
CYP3A5*3 and CYP3A4*22 Cluster Polymorphism Effects on LCP-Tac Tacrolimus Exposure: Population Pharmacokinetic Approach

 , , ,
, , ,
Abstract
:1. Introduction
2. Methods
2.1. Study Design
2.2. Tacrolimus Measurement and Data Recording
2.3. Genotyping
2.4. Statistical Analysis
2.5. Population Pharmacokinetic Analysis
- Base Model Development
- Covariate Model
2.6. Model Evaluation and Internal Validation
3. Results
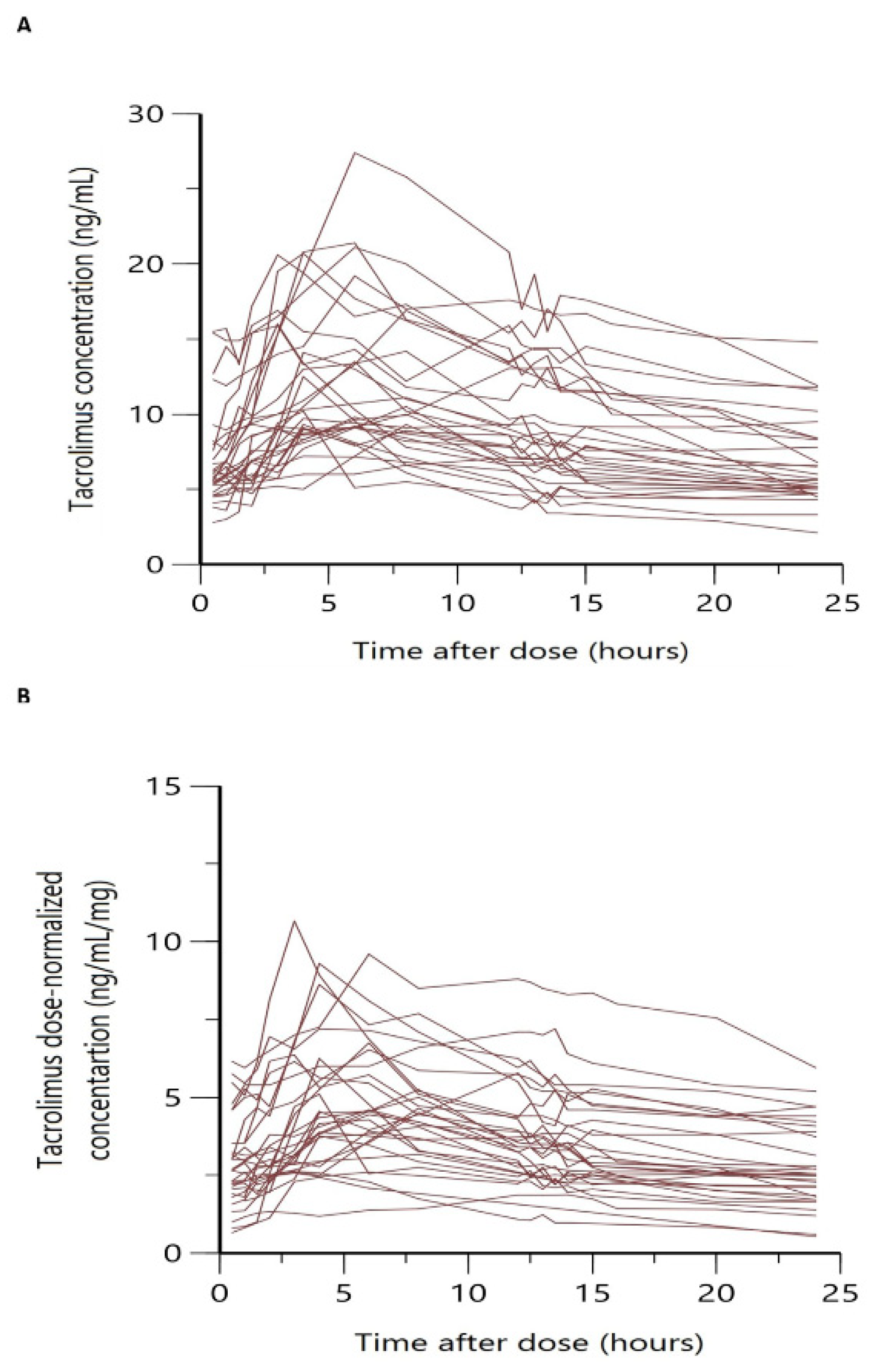
3.1. Patient Characteristics and Datasets
3.2. Population PK Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bowman, L.J.; Brennan, D.C. The role of tacrolimus in renal transplantation. Expert Opin. Pharmacother. 2008, 9, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Kamel, M.; Kadian, M.; Srinivas, T.; Taber, D.; Salas, M.A.P. Tacrolimus confers lower acute rejection rates and better renal allograft survival compared to cyclosporine. World J. Transplant. 2016, 6, 697. [Google Scholar] [CrossRef] [PubMed]
- Venkataramanan, R.; Swaminathan, A.; Prasad, T.; Jain, A.; Zuckerman, S.; Warty, V.; McMichael, J.; Lever, J.; Burckart, G.; Starzl, T. Clinical Pharmacokinetics of Tacrolimus. Clin. Pharmacokinet. 1995, 29, 404–430. [Google Scholar] [CrossRef]
- Staatz, C.E.; Tett, S.E. Clinical pharmacokinetics and pharmacodynamics of tacrolimus in solid organ transplantation. Clin. Pharmacokinet. 2004, 43, 623–653. [Google Scholar] [CrossRef] [PubMed]
- Kuypers, D.R.J. Intrapatient Variability of Tacrolimus Exposure in Solid Organ Transplantation: A Novel Marker for Clinical Outcome. Clin. Pharmacol. Ther. 2020, 107, 347–358. [Google Scholar] [CrossRef]
- Neylan, J.F. Effect of race and immunosuppression in renal transplantation: Three- year survival results from a US multicenter, randomized trial. Transplant. Proc. 1998, 30, 1355–1358. [Google Scholar] [CrossRef]
- Staatz, C.E.; Goodman, L.K.; Tett, S.E. Effect of CYP3A and ABCB1 single nucleotide polymorphisms on the pharmacokinetics and pharmacodynamics of calcineurin inhibitors: Part I. Clin. Pharmacokinet. 2010, 49, 141–175. [Google Scholar] [CrossRef]
- Bekersky, I.; Dressler, D.; Mekki, Q.A. Effect of low- and high-fat meals on tacrolimus absorption following 5 mg single oral doses to healthy human subjects. J. Clin. Pharmacol. 2001, 41, 176–182. [Google Scholar] [CrossRef]
- Tuteja, S.; Alloway, R.R.; Johnson, J.A.; Gaber, A.O. The effect of gut metabolism on tacrolimus bioavailability in renal transplant recipients. Transplantation 2001, 71, 1303–1307. [Google Scholar] [CrossRef]
