

Mechanism for Stabilizing an Amorphous Drug Using Amino Acids within Co-Amorphous Blends


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Ball Milling
2.2.2. Raman Spectroscopy
3. Results
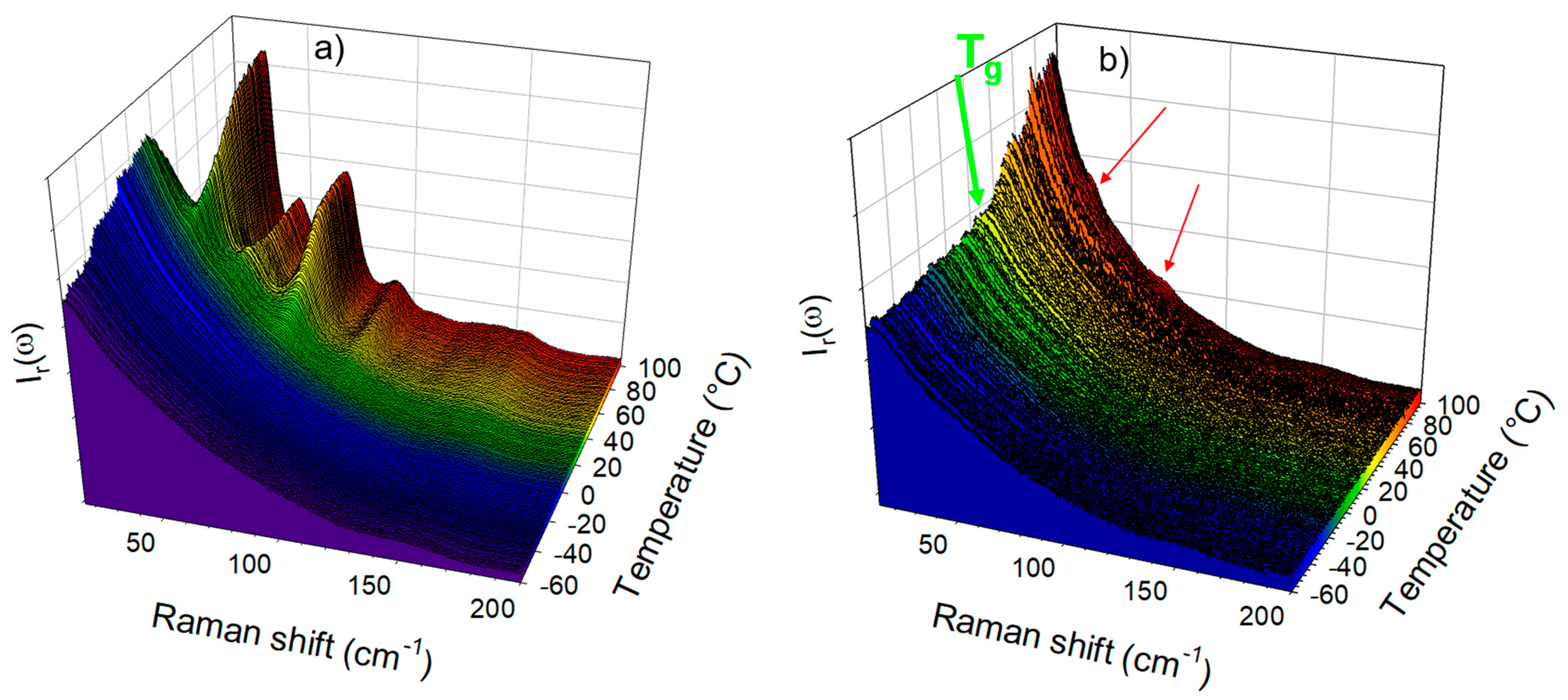
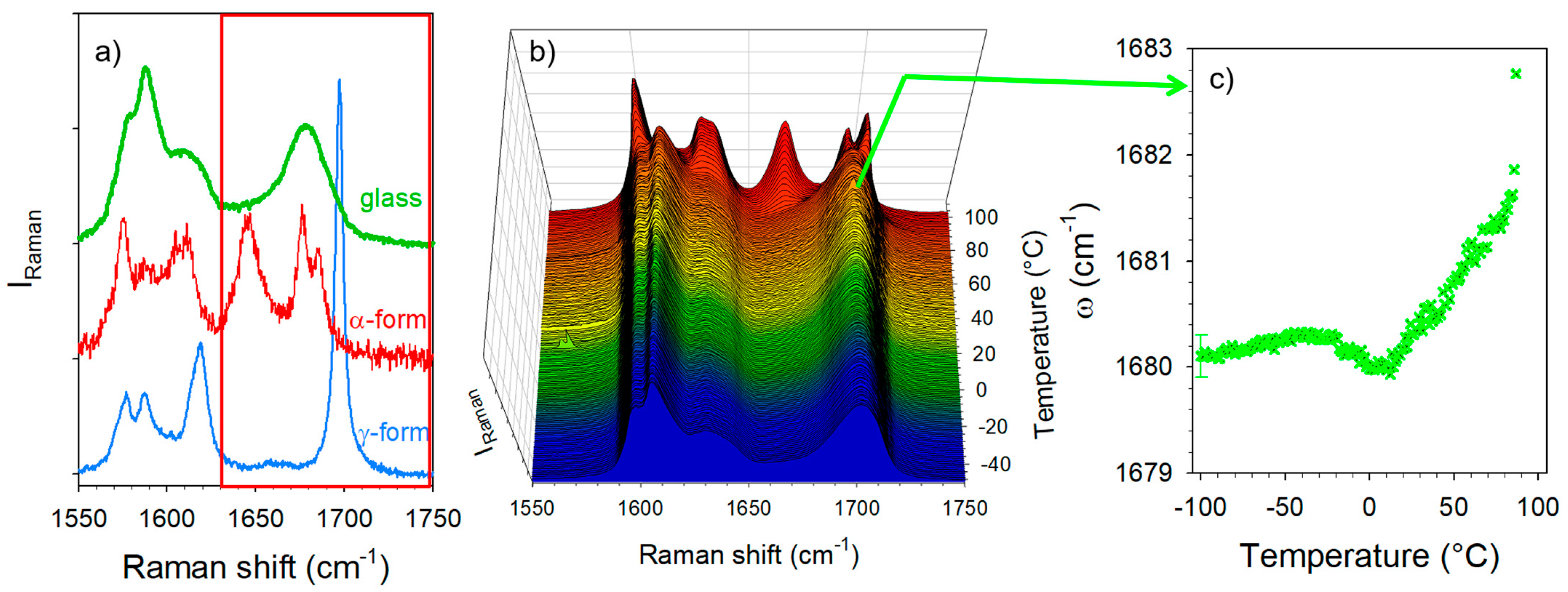
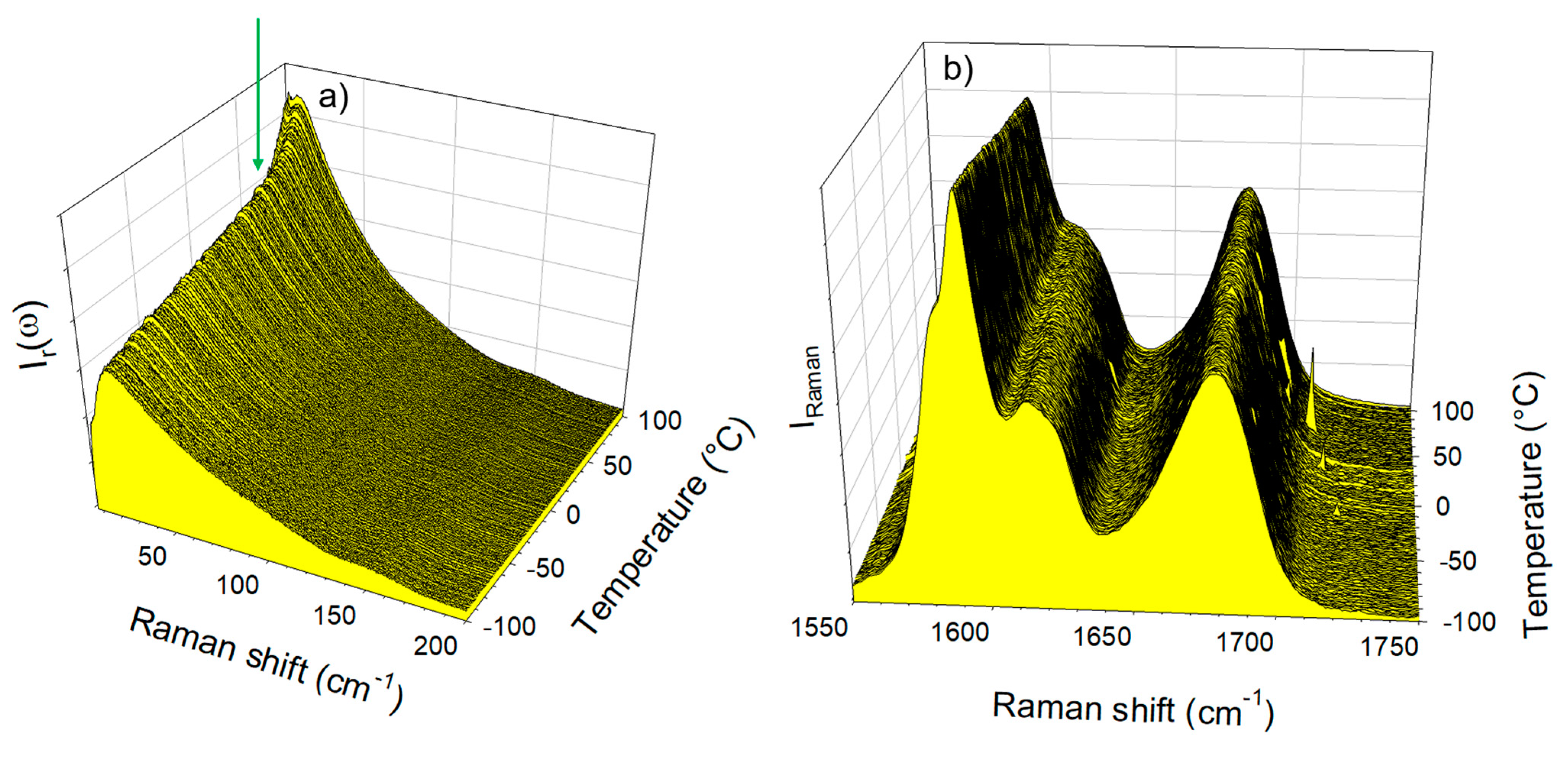
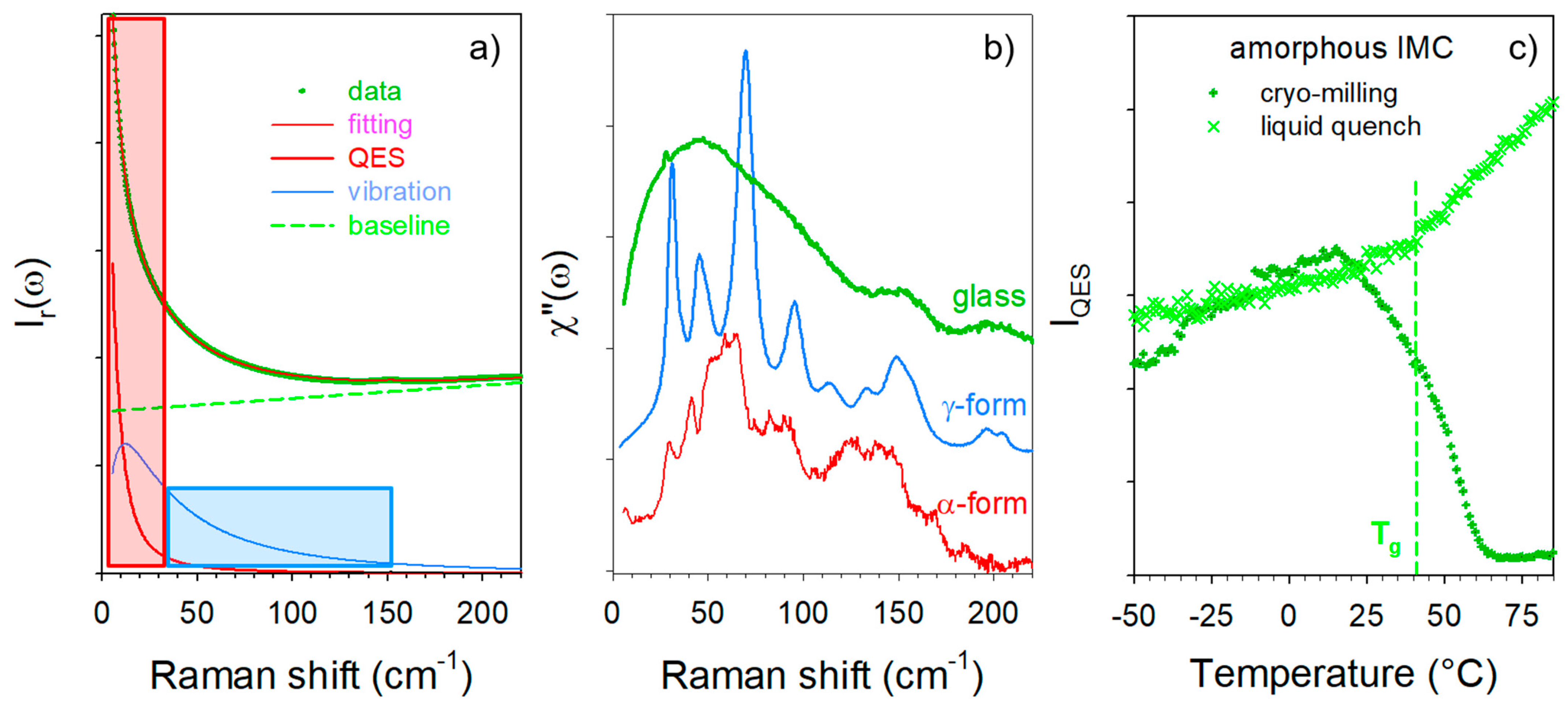
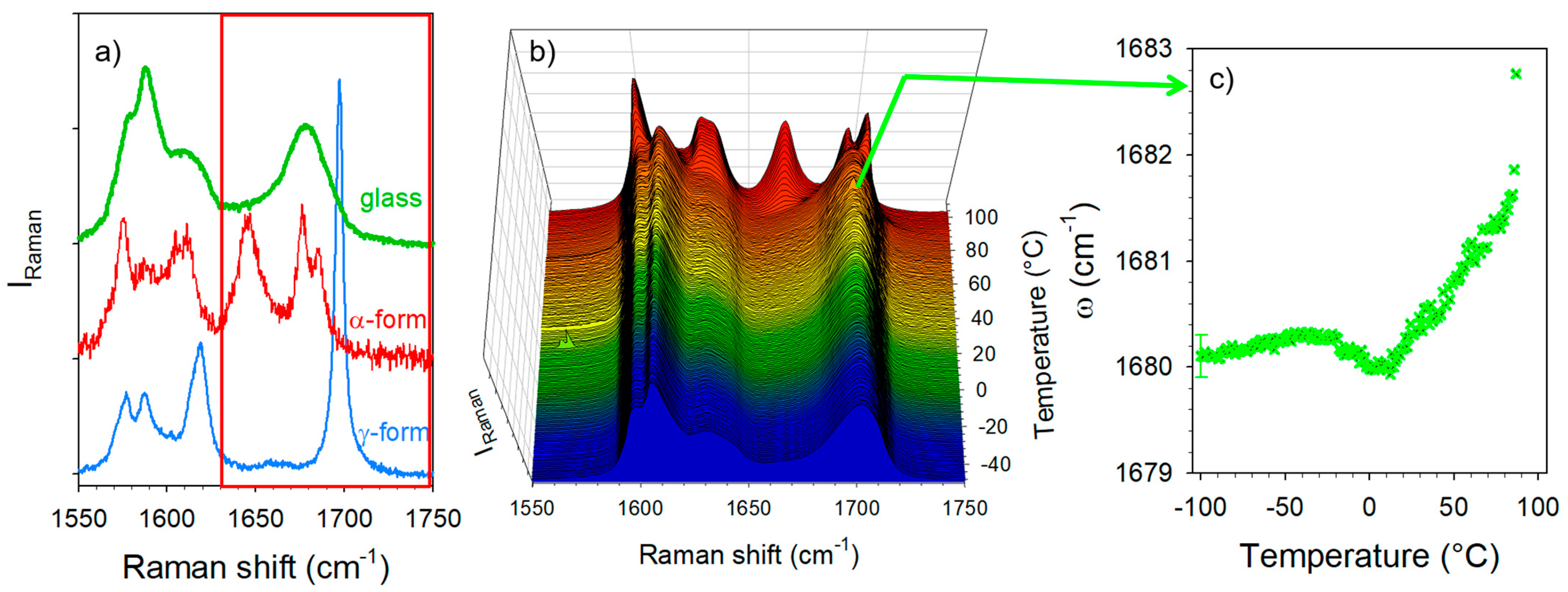
3.1. Analysis of Amorphous IMC upon Heating
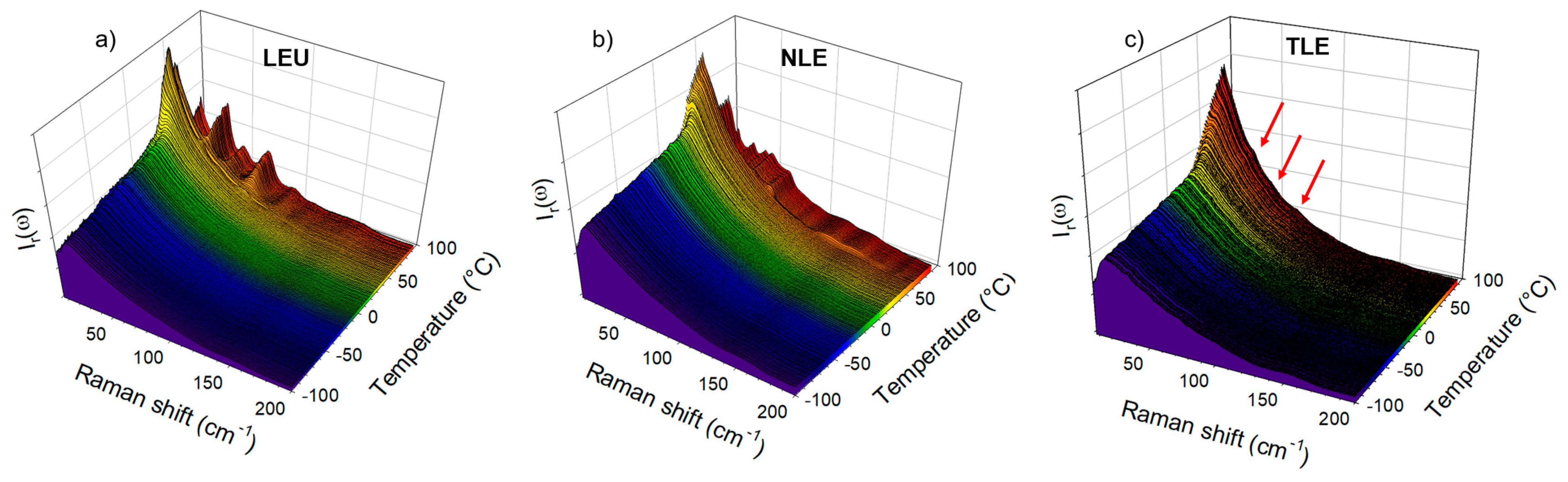
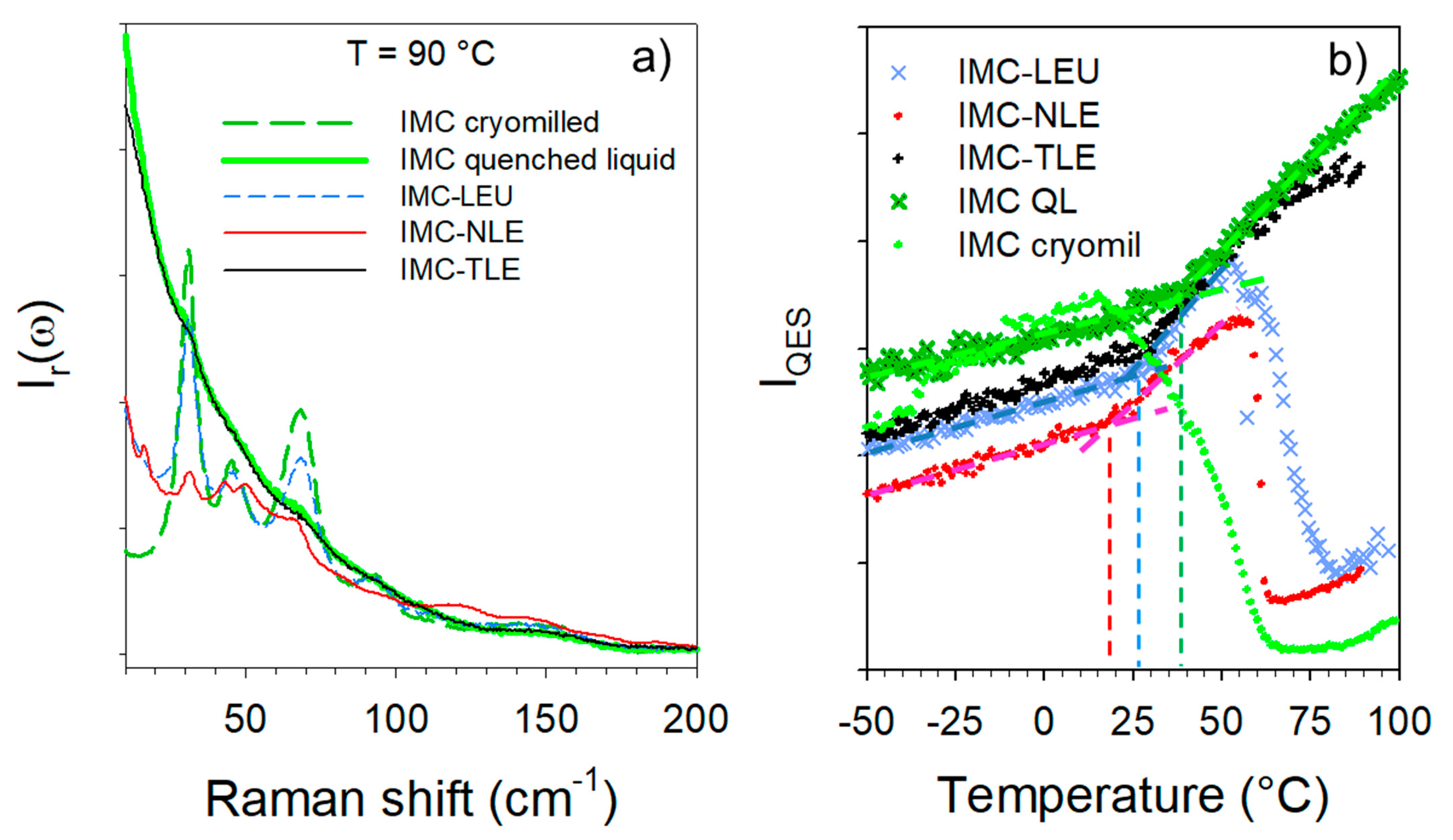
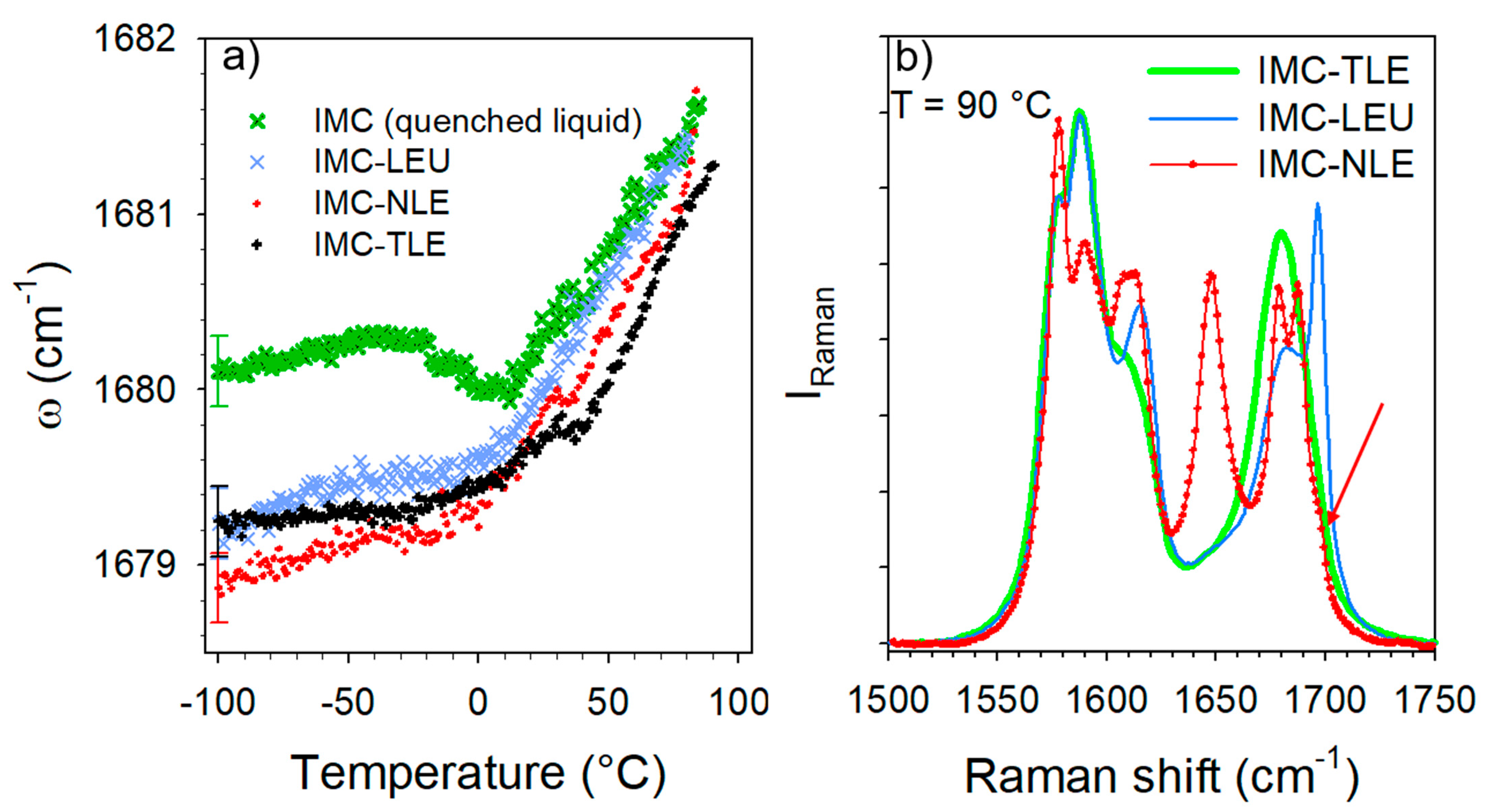
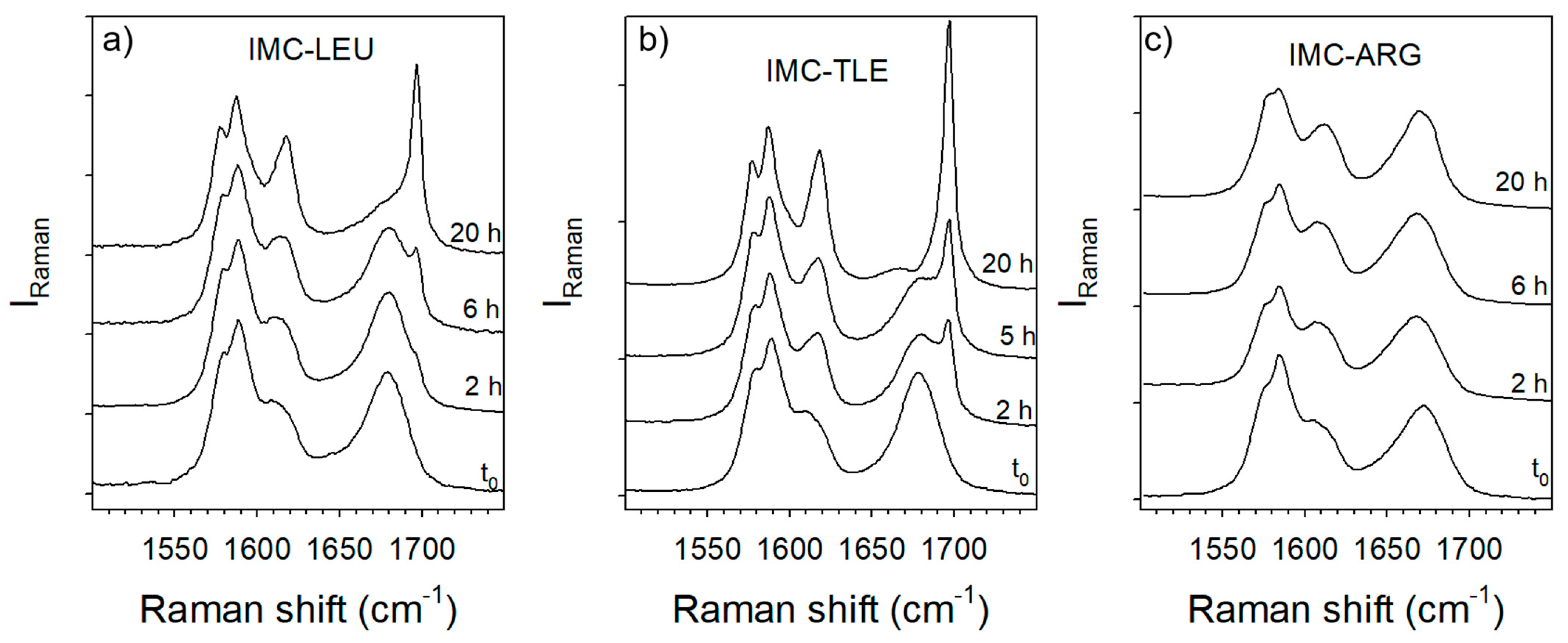
3.2. Co-Amorphous Formulations of IMC with LEU, NLE and TLE
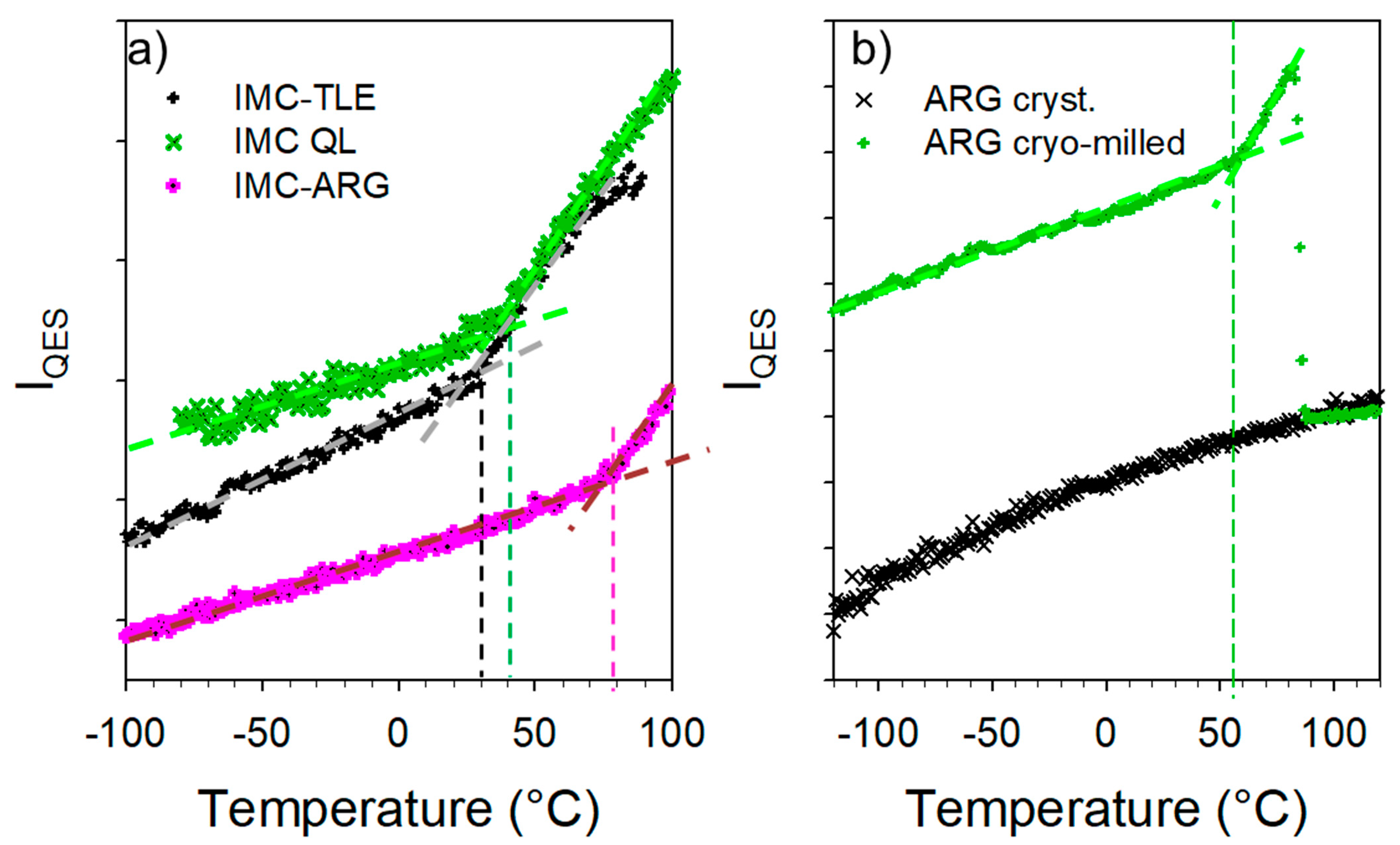
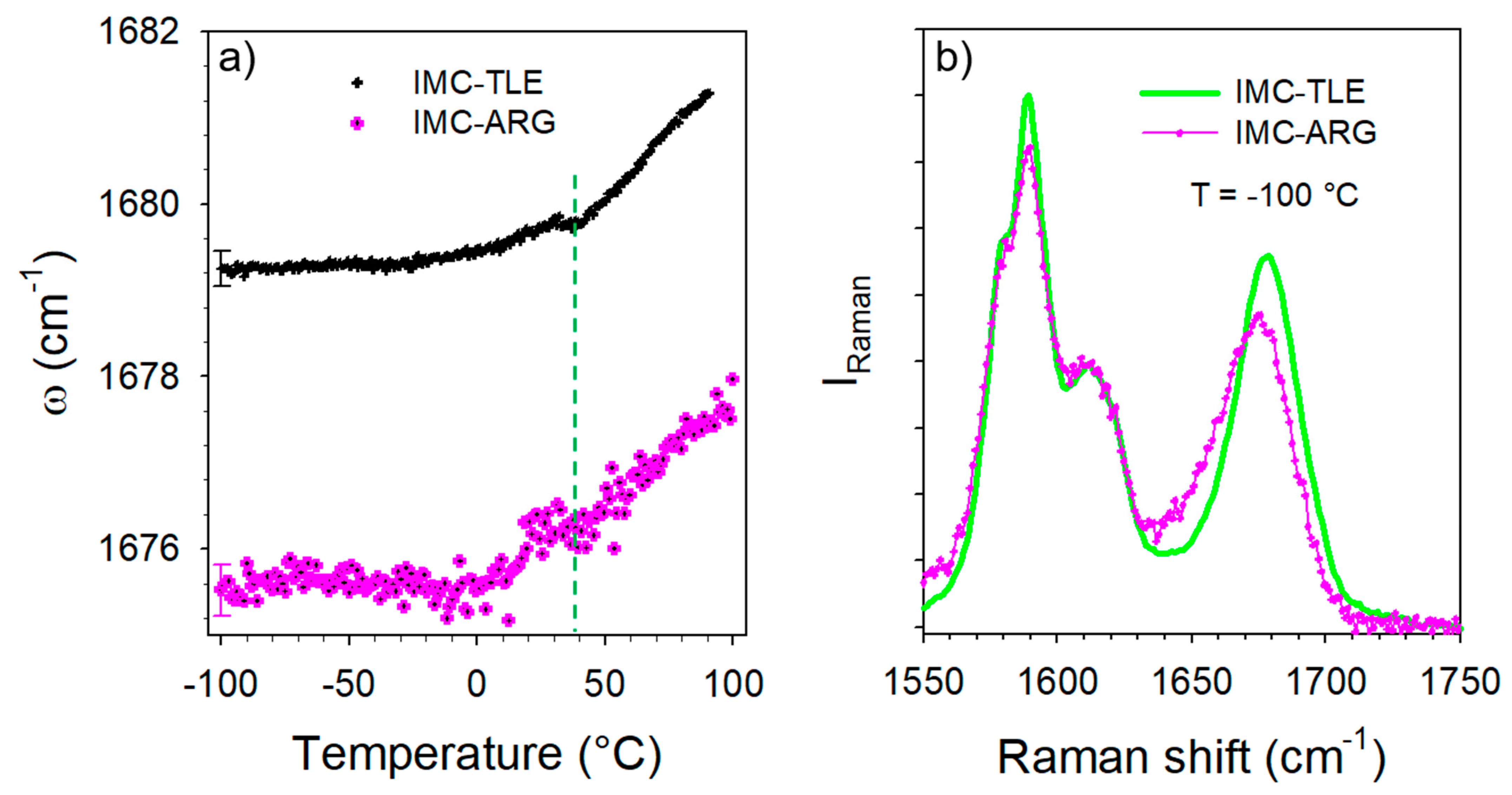
3.3. Co-Amorphous IMC–ARG Formulation
3.4. Hydration Stability
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rumondor, A.C.F.; Dhareshwar, S.S.; Kesisoglou, F. Amorphous solid dispersions or prodrugs: Complementary strategies to increase drug absorption. J. Pharm. Sci. 2015, 105, 2498–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grohganz, H.; Priemel, P.A.; Lobmann, K.; Nielsen, L.; Laitinen, R.; Mullertz, A.; Van den Mooter, G.; Rades, T. Refining stability and dissolution rate of amorphous drug formulations. Expert Opin. Drug Deliv. 