
Antiviral Peptide-Based Conjugates: State of the Art and Future Perspectives



Abstract
1. Introduction
2. PDCs and Antiviral Cargoes
3. PDC Design Considerations
- (i)
- A robust enough biological rationale endorsing the combination of the two (peptide + antiviral) or three (peptide + antiviral drug + linker) components of the conjugate is desirable.
- (ii)
- A CPP moiety should be chosen that warrants tissue-specific delivery and hence reduces off-target adverse effects. This ideal scenario is very often ignored, as can be seen in Table 2, where typical CPP sequences with broad spectra of membrane permeability are the ones used in the design of the PDCs.
- (iii)
- Antiviral cargoes active at concentrations commensurate with those of the CPP are desirable. Peptides can act at rather low (e.g., nM) concentrations not met by antiviral molecules. These, in turn, can bind plasma proteins (e.g., albumin) with high affinities that can significantly alter pharmacokinetics and/or pharmacodynamics of the conjugate. Those issues should ideally be considered and harmonized.
- (iv)
- For linker-containing conjugates, the linker should if possible be chosen while bearing in mind factors such as the desirable circulation time for the conjugate to reach its target or the specific location where the drug needs to be released.
- (v)
- Another important consideration is the position where the payload is placed. While the N-terminus of the CPP—elongated or not via an intervening spacer unit—is rather usual, alternative approaches, e.g., by way of an extra residue (often Lys or Cys) at either (N- or C-) end of the proper CPP sequence are also favored. Recent work has shown that whichever of these attachment modes is used can have a significant impact on conjugate performance [82,83].
- (vi)
- The conjugate end-product should ideally be non-cytotoxic, non-immunogenic and have minimal interference (hence adverse reactions) with other drugs in multi-therapy schedules.
- (vii)
- Finally, uptake mechanisms ensuring successful release of the antiviral drug from the PDC need to be elucidated. CPP internalization (with or without cargo) is a complex process with multiple factors (positive charge, amphipathicity, folding ability, cargo structure or cell internalization, through active (energy-dependent) or passive (energy-independent) penetration pathways) influencing the peptide–membrane interaction, which is crucial for successful outcomes [86,87,88,89,90].
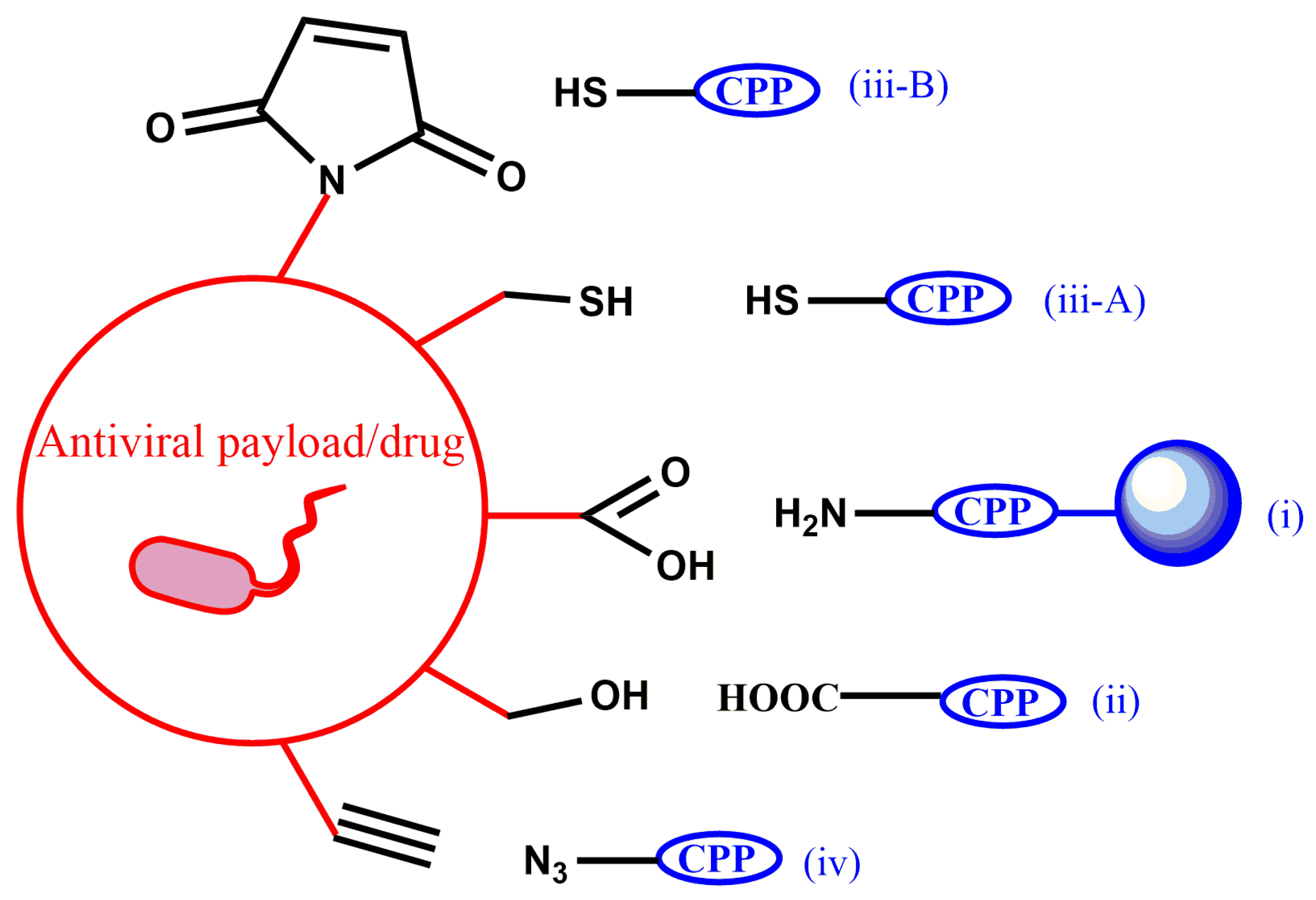
4. Conjugation Chemistry
4.1. Conjugation Based on Amide Bond Formation
4.2. Conjugation Based on Ester Bond Formation
4.3. Conjugation Based on Thiol Chemistry
4.4. Conjugation Based on Click Chemistry
5. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roychoudhury, S.; Das, A.; Sengupta, P.; Dutta, S.; Roychoudhury, S.; Choudhury, A.P.; Ahmed, A.B.F.; Bhattacharjee, S.; Slama, P. Viral Pandemics of the Last Four Decades: Pathophysiology, Health Impacts and Perspectives. Int. J. Environ. Res. Pub. Health 2020, 17, 9411. [Google Scholar] [CrossRef] [PubMed]
- Falcinelli, S.D.; Chertow, D.S.; Kindrachuk, J. Integration of Global Analyses of Host Molecular Responses with Clinical Data to Evaluate Pathogenesis and Advance Therapies for Emerging and Re-Emerging Viral Infections. ACS Infect. Dis. 2016, 2, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Ka-Wai Hui, E. Reasons for the Increase in Emerging and Re-Emerging Viral Infectious Diseases. Microbes. Infect. 2006, 8, 905–916. [Google Scholar] [CrossRef]
- Nováková, L.; Pavlík, J.; Chrenková, L.; Martinec, O.; Červený, L. Current Antiviral Drugs and Their Analysis in Biological Materials—Part II: Antivirals against Hepatitis and HIV Viruses. J. Pharm. Biomed. Anal. 2018, 147, 378–399. [Google Scholar] [CrossRef]
- Szunerits, S.; Barras, A.; Khanal, M.; Pagneux, Q.; Boukherroub, R. Nanostructures for the Inhibition of Viral Infections. Molecules 2015, 20, 14051–14081. [Google Scholar] [CrossRef]
- Irwin, K.K.; Renzette, N.; Kowalik, T.F.; Jensen, J.D. Antiviral Drug Resistance as an Adaptive Process. Virus Evol. 2016, 2, vew014. [Google Scholar] [CrossRef] [PubMed]
- de Clercq, E.; Li, G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef]
- Lou, Z.; Sun, Y.; Rao, Z. Current Progress in Antiviral Strategies. Trends Pharmacol. Sci. 2014, 35, 86–102. [Google Scholar] [CrossRef]
- Divyashree, M.; Mani, M.K.; Reddy, D.; Kumavath, R.; Ghosh, P.; Azevedo, V.; Barh, D. Clinical Applications of Antimicrobial Peptides (AMPs): Where Do We Stand Now? Protein Pept. Lett. 2020, 27, 120–134. [Google Scholar] [CrossRef]
