031. [Google Scholar] [CrossRef] [PubMed]
- Koningsveld, R.; Stockmayer, W.H.; Nies, E. Polymer Phase Diagrams: A Textbook; Oxford University Press: Oxford, UK, 2008; ISBN 9780198556343. [Google Scholar]
- Wilkie, C.A. Polymer Blends Handbook, 2nd ed.; Springer: Dordrecht, The Netherlands, 2014; ISBN 9789400760639. [Google Scholar]
- Potter, C.B.; Davis, M.T.; Albadarin, A.B.; Walker, G.M. Investigation of the Dependence of the Flory-Huggins Interaction Parameter on Temperature and Composition in a Drug-Polymer System. Mol. Pharm. 2018, 15, 5327–5335. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.; Nyamweya, N.; Gönen, S.; Martínez-Miranda, L.J.; Hoag, S.W. Influence of various drugs on the glass transition temperature of poly(vinylpyrrolidone): A thermodynamic and spectroscopic investigation. Int. J. Pharm. 2001, 225, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Wolbert, F.; Fahrig, I.-K.; Gottschalk, T.; Luebbert, C.; Thommes, M.; Sadowski, G. Factors influencing the crystallization-onset time of metastable ASDs. Pharmaceutics 2022, 14, 269. [Google Scholar] [CrossRef] [PubMed]
- Six, K.; Verreck, G.; Peeters, J.; Brewster, M.; van den Mooter, G. Increased physical stability and improved dissolution properties of itraconazole, a class II drug, by solid dispersions that combine fast- and slow-dissolving polymers. J. Pharm. Sci. 2004, 93, 124–131. [Google Scholar] [CrossRef]
- Zhou, D.; Zhang, G.G.Z.; Law, D.; Grant, D.J.W.; Schmitt, E.A. Thermodynamics, molecular mobility and crystallization kinetics of amorphous griseofulvin. Mol. Pharm. 2008, 5, 927–936. [Google Scholar] [CrossRef]
- Dos Santos, J.; Da Silva, G.S.; Velho, M.C.; Beck, R.C.R. Eudragit®: A Versatile Family of Polymers for Hot Melt Extrusion and 3D Printing Processes in Pharmaceutics. Pharmaceutics 2021, 13, 1424. [Google Scholar] [CrossRef]
- Gottschalk, T.; Özbay, C.; Feuerbach, T.; Thommes, M. Predicting Throughput and Melt Temperature in Pharmaceutical Hot Melt Extrusion. Pharmaceutics 2022, 14, 1757. [Google Scholar] [CrossRef]
- Altamimi, M.A.; Neau, S.H. Use of the Flory–Huggins theory to predict the solubility of nifedipine and sulfamethoxazole in the triblock, graft copolymer Soluplus. Drug Dev. Ind. Pharm. 2016, 42, 446–455. [Google Scholar] [CrossRef]
- Höhne, G.; Hemminger, W.; Flammersheim, H.-J. Differential Scanning Calorimetry: An Introduction for Practitioners; 2nd Revised and Enlarged ed.; Springer: Berlin/Heidelberg, Germany, 2003; ISBN 9783662067109. [Google Scholar]
- Moseson, D.E.; Taylor, L.S. The application of temperature-composition phase diagrams for hot melt extrusion processing of amorphous solid dispersions to prevent residual crystallinity. Int. J. Pharm. 2018, 553, 454–466. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.; Taylor, J.S. Ideal copolymers and the second-order transitions of synthetic rubbers. i. non-crystalline copolymers. J. Appl. Chem. 1952, 2, 493–500. [Google Scholar] [CrossRef]
- Simha, R.; Boyer, R.F. On a General Relation Involving the Glass Temperature and Coefficients of Expansion of Polymers. J. Chem. Phys. 1962, 37, 1003–1007. [Google Scholar] [CrossRef]
- Katkov, I.I.; Levine, F. Prediction of the glass transition temperature of water solutions: Comparison of different models. Cryobiology 2004, 49, 62–82. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Xu, P.; Ashour, E.A.; Repka, M.A. Prediction and Construction of Drug-Polymer Binary System Thermodynamic Phase Diagram in Amorphous Solid Dispersions (ASDs). AAPS PharmSciTech 2022, 23, 169. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, M.I.; Pratt, L.R.; Paulaitis, M.E. Concentration dependence of the Flory-Huggins interaction parameter in aqueous solutions of capped PEO chains. J. Chem. Phys. 2014, 141, 244908. [Google Scholar] [CrossRef]