2014, 11, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Grohganz, H.; Lobmann, K.; Rades, T.; Hempel, N.-J. Co-Amorphous Drug Formulations in Numbers: Recent Advances in Co-Amorphous Drug Formulations with Focus on Co-Formability, Molar Ratio, Preparation Methods, Physical Stability, In Vitro and In Vivo Performance, and New Formulation Strategies. Pharmaceutics 2021, 13, 389. [Google Scholar] [CrossRef] [PubMed]
- Kasten, G.; Grohganz, H.; Rades, T.; Lobmann, K. Development of a screening method for co-amorphous formulations of drugs and amino acids. Eur. J. Pharm. Sci. 2021, 95, 28–35. [Google Scholar] [CrossRef]
- Mishra, J.; Rades, T.; Lobmann, K.; Grohganz, H. Influence of solvent composition on the performance of spray-dried co-amorphous formulations. Pharmaceutics 2018, 10, 47. [Google Scholar] [CrossRef] [Green Version]
- Ueda, H.; Muranushi, N.; Sakuma, S.; Ida, Y.; Endoh, T.; Kadota, K.; Tozuka, Y. A strategy for co-former selection to design stable co-amorphous formations based on phisicochemical properties of non-steroidal inflammatory drugs. Pharm. Res. 2016, 33, 1018–1029. [Google Scholar] [CrossRef]
