

Microwave-Assisted Freeze–Drying: Impact of Microwave Radiation on the Quality of High-Concentration Antibody Formulations

Abstract
:1. Introduction
2. Materials and Methods
2.1. Proteins and Chemicals
2.2. Preparation of the Formulations
2.3. Freeze-Drying Process
2.4. Karl–Fischer Titration
2.5. Brunauer–Emmet–Teller (BET) Krypton Gas Adsorption
2.6. Scanning Electron Microscopy (SEM)
2.7. Experiments with the Microwave Oven
2.8. Reconstitution of the Lyophilizates
2.9. Size-Exclusion Chromatography (SEC)
2.10. Cation-Exchange Chromatography (IEX)
2.11. Flow Imaging Microscopy
3. Results and Discussion
3.1. Substitution of Sugar by an Antibody
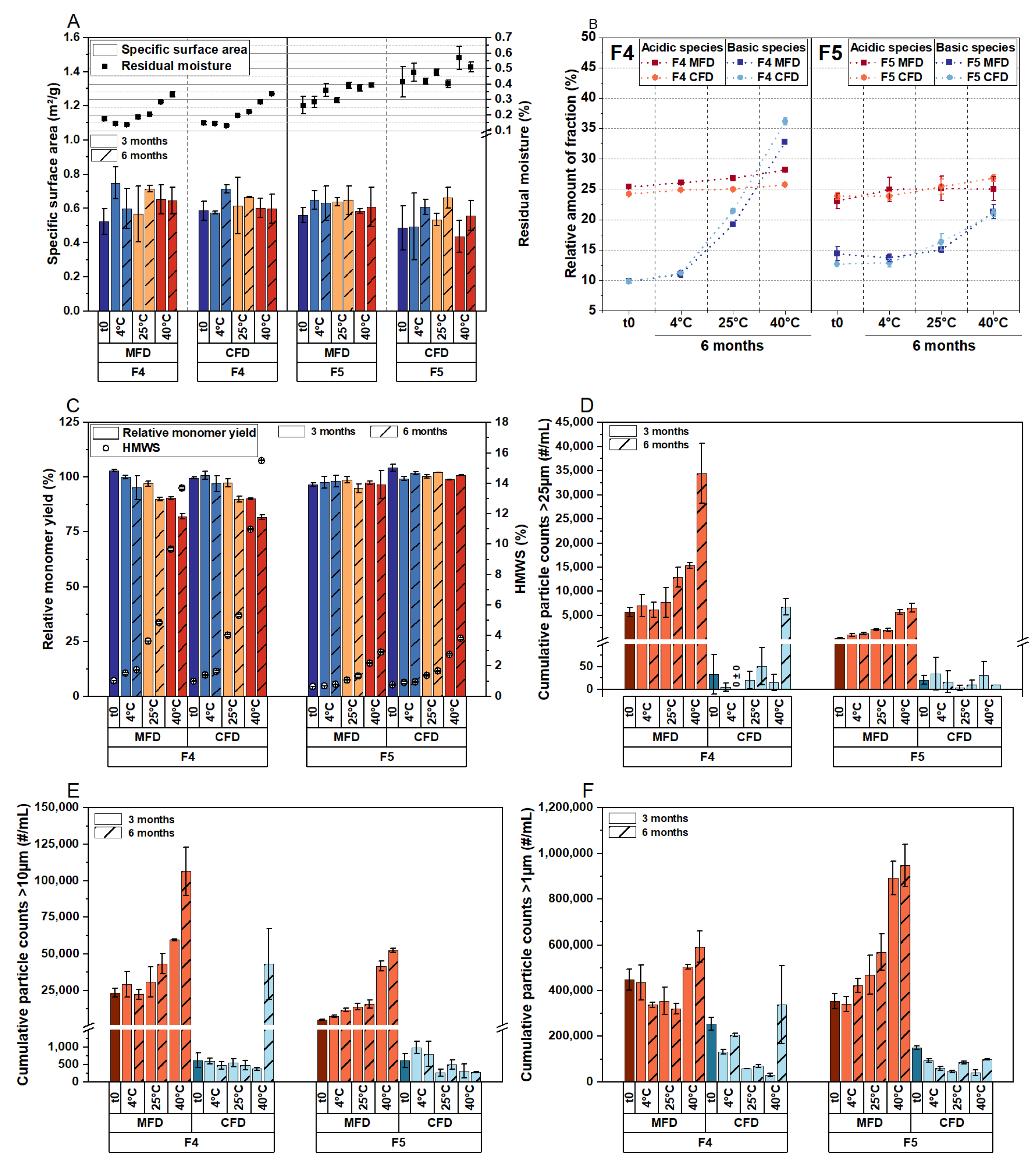
3.2. Comparison of Critical Quality Attributes of a Highly Concentrated mAb Formulation following MFD and CFD
3.3. Effect of Thermal History and Investigation of Two Other Proteins in MFD
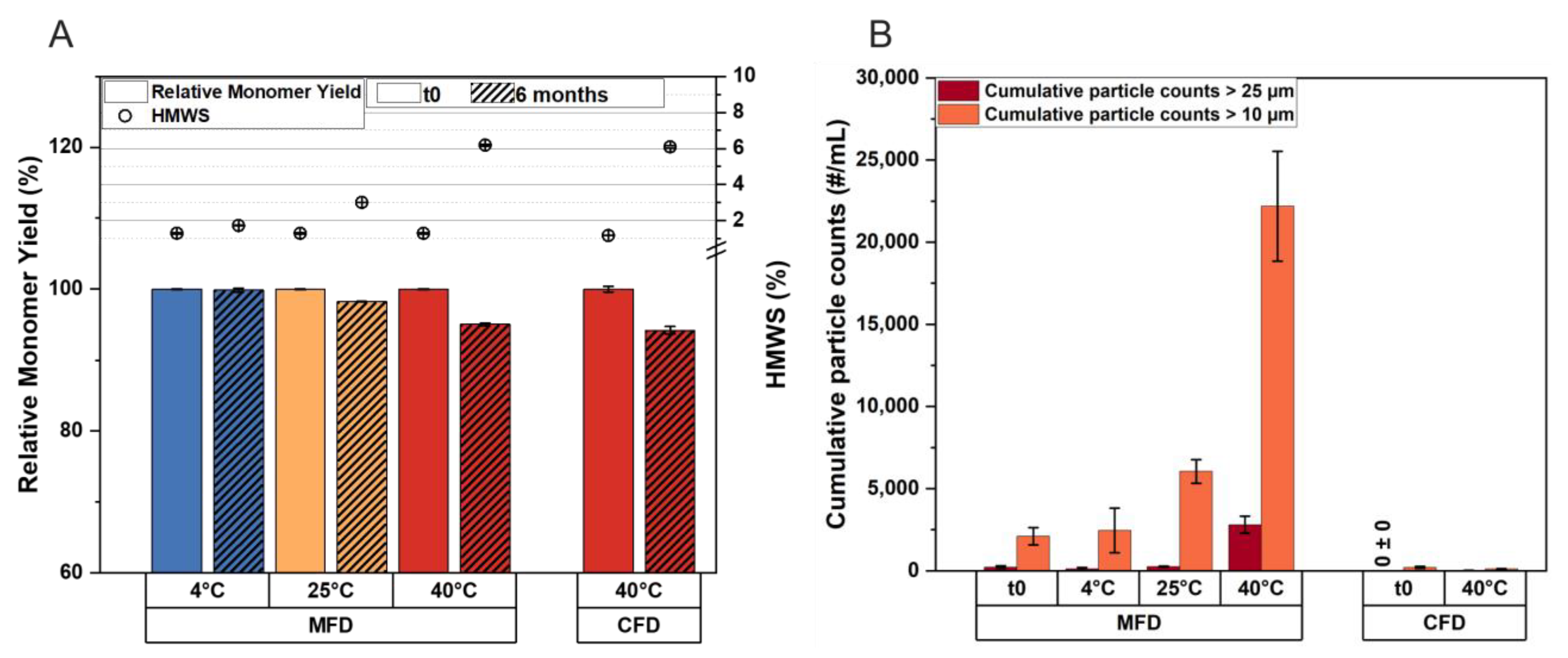
3.4. The Critical Timeframe That Leads to Protein Aggregation during MFD
3.5. Effect of Residual Moisture, Cooling, and the Source of Energy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ghosh, I.; Gutka, H.; Krause, M.E.; Clemens, R.; Kashi, R.S. A systematic review of commercial high concentration antibody drug products approved in the US: Formulation composition, dosage form design and primary packaging considerations. mAbs 2023, 15, 2205540. [Google Scholar] [CrossRef] [PubMed]
- Mieczkowski, C.A. The Evolution of Commercial Antibody Formulations. J. Pharm. Sci. 2023, 112, 1801–1810. [Google Scholar] [CrossRef]
- Wang, W.; Singh, S.; Zeng, D.L.; King, K.; Nema, S. Antibody Structure, Instability, and Formulation. J. Pharm. Sci. 2007, 96, 1–26. [Google Scholar] [CrossRef]
- Carpenter, J.F.; Pikal, M.J.; Chang, B.S.; Randolph, T.W. Rational Design of Stable Lyophilized Protein Formulations: Some Practical Advice. Pharm. Res. 1997, 14, 969–975. [Google Scholar] [CrossRef]
- Wang, W. Lyophilization and development of solid protein pharmaceuticals. Int. J. Pharm. 2000, 203, 1–60. [Google Scholar] [CrossRef]
- Patel, S.M.; Pikal, M.J. Lyophilization process design space. J. Pharm. Sci. 2013, 102, 3883–3887. [Google Scholar] [CrossRef] [PubMed]