- Jhong, J.-H.; Chi, Y.-H.; Li, W.-C.; Lin, T.-H.; Huang, K.-Y.; Lee, T.-Y. DbAMP: An Integrated Resource for Exploring Antimicrobial Peptides with Functional Activities and Physicochemical Properties on Transcriptome and Proteome Data. Nucleic Acids Res. 2019, 47, D285–D297. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic Peptides: Historical Perspectives, Current Development Trends, and Future Directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Järver, P.; Mäger, I.; Langel, Ü. In Vivo Biodistribution and Efficacy of Peptide Mediated Delivery. Trends Pharmacol. Sci. 2010, 31, 528–535. [Google Scholar] [CrossRef]
- Schwarze, S.R.; Hruska, K.A.; Dowdy, S.F. Protein Transduction: Unrestricted Delivery into All Cells? Trends Cell Biol. 2000, 10, 290–295. [Google Scholar] [CrossRef]
- Agrawal, P.; Bhalla, S.; Usmani, S.S.; Singh, S.; Chaudhary, K.; Raghava, G.P.S.; Gautam, A. CPPsite 2.0: A Repository of Experimentally Validated Cell-Penetrating Peptides. Nucleic Acids Res. 2016, 44, D1098–D1103. [Google Scholar] [CrossRef]
- Frankel, A.D.; Pabo, C.O. Cellular Uptake of the Tat Protein from Human Immunodeficiency Virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Vivès, E.; Brodin, P.; Lebleu, B. A Truncated HIV-1 Tat Protein Basic Domain Rapidly Translocates through the Plasma Membrane and Accumulates in the Cell Nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef]
- Pooga, M.; Hällbrink, M.; Zorko, M.; Langel, Ü. Cell Penetration by Transportan. FASEB J. 1998, 12, 67–77. [Google Scholar] [CrossRef]
- Lindgren, M.; Hällbrink, M.; Prochiantz, A.; Langel, Ü. Cell-Penetrating Peptides. Trends Pharmacol. Sci. 2000, 21, 99–103. [Google Scholar] [CrossRef]
- Derakhshankhah, H.; Jafari, S. Cell Penetrating Peptides: A Concise Review with Emphasis on Biomedical Applications. Biomed. Pharmacother. 2018, 108, 1090–1096. [Google Scholar] [CrossRef]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef]
- Copolovici, D.M.; Langel, K.; Eriste, E.; Langel, Ü. Cell-Penetrating Peptides: Design, Synthesis, and Applications. ACS Nano 2014, 8, 1972–1994. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Bi, Y.; Zhang, H.; Dong, S.; Teng, L.; Lee, R.J.; Yang, Z. Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front. Pharmacol. 2020, 11, 697. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, J.; Nilvebrant, J.; Nygren, P.-Å.; Lehmann, F. Progress and Future Directions with Peptide-Drug Conjugates for Targeted Cancer Therapy. Molecules 2021, 26, 6042. [Google Scholar] [CrossRef] [PubMed]
- Hoppenz, P.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide-Drug Conjugates and Their Targets in Advanced Cancer Therapies. Front. Chem. 2020, 8, 571. [Google Scholar] [CrossRef]
- al Shaer, D.; al Musaimi, O.; Albericio, F.; de la Torre, B.G. 2021 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2022, 15, 222. [Google Scholar] [CrossRef]
- He, R.; Finan, B.; Mayer, J.P.; DiMarchi, R.D. Peptide Conjugates with Small Molecules Designed to Enhance Efficacy and Safety. Molecules 2019, 24, 1855. [Google Scholar] [CrossRef]
- Balogh, B.; Ivánczi, M.; Nizami, B.; Beke-Somfai, T.; Mándity, I.M. ConjuPepDB: A Database of Peptide–Drug Conjugates. Nucleic Acids Res. 2021, 49, D1102–D1112. [Google Scholar] [CrossRef]
- Liang, G.; Wang, H.; Chong, H.; Cheng, S.; Jiang, X.; He, Y.; Wang, C.; Liu, K. An Effective Conjugation Strategy for Designing Short Peptide-Based HIV-1 Fusion Inhibitors. Org. Biomol. Chem. 2016, 14, 7875–7882. [Google Scholar] [CrossRef]
- Wang, C.; Shi, W.; Cai, L.; Lu, L.; Wang, Q.; Zhang, T.; Li, J.; Zhang, Z.; Wang, K.; Xu, L.; et al. Design, Synthesis, and Biological Evaluation of Highly Potent Small Molecule–Peptide Conjugates as New HIV-1 Fusion Inhibitors. J. Med. Chem. 2013, 56, 2527–2539. [Google Scholar] [CrossRef]
- Zhou, L.; Thakur, C.S.; Molinaro, R.J.; Paranjape, J.M.; Hoppes, R.; Jeang, K.-T.; Silverman, R.H.; Torrence, P.F. Delivery of 2-5A Cargo into Living Cells Using the Tat Cell Penetrating Peptide: 2-5A-Tat. Bioorg. Med. Chem. 2006, 14, 7862–7874. [Google Scholar] [CrossRef]
- García-Aparicio, C.; Diez-Torrubia, A.; Balzarini, J.; Lambeir, A.-M.; Velázquez, S.; Camarasa, M.-J. Efficient Conversion of Tetrapeptide-Based TSAO Prodrugs to the Parent Drug by Dipeptidyl-Peptidase IV (DPPIV/CD26). Antivir. Res. 2007, 76, 130–139. [Google Scholar] [CrossRef]
- Diez-Torrubia, A.; Cabrera, S.; de Castro, S.; García-Aparicio, C.; Mulder, G.; de Meester, I.; Camarasa, M.-J.; Balzarini, J.; Velázquez, S. Novel Water-Soluble Prodrugs of Acyclovir Cleavable by the Dipeptidyl-Peptidase IV (DPP IV/CD26) Enzyme. Eur. J. Med. Chem. 2013, 70, 456–468. [Google Scholar] [CrossRef]
- Liotard, J.-F.; Mehiri, M.; di Giorgio, A.; Boggetto, N.; Reboud-Ravaux, M.; Aubertin, A.-M.; Condom, R.; Patino, N. AZT and AZT-Monophosphate Prodrugs Incorporating HIV-Protease Substrate Fragment: Synthesis and Evaluation as Specific Drug Delivery Systems. Antivir. Chem. Chemother. 2006, 17, 193–213. [Google Scholar] [CrossRef]
- Nitsche, C.; Schreier, V.N.; Behnam, M.A.M.; Kumar, A.; Bartenschlager, R.; Klein, C.D. Thiazolidinone–Peptide Hybrids as Dengue Virus Protease Inhibitors with Antiviral Activity in Cell Culture. J. Med. Chem. 2013, 56, 8389–8403. [Google Scholar] [CrossRef]