- Kissi, E.O.; Kasten, G.; Lobmann, K.; Rades, T.; Grohganz, H. The Role of Glass Transition Temperatures in Coamorphous Drug−Amino Acid Formulations. Mol. Pharm. 2018, 15, 4247–4256. [Google Scholar] [CrossRef]
- Andronis, V.; Zografi, G. Crystal nucleation and growth of indomethacin polymorphs from the amorphous state. J. Non-Cryst. Solids 2000, 271, 236–248. [Google Scholar] [CrossRef]
- Chen, X.; Morris, K.R.; Griesser, U.J.; Byrn, S.R.; Stowell, J.G. Reactivity differences of indomethacin solid forms with ammonia gas. J. Am. Chem. Soc. 2002, 124, 15012–15019. [Google Scholar] [CrossRef]
- Yoshioka, M.; Hancock, B.C.; Zografi, G. Crystallization of indomethacin from the amorphous state below and above its glass transition temperature. J. Pharm. Sci. 1994, 83, 1700–1705. [Google Scholar] [CrossRef]
- Kaneniwa, N.; Otsuka, M.; Hayashi, T. Physicochemical characterization of indomethacin polymorphs and the transformation kinetics in ethanol. Chem. Pharm. Bull. 1985, 33, 3447–3455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hédoux, A. Recent developments in the Raman and infrared investigations of amorphous pharmaceuticals and protein formulations: A review. Adv. Drug Deliv. Rev. 2016, 100, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Hédoux, A.; Guinet, Y.; Descamps, M. The contribution of Raman spectroscopy to the analysis of phase transformations in pharmaceutical compounds. Int. J. Pharm. 