
Physiologically Based Pharmacokinetic Modelling of Cabotegravir Microarray Patches in Rats and Humans

 ,
,  ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human and Rat Physiological Parameters
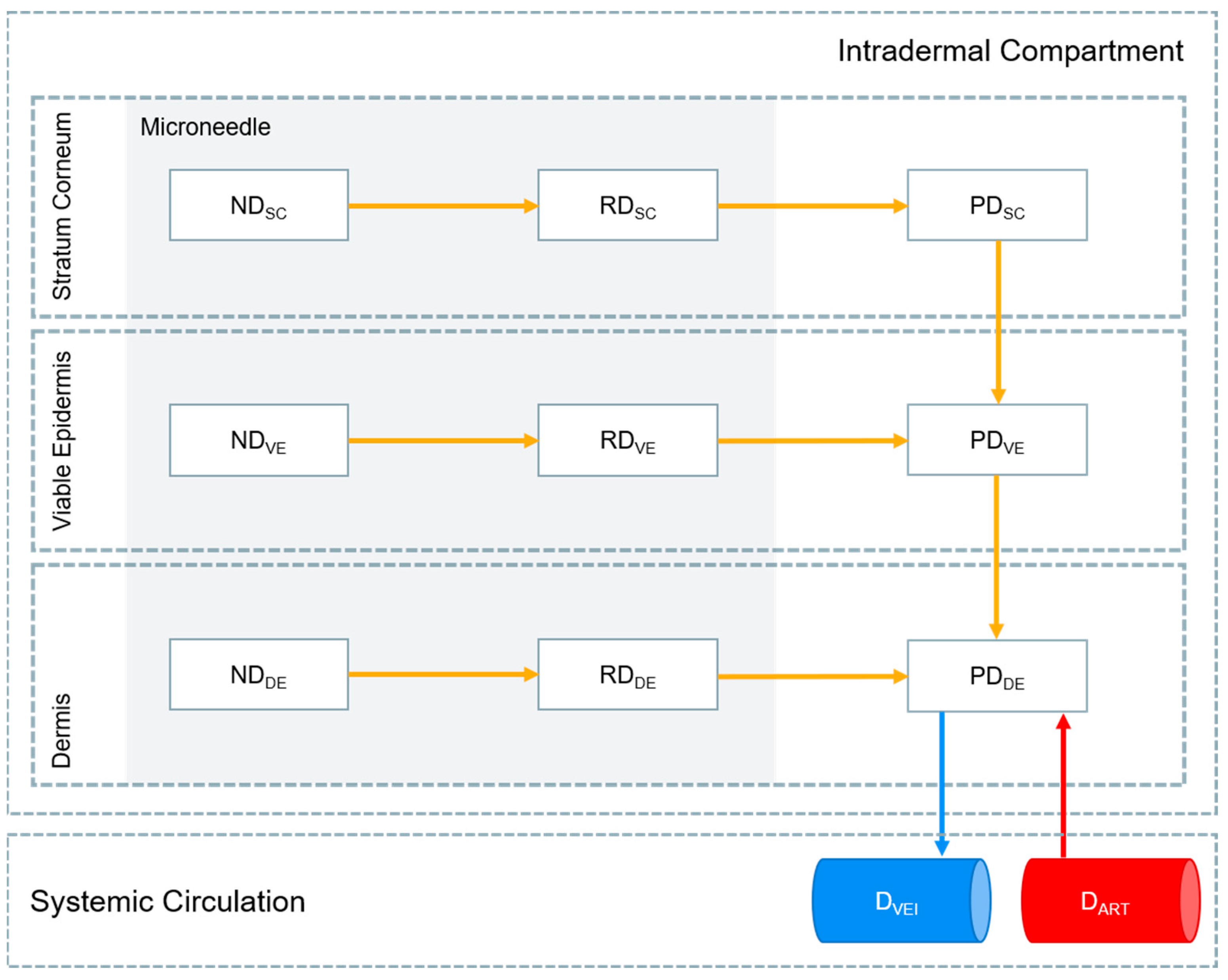
2.2. MAP Intradermal PBPK Model
2.3. PBPK Model Verification
2.4. Prediction of Cabotegravir MAP Pharmacokinetics in Humans
3. Results
3.1. PBPK Model Verification
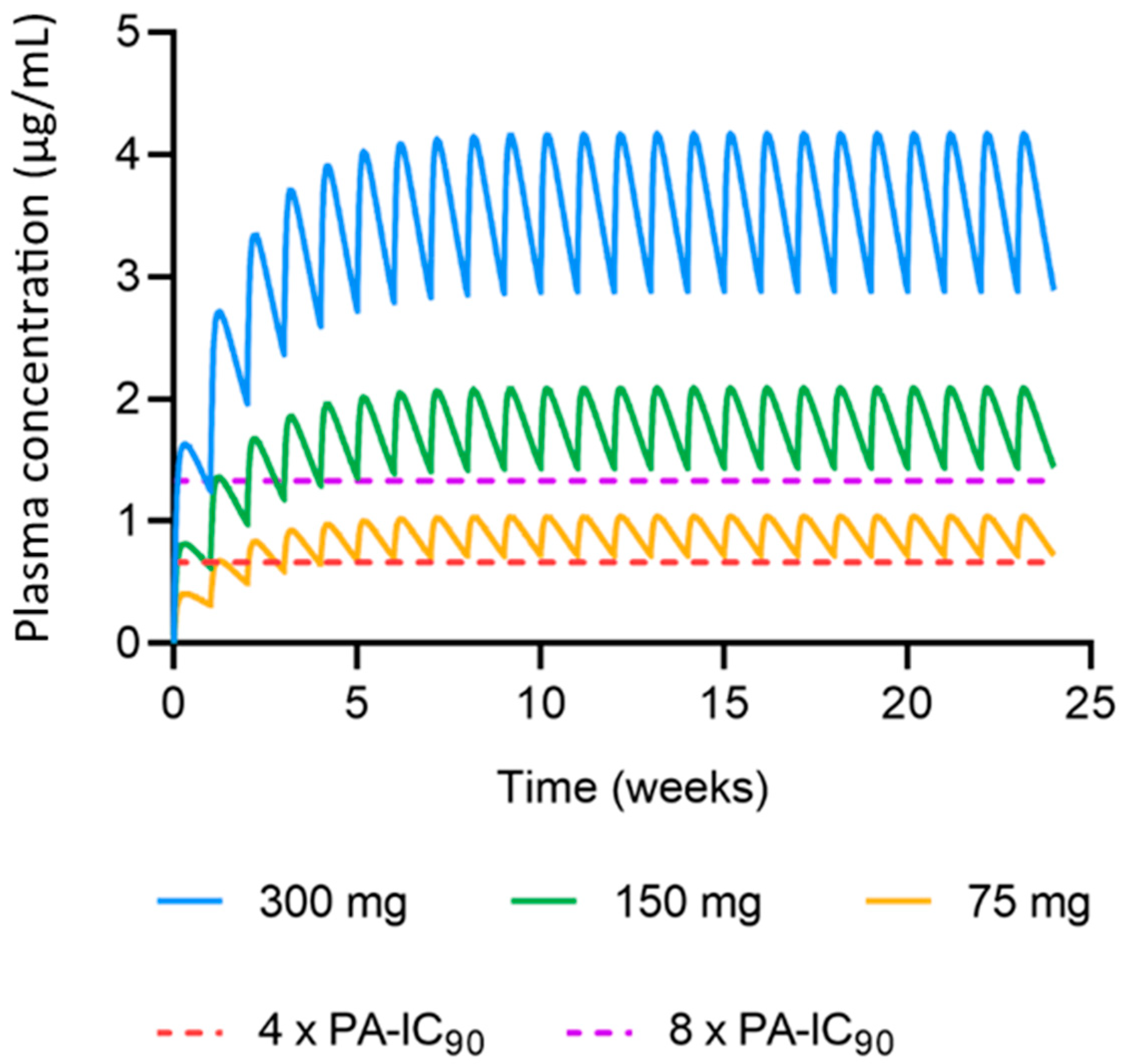
3.2. Predicted Cabotegravir MAP Pharmacokinetics in Humans
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, W.; Li, S.; Fan, X.; Prausnitz, M.R. Microneedle patch designs to increase dose administered to human subjects. J. Control. Release 2021, 339, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Jin, S.G. Microneedle for transdermal drug delivery: Current trends and fabrication. J. Pharm. Investig. 2021, 51, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Dugam, S.; Tade, R.; Dhole, R.; Nangare, S. Emerging era of microneedle array for pharmaceutical and biomedical applications: Recent advances and toxicological perspectives. Future J. Pharm. Sci. 2021, 7, 19. [Google Scholar] [CrossRef]
- Vora, L.K.; Moffatt, K.; Tekko, I.A.; Paredes, A.J.; Volpe-Zanutto, F.; Mishra, D.; Peng, K.; Raj Singh Thakur, R.; Donnelly, R.F. Microneedle array systems for long-acting drug delivery. Eur. J. Pharm. Biopharm. 2021, 159, 44–76. [Google Scholar] [CrossRef] [PubMed]
- Paredes, A.J.; Ramöller, I.K.; McKenna, P.E.; Abbate, M.T.A.; Volpe-Zanutto, F.; Vora, L.K.; Kilbourne-Brook, M.; Jarrahian, C.; Moffatt, K.; Zhang, C.; et al. Microarray patches: Breaking down the barriers to contraceptive care and HIV prevention for women across the globe. Adv. Drug Deliv. Rev. 2021, 173, 331–348. [Google Scholar] [CrossRef] [PubMed]
- Scarsi, K.K.; Swindells, S. The Promise of Improved Adherence with Long-Acting Antiretroviral Therapy: What Are the Data? J. Int. Assoc. Provid. AIDS Care 2021, 20, 23259582211009011. [Google Scholar] [CrossRef] [PubMed]
- UNAIDS. Global HIV & AIDS Statistics—2020 Fact Sheet. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 17 February 2021).