- Han, S.S.; Kim, D.H.; Lee, S.M.; Han, N.Y.; Oh, J.M.; Ha, J.; Kim, Y.S. Pharmacokinetics of tacrolimus according to body composition in recipients of kidney transplants. Kidney Res. Clin. Pract. 2012, 31, 157. [Google Scholar] [CrossRef]
- Seegars, M.B.; Tooze, J.; Isom, S.; Anders, B.; Kennedy, L.; Rodriguez, C. Tacrolimus Levels and Correlation to Age and Weight Calculation. Biol. Blood Marrow Transplant. 2018, 24, S337. [Google Scholar] [CrossRef]
- Katari, S.R.; Magnone, M.; Shapiro, R.; Jordan, M.; Scantlebury, V.; Vivas, C.; Gritsch, A.; Mccauley, J.; Starzl, T.; Demetris, A.J.; et al. Clinical features of acute reversible tacrolimus (FK 506) nephrotoxicity in kidney transplant recipients. Clin. Transplant. 1997, 11, 237. [Google Scholar] [PubMed]
- Weng, L.C.; Chiang, Y.J.; Lin, M.H.; Hsieh, C.Y.; Lin, S.C.; Wei, T.Y.; Chou, H.F. Association Between Use of FK506 and Prevalence of Post-transplantation Diabetes Mellitus in Kidney Transplant Patients. Transplant. Proc. 2014, 46, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Sierra-Hidalgo, F.; Martínez-Salio, A.; Moreno-García, S.; De Pablo-Fernández, E.; Correas-Callero, E.; Ruiz-Morales, J. Akinetic mutism induced by tacrolimus. Clin. Neuropharmacol. 2009, 32, 293–294. [Google Scholar] [CrossRef] [PubMed]
- Wallemacq, P.E.; Furlan, V.; Möller, A.; Schäfer, A.; Stadler, P.; Firdaous, I.; Taburet, A.M.; Reding, R.; Clement De Clety, S.; De Ville De Goyet, J.; et al. Pharmacokinetics of tacrolimus (FK506) in paediatric liver transplant recipients. Eur. J. Drug Metab. Pharmacokinet. 1998, 23, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, S.; Nigro, V.; Weinberg, J.; Woodle, E.S.; Alloway, R.R. A Steady-State Head-to-Head Pharmacokinetic Comparison of All FK-506 (Tacrolimus) Formulations (ASTCOFF): An Open-Label, Prospective, Randomized, Two-Arm, Three-Period Crossover Study. Am. J. Transplant. 2017, 17, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Gaber, A.O.; Alloway, R.R.; Bodziak, K.; Kaplan, B.; Bunnapradist, S. Conversion from twice-daily tacrolimus capsules to once-daily extended-release tacrolimus (LCPT): A phase 2 trial of stable renal transplant recipients. Transplantation 2013, 96, 191–197. [Google Scholar] [CrossRef]