2011, 417, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Hédoux, A.; Guinet, Y.; Derollez, P.; Dudognon, E.; Correia, N. Raman spectroscopy of racemic ibuprofen: Evidence of molecular disorder in phase II. Int. J. Pharm. 2011, 421, 45–52. [Google Scholar] [CrossRef]
- Sokolov, A.P.; Rossler, E.; Kisliuk, A.; Quitmann, D. Dynamics of strong and fragile glass formers: Differences and correlation with low-temperature properties. Phys. Rev. Lett. 1993, 71, 2062–2065. [Google Scholar] [CrossRef]
- Hédoux, A.; Decroix, A.-A.; Guinet, Y.; Paccou, L.; Derollez, P.; Descamps, M. Low- and high-frequency investigations on caffeine: Polymorphism, disorder and phase transformation. J. Phys. Chem. B 2011, 115, 5746–5753. [Google Scholar] [CrossRef]
- Denicourt, T.; Hedoux, A.; Guinet, Y.; Willart, J.F.; Descamps, M. Raman scattering investigations of the stable and metastable phases of cyanoadamantane glassy crystal. J. Phys. Chem. B 2003, 107, 8629–8636. [Google Scholar] [CrossRef]
- Malfait, B.; Paccou, L.; Derollez, P.; Guinet, Y.; Hedoux, A. Capabilities of low-wavenumber Raman spectroscopy for analyzing the mechanism of devitrification of molecular glasses. J. Raman Spectrosc. 2019, 50, 1027–1033. [Google Scholar] [CrossRef]
