

CD13-Mediated Pegylated Carboxymethyl Chitosan-Capped Mesoporous Silica Nanoparticles for Enhancing the Therapeutic Efficacy of Hepatocellular Carcinoma


Abstract
:1. Introduction
2. Materials and Methods
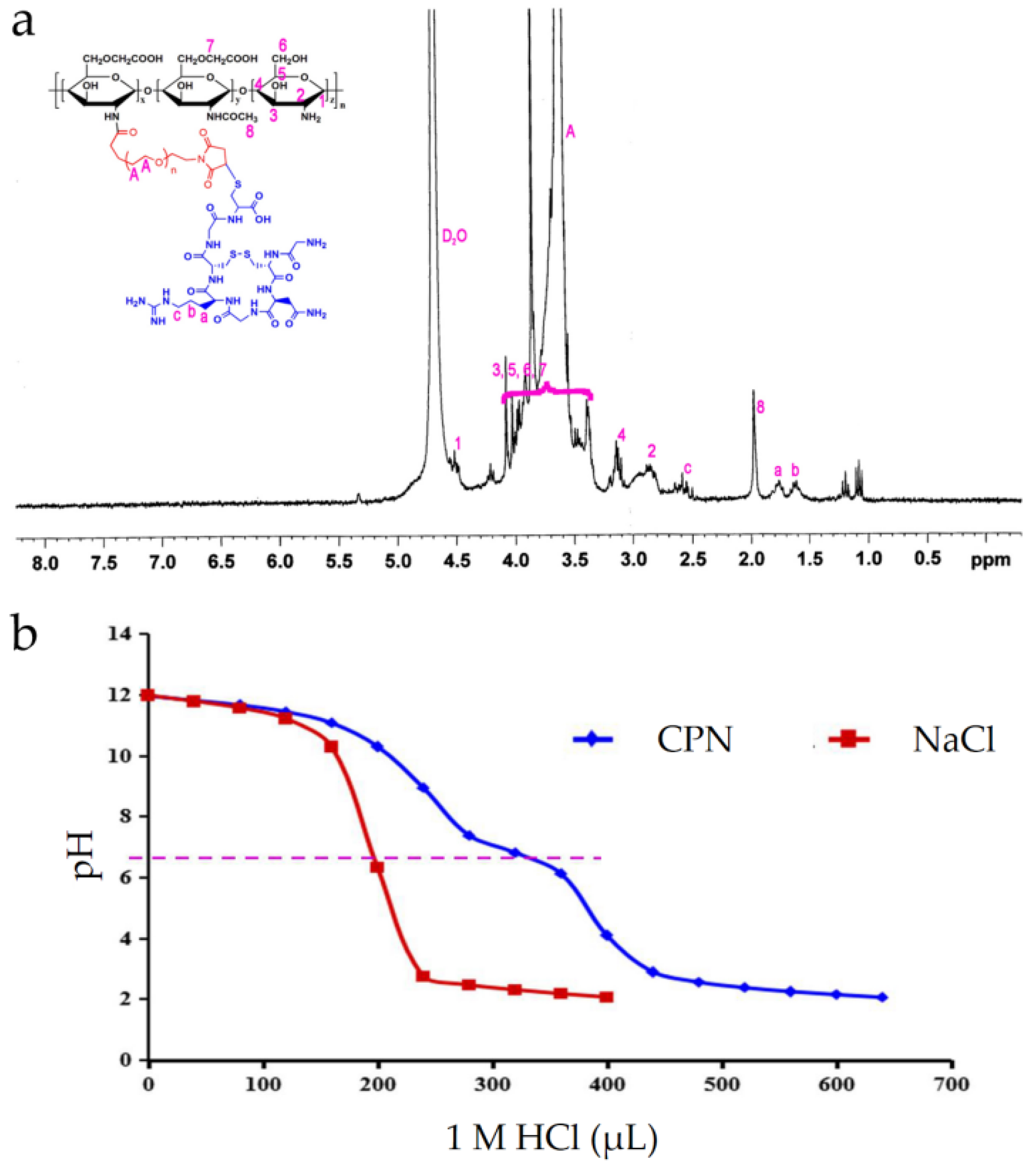
2.1. Synthesis of CPN Conjugates
2.2. Acid-Base Titration of CPN
2.3. Preparation of MSN-NH2 and FITC-MSN (MCM-41 Type)
2.4. Preparation of DOX/MSN-CPN
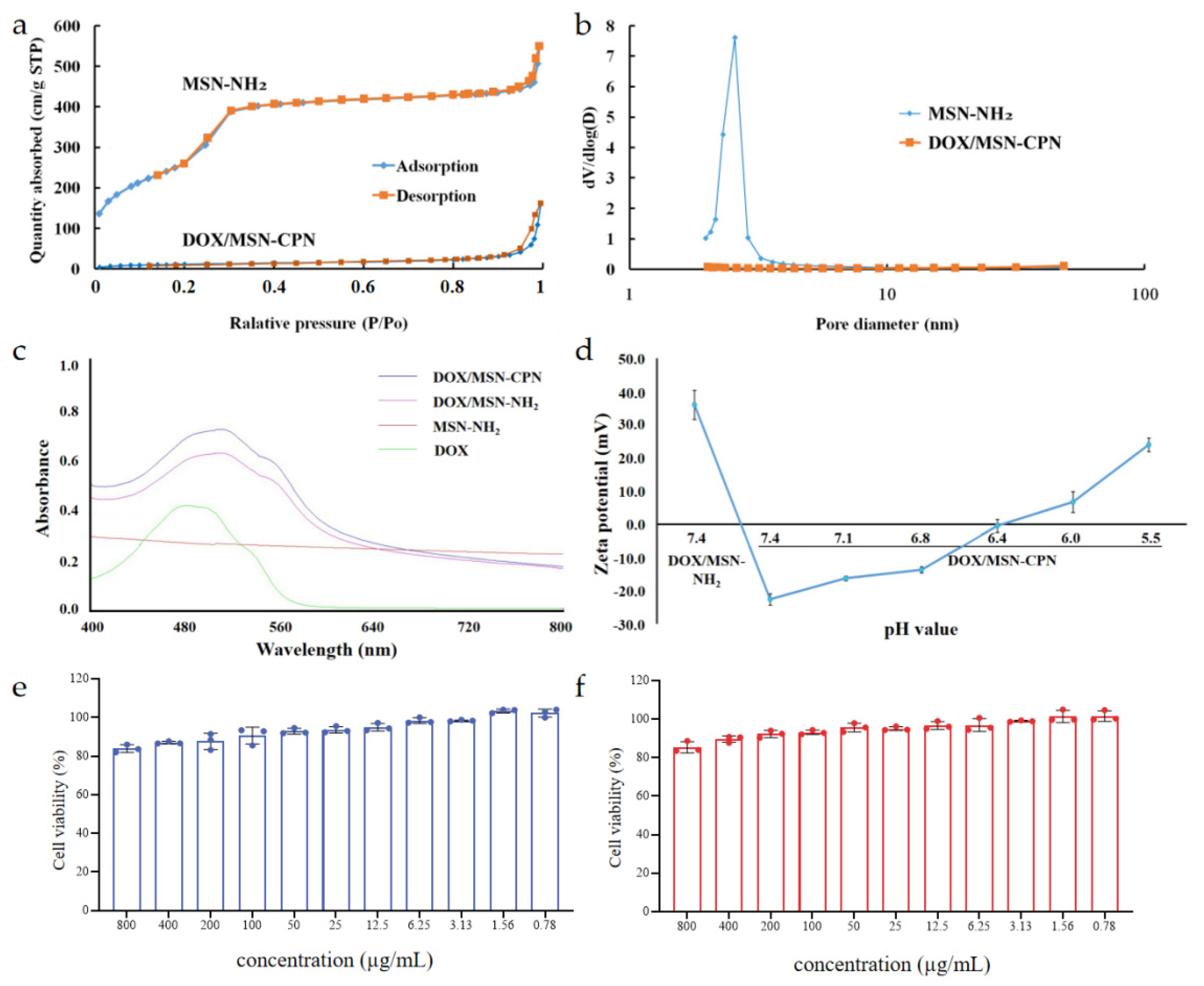
2.5. Physicochemical Characterization
EE (%) = (Wtotal drug input − Wtotal drug in supernatant)/Wtotal drug input × 100%