- NIH. FDA-Approved HIV Medicines. Available online: https://hivinfo.nih.gov/understanding-hiv/fact-sheets/fda-approved-hiv-medicines (accessed on 18 February 2021).
- WHO. Update of Recommendations on First—And Second-Line Antiretroviral Regimens; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Menéndez-Arias, L.; Martín-Alonso, S.; Frutos-Beltrán, E. An Update on Antiretroviral Therapy. In Antiviral Drug Discovery and Development; Liu, X., Zhan, P., Menéndez-Arias, L., Poongavanam, V., Eds.; Springer: Singapore, 2021. [Google Scholar]
- Maartens, G.; Celum, C.; Lewin, S.R. HIV infection: Epidemiology, pathogenesis, treatment, and prevention. Lancet 2014, 384, 258–271. [Google Scholar] [CrossRef]
- CDC. Pre-Exposure Prophylaxis (PrEP) and Post-Exposure Prophylaxis (PEP). Available online: https://www.cdc.gov/hiv/risk/prep/index.html (accessed on 18 February 2021).
- Cobb, D.A.; Smith, N.A.; Edagwa, B.J.; McMillan, J.M. Long-acting approaches for delivery of antiretroviral drugs for prevention and treatment of HIV: A review of recent research. Expert Opin. Drug Deliv. 2020, 17, 1227–1238. [Google Scholar] [CrossRef]
- FDA. FDA Approves First Extended-Release, Injectable Drug Regimen for Adults Living with HIV. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-extended-release-injectable-drug-regimen-adults-living-hiv (accessed on 19 February 2021).
- Overton, E.T.; Richmond, G.; Rizzardini, G.; Jaeger, H.; Orrell, C.; Nagimova, F.; Bredeek, F.; Garcia Deltoro, M.; Swindells, S.; Andrade-Villanueva, J.F.; et al. Long-acting cabotegravir and rilpivirine dosed every 2 months in adults with HIV-1 infection (ATLAS-2M), 48-week results: A randomised, multicentre, open-label, phase 3b, non-inferiority study. Lancet 2021, 396, 1994–2005. [Google Scholar] [CrossRef]
- Jaeger, H.; Overton, E.T.; Richmond, G.; Rizzardini, G.; Andrade-Villanueva, J.F.; Mngqibisa, R.; Hermida, A.O.; Thalme, A.; Belonosova, E.; Ajana, F.; et al. Long-acting cabotegravir and rilpivirine dosed every 2 months in adults with HIV-1 infection (ATLAS-2M), 96-week results: A randomised, multicentre, open-label, phase 3b, non-inferiority study. Lancet HIV 2021, 8, e679–e689. [Google Scholar] [CrossRef]