- Malinovski, V.K.; Novikov, V.N.; Sokolov, A.P. Log-normal spectrum of low-energy vibrational excitations in glasses. Phys. Lett. A 1991, 153, 63–66. [Google Scholar] [CrossRef]
- Shuker, R.; Gammon, R. Raman-scattering selection-rule breaking and the density of states in amorphous materials. Phys. Rev. Lett. 1970, 25, 222–225. [Google Scholar] [CrossRef]
- Galeener, F.L.; Sen, P.N. Theory for the first-order vibrational spectra of disordered solids. Phys. Rev. B 1978, 17, 1928–1933. [Google Scholar] [CrossRef]
- Hédoux, A.; Paccou, L.; Guinet, Y.; Willart, J.-F.; Descamps, M. Using the low-frequency Raman spectroscopy to analyze the crystallization of amorphous indomethacin. Eur. J. Pharm. Sci. 2009, 38, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Taylor, L.S.; Zografi, G. The quantitative analysis of crystallinity using FT-Raman spectroscopy. Pharm. Res. 1998, 15, 755–761. [Google Scholar] [CrossRef]
- Taylor, L.S.; Zografi, G. Spectroscopic characterization of interactions between PVP and indomethacin in amorphous molecular dispersions. Pharm. Res. 1997, 14, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Okumura, T.; Otsuka, M. Evaluation of