- Trofe-Clark, J.; Brennan, D.C.; West-Thielke, P.; Milone, M.C.; Lim, M.A.; Neubauer, R.; Nigro, V.; Bloom, R.D. Results of ASERTAA, a Randomized Prospective Crossover Pharmacogenetic Study of Immediate-Release Versus Extended-Release Tacrolimus in African American Kidney Transplant Recipients. Am. J. Kidney Dis. 2018, 71, 315–326. [Google Scholar] [CrossRef]
- Staatz, C.E.; Tett, S.E. Clinical Pharmacokinetics of Once-Daily Tacrolimus in Solid-Organ Transplant Patients. Clin. Pharmacokinet. 2015, 54, 993–1025. [Google Scholar] [CrossRef]
- Budde, K.; Bunnapradist, S.; Grinyo, J.M.; Ciechanowski, K.; Denny, J.E.; Silva, H.T.; Rostaing, L. Novel Once-Daily Extended-Release Tacrolimus (LCPT) Versus Twice-Daily Tacrolimus in De Novo Kidney Transplants: One-Year Results of Phase III, Double-Blind, Randomized Trial. Am. J. Transplant. 2014, 14, 2796–2806. [Google Scholar] [CrossRef]
- Thörn, M.; Finnström, N.; Lundgren, S.; Rane, A.; Lööf, L. Cytochromes P450 and MDR1 mRNA expression along the human gastrointestinal tract. Br. J. Clin. Pharmacol. 2005, 60, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Birdwell, K.A.; Decker, B.; Barbarino, J.M.; Peterson, J.F.; Stein, C.M.; Sadee, W.; Wang, D.; Vinks, A.A.; He, Y.; Swen, J.J.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines for CYP3A5 Genotype and Tacrolimus Dosing. Clin. Pharmacol. Ther. 2015, 98, 19–24. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, Z.H.; Zheng, J.M.; Chen, Z.H.; Tang, Z.; Chen, J.S.; Li, L.S. Influence of CYP3A5 and MDR1 polymorphisms on tacrolimus concentration in the early stage after renal transplantation. Clin. Transplant. 2005, 19, 638–643. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, H.; De Loor, H.; Verbeke, K.; Vanrenterghem, Y.; Kuypers, D.R. In vivo CYP3A4 activity, CYP3A5 genotype, and hematocrit predict tacrolimus dose requirements and clearance in renal transplant patients. Clin. Pharmacol. Ther. 2012, 92, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Capron, A.; Mourad, M.; De Meyer, M.; De Pauw, L.; Eddour, D.C.; Latinne, D.; Elens, L.; Haufroid, V.; Wallemacq, P. CYP3A5 and ABCB1 polymorphisms influence tacrolimus concentrations in peripheral blood mononuclear cells after renal transplantation. Pharmacogenomics 2010, 11, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Pallet, N.; Jannot, A.S.; El Bahri, M.; Etienne, I.; Buchler, M.; De Ligny, B.H.; Choukroun, G.; Colosio, C.; Thierry, A.; Vigneau, C.; et al. Kidney Transplant Recipients Carrying the CYP3A4*22 Allelic Variant Have Reduced Tacrolimus Clearance and Often Reach Supratherapeutic Tacrolimus Concentrations. Am. J. Transplant. 2015, 15, 800–805. [Google Scholar] [CrossRef]