- Pau, A.K.; George, J.M. Antiretroviral therapy: Current drugs. Infect. Dis. Clin. N. Am. 2014, 28, 371–402. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gu, S.-X.; He, Q.; Fan, R. Advances in the development of HIV integrase strand transfer inhibitors. Eur. J. Med. Chem. 2021, 225, 113787. [Google Scholar] [CrossRef]
- Hodge, D.; Back, D.J.; Gibbons, S.; Khoo, S.H.; Marzolini, C. Pharmacokinetics and Drug-Drug Interactions of Long-Acting Intramuscular Cabotegravir and Rilpivirine. Clin. Pharmacokinet. 2021, 60, 835–853. [Google Scholar] [CrossRef] [PubMed]
- Landovitz, R.J.; Donnell, D.; Clement, M.E.; Hanscom, B.; Cottle, L.; Coelho, L.; Cabello, R.; Chariyalertsak, S.; Dunne, E.F.; Frank, I.; et al. Cabotegravir for HIV Prevention in Cisgender Men and Transgender Women. N. Engl. J. Med. 2021, 385, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Engelman, K.D.; Engelman, A.N. Long-Acting Cabotegravir for HIV/AIDS Prophylaxis. Biochemistry 2021, 60, 1731–1740. [Google Scholar] [CrossRef]
- Lazarus, G.; Wangsaputra, V.K.; Christianto; Louisa, M.; Soetikno, V.; Hamers, R.L. Safety and Pharmacokinetic Profiles of Long-Acting Injectable Antiretroviral Drugs for HIV-1 Pre-Exposure Prophylaxis: A Systematic Review and Meta-analysis of Randomized Trials. Front. Pharmacol. 2021, 12, 664875. [Google Scholar] [CrossRef]
- Bares, S.H.; Scarsi, K.K. A new paradigm for antiretroviral delivery: Long-acting cabotegravir and rilpivirine for the treatment and prevention of HIV. Curr. Opin. HIV AIDS 2022, 17, 22–31. [Google Scholar] [CrossRef]
- Cambou, M.C.; Landovitz, R.J. Challenges and Opportunities for Preexposure Prophylaxis. Top. Antivir. Med. 2021, 29, 399–406. [Google Scholar]
- Tekko, I.A.; Vora, L.K.; Volpe-Zanutto, F.; Moffatt, K.; Jarrahian, C.; McCarthy, H.O.; Donnelly, R.F. Novel Bilayer Microarray Patch-Assisted Long-Acting Micro-Depot Cabotegravir Intradermal Delivery for HIV Pre-Exposure Prophylaxis. Adv. Funct. Mater. 2022, 32, 2106999. [Google Scholar] [CrossRef]