- Anderson, B.D. Predicting Solubility/Miscibility in Amorphous Dispersions: It Is Time to Move Beyond Regular Solution Theories. J. Pharm. Sci. 2018, 107, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Ehrenstein, G.W.; Riedel, G.; Trawiel, P. Thermal Analysis of Plastics: Theory and Practice; Hanser Verlag: München, Germany, 2004; ISBN 9783446434141. [Google Scholar]
- Tu, W.; Knapik-Kowalczuk, J.; Chmiel, K.; Paluch, M. Glass Transition Dynamics and Physical Stability of Amorphous Griseofulvin in Binary Mixtures with Low-Tg Excipients. Mol. Pharm. 2019, 16, 3626–3635. [Google Scholar] [CrossRef] [PubMed]
- Winck, J.; Daalmann, M.; Berghaus, A.; Thommes, M. In-line monitoring of solid dispersion preparation in small scale extrusion based on UV-vis spectroscopy. Pharm. Dev. Technol. 2022, 27, 1009–1015. [Google Scholar] [CrossRef]
- Tao, J.; Sun, Y.; Zhang, G.G.Z.; Yu, L. Solubility of small-molecule crystals in polymers: D-mannitol in PVP, indomethacin in PVP/VA, and nifedipine in PVP/VA. Pharm. Res. 2009, 26, 855–864. [Google Scholar] [CrossRef]
- Winck, J.; Gottschalk, T.; Thommes, M. Predicting Residence Time and Melt Temperature in Pharmaceutical Hot Melt Extrusion. Pharmaceutics 2023, 15, 1417. [Google Scholar] [CrossRef] [PubMed]
- Andrews, G.P.; Qian, K.; Jacobs, E.; Jones, D.S.; Tian, Y. High drug loading nanosized amorphous solid dispersion (NASD) with enhanced in vitro solubility and permeability: Benchmarking conventional ASD. Int. J. Pharm. 2023, 632, 122551. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substance | |||||
|---|---|---|---|---|---|
| ACE | 151.2 a | 1.29 b | 179.2 | 22.5 | 170.0 |
| ITR | 705.6 c | 1.27 d | 85.8 | 57.6 | 164.9 |
| GRI | 352.8 c | 1.42 e | 103.8 | 89.8 | 220.5 |
| bBMA | 47 000 f | 1.09 g | – | 53.6 | – |
| SOL | 118 000 h | 1.08 g | – | 78.2 | – |
| PVPVA | 57 500 h | 1.19 g | – | 109.0 | – |
| Formulation | Empirical | |||||
|---|---|---|---|---|---|---|
| ACE/bBMA | 59.01 | −0.5662 | 0.4210 | 0.7025 | −0.9608 | 0 |
| ACE/SOL | 198.21 | 2.1004 | −0.0047 | 0.2893 | −1.0933 | 1 |
| ACE/PVPVA | 316.10 | 3.9999 | −0.4130 | 0.1379 | −2.8219 | 1 |
| ITR/bBMA | 11.65 | −0.7288 | 0.3714 | 1.2006 | −2.7956 | 0 |
| ITR/SOL | 73.99 | −0.1340 | −0.2013 | 0.1846 | −1.3103 | 0 |
| ITR/PVPVA | 49.59 | −0.9585 | −0.2407 | 0.5347 | −2.4495 | 0 |
| GRI/bBMA | 111.32 | −11.0231 | −1.2948 | 0.5949 | −6.9489 | 0 |
| GRI/SOL | 115.47 | 2.6304 | 0.5522 | 0.2617 | 1.0764 | 0 |
| GRI/PVPVA | 131.77 | 1.9122 | 0.3252 | 0.0419 | 0.9730 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klueppelberg, J.; Handge, U.A.; Thommes, M.; Winck, J. Composition Dependency of the Flory–Huggins Interaction Parameter in Drug–Polymer Phase Behavior. Pharmaceutics 2023, 15, 2650. https://doi.org/10.3390/pharmaceutics15122650
Klueppelberg J, Handge UA, Thommes M, Winck J. Composition Dependency of the Flory–Huggins Interaction Parameter in Drug–Polymer Phase Behavior. Pharmaceutics. 2023; 15(12):2650. https://doi.org/10.3390/pharmaceutics15122650
Chicago/Turabian StyleKlueppelberg, Jana, Ulrich A. Handge, Markus Thommes, and Judith Winck. 2023. "Composition Dependency of the Flory–Huggins Interaction Parameter in Drug–Polymer Phase Behavior" Pharmaceutics 15, no. 12: 2650. https://doi.org/10.3390/pharmaceutics15122650
APA StyleKlueppelberg, J., Handge, U. A., Thommes, M., & Winck, J. (2023). Composition Dependency of the Flory–Huggins Interaction Parameter in Drug–Polymer Phase Behavior. Pharmaceutics, 15(12), 2650. https://doi.org/10.3390/pharmaceutics15122650