- Abdel-Kahaar, E.; Winter, S.; Tremmel, R.; Schaeffeler, E.; Olbricht, C.J.; Wieland, E.; Schwab, M.; Shipkova, M.; Jaeger, S.U. The impact of CYP3A4*22 on tacrolimus pharmacokinetics and outcome in clinical practice at a single kidney transplant center. Front. Genet. 2019, 10, 871. [Google Scholar] [CrossRef]
- Tang, J.T.; Andrews, L.M.; Van Gelder, T.; Shi, Y.Y.; Van Schaik, R.H.N.; Wang, L.L.; Hesselink, D.A. Pharmacogenetic aspects of the use of tacrolimus in renal transplantation: Recent developments and ethnic considerations. Expert Opin. Drug Metab. Toxicol. 2016, 12, 555–565. [Google Scholar] [CrossRef]
- Brooks, E.; Tett, S.E.; Isbel, N.M.; Staatz, C.E. Population Pharmacokinetic Modelling and Bayesian Estimation of Tacrolimus Exposure: Is this Clinically Useful for Dosage Prediction Yet? Clin. Pharmacokinet. 2016, 55, 1295–1335. [Google Scholar] [CrossRef]
- Woillard, J.B.; Mourad, M.; Neely, M.; Capron, A.; van Schaik, R.H.; van Gelder, T.; Lloberas, N.; Hesselink, D.A.; Marquet, P.; Haufroid, V.; et al. Tacrolimus updated guidelines through popPK modeling: How to benefit more from CYP3A pre-emptive genotyping prior to kidney transplantation. Front. Pharmacol. 2017, 8, 358. [Google Scholar] [CrossRef]
- Andrews, L.M.; Hesselink, D.A.; van Schaik, R.H.N.; van Gelder, T.; de Fijter, J.W.; Lloberas, N.; Elens, L.; Moes, D.J.A.R.; de Winter, B.C.M. A population pharmacokinetic model to predict the individual starting dose of tacrolimus in adult renal transplant recipients. Br. J. Clin. Pharmacol. 2019, 85, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Andreu, F.; Colom, H.; Elens, L.; van Gelder, T.; van Schaik, R.H.N.; Hesselink, D.A.; Bestard, O.; Torras, J.; Cruzado, J.M.; Grinyó, J.M.; et al. A New CYP3A5*3 and CYP3A4*22 Cluster Influencing Tacrolimus Target Concentrations: A Population Approach. Clin. Pharmacokinet. 2017, 56, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Andreu, F.; Colom, H.; Grinyó, J.M.; Torras, J.; Cruzado, J.M.; Lloberas, N. Development of a population PK model of tacrolimus for adaptive dosage control in stable kidney transplant patients. Ther. Drug Monit. 2015, 37, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Woillard, J.B.; Saint-Marcoux, F.; Debord, J.; Åsberg, A. Pharmacokinetic models to assist the prescriber in choosing the best tacrolimus dose. Pharmacol. Res. 2018, 130, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Henin, E.; Govoni, M.; Cella, M.; Laveille, C.; Piotti, G. Therapeutic Drug Monitoring Strategies for Envarsus in De Novo Kidney Transplant Patients Using Population Modelling and Simulations. Adv. Ther. 2021, 38, 5317–5332. [Google Scholar] [CrossRef] [PubMed]