the microcrystallinity of a drug substance, indomethacin, in a pharmaceutical model tablet by chemometric FT-Raman spectroscopy. Pharm. Res. 2005, 22, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.G.; Wartewig, S.; Picker, K.M. Polyethylene oxides: Protection potential against polymorphic transitions of drugs? J. Raman Spectrosc. 2004, 35, 360–367. [Google Scholar] [CrossRef]
- Matsumoto, T.; Zografi, G. Physical properties of solid molecular dispersions of indomethacin with poly(vinylpyrrolidone) and poly(vinylpyrrolidone-co-vinyl-acetate) in relation to indomethacin crystallization. Pharm. Res. 1999, 16, 1722–1728. [Google Scholar] [CrossRef] [PubMed]
- Guinet, Y.; Paccou, L.; Danede, F.; Willart, J.F.; Derollez, P.; Hédoux, A. Comparison of amorphous states prepared by melt-quenching and cryomilling polymorphs of carbamazépine. Int. J. Pharm. 2016, 509, 305–313. [Google Scholar] [CrossRef]
- Guinet, Y.; Paccou, L.; Danede, F.; Derollez, P.; Hédoux, A. Structural description of the marketed form of valsartan: A crystalline mesophase characterized by nanocrystals and conformational disorder. Int. J. Pharm. 2017, 526, 209–216. [Google Scholar] [CrossRef]