- Liu, N.; Zhang, Y.; Lei, Y.; Wang, R.; Zhan, M.; Liu, J.; An, Y.; Zhou, Y.; Zhan, J.; Yin, F.; et al. Design and Evaluation of a Novel Peptide–Drug Conjugate Covalently Targeting SARS-CoV-2 Papain-like Protease. J. Med. Chem. 2022, 65, 876–884. [Google Scholar] [CrossRef]
- Lan, Q.; Wang, C.; Zhou, J.; Wang, L.; Jiao, F.; Zhang, Y.; Cai, Y.; Lu, L.; Xia, S.; Jiang, S. 25-Hydroxycholesterol-Conjugated EK1 Peptide with Potent and Broad-Spectrum Inhibitory Activity against SARS-CoV-2, Its Variants of Concern, and Other Human Coronaviruses. Int. J. Mol. Sci. 2021, 22, 11869. [Google Scholar] [CrossRef]
- Lan, Q.; Chan, J.F.-W.; Xu, W.; Wang, L.; Jiao, F.; Zhang, G.; Pu, J.; Zhou, J.; Xia, S.; Lu, L.; et al. A Palmitic Acid-Conjugated, Peptide-Based Pan-CoV Fusion Inhibitor Potently Inhibits Infection of SARS-CoV-2 Omicron and Other Variants of Concern. Viruses 2022, 14, 549. [Google Scholar] [CrossRef]
- Rosenke, K.; Leventhal, S.; Moulton, H.M.; Hatlevig, S.; Hawman, D.; Feldmann, H.; Stein, D.A. Inhibition of SARS-CoV-2 in Vero Cell Cultures by Peptide-Conjugated Morpholino Oligomers. J. Antimicrob. Chemother. 2021, 76, 413–417. [Google Scholar] [CrossRef]
- Deas, T.S.; Binduga-Gajewska, I.; Tilgner, M.; Ren, P.; Stein, D.A.; Moulton, H.M.; Iversen, P.L.; Kauffman, E.B.; Kramer, L.D.; Shi, P.-Y. Inhibition of Flavivirus Infections by Antisense Oligomers Specifically Suppressing Viral Translation and RNA Replication. J. Virol. 2005, 79, 4599–4609. [Google Scholar] [CrossRef]
- Deas, T.S.; Bennett, C.J.; Jones, S.A.; Tilgner, M.; Ren, P.; Behr, M.J.; Stein, D.A.; Iversen, P.L.; Kramer, L.D.; Bernard, K.A.; et al. In Vitro Resistance Selection and In Vivo Efficacy of Morpholino Oligomers against West Nile Virus. Antimicrob. Agents Chemother. 2007, 51, 2470–2482. [Google Scholar] [CrossRef]
- Stone, J.K.; Rijnbrand, R.; Stein, D.A.; Ma, Y.; Yang, Y.; Iversen, P.L.; Andino, R. A Morpholino Oligomer Targeting Highly Conserved Internal Ribosome Entry Site Sequence Is Able To Inhibit Multiple Species of Picornavirus. Antimicrob. Agents Chemother. 2008, 52, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Burrer, R.; Neuman, B.W.; Ting, J.P.C.; Stein, D.A.; Moulton, H.M.; Iversen, P.L.; Kuhn, P.; Buchmeier, M.J. Antiviral Effects of Antisense Morpholino Oligomers in Murine Coronavirus Infection Models. J. Virol. 2007, 81, 5637–5648. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Stein, D.A.; Lim, T.; Qiu, D.; Coughlin, S.; Liu, Z.; Wang, Y.; Blouch, R.; Moulton, H.M.; Iversen, P.L.; et al. Inhibition of Coxsackievirus B3 in Cell Cultures and in Mice by Peptide-Conjugated Morpholino Oligomers Targeting the Internal Ribosome Entry Site. J. Virol. 2006, 80, 11510–11519. [Google Scholar] [CrossRef] [PubMed]
- Paessler, S.; Rijnbrand, R.; Stein, D.A.; Ni, H.; Yun, N.E.; Dziuba, N.; Borisevich, V.; Seregin, A.; Ma, Y.; Blouch, R.; et al. Inhibition of Alphavirus Infection in Cell Culture and in Mice with Antisense Morpholino Oligomers. Virology 2008, 376, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.-H.; Stein, D.A.; Guerrero-Plata, A.; Liao, S.-L.; Ivanciuc, T.; Hong, C.; Iversen, P.L.; Casola, A.; Garofalo, R.P. Inhibition of Respiratory Syncytial Virus Infections With Morpholino Oligomers in Cell Cultures and in Mice. Mol. Ther. 2008, 16, 1120–1128. [Google Scholar] [CrossRef]
- Sleeman, K.; Stein, D.A.; Tamin, A.; Reddish, M.; Iversen, P.L.; Rota, P.A. Inhibition of Measles Virus Infections in Cell Cultures by Peptide-Conjugated Morpholino Oligomers. Virus Res. 2009, 140, 49–56. [Google Scholar] [CrossRef]
- Ge, Q.; Pastey, M.; Kobasa, D.; Puthavathana, P.; Lupfer, C.; Bestwick, R.K.; Iversen, P.L.; Chen, J.; Stein, D.A. Inhibition of Multiple Subtypes of Influenza A Virus in Cell Cultures with Morpholino Oligomers. Antimicrob. Agents Chemother. 2006, 50, 3724–3733. [Google Scholar] [CrossRef]
- Gabriel, G.; Nordmann, A.; Stein, D.A.; Iversen, P.L.; Klenk, H.-D. Morpholino Oligomers Targeting the PB1 and NP Genes Enhance the Survival of Mice Infected with Highly Pathogenic Influenza A H7N7 Virus. J. Gen. Virol. 2008, 89, 939–948. [Google Scholar] [CrossRef]
- Lupfer, C.; Stein, D.A.; Mourich, D.v.; Tepper, S.E.; Iversen, P.L.; Pastey, M. Inhibition of Influenza A H3N8 Virus Infections in Mice by Morpholino Oligomers. Arch. Virol. 2008, 153, 929–937. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Bonaparte, R.S.; Patel, D.; Stein, D.A.; Iversen, P.L. Blockade of Viral Interleukin-6 Expression of Kaposi’s Sarcoma–Associated Herpesvirus. Mol. Cancer Ther. 2008, 7, 712–720. [Google Scholar] [CrossRef]
- Moerdyk-Schauwecker, M.; Stein, D.A.; Eide, K.; Blouch, R.E.; Bildfell, R.; Iversen, P.; Jin, L. Inhibition of HSV-1 Ocular Infection with Morpholino Oligomers Targeting ICP0 and ICP27. Antivir. Res. 2009, 84, 131–141. [Google Scholar] [CrossRef]