- Rajoli, R.K.R.; Flexner, C.; Chiong, J.; Owen, A.; Donnelly, R.F.; Larraneta, E.; Siccardi, M. Modelling the intradermal delivery of microneedle array patches for long-acting antiretrovirals using PBPK. Eur. J. Pharm. Biopharm. 2019, 144, 101–109. [Google Scholar] [CrossRef]
- National Center for Health Statistics. Anthropometric Reference Data for Children and Adults: United States, 2011–2014; National Center for Health Statistics: Washington, DC, USA, 2016.
- Bosgra, S.; van Eijkeren, J.; Bos, P.; Zeilmaker, M.; Slob, W. An improved model to predict physiologically based model parameters and their inter-individual variability from anthropometry. Crit. Rev. Toxicol. 2012, 42, 751–767. [Google Scholar] [CrossRef] [PubMed]
- Shuter, B.; Aslani, A. Body surface area: Du bois and du bois revisited. Eur. J. Appl. Physiol. 2000, 82, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.P.; Delp, M.D.; Lindstedt, S.L.; Rhomberg, L.R.; Beliles, R.P. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol. Ind. Health 1997, 13, 407–484. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.A. Physiologically-Based Pharmacokinetic (PBPK) Modeling and Simulations: Principles, Methods, and Applications in the Pharmaceutical Industry, 1st ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Poulin, P.; Theil, F.P. Prediction of pharmacokinetics prior to in vivo studies. 1. mechanism-based prediction of volume of distribution. J. Pharm. Sci. 2002, 91, 129–156. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.A. Evaluation of a generic physiologically based pharmacokinetic model for lineshape analysis. Clin. Pharmacokinet. 2008, 47, 261–275. [Google Scholar] [CrossRef]
- Gajewska, M.; Worth, A.; Urani, C.; Briesen, H.; Schramm, K.W. Application of physiologically-based toxicokinetic modelling in oral-to-dermal extrapolation of threshold doses of cosmetic ingredients. Toxicol. Lett. 2014, 227, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Borchers, H.W. pracma: Practical Numerical Math Functions. R Package. Version 2.2.9. Available online: https://cran.r-project.org/web/packages/pracma/index.html (accessed on 17 February 2021).