- Kasper, J.C.; Winter, G.; Friess, W. Recent advances and further challenges in lyophilization. Eur. J. Pharm. Biopharm. 2013, 85, 162–169. [Google Scholar] [CrossRef]
- Haeuser, C.; Goldbach, P.; Huwyler, J.; Friess, W.; Allmendinger, A. Be Aggressive! Amorphous Excipients Enabling Single-Step Freeze-Drying of Monoclonal Antibody Formulations. Pharmaceutics 2019, 11, 616. [Google Scholar] [CrossRef] [PubMed]
- Teagarden, D.L.; Baker, D.S. Practical aspects of lyophilization using non-aqueous co-solvent systems. Eur. J. Pharm. Sci. 2002, 15, 115–133. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, L.; Van Bockstal, P.J.; Corver, J.; Vervaet, C.; Remon, J.P.; De Beer, T. Evaluation of spin freezing versus conventional freezing as part of a continuous pharmaceutical freeze-drying concept for unit doses. Int. J. Pharm. 2015, 496, 75–85. [Google Scholar] [CrossRef]
- Capozzi, L.C.; Trout, B.L.; Pisano, R. From Batch to Continuous: Freeze-Drying of Suspended Vials for Pharmaceuticals in Unit-Doses. Ind. Eng. Chem. Res. 2019, 58, 1635–1649. [Google Scholar] [CrossRef]
- Ambros, S.; Bauer, S.A.W.; Shylkina, L.; Foerst, P.; Kulozik, U. Microwave-Vacuum Drying of Lactic Acid Bacteria: Influence of Process Parameters on Survival and Acidification Activity. Food Bioprocess Technol. 2016, 9, 1901–1911. [Google Scholar] [CrossRef]
- Bhambhani, A.; Stanbro, J.; Roth, D.; Sullivan, E.; Jones, M.; Evans, R.; Blue, J. Evaluation of Microwave Vacuum Drying as an Alternative to Freeze-Drying of Biologics and Vaccines: The Power of Simple Modeling to Identify a Mechanism for Faster Drying Times Achieved with Microwave. AAPS PharmSciTech 2021, 22, 52. [Google Scholar] [CrossRef]
- Abdelraheem, A.; Tukra, R.; Kazarin, P.; Sinanis, M.D.; Topp, E.M.; Alexeenko, A.; Peroulis, D. Statistical electromagnetics for industrial pharmaceutical lyophilization. PNAS Nexus 2022, 1, pgac052. [Google Scholar] [CrossRef] [PubMed]
- Gitter, J.H.; Geidobler, R.; Presser, I.; Winter, G. Significant Drying Time Reduction Using Microwave-Assisted Freeze-Drying for a Monoclonal Antibody. J. Pharm. Sci. 2018, 107, 2538–2543. [Google Scholar] [CrossRef] [PubMed]