- Kinney, R.M.; Huang, C.Y.-H.; Rose, B.C.; Kroeker, A.D.; Dreher, T.W.; Iversen, P.L.; Stein, D.A. Inhibition of Dengue Virus Serotypes 1 to 4 in Vero Cell Cultures with Morpholino Oligomers. J. Virol. 2005, 79, 5116–5128. [Google Scholar] [CrossRef]
- Holden, K.L.; Stein, D.A.; Pierson, T.C.; Ahmed, A.A.; Clyde, K.; Iversen, P.L.; Harris, E. Inhibition of Dengue Virus Translation and RNA Synthesis by a Morpholino Oligomer Targeted to the Top of the Terminal 3′ Stem–Loop Structure. Virology 2006, 344, 439–452. [Google Scholar] [CrossRef]
- Stein, D.A.; Huang, C.Y.-H.; Silengo, S.; Amantana, A.; Crumley, S.; Blouch, R.E.; Iversen, P.L.; Kinney, R.M. Treatment of AG129 Mice with Antisense Morpholino Oligomers Increases Survival Time Following Challenge with Dengue 2 Virus. J. Antimicrob. Chemother. 2008, 62, 555–565. [Google Scholar] [CrossRef]
- Neuman, B.W.; Stein, D.A.; Kroeker, A.D.; Churchill, M.J.; Kim, A.M.; Kuhn, P.; Dawson, P.; Moulton, H.M.; Bestwick, R.K.; Iversen, P.L.; et al. Inhibition, Escape, and Attenuated Growth of Severe Acute Respiratory Syndrome Coronavirus Treated with Antisense Morpholino Oligomers. J. Virol. 2005, 79, 9665–9676. [Google Scholar] [CrossRef]
- van den Born, E.; Stein, D.A.; Iversen, P.L.; Snijder, E.J. Antiviral Activity of Morpholino Oligomers Designed to Block Various Aspects of Equine Arteritis Virus Amplification in Cell Culture. J. Gen. Virol. 2005, 86, 3081–3090. [Google Scholar] [CrossRef]
- Vagnozzi, A.; Stein, D.A.; Iversen, P.L.; Rieder, E. Inhibition of Foot-and-Mouth Disease Virus Infections in Cell Cultures with Antisense Morpholino Oligomers. J. Virol. 2007, 81, 11669–11680. [Google Scholar] [CrossRef]
- Neuman, B.W.; Stein, D.A.; Kroeker, A.D.; Paulino, A.D.; Moulton, H.M.; Iversen, P.L.; Buchmeier, M.J. Antisense Morpholino-Oligomers Directed against the 5′ End of the Genome Inhibit Coronavirus Proliferation and Growth. J. Virol. 2004, 78, 5891–5899. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Stein, D.A.; Fan, S.-M.; Wang, K.-Y.; Kroeker, A.D.; Meng, X.-J.; Iversen, P.L.; Matson, D.O. Suppression of Porcine Reproductive and Respiratory Syndrome Virus Replication by Morpholino Antisense Oligomers. Vet. Microbiol. 2006, 117, 117–129. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Wang, K.-Y.; Stein, D.A.; Patel, D.; Watkins, R.; Moulton, H.M.; Iversen, P.L.; Matson, D.O. Inhibition of Replication and Transcription Activator and Latency-Associated Nuclear Antigen of Kaposi’s Sarcoma-Associated Herpesvirus by Morpholino Oligomers. Antivir. Res. 2007, 73, 12–23. [Google Scholar] [CrossRef]
- Swenson, D.L.; Warfield, K.L.; Warren, T.K.; Lovejoy, C.; Hassinger, J.N.; Ruthel, G.; Blouch, R.E.; Moulton, H.M.; Weller, D.D.; Iversen, P.L.; et al. Chemical Modifications of Antisense Morpholino Oligomers Enhance Their Efficacy against Ebola Virus Infection. Antimicrob. Agents Chemother. 2009, 53, 2089–2099. [Google Scholar] [CrossRef] [PubMed]
- Enterlein, S.; Warfield, K.L.; Swenson, D.L.; Stein, D.A.; Smith, J.L.; Gamble, C.S.; Kroeker, A.D.; Iversen, P.L.; Bavari, S.; Mühlberger, E. VP35 Knockdown Inhibits Ebola Virus Amplification and Protects against Lethal Infection in Mice. Antimicrob. Agents Chemother. 2006, 50, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, N.; Basu, A.; Palumbo, P.; Myers, R.L.; Pandey, V.N. Anti-TAR Polyamide Nucleotide Analog Conjugated with a Membrane-Permeating Peptide Inhibits Human Immunodeficiency Virus Type 1 Production. J. Virol. 2002, 76, 3881–3891. [Google Scholar] [CrossRef] [PubMed]
- Chaubey, B.; Tripathi, S.; Ganguly, S.; Harris, D.; Casale, R.A.; Pandey, V.N. A PNA-Transportan Conjugate Targeted to the TAR Region of the HIV-1 Genome Exhibits Both Antiviral and Virucidal Properties. Virology 2005, 331, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S. Anti-HIV-1 Activity of Anti-TAR Polyamide Nucleic Acid Conjugated with Various Membrane Transducing Peptides. Nucleic Acids Res. 2005, 33, 4345–4356. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Chaubey, B.; Barton, B.E.; Pandey, V.N. Anti HIV-1 Virucidal Activity of Polyamide Nucleic Acid-Membrane Transducing Peptide Conjugates Targeted to Primer Binding Site of HIV-1 Genome. Virology 2007, 363, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.J. Cell-Penetrating Peptide Conjugates of Peptide Nucleic Acids (PNA) as Inhibitors of HIV-1 Tat-Dependent Trans-Activation in Cells. Nucleic Acids Res. 2005, 33, 6837–6849. [Google Scholar] [CrossRef]
- Upadhyay, A.; Ponzio, N.M.; Pandey, V.N. Immunological Response to Peptide Nucleic Acid and Its Peptide Conjugate Targeted to Transactivation Response (TAR) Region of HIV-1 RNA Genome. Oligonucleotides 2008, 18, 329–335. [Google Scholar] [CrossRef]