- DrugBank. Cabotegravir. Available online: https://go.drugbank.com/drugs/DB11751 (accessed on 20 December 2021).
- ViiV-Healthcare. Product Monograph Including Patient Medication Information: Vocabria & Cabenuva; ViiV-Healthcare: Brentford, UK, 2020. [Google Scholar]
- Markowitz, M.; Frank, I.; Grant, R.M.; Mayer, K.H.; Elion, R.; Goldstein, D.; Fisher, C.; Sobieszczyk, M.E.; Gallant, J.E.; Van Tieu, H.; et al. Safety and tolerability of long-acting cabotegravir injections in HIV-uninfected men (ECLAIR): A multicentre, double-blind, randomised, placebo-controlled, phase 2a trial. Lancet HIV 2017, 4, e331–e340. [Google Scholar] [CrossRef]
- Rajoli, R.K.R.; Back, D.J.; Rannard, S.; Meyers, C.F.; Flexner, C.; Owen, A.; Siccardi, M. In Silico Dose Prediction for Long-Acting Rilpivirine and Cabotegravir Administration to Children and Adolescents. Clin. Pharmacokinet. 2018, 57, 255–266. [Google Scholar] [CrossRef]
- Kattou, P.; Lian, G.; Glavin, S.; Sorrell, I.; Chen, T. Development of a Two-Dimensional Model for Predicting Transdermal Permeation with the Follicular Pathway: Demonstration with a Caffeine Study. Pharm. Res. 2017, 34, 2036–2048. [Google Scholar] [CrossRef]
- Wei, J.C.J.; Edwards, G.A.; Martin, D.J.; Huang, H.; Crichton, M.L.; Kendall, M.A.F. Allometric scaling of skin thickness, elasticity, viscoelasticity to mass for micro-medical device translation: From mice, rats, rabbits, pigs to humans. Sci. Rep. 2017, 7, 15885. [Google Scholar] [CrossRef]
- Spreen, W.; Ford, S.L.; Chen, S.; Wilfret, D.; Margolis, D.; Gould, E.; Piscitelli, S. GSK1265744 pharmacokinetics in plasma and tissue after single-dose long-acting injectable administration in healthy subjects. J. Acquir. Immune Defic. Syndr. 2014, 67, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Shebley, M.; Sandhu, P.; Emami Riedmaier, A.; Jamei, M.; Narayanan, R.; Patel, A.; Peters, S.A.; Reddy, V.P.; Zheng, M.; de Zwart, L.; et al. Physiologically based pharmacokinetic model qualification and reporting procedures for regulatory submissions: A consortium perspective. Clin. Pharmacol. Ther. 2018, 104, 88–110. [Google Scholar] [CrossRef] [PubMed]
- Riley, R.J.; McGinnity, D.F.; Austin, R.P. A unified model for predicting human hepatic, metabolic clearance from in vitro intrinsic clearance data in hepatocytes and microsomes. Drug Metab. Dispos. 2005, 33, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Wannamaker, P.; Lu, H.; Zhu, B.; Wang, M.; Paff, M.; Spreen, W.R.; Ford, S.L. Safety, Tolerability, Pharmacokinetics, and Acceptability of Oral and Long-Acting Cabotegravir in HIV-Negative Chinese Men. Antimicrob. Agents Chemother. 2022, 66, e0205721. [Google Scholar] [CrossRef] [PubMed]
- Kinvig, H. Integrated Experimental and Computational Approaches for the Prediction of Drug-Drug Interactions. Ph.D. Thesis, University of Liverpool, Liverpool, UK, 2021. [Google Scholar]
- Exposure Assessment Group. Dermal Exposure Assessment: Principles and Applications; US Environmental Protection Agency, Office of Health and Environmental Assessment: Washington, DC, USA, 1992.