- Martial, L.C.; Biewenga, M.; Ruijter, B.N.; Keizer, R.; Swen, J.J.; van Hoek, B.; Moes, D.J.A.R. Population pharmacokinetics and genetics of oral meltdose tacrolimus (Envarsus) in stable adult liver transplant recipients. Br. J. Clin. Pharmacol. 2021, 87, 4262–4272. [Google Scholar] [CrossRef]
- Woillard, J.B.; Debord, J.; Monchaud, C.; Saint-Marcoux, F.; Marquet, P. Population Pharmacokinetics and Bayesian Estimators for Refined Dose Adjustment of a New Tacrolimus Formulation in Kidney and Liver Transplant Patients. Clin. Pharmacokinet. 2017, 56, 1491–1498. [Google Scholar] [CrossRef]
- Rigo-Bonnin, R.; Arbiol-Roca, A.; de Aledo-Castillo, J.M.G.; Alía, P. Simultaneous Measurement of Cyclosporine A, Everolimus, Sirolimus and Tacrolimus Concentrations in Human Blood by UPLC–MS/MS. Chromatographia 2015, 78, 1459–1474. [Google Scholar] [CrossRef]
- Savic, R.M.; Jonker, D.M.; Kerbusch, T.; Karlsson, M.O. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J. Pharmacokinet. Pharmacodyn. 2007, 34, 711–726. [Google Scholar] [CrossRef]
- Karlsson, M.O.; Sheiner, L.B. The importance of modeling interoccasion variability in population pharmacokinetic analyses. J. Pharmacokinet. Biopharm. 1993, 21, 735–750. [Google Scholar] [CrossRef]
- Yamaoka, K.; Nakagawa, T.; Uno, T. Application of Akaike’s information criterion (AIC) in the evaluation of linear pharmacokinetic equations. J. Pharmacokinet. Biopharm. 1978, 6, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Savic, R.M.; Karlsson, M.O. Importance of Shrinkage in Empirical Bayes Estimates for Diagnostics: Problems and Solutions. AAPS J. 2009, 11, 558. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, E.N.; Karlsson, M.O. Automated covariate model building within NONMEM. Pharm. Res. 1998, 15, 1463–1468. [Google Scholar] [CrossRef] [PubMed]
- Bergstrand, M.; Hooker, A.C.; Wallin, J.E.; Karlsson, M.O. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011, 13, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Comets, E.; Brendel, K.; Mentré, F. Computing normalised prediction distribution errors to evaluate nonlinear mixed-effect models: The npde add-on package for R. Comput. Methods Programs Biomed. 2008, 90, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Nigro, V.; Glicklich, A.; Weinberg, J. Improved Bioavailability of MELTDOSE Once-Daily Formulation of Tacrolimus (LCP-Tacro) with Controlled Agglomeration Allows for Consistent Absorption over 24 Hrs: A Scintigraphic and Pharmacokinetic Evaluation. Am. J. Transplant. 2013, 13 (Suppl. S5), B1034. [Google Scholar]