- Chaubey, B.; Tripathi, S.; Pandey, V.N. Single Acute-Dose and Repeat-Doses Toxicity of Anti-HIV-1 PNA TAR –Penetratin Conjugate after Intraperitoneal Administration to Mice. Oligonucleotides 2008, 18, 9–20. [Google Scholar] [CrossRef]
- Ganguly, S.; Chaubey, B.; Tripathi, S.; Upadhyay, A.; Neti, P.V.S.V.; Howell, R.W.; Pandey, V.N. Pharmacokinetic Analysis of Polyamide Nucleic-Acid-Cell Penetrating Peptide Conjugates Targeted against HIV-1 Transactivation Response Element. Oligonucleotides 2008, 18, 277–286. [Google Scholar] [CrossRef]
- Yoo, J.-S.; Kim, C.-M.; Kim, J.-H.; Kim, J.-Y.; Oh, J.-W. Inhibition of Japanese Encephalitis Virus Replication by Peptide Nucleic Acids Targeting Cis-Acting Elements on the plus- and Minus-Strands of Viral RNA. Antivir. Res. 2009, 82, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Han, S.; Hong, W.; Lang, Y.; Li, F.; Liu, Y.; Li, Z.; Wu, Y.; Li, W.; Zhang, X.; et al. A Tat-Conjugated Peptide Nucleic Acid Tat-PNA-DR Inhibits Hepatitis B Virus Replication In Vitro and In Vivo by Targeting LTR Direct Repeats of HBV RNA. Mol. Ther. Nucleic Acids 2016, 5, e295. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.-G.; Shim, S.-B.; Moon, J.-E.; Kim, J.-H.; Kim, S.-J.; Oh, J.-W. Interference of Hepatitis C Virus Replication in Cell Culture by Antisense Peptide Nucleic Acids Targeting the X-RNA. J. Viral Hepat. 2011, 18, e298–e306. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.-G.; Lee, W.; Choi, J.-K.; Kim, S.-J.; Plant, E.P.; Almazán, F.; Taylor, D.R.; Enjuanes, L.; Oh, J.-W. Interference of Ribosomal Frameshifting by Antisense Peptide Nucleic Acids Suppresses SARS Coronavirus Replication. Antivir. Res. 2011, 91, 1–10. [Google Scholar] [CrossRef]
- Meng, S.; Wei, B.; Xu, R.; Zhang, K.; Wang, L.; Zhang, R.; Li, J. TAT Peptides Mediated Small Interfering RNA Delivery to Huh-7 Cells and Efficiently Inhibited Hepatitis C Virus RNA Replication. Intervirology 2009, 52, 135–140. [Google Scholar] [CrossRef]
- Kumar, P.; Ban, H.-S.; Kim, S.-S.; Wu, H.; Pearson, T.; Greiner, D.L.; Laouar, A.; Yao, J.; Haridas, V.; Habiro, K.; et al. T Cell-Specific SiRNA Delivery Suppresses HIV-1 Infection in Humanized Mice. Cell 2008, 134, 577–586. [Google Scholar] [CrossRef]
- Bivalkar-Mehla, S.; Mehla, R.; Chauhan, A. Chimeric Peptide-Mediated SiRNA Transduction to Inhibit HIV-1 Infection. J. Drug Target 2017, 25, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ren, W.; Liu, Q.; Tan, Z.; Li, J.; Tong, C. Transportan-Derived Cell-Penetrating Peptide Delivers SiRNA to Inhibit Replication of Influenza Virus in Vivo. Drug Des. Devel. Ther. 2019, 13, 1059. [Google Scholar] [CrossRef]
- Mino, T.; Mori, T.; Aoyama, Y.; Sera, T. Cell-Permeable Artificial Zinc-Finger Proteins as Potent Antiviral Drugs for Human Papillomaviruses. Arch. Virol. 2008, 153, 1291–1298. [Google Scholar] [CrossRef]
- Chu, X.; Wu, B.; Fan, H.; Hou, J.; Hao, J.; Hu, J.; Wang, B.; Liu, G.; Li, C.; Meng, S. PTD-Fused P53 as a Potential Antiviral Agent Directly Suppresses HBV Transcription and Expression. Antivir. Res. 2016, 127, 41–49. [Google Scholar] [CrossRef]
- Jung, H.; Oh, J.; Lee, H. Cell-Penetrating Mx1 Enhances Anti-Viral Resistance against Mucosal Influenza Viral Infection. Viruses 2019, 11, 109. [Google Scholar] [CrossRef] [PubMed]
- Mendonça, D.A.; Bakker, M.; Cruz-Oliveira, C.; Neves, V.; Jiménez, M.A.; Defaus, S.; Cavaco, M.; Veiga, A.S.; Cadima-Couto, I.; Castanho, M.A.R.B.; et al. Penetrating the Blood-Brain Barrier with New Peptide–Porphyrin Conjugates Having Anti-HIV Activity. Bioconjug. Chem. 2021, 32, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Todorovski, T.; Mendonça, D.A.; Fernandes-Siqueira, L.O.; Cruz-Oliveira, C.; Guida, G.; Valle, J.; Cavaco, M.; Limas, F.I.V.; Neves, V.; Cadima-Couto, Í.; et al. Targeting Zika Virus with New Brain- and Placenta-Crossing Peptide–Porphyrin Conjugates. Pharmaceutics 2022, 14, 738. [Google Scholar] [CrossRef] [PubMed]
- Saarbach, J.; Sabale, P.M.; Winssinger, N. Peptide Nucleic Acid (PNA) and Its Applications in Chemical Biology, Diagnostics, and Therapeutics. Curr. Opin. Chem. Biol. 2019, 52, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; McQuistan, T.J.; Stanek, J.W.; Summerton, J.E.; Mata, J.E.; Squier, T.C. Detection of Unique Ebola Virus Oligonucleotides Using Fluorescently-Labeled Phosphorodiamidate Morpholino Oligonucleotide Probe Pairs. Anal. Biochem. 2018, 557, 84–90. [Google Scholar] [CrossRef]
- Pärn, K.; Eriste, E.; Langel, Ü. The Antimicrobial and Antiviral Applications of Cell-Penetrating Peptides. Methods Mol. Biol. 2015, 1324, 223–245. [Google Scholar]
- Sadiq, I.Z.; Muhammad, A.; Mada, S.B.; Ibrahim, B.; Umar, U.A. Biotherapeutic Effect of Cell-Penetrating Peptides against Microbial Agents: A Review. Tissue Barriers 2022, 10, 1995285. [Google Scholar] [CrossRef]
- Kalafatovic, D.; Giralt, E. Cell-Penetrating Peptides: Design Strategies beyond Primary Structure and Amphipathicity. Molecules 2017, 22, 1929. [Google Scholar] [CrossRef]