- Otberg, N.; Patzelt, A.; Rasulev, U.; Hagemeister, T.; Linscheid, M.; Sinkgraven, R.; Sterry, W.; Lademann, J. The role of hair follicles in the percutaneous absorption of caffeine. Br. J. Clin. Pharmacol. 2008, 65, 488–492. [Google Scholar] [CrossRef]
- Supersaxo, A.; Hein, W.R.; Steffen, H. Effect of molecular weight on the lymphatic absorption of water-soluble compounds following subcutaneous administration. Pharm. Res. 1990, 7, 167–169. [Google Scholar] [CrossRef]
- Ibrahim, R.; Nitsche, J.M.; Kasting, G.B. Dermal clearance model for epidermal bioavailability calculations. J. Pharm. Sci. 2012, 101, 2094–2108. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Cabotegravir | Reference |
|---|---|---|
| Molecular Weight (g/mol) | 405.358 | [36] |
| HBD | 2 | [36] |
| Log PO:W | 1.04 | [36] |
| pKa | 10.04, −0.7 | [36] |
| Protein Binding (%) | 99.8 | [37] |
| PSA (Å2) | 99.18 | [36] |
| R | 0.5 | [37] |
| Bioavailability (%) | 20 | [25] |
| CL/FIM,human (L/h) | 0.197 | [38] |
| Knp,IM,human (h−1) | 4.54 × 10−4 | [39] |
| PCSC/W | 0.304 * | [26] |
| PCVE/W | 0.106 ** | [26] |
| PCDE/W | 0.106 ** | [26,40] |
| PCSC/VE | 3.993 * | [26] |
| PCVE/DE | 1 ** | [40] |
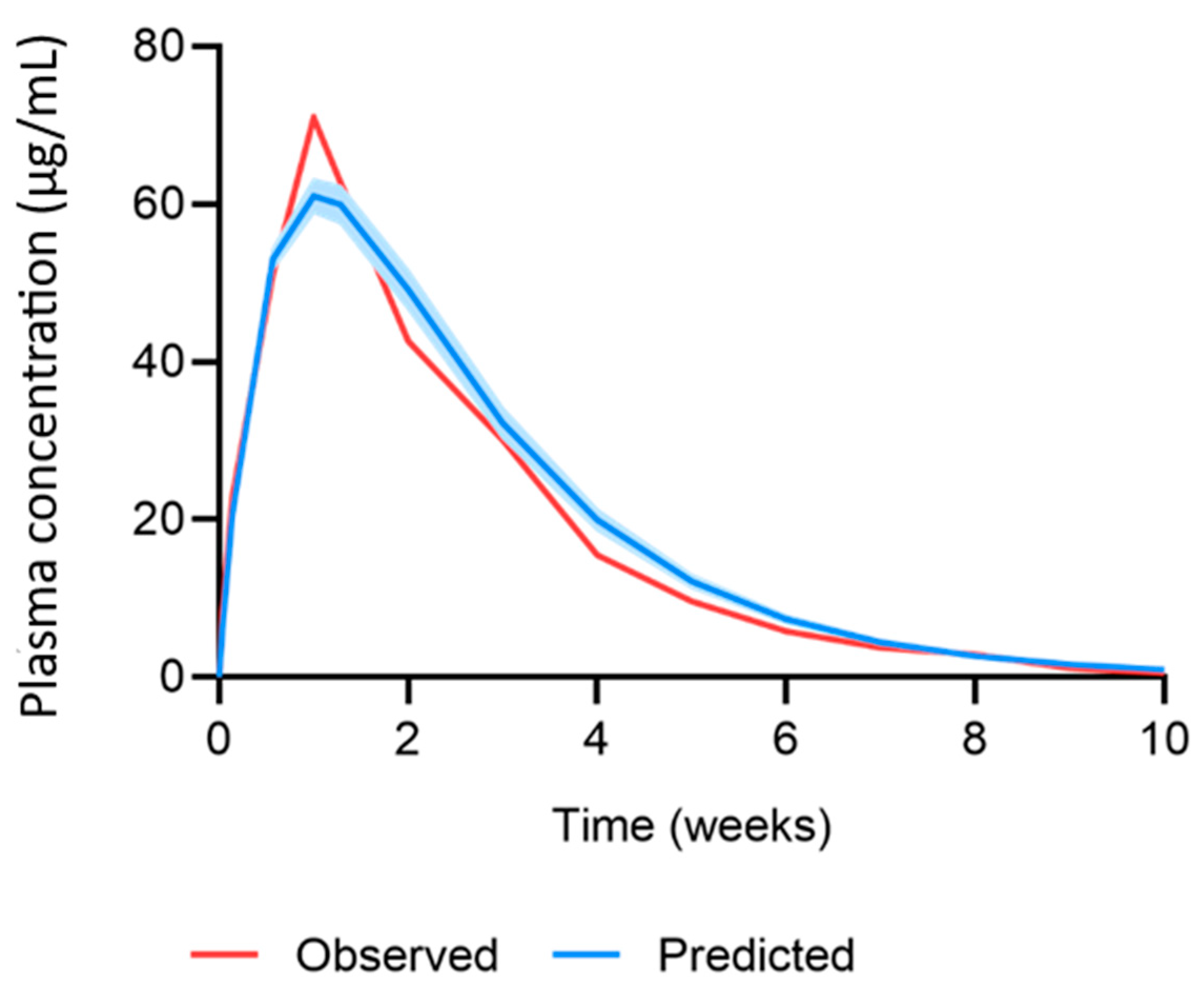
| Parameter | Observed | Predicted | Ratio | AAFE |