- Gitter, J.H.; Geidobler, R.; Presser, I.; Winter, G. Microwave-Assisted Freeze-Drying of Monoclonal Antibodies: Product Quality Aspects and Storage Stability. Pharmaceutics 2019, 11, 674. [Google Scholar] [CrossRef]
- Härdter, N.; Geidobler, R.; Presser, I.; Winter, G. Accelerated Production of Biopharmaceuticals via Microwave-Assisted Freeze-Drying (MFD). Pharmaceutics 2023, 15, 1342. [Google Scholar] [CrossRef] [PubMed]
- Thostenson, E.T.; Chou, T.W. Microwave processing: Fundamentals and applications. Compos. Part A Appl. Sci. Manuf. 1999, 30, 1055–1071. [Google Scholar] [CrossRef]
- Metaxas, A.C. Microwave heating. Power Eng. J. 1991, 5, 237–247. [Google Scholar] [CrossRef]
- Meredith, R.J. Engineers’ Handbook of Industrial Microwave Heating; The Institution of Electrical Engineers: London, UK, 1998. [Google Scholar]
- Durance, T.; Noorbakhsh, R.; Sandberg, G.; Sáenz-Garza, N. Microwave Drying of Pharmaceuticals. In Drying Technologies for Biotechnologies and Pharmaceutical Applications; Ohtake, S., Izutsu, K.-I., Lechuga-Ballesteros, D., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2020; pp. 239–255. ISBN 9783527341122. [Google Scholar]
- Wang, S.S.; Yan, Y.; Ho, K. US FDA-approved therapeutic antibodies with high-concentration formulation: Summaries and perspectives. Antib. Ther. 2021, 4, 262–272. [Google Scholar] [CrossRef]
- Shire, S.J.; Shahrokh, Z.; Liu, J. Challenges in the development of high protein concentration formulations. J. Pharm. Sci. 2004, 93, 1390–1402. [Google Scholar] [CrossRef] [PubMed]
- Svilenov, H.; Winter, G. The ReFOLD assay for protein formulation studies and prediction of protein aggregation during long-term storage. Eur. J. Pharm. Biopharm. 2019, 137, 131–139. [Google Scholar] [CrossRef]
- Depaz, R.A.; Pansare, S.; Patel, S.M. Freeze-Drying above the Glass Transition Temperature in Amorphous Protein Formulations while Maintaining Product Quality and Improving Process Efficiency. J. Pharm. Sci. 2016, 105, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Haeuser, C.; Goldbach, P.; Huwyler, J.; Friess, W.; Allmendinger, A. Excipients for Room Temperature Stable Freeze-Dried Monoclonal Antibody Formulations. J. Pharm. Sci. 2020, 109, 807–817. [Google Scholar] [CrossRef]