- Latreche, M.; Willart, J.F.; Paccou, L.; Guinet, Y.; Hédoux, A. Polymorphism versus devitrification mechanism: Low-wavenumber Raman investigations in sulindac. Int. J. Pharm. 2019, 567, 118476. [Google Scholar] [CrossRef]
- Otsuka, M.; Kaneniwa, N. A kinetic study of the crystallization process of noncrystalline indomethacin under isothermal conditions. Chem. Pharm. Bull. 1988, 36, 4026–4032. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Yu, L. Surface Crystallization of Indomethacin Below T (g). Pharm. Res. 2006, 23, 2350–2355. [Google Scholar] [CrossRef]
- Hedoux, A.; Guinet, Y.; Capet, F.; Paccou, L.; Descamps, M. Evidence for a high-density amorphous form in indomethacin from Raman scattering investigations. Phys. Rev. B 2008, 77, 094205. [Google Scholar] [CrossRef]
- Izutsu, K.; Fujimaki, Y.; Kuwabara, A.; Aoyagi, N. Effect of counterions on the physical properties of L-arginine in frozen solutions and freeze-dried solids. Int. J. Pharm. 2005, 301, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.T.; Lobmann, K.; Rades, T.; Grohganz, H. Improving Co-Amorphous Drug Formulations by the Addition of the Highly Water Soluble Amino Acid, Proline. Pharmaceutics 2014, 6, 416–435. [Google Scholar] [CrossRef] [PubMed]