|---|---|---|---|---|
| AUC0–28 (µg·day/mL) | 1268.1 ± 290.3 | 1431.1 ± 67.9 | 1.13 | 1.13 |
| Cmax (µg/mL) | 74.5 ± 11.6 | 61.1 ± 2.1 | 0.82 | 1.22 |
| C28 (µg/mL) | 14.5 ± 1.5 | 20 ± 1.3 | 1.38 | 1.38 |
| Profile | - | - | 1.10 | - |
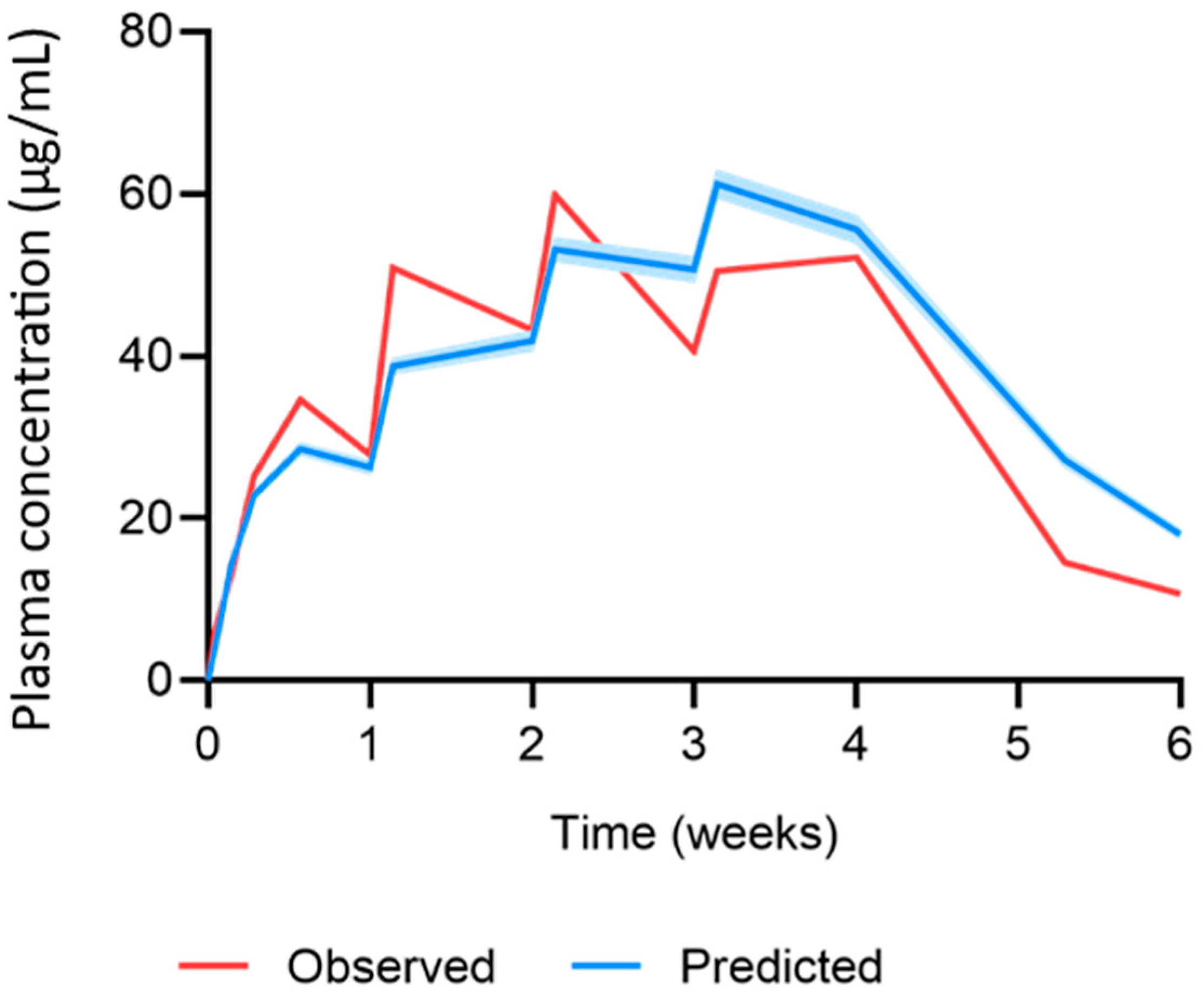
| Dose (mg) | Parameter | Observed | Predicted | Ratio | AAFE |
|---|---|---|---|---|---|
| 11.72 (single dose) | AUC0–28 (µg·day/mL) | 291.1 ± 23.4 | 304.6 ± 7.6 | 1.05 | 1.05 |
| Cmax (µg/mL) | 18.1 ± 2.3 | 20.7 ± 0.5 | 1.15 | 1.15 | |
| C28 (µg/mL) | 3.1 ± 1.1 | 3.3 ± 0.09 | 1.06 | 1.06 | |
| Profile | - | - | - | 1.12 | |
| 11.72 (once-weekly dose) | Cmax-1 (µg/mL) | 35.1 ± 4.6 | 28.7 ± 0.7 | 0.82 | 1.22 |
| Cmax-ss (µg/mL) | 60.3 ± 10.7 | 67.4 ± 1.8 | 1.12 | 1.12 | |
| C42 (µg/mL) | 11.2 ± 2.2 | 17.9 ± 0.6 | 1.60 | 1.60 | |
| Profile | - | - | - | 1.04 |
| Parameter | Observed | Predicted | Ratio | AAFE |
|---|---|---|---|---|
| AUC0–w4 (µg·h/mL) | 1497 | 1993.7 ± 48.3 | 1.33 | 1.33 |
| AUC0–w12 (µg·h/mL) | 3851 | 4792.7 ± 65 | 1.24 | 1.24 |
| Cmax (µg/mL) | 3.3 | 3.4 ± 0.06 | 1.02 | 1.02 |
| Cw4 (µg/mL) | 2 | 2.8 ± 0.02 | 1.39 | 1.39 |