- Härdter, N.; Geidobler, R.; Presser, I.; Winter, G.; (Ludwig-Maximilians-Universität München, Munich, Germany). Personal communication/observation, 2020.
- Khawli, L.A.; Goswami, S.; Hutchinson, R.; Kwong, Z.W.; Yang, J.; Wang, X.; Yao, Z.; Sreedhara, A.; Cano, T.; Tesar, D.; et al. Charge variants in IgG1: Isolation, characterization, in vitro binding properties and pharmacokinetics in rats. mAbs 2010, 2, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Cleland, J.L.; Lam, X.; Kendrick, B.; Yang, J.; Yang, T.H.; Overcashier, D.; Brooks, D.; Hsu, C.; Carpenter, J.F. A specific molar ratio of stabilizer to protein is required for storage stability of a lyophilized monoclonal antibody. J. Pharm. Sci. 2001, 90, 310–321. [Google Scholar] [CrossRef]
- Park, J.; Cho, J.H.; Braatz, R.D. Mathematical modeling and analysis of microwave-assisted freeze-drying in biopharmaceutical applications. Comput. Chem. Eng. 2021, 153, 107412. [Google Scholar] [CrossRef]
- Kelen, Á.; Ress, S.; Nagy, T.; Pallai, E.; Pintye-Hódi, K. Mapping of temperature distribution in pharmaceutical microwave vacuum drying. Powder Technol. 2006, 162, 133–137. [Google Scholar] [CrossRef]
- Lo Presti, K.; Frieß, W. Adjustment of specific residual moisture levels in completely freeze-dried protein formulations by controlled spiking of small water volumes. Eur. J. Pharm. Biopharm. 2021, 169, 292–296. [Google Scholar] [CrossRef]
- Patel, S.M.; Doen, T.; Pikal, M.J. Determination of End Point of Primary Drying in Freeze-Drying Process Control. AAPS PharmSciTech 2010, 11, 73–84. [Google Scholar] [CrossRef]
- Willmann, M. Stabilisierung von Pharmazeutischen Proteinlösungen Durch Vakuumtrocknung; Verfahrenstechnische Optimierung Verschiedener Vakuumtrocknungsverfahren, Untersuchung von Aggregations-Phänomenen und Evaluierung von Hilfsstoffen. Ph.D. Thesis, Ludwig-Maximilians-Universität München, München, Germany, 2003. [Google Scholar]