- Gallo, M.; Defaus, S.; Andreu, D. 1988–2018: Thirty Years of Drug Smuggling at the Nano Scale. Challenges and Opportunities of Cell-Penetrating Peptides in Biomedical Research. Arch. Biochem. Biophys. 2019, 661, 74–86. [Google Scholar] [CrossRef]
- Sánchez-Navarro, M.; Giralt, E. Peptide Shuttles for Blood–Brain Barrier Drug Delivery. Pharmaceutics 2022, 14, 1874. [Google Scholar] [CrossRef]
- Kalafatovic, D.; Mauša, G.; Todorovski, T.; Giralt, E. Algorithm-Supported, Mass and Sequence Diversity-Oriented Random Peptide Library Design. J. Cheminform. 2019, 11, 25. [Google Scholar] [CrossRef]
- Otović, E.; Njirjak, M.; Kalafatovic, D.; Mauša, G. Sequential Properties Representation Scheme for Recurrent Neural Network-Based Prediction of Therapeutic Peptides. J. Chem. Inf. Model 2022, 62, 2961–2972. [Google Scholar] [CrossRef]
- Torrent, M.; di Tommaso, P.; Pulido, D.; Nogués, M.V.; Notredame, C.; Boix, E.; Andreu, D. AMPA: An Automated Web Server for Prediction of Protein Antimicrobial Regions. Bioinformatics 2012, 28, 130–131. [Google Scholar] [CrossRef]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide Therapeutics: Current Status and Future Directions. Drug. Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef]
- Kaspar, A.A.; Reichert, J.M. Future Directions for Peptide Therapeutics Development. Drug. Discov. Today 2013, 18, 807–817. [Google Scholar] [CrossRef]
- Böhme, D.; Beck-Sickinger, A.G. Controlling Toxicity of Peptide-Drug Conjugates by Different Chemical Linker Structures. Chem. Med. Chem. 2015, 10, 804–814. [Google Scholar] [CrossRef]
- Mahesh, S.; Tang, K.-C.; Raj, M. Amide Bond Activation of Biological Molecules. Molecules 2018, 23, 2615. [Google Scholar] [CrossRef]
- Finan, B.; Yang, B.; Ottaway, N.; Stemmer, K.; Müller, T.D.; Yi, C.-X.; Habegger, K.; Schriever, S.C.; García-Cáceres, C.; Kabra, D.G.; et al. Targeted Estrogen Delivery Reverses the Metabolic Syndrome. Nat. Med. 2012, 18, 1847–1856. [Google Scholar] [CrossRef]
- Lambeth, T.R.; Dai, Z.; Zhang, Y.; Julian, R.R. A Two-Trick Pony: Lysosomal Protease Cathepsin B Possesses Surprising Ligase Activity. RSC Chem. Biol. 2021, 2, 606–611. [Google Scholar] [CrossRef]
- Tugyi, R.; Mezõ, G.; Gitta, S.; Fellinger, E.; Andreu, D.; Hudecz, F. Effect of Conjugation with Polypeptide Carrier on the Enzymatic Degradation of Herpes Simplex Virus Glycoprotein D Derived Epitope Peptide. Bioconjug. Chem. 2008, 19, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Marqués-Gallego, P.; de Kroon, A.I.P.M. Ligation Strategies for Targeting Liposomal Nanocarriers. Biomed. Res. Int. 2014, 2014, 129458. [Google Scholar] [CrossRef] [PubMed]
- Spicer, C.D.; Jumeaux, C.; Gupta, B.; Stevens, M.M. Peptide and Protein Nanoparticle Conjugates: Versatile Platforms for Biomedical Applications. Chem. Soc. Rev. 2018, 47, 3574–3620. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, C.; Behnam, M.A.M.; Steuer, C.; Klein, C.D. Retro Peptide-Hybrids as Selective Inhibitors of the Dengue Virus NS2B-NS3 Protease. Antivir. Res. 2012, 94, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Moulton, H.M.; Nelson, M.H.; Hatlevig, S.A.; Reddy, M.T.; Iversen, P.L. Cellular Uptake of Antisense Morpholino Oligomers Conjugated to Arginine-Rich Peptides. Bioconjug. Chem. 2004, 15, 290–299. [Google Scholar] [CrossRef]
- Abes, S.; Moulton, H.M.; Clair, P.; Prevot, P.; Youngblood, D.S.; Wu, R.P.; Iversen, P.L.; Lebleu, B. Vectorization of Morpholino Oligomers by the (R-Ahx-R)4 Peptide Allows Efficient Splicing Correction in the Absence of Endosomolytic Agents. J. Control. Release 2006, 116, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Stenzel, M.H. Bioconjugation Using Thiols: Old Chemistry Rediscovered to Connect Polymers with Nature’s Building Blocks. ACS Macro Lett. 2013, 2, 14–18. [Google Scholar] [CrossRef]
- Fishkin, N.; Maloney, E.K.; Chari, R.V.J.; Singh, R. A Novel Pathway for Maytansinoid Release from Thioether Linked Antibody–Drug Conjugates (ADCs) under Oxidative Conditions. Chem. Commun. 2011, 47, 10752–10754. [Google Scholar] [CrossRef]
- Baldwin, A.D.; Kiick, K.L. Tunable Degradation of Maleimide–Thiol Adducts in Reducing Environments. Bioconjug. Chem. 2011, 22, 1946–1953. [Google Scholar] [CrossRef]
- Andrieu, J.; Re, F.; Russo, L.; Nicotra, F. Phage-Displayed Peptides Targeting Specific Tissues and Organs. J. Drug Target 2019, 27, 555–565. [Google Scholar] [CrossRef]