| Profile | - | - | - | 1.23 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinvig, H.; Rajoli, R.K.R.; Pertinez, H.; Vora, L.K.; Volpe-Zanutto, F.; Donnelly, R.F.; Rannard, S.; Flexner, C.; Siccardi, M.; Owen, A. Physiologically Based Pharmacokinetic Modelling of Cabotegravir Microarray Patches in Rats and Humans. Pharmaceutics 2023, 15, 2709. https://doi.org/10.3390/pharmaceutics15122709
Kinvig H, Rajoli RKR, Pertinez H, Vora LK, Volpe-Zanutto F, Donnelly RF, Rannard S, Flexner C, Siccardi M, Owen A. Physiologically Based Pharmacokinetic Modelling of Cabotegravir Microarray Patches in Rats and Humans. Pharmaceutics. 2023; 15(12):2709. https://doi.org/10.3390/pharmaceutics15122709
Chicago/Turabian StyleKinvig, Hannah, Rajith K. R. Rajoli, Henry Pertinez, Lalitkumar K. Vora, Fabiana Volpe-Zanutto, Ryan F. Donnelly, Steve Rannard, Charles Flexner, Marco Siccardi, and Andrew Owen. 2023. "Physiologically Based Pharmacokinetic Modelling of Cabotegravir Microarray Patches in Rats and Humans" Pharmaceutics 15, no. 12: 2709. https://doi.org/10.3390/pharmaceutics15122709
APA StyleKinvig, H., Rajoli, R. K. R., Pertinez, H., Vora, L. K., Volpe-Zanutto, F., Donnelly, R. F., Rannard, S., Flexner, C., Siccardi, M., & Owen, A. (2023). Physiologically Based Pharmacokinetic Modelling of Cabotegravir Microarray Patches in Rats and Humans. Pharmaceutics, 15(12), 2709. https://doi.org/10.3390/pharmaceutics15122709