2.6. In Vitro pH-Sensitivity
2.7. In Vitro Biocompatibility
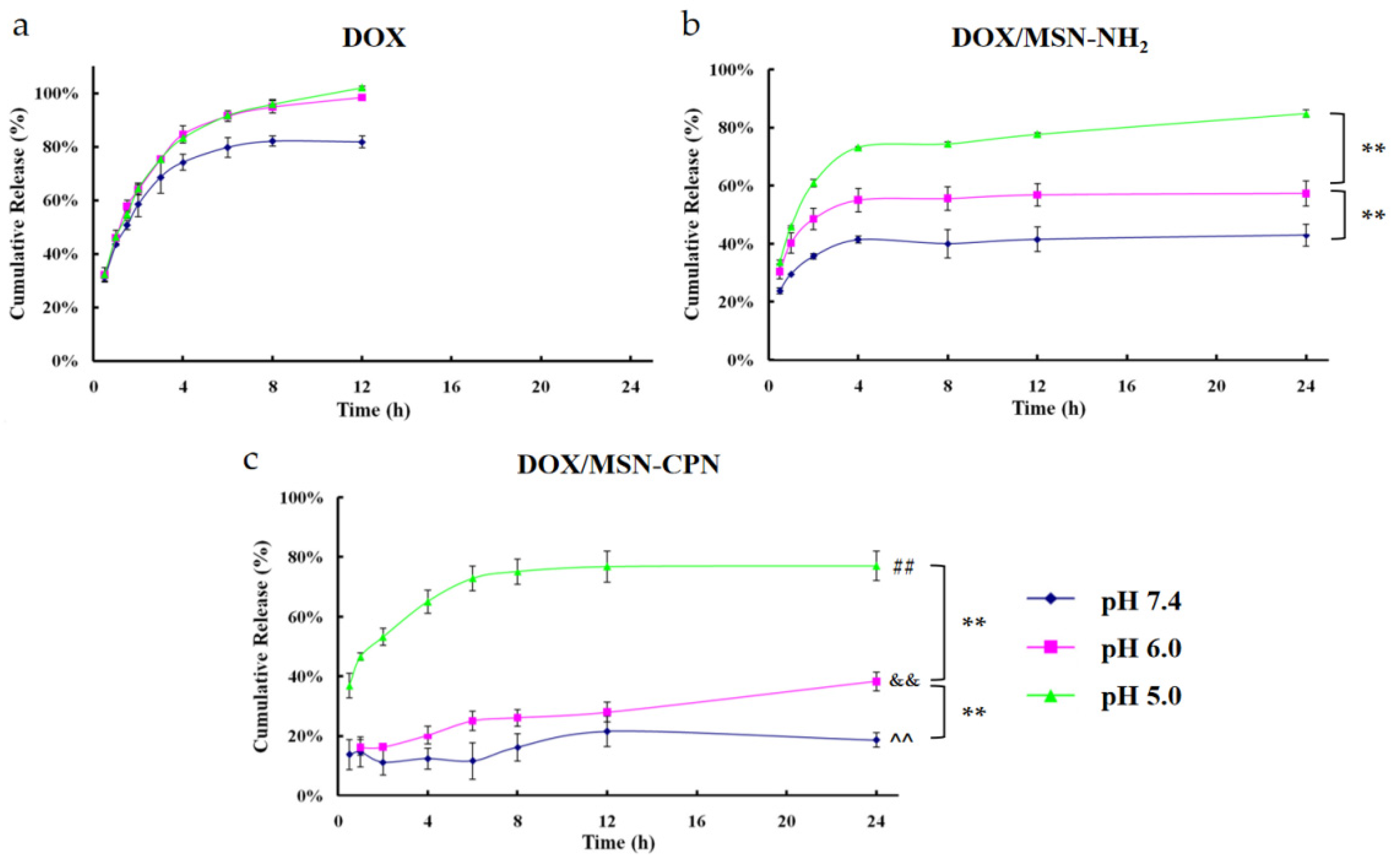
2.8. In Vitro Drug Release
2.9. Cell Culture
2.10. In Vitro Cell Uptake of DOX/MSN-CPN
2.11. In Vitro Cell Uptake of FITC-MSN-CPN
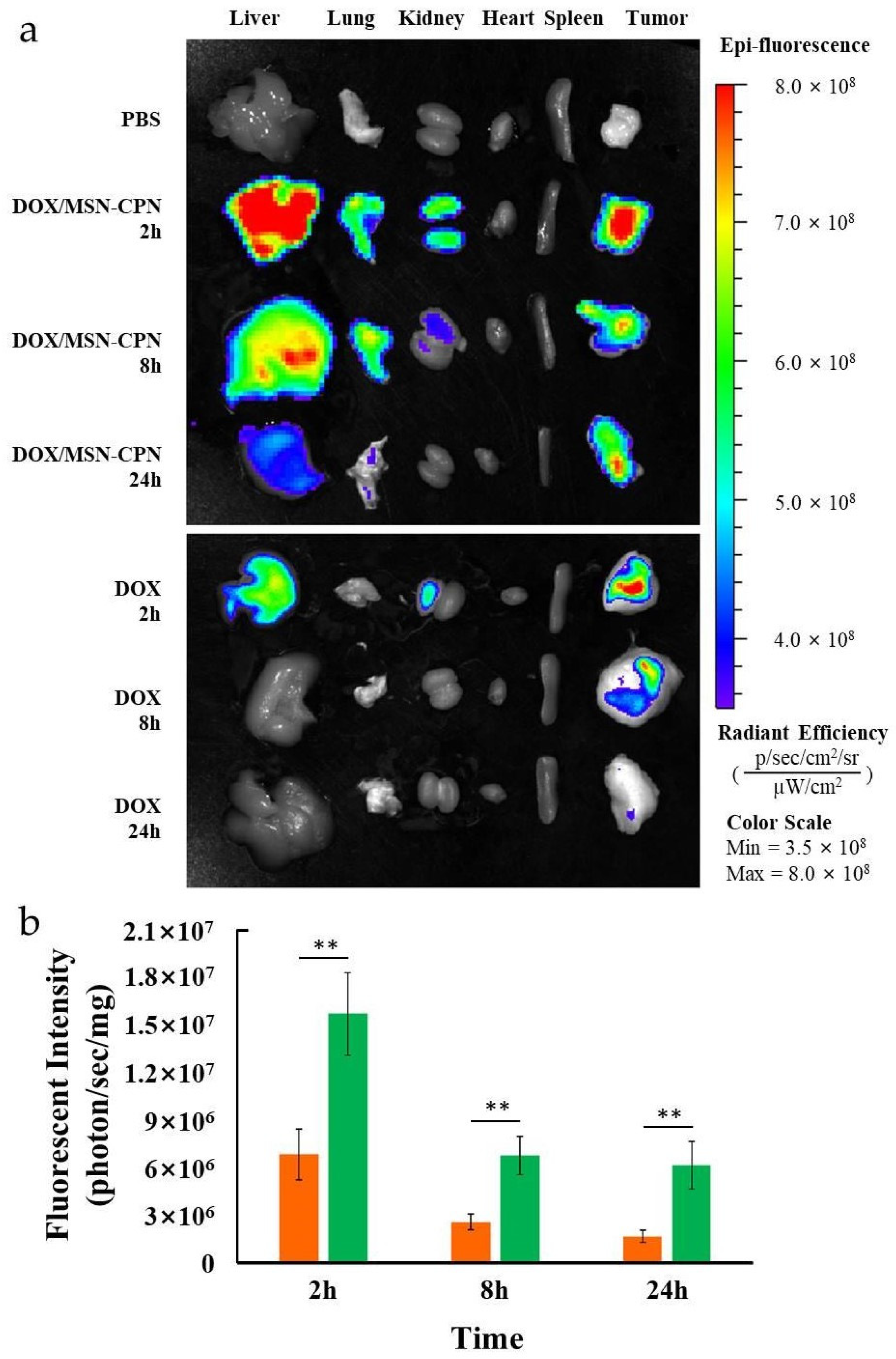
2.12. Ex Vivo Animal Imaging
2.13. In Vivo Tumor Inhibition
2.14. Histological Analysis
2.15. Statistical Analysis
3. Results and Discussion
3.1. Synthesis of CPN
3.2. Acid–Base Titration of CPN
3.3. Characterization of the DOX/MSN-CPN