- Guinet, Y.; Paccou, L.; Derollez, P.; Danède, F.; Hédoux, A. Identification of incommensurability in L-leucine: Can lattice instabbilities be considered as general phenomena in hydrophobic amino acids. J. Chem. Phys. 2022, 24, 27023–27030. [Google Scholar] [CrossRef]
- Gorbitz, C.H.; Karen, P.; Dusek, M.; Petricek, V. An exceptional series of phase transitions in hydrophobic amino acids with linear side chains. IUCrJ 2016, 3, 341–353. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guinet, Y.; Paccou, L.; Hédoux, A. Mechanism for Stabilizing an Amorphous Drug Using Amino Acids within Co-Amorphous Blends. Pharmaceutics 2023, 15, 337. https://doi.org/10.3390/pharmaceutics15020337
Guinet Y, Paccou L, Hédoux A. Mechanism for Stabilizing an Amorphous Drug Using Amino Acids within Co-Amorphous Blends. Pharmaceutics. 2023; 15(2):337. https://doi.org/10.3390/pharmaceutics15020337
Chicago/Turabian StyleGuinet, Yannick, Laurent Paccou, and Alain Hédoux. 2023. "Mechanism for Stabilizing an Amorphous Drug Using Amino Acids within Co-Amorphous Blends" Pharmaceutics 15, no. 2: 337. https://doi.org/10.3390/pharmaceutics15020337
APA StyleGuinet, Y., Paccou, L., & Hédoux, A. (2023). Mechanism for Stabilizing an Amorphous Drug Using Amino Acids within Co-Amorphous Blends. Pharmaceutics, 15(2), 337. https://doi.org/10.3390/pharmaceutics15020337