- Jiskoot, W.; Hawe, A.; Menzen, T.; Volkin, D.B.; Crommelin, D.J.A. Ongoing Challenges to Develop High Concentration Monoclonal Antibody-based Formulations for Subcutaneous Administration: Quo Vadis? J. Pharm. Sci. 2022, 111, 681–867. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.S.; Patel, S.M.; Bogner, R.H. Reconstitution Time for Highly Concentrated Lyophilized Proteins: Role of Formulation and Protein. J. Pharm. Sci. 2020, 109, 2975–2985. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation Number | Protein Conc. (g/L) | Sucrose (%) | Trehalose (%) | Mannitol (%) | Methionine (mM) | PS 20 (%) | pH | ||
|---|---|---|---|---|---|---|---|---|---|
| LMU1 | LMU2 | G-CSF | |||||||
| F1 | 10 | 8 | 0.040 | 5.5 | |||||
| F2 | 30 | 6 | 0.040 | 5.5 | |||||
| F3 | 50 | 4 | 0.040 | 5.5 | |||||
| F4 | 70 | 2 | 0.040 | 5.5 | |||||
| F5 | 100 | 8 | 0.040 | 5.5 | |||||
| F6 | 21 | 1.9 | 0.009 | 6.0 | |||||
| F7 | 0.5 | 1 | 4 | 20 | 0.010 | 4.0 | |||
| Drying Process | Step | Ts (°C) | Pc (mbar) | Hold Time (h) | Ramp Toward Step (K/min) | MW Application (W) |
|---|---|---|---|---|---|---|
| P1 | 1 | −15 | 0.05 | * | 1.0 | 2 × 90 ** |
| 2 | 30 | 0.05 | 6 | 1.0 | 2 × 90 **/† | |
| P2 | 1 | 30 | 0.05 | * | 0.2 | |
| P3 | 1 | 30 | 0.05 | * | 0.2 | 2 × 90 ‡ |
| P4 | 1 | 10 | 0.05 | * | 0.2 | 2 × 90 § |
| 2 | 30 | 0.05 | 4 | 1.0 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Härdter, N.; Geidobler, R.; Presser, I.; Winter, G. Microwave-Assisted Freeze–Drying: Impact of Microwave Radiation on the Quality of High-Concentration Antibody Formulations. Pharmaceutics 2023, 15, 2783. https://doi.org/10.3390/pharmaceutics15122783
Härdter N, Geidobler R, Presser I, Winter G. Microwave-Assisted Freeze–Drying: Impact of Microwave Radiation on the Quality of High-Concentration Antibody Formulations. Pharmaceutics. 2023; 15(12):2783. https://doi.org/10.3390/pharmaceutics15122783
Chicago/Turabian StyleHärdter, Nicole, Raimund Geidobler, Ingo Presser, and Gerhard Winter. 2023. "Microwave-Assisted Freeze–Drying: Impact of Microwave Radiation on the Quality of High-Concentration Antibody Formulations" Pharmaceutics 15, no. 12: 2783. https://doi.org/10.3390/pharmaceutics15122783
APA StyleHärdter, N., Geidobler, R., Presser, I., & Winter, G. (2023). Microwave-Assisted Freeze–Drying: Impact of Microwave Radiation on the Quality of High-Concentration Antibody Formulations. Pharmaceutics, 15(12), 2783. https://doi.org/10.3390/pharmaceutics15122783