- Saw, P.E.; Song, E.-W. Phage Display Screening of Therapeutic Peptide for Cancer Targeting and Therapy. Protein Cell 2019, 10, 787–807. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-H.; Liu, I.-J.; Lu, R.-M.; Wu, H.-C. Advancement and Applications of Peptide Phage Display Technology in Biomedical Science. J. Biomed. Sci. 2016, 23, 8. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Sun, Y.; Lee, R.J.; Wang, G.; Zheng, X.; Zhang, H.; Fu, Y.; Yan, G.; Wang, Y.; Deng, W.; et al. Folate Receptor-Targeted Albumin Nanoparticles Based on Microfluidic Technology to Deliver Cabazitaxel. Cancers 2019, 11, 1571. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, S.; Chai, H.; Yang, Z.; Lee, R.J.; Liao, W.; Teng, L. A Novel Isoquinoline Derivative Anticancer Agent and Its Targeted Delivery to Tumor Cells Using Transferrin-Conjugated Liposomes. PLoS ONE 2015, 10, e0136649. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Shin, M.C.; Liang, Q.; He, H.; Yang, V.C. 15 Years of ATTEMPTS: A Macromolecular Drug Delivery System Based on the CPP-Mediated Intracellular Drug Delivery and Antibody Targeting. J. Control. Release 2015, 205, 58–69. [Google Scholar] [CrossRef]
- Orange, J.S.; May, M.J. Cell Penetrating Peptide Inhibitors of Nuclear Factor-Kappa B. Cellular and Molecular Life Sci. 2008, 65, 3564–3591. [Google Scholar] [CrossRef]
- Wang, F.; Wang, Y.; Zhang, X.; Zhang, W.; Guo, S.; Jin, F. Recent Progress of Cell-Penetrating Peptides as New Carriers for Intracellular Cargo Delivery. J. Control. Release 2014, 174, 126–136. [Google Scholar] [CrossRef]
- Kristensen, M.; Birch, D.; Mørck Nielsen, H. Applications and Challenges for Use of Cell-Penetrating Peptides as Delivery Vectors for Peptide and Protein Cargos. Int. J. Mol. Sci. 2016, 17, 185. [Google Scholar] [CrossRef]
- Gavriel, A.G.; Sambrook, M.R.; Russell, A.T.; Hayes, W. Recent Advances in Self-Immolative Linkers and Their Applications in Polymeric Reporting Systems. Polym. Chem. 2022, 13, 3188–3269. [Google Scholar] [CrossRef]
- Dal Corso, A.; Arosio, S.; Arrighetti, N.; Perego, P.; Belvisi, L.; Pignataro, L.; Gennari, C. A Trifunctional Self-Immolative Spacer Enables Drug Release with Two Non-Sequential Enzymatic Cleavages. Chem. Commun. 2021, 57, 7778–7781. [Google Scholar] [CrossRef]
- Viru, L.; Heller, G.; Lehto, T.; Pärn, K.; el Andaloussi, S.; Langel, Ü.; Merits, A. Novel Viral Vectors Utilizing Intron Splice-Switching to Activate Genome Rescue, Expression and Replication in Targeted Cells. Virol. J. 2011, 8, 243. [Google Scholar] [CrossRef] [PubMed]




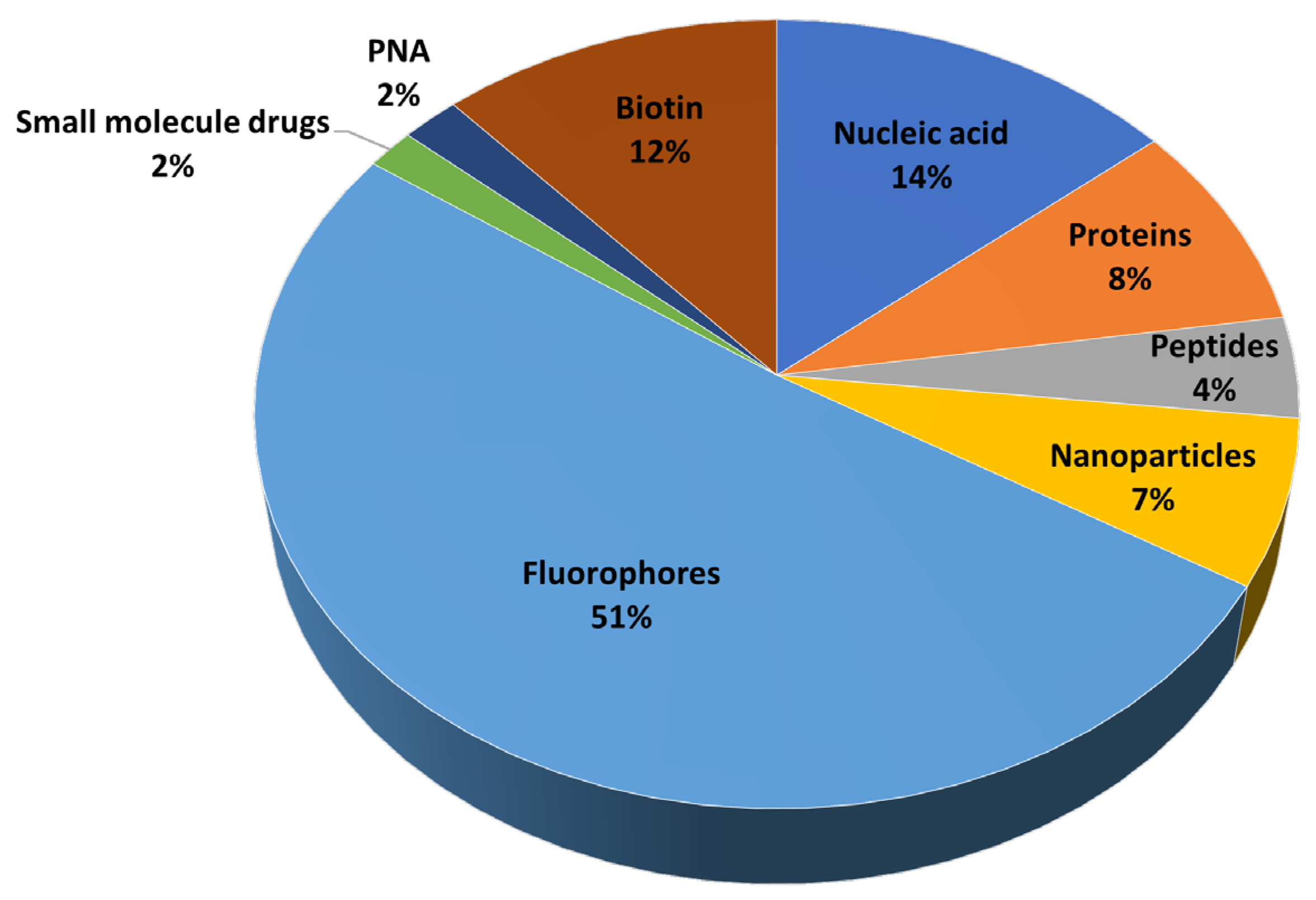{kind=link}
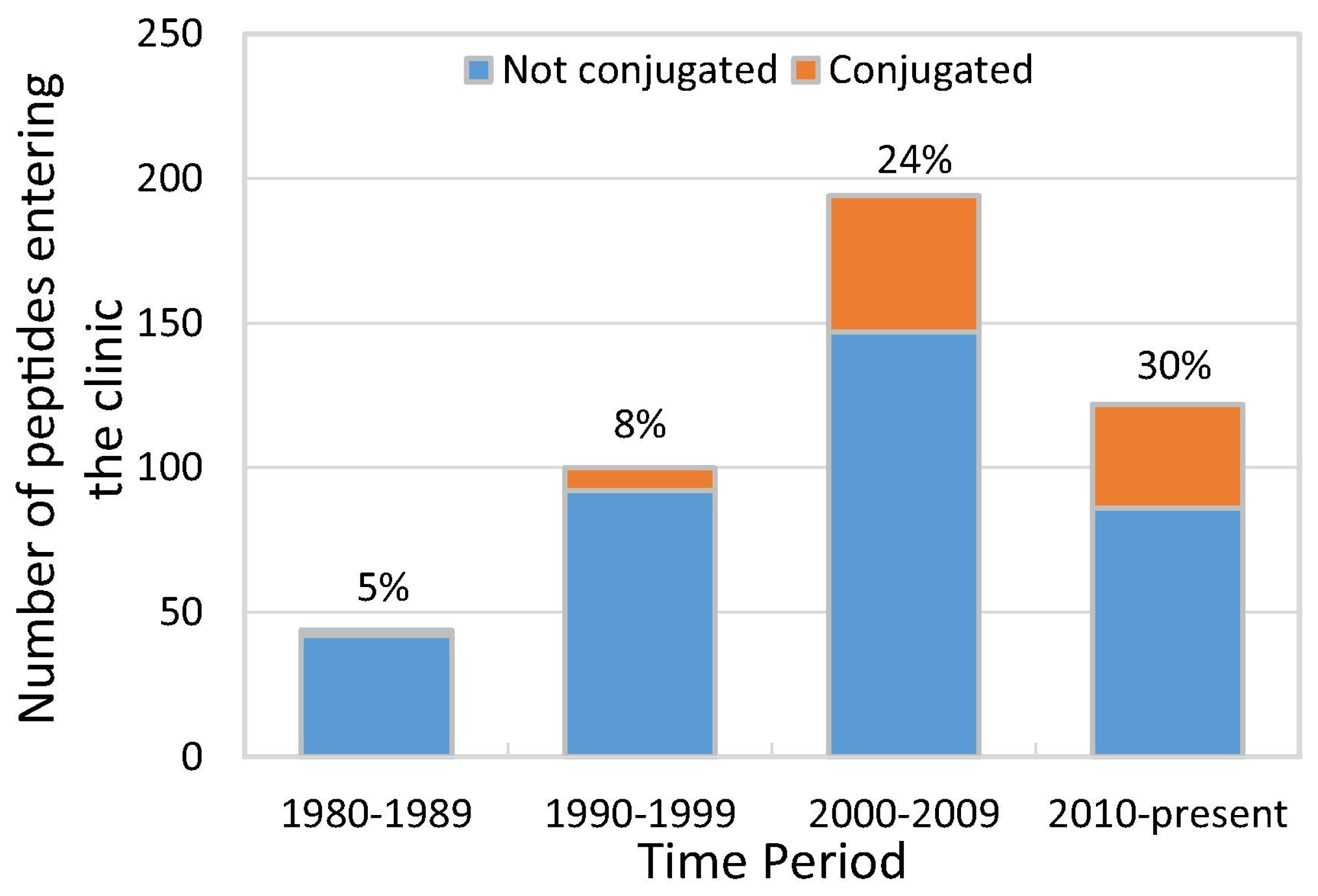{kind=link}
{kind=link}
| Total Numbers | Percentage (%) a | ||
|---|---|---|---|
| Sequence type | Linear | 1753 | 94.5 |
| Cyclic | 102 | 5.5 | |
| Peptide class | Cationic | 714 | 38.5 |
| Amphipathic | 391 | 21.1 | |
| Origin | Protein | 774 | 41.7 |
| Synthetic | 1017 | 54.8 | |
| Chimeric | 64 | 3.5 | |
| Chirality | L | 1564 | 84.3 |
| D | 63 | 3.4 | |
| Mixed | 32 | 1.7 | |
| Modified | 110 | 5.9 | |
| Length | Up to 5 AA | 60 | 3.2 |
| 6–10 AA | 384 | 20.7 | |
| 11–15 AA | 550 | 29.6 | |
| 16–20 AA | 446 | 24.1 | |
| 21–30 AA | 320 | 17.3 | |
| >30 AA | 95 | 5.1 | |
| Entry | Antiviral Cargo | CPP | Conjugation Chemistry | Targeted Virus | Experimental System | Literature |
|---|---|---|---|---|---|---|
| 1 | Indole | βAla-EYAARIEALIRAAQEQQEKNEAALRE | Click chemistry | HIV-1 | Cell culture (HL2/3 and MT-2 cells) | [28] |
| 2 | N-carboxyphenylpyrrole derivative (Gls) | |||||
| 3 | Indole | βAla-EYAARIEALIRAAQEQQKKNEE | ||||
| 4 | N-carboxyphenylpyrrole derivative (Gls) | |||||
| 5 | Indole | βAla-EYAARIEALIRAAQEQQKK | ||||
| 6 | N-carboxyphenylpyrrole derivative (Gls) | |||||
| 7 | Carboxymethyl derivative of N-(3-carboxy-4-hydroxyphenyl)-2,5-dimethylpyrrole (Aoc) | βAla-NNYTSLIHSLIEESQNQQEKNEQELL | Amide bond formation | HIV-1 | Cell culture (HL2/3 and MT-2 cells) | [29] |
| 8 | Carboxymethyl derivative of N-(4-carboxy-3-hydroxyphenyl)-2,5-dimethylpyrrole (Noc) | |||||
| 9 | 2-5A 2′ 5′-phosphodiester linker oligoadenylate | GGRRKKRRQRRR (HIV-Tat) | Click chemistry | HIV | Cell culture (HeLa M cells) | [30] |
| 10 | 2-5A 2′ 5′-phosphodiester linker oligoadenylate | CGGRKKRRQRRR (HIV-Tat) | Thiol-chloroacetyl ligation | |||
| 11 | N-3 aminopropyl TSAO-T | VAVP | Amide bond formation | HIV-1 | Cell culture (Human T lymphocytic CEM and MT-4 cells) | [31] |
| 12 | VAVA | |||||
| 13 | KPDP | |||||
| 14 | Acyclovir | VPVP | Amide bond formation | HSV-1, HSV-2 | Cell culture (HEL cells) | [32] |
| 15 | VPV | Ester bond formation | ||||
| 16 | Zidovudine (AZT) | Boc-FP; Boc-NFP; Boc-FPI; Boc-NFPI; Fmoc-FP; Fmoc-NFP; Fmoc-FPI; Fmoc-NFPI; Z-FP; Z-NFP; Z-FPI; Z-NFPI; Qnc-FP; Qnc-NFP; Qnc-FPI; Qnc-NFPI | Ester bond formation | HIV-1 | Cell culture (CEM-SS TK+, CEM-SS TK- and MT-4 cells) | [33] |
| 17 | Zidovudine monophosphate (AZT-MT) | FP-OMe; FPI-OMe; NFP-OMe; NFPI-OMe; AFP-OMe; AFPI-OMe; ANFP-OMe; ANFPI-OMe | Phosphoramidate bond formation | |||
| 18 | Rhodanine b | Arg-Lys-Nle | Amide bond formation | Dengue virus, West Nile fever virus | Cell culture (Huh-7 cells) | [34] |
| 19 | Thiazolidinedione a | |||||
| 20 | GRL0617 (C20H20N2O) | ECLRGM (cyclic) | Amide bond formation | SARS-CoV-2 | Cell culture (Human kidney cells 293T; Human lung adenocarcinoma A549 cells; HCT116 cells) | [35] |