- Kamdem, L.K.; Streit, F.; Zanger, U.M.; Brockmöller, J.; Oellerich, M.; Armstrong, V.W.; Wojnowski, L. Contribution of CYP3A5 to the in vitro hepatic clearance of tacrolimus. Clin. Chem. 2005, 51, 1374–1381. [Google Scholar] [CrossRef]
- Undre, N.A. Pharmacokinetics of tacrolimus-based combination therapies. Nephrol. Dial. Transplant. 2003, 18 (Suppl. S1), i12–i15. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | All Patients | Full PK Profile | Sparse Sampling (C0) |
|---|---|---|---|
| No. of Patients | 98 | 30 | 68 |
| No. of Sampling | 655 | 480 | 175 |
| Gender (Male/Female) | 68/30 | 20/10 | 45/23 |
| Weight (kg) | 74.73 (65–81.13) | 72.82 (63.75–80) | 75.36 (65.5–88) |
| Age (Years) | 56 (46–68) | 57 (48–67) | 56 (45–68) |
| BMI (kg·m−2) | 26.33 (22.94–28.94) | 26.44 (23.44–29.70) | 26.28 (22.89–28.64) |
| HTC (%) | 40.53 (37.4–44.0) | 39.7 (37.1–41.9) | 40.64 (37.42–44.0) |
| GF (mL·min−1) | 47.72 (36–58) | 54.86 (45.5–67) | 46.7 (36–57) |
| CR (μmol·L−1) | 146.9 (116–163) | 130.47 (106.5–149) | 149 (118–164) |
| CYP3A5 Genotype | |||
| *1/*1 | 4 (4.1%) | 1 (3.3%) | 3 (4.5%) |
| *1/*3 | 17 (17.3%) | 9 (30%) | 8 (11.5%) |
| *3/*3 | 77 (78.6%) | 20 (66.7%) | 57 (84%) |
| CYP3A4 Genotype | |||
| *1/*1 | 86 (86.7%) | 28 (93.3%) | 58 (84%) |
| *1/*22 | 12 (13.3%) | 2 (6.7%) | 10(16%) |
| Cluster | |||
| HM | 19 (19.4%) | 10 (33%) | 9 (13.25%) |
| IM | 68 (69.4%) | 18 (60%) | 50 (73.5%) |
| PM | 11 (11.2%) | 2 (7%) | 9 (13.25%) |
| ABCB1 Genotype | |||
| *T/*T | 21 (21%) | 9 (30%) | 12 (18%) |
| *C/*T | 46 (47%) | 12 (40%) | 34 (50%) |
| *C/*C | 31 (32%) | 9 (30%) | 22 (32%) |
| Genotype Group | Dose (mg·day−1) | C0 | N | AUC | N | C0/D | p-Value * | AUC/D | p-Value z |
|---|---|---|---|---|---|---|---|---|---|
| (ng·mL−1) | (ng·h·mL−1) | ||||||||
| CYP3A5 | |||||||||
| CYP3A5 *1/*1, *1/*3 | 5 (2–12) | 6.14 (5.47–6.90) | 46 | 252 (180–353) | 10 | 1.22 (1.07–1.42) | <0.001 | 51 (41–64) | <0.001 |
| CYP3A5 *3/*3 | 2 (0.5–8) | 6.4 (6.06–6.75) | 187 | 191 (165–223) | 20 | 3.08 (2.94–3.44) | 95 (81–110) | ||
| CYP3A4 | |||||||||