| EMLRGC (cyclic) | ||||||
| 21 | 25-Hydroxycholesterol (25-HC) | SLDQINVTFLDLEYEMKKLEEAIKKLEESYIDLKELGSGSG | Amide bond formation through linker | SARS-CoV-2 | Human kidney 293T cells; Huh-7 cells; RD cells; Caco2 cells | [36,37] |
| Palmitic acid (C16) | ||||||
| 22 | PMO | (RAhx c R)4 | Amide bond formation through linker | SARS-CoV-2 | Vero-E6 cells | [38] |
| 23 | PMO | (RAhxR)4-Ahx-βAla | Amide bond formation through linker; thioether bond formation through linker; | West Nile fever virus, Japanese encephalitis virus, St. Louis encephalitis virus, Coxsackievirus B2, Coxsackievirus B3, poliovirus 1, human rhinovirus 14, mouse hepatitis virus, Venezuelan equine encephalitis virus, respiratory syncytial virus, measles virus, influenza A virus, Kaposi’s sarcoma-associated herpesvirus, herpesvirus type 1 | Cell culture (KSHV-infected BC-1 and BCBL-1 cells; MDCK cells; Vero or Vero/hSLAM cells; HeLa and HL-1 cells; BHK-21 cells), in vivo mouse infection model | [39,40,41,42,43,44,45,46,47,48,49,50,51] |
| 24 | PMO | RRRRRFFRRRRC; RRRRRRRRRFFC; (RAhxR)4-Ahx-βAla | Amide bond formation through linker; thioether bond formation through linker | Dengue virus | Cell culture (Vero and BHK-21 cells), in vivo mouse infection model | [52,53,54] |
| 25 | PMO | RRRRRFFRRRRC; RRRRRRRRRFFC | Thioether bond formation through linker; | SARS-CoV1 | Cell culture (Vero-E6 cells) | [55] |
| 26 | PMO | RRRRRRRRRFFC | Thioether bond formation through linker | Equine arteritis virus, foot-and-mouth disease virus, poliovirus 1, human rhinovirus 14, coxsackievirus B2, Mouse hepatitis virus, Sindbis virus | Cell culture (BHK-21 and Vero cells; DBT cells; HeLa cells; Vero-E6 cells) | [41,44,56,57,58] |
| 27 | PMO | RRRRRFFRRRRC | Thioether bond formation through linker | Influenza A virus, porcine reproductive and respiratory syndrome virus, Kaposis sarcoma-associated herpesvirus | Cell culture (ATCC CRL11171 cell line; BC-1 and BCBL-1 cells; MDCK cells) | [47,50,59,60] |
| 28 | PMO | RRRRRRRRRFFC; (RAhxR)4-Ahx-βAla; (RβAla)8βAla; (RAhx)n=2-8βAla | Amide bond formation through linker; thioether bond formation through linker | Ebola virus | Cell culture (Vero and Vero-E6 cells) in vivo mouse infection model | [61,62] |
| 29 | PNA | CGWTLNSAGYLLGKINLKALAALAKKIL; (Npys) d GWTLNSAGYLLGKINLKALAALAKKIL; RQIKIWFQNRRMKWKK; GRKKRRQRRRPPQ; GWYLNSAGYLLGK(Cys)INLKALAALAKKIL; AGYLLGK(Cys)INLKALAALAKKIL; GWYLNSAGYLLGK(Cys)INLKALAAL; GRKKRRQRRRP; GWTLNSAGYLLGKINLKALAALAKKIL; GWYLNSAGYLLGKINLKALAALAKKIL; PKKKRKV; GRKKRRQRRRPC; RQIKIWFQNRRMKWKKGGC; RRRRRRRRRFFC; RRRRRRRQIKIWFQNRRMKWKKGGC | Disulfide bridge formation; amide bond formation through linker | HIV-1 | Cell culture (HeLa cells; 293T cells; CEM CD4+ cells; Jurkat T-cell lymphocites; Vero and Vero E6 cells). in vivo mouse infection model | [63,64,65,66,67,68,69,70] |
| 30 | PNA | GRKKRRQRRRPPQ; GRKKRRQRRRPPC; YGRRRRRRRRR; RKKRRQRRR | Amide bond formation through linker; thiol-maleimide bond formation | Japanese encephalitis virus, hepatitis B virus, hepatitis C virus, SARS-CoV | Cell culture (Huh7 cells; Vero and BHK-21 cells; HepG2.2.1.5, HepG2 and L-02 cells), in vivo mouse infection model | [71,72,73,74] |
| 31 | siRNA | CYGRKKRRQRRR; RRRRRRRRR; KETWWETWWTEWSQPGRKKRRQRRR; GWTLNSAGYLLGKINLKALAALAKKILrrrrrrrrr e | Disulfide bridge formation; thiol-maleimide bond formation; non-covalent complex formation | Hepatitis C virus, HIV-1 influenza virus | Cell culture (Huh7 cells; MDCK and A549 cells; MDM cells), in vivo mouse infection model | [75,76,77,78] |
| 32 | Protein | RRRRRRRRR; YGRKKRRQRRR | Cell expression | Human papilloma virus type 18, hepatitis B virus, mucosal influenza | Cell culture (MDCK cells; Huh7 and HepG2.2.1.5; human cell line 293H), in vivo mouse infection model | [79,80,81] |
| 33 | Porphyrin | AGILKRW AGILKRWK VQQLTKRFSL VQQLTKRFSLK SGTQEEY SGTQEEYK | Amide bond formation | HIV-1 Zika virus | Cell culture (Vero and TZM-bl cells) | [82,83] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Todorovski, T.; Kalafatovic, D.; Andreu, D. Antiviral Peptide-Based Conjugates: State of the Art and Future Perspectives. Pharmaceutics 2023, 15, 357. https://doi.org/10.3390/pharmaceutics15020357
Todorovski T, Kalafatovic D, Andreu D. Antiviral Peptide-Based Conjugates: State of the Art and Future Perspectives. Pharmaceutics. 2023; 15(2):357. https://doi.org/10.3390/pharmaceutics15020357
Chicago/Turabian StyleTodorovski, Toni, Daniela Kalafatovic, and David Andreu. 2023. "Antiviral Peptide-Based Conjugates: State of the Art and Future Perspectives" Pharmaceutics 15, no. 2: 357. https://doi.org/10.3390/pharmaceutics15020357
APA StyleTodorovski, T., Kalafatovic, D., & Andreu, D. (2023). Antiviral Peptide-Based Conjugates: State of the Art and Future Perspectives. Pharmaceutics, 15(2), 357. https://doi.org/10.3390/pharmaceutics15020357