| CYP3A4 *1/*1 | 2.5 (0.75–12) | 6.19 (5.86–6.54) | 180 | 207 (178–241) | 28 | 2.42 (2.23–2.67) | <0.05 | 76 (64–91) | 0.2410 |
| CYP3A4 *1/*22 | 1.75 (0.5–8) | 7.05 (5.71–7.20) | 53 | 198 (99–398) | 2 | 3.01 (2.80–4.15) | 122 (76–194) | ||
| CLUSTER | |||||||||
| High-Metabolizer | 3 (2–12) | 6.16 (5.47–6.93) | 46 | 252 (180–353) | 10 | 1.23 (1.06–1.42) | <0.001 # | 51 (41–64) | <0.001 # |
| <0.001 & | <0.05 & | ||||||||
| Intermediate-Metabolizer | 2 (0.75–8) | 6.16 (5.80–6.54) | 141 | 190 (161–224) | 18 | 2.91 (2.67–3.18) | <0.05 $ | 92 (80–106) | 0.5756 $ |
| Poor-Metabolizer | 1.75 (0.5–8) | 7.3 (6.60–8.10) | 46 | 198 (99–398) | 2 | 4.04 (0.48–4.1) | 122 (76–194) | ||
| ABCB1 | |||||||||
| ABCB1 *C/*T, *C/*C | 2 (0.5–12) | 6.25 (5.94–6.63) | 181 | 203 (170–242) | 21 | 2.58 (2.33–2.85) | 0.3073 | 78 (64–95) | 0.4537 |
| ABCB1 *T/*T | 2.125 (1–8) | 6.4 (5.79–7.30) | 52 | 213 (163–279) | 9 | 2.81 (2.48–3.19) | 83 (62–112) |
| Parameter | Value (RSE %) | IIV % (RSE %) |
|---|---|---|
| CL/FHM (L·h−1) | 19.6 (10) | 37.9 (17.9) |
| CL/FIM (L·h−1) | 10.6 (5.2) | - |
| CL/FPM (L·h−1) | 7.37 (11.9) | - |
| Vc/F (L) | 169 (17.2) | 70 (41.4) |
| CLD/F (L·h−1) | 37.6 (13.5) | - |
| Ka (h−1) | 0.72 (33.2) | - |
| Vp (L) | 460 (27.8) | 75 (44.3) |
| MTT (h) | 2.91 (15.5) | 54.6 (37.1) |
| NN | 2 FIX | - |
| IOV (%) | 44.8 (27.4) | - |
| RE (%) | 9.67 (8) | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammed Ali, Z.; Meertens, M.; Fernández, B.; Fontova, P.; Vidal-Alabró, A.; Rigo-Bonnin, R.; Melilli, E.; Cruzado, J.M.; Grinyó, J.M.; Colom, H.; et al. CYP3A5*3 and CYP3A4*22 Cluster Polymorphism Effects on LCP-Tac Tacrolimus Exposure: Population Pharmacokinetic Approach. Pharmaceutics 2023, 15, 2699. https://doi.org/10.3390/pharmaceutics15122699
Mohammed Ali Z, Meertens M, Fernández B, Fontova P, Vidal-Alabró A, Rigo-Bonnin R, Melilli E, Cruzado JM, Grinyó JM, Colom H, et al. CYP3A5*3 and CYP3A4*22 Cluster Polymorphism Effects on LCP-Tac Tacrolimus Exposure: Population Pharmacokinetic Approach. Pharmaceutics. 2023; 15(12):2699. https://doi.org/10.3390/pharmaceutics15122699
Chicago/Turabian StyleMohammed Ali, Zeyar, Marinda Meertens, Beatriz Fernández, Pere Fontova, Anna Vidal-Alabró, Raul Rigo-Bonnin, Edoardo Melilli, Josep M. Cruzado, Josep M. Grinyó, Helena Colom, and et al. 2023. "CYP3A5*3 and CYP3A4*22 Cluster Polymorphism Effects on LCP-Tac Tacrolimus Exposure: Population Pharmacokinetic Approach" Pharmaceutics 15, no. 12: 2699. https://doi.org/10.3390/pharmaceutics15122699