3.4. In Vitro pH-Sensitivity
3.5. In Vitro Biocompatibility
3.6. In Vitro Drug Release
3.7. In Vitro Cell Uptake
3.8. Ex Vivo Animal Imaging
3.9. In Vivo Tumor Inhibition
3.10. Histological Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, P.; Li, Z.; Feng, X.; Lv, C.; Zhang, H.; Xiao, H.; Ding, J.; Chen, X. Evaluation of Polymer Nanoformulations in Hepatoma Therapy by Established Rodent Models. Theranostics 2019, 9, 1426–1452. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhai, Y.; Cai, Y.; Zhao, Y.; Li, Y. Nanomedicine-based immunotherapy for the treatment of cancer metastasis. Adv. Mater. 2019, 31, 1904156. [Google Scholar] [CrossRef] [PubMed]
- Mu, W.; Chu, Q.; Liu, Y.; Zhang, N. A review on nano-based drug delivery system for cancer chemoimmunotherapy. Nano-Micro Lett. 2020, 12, 142. [Google Scholar] [CrossRef] [PubMed]
- Lammers, T.; Kiessling, F.; Ashford, M.; Hennink, W.; Crommelin, D.; Storm, G. Cancer nanomedicine: Is targeting our target? Nat. Rev. Mater. 2016, 1, 16069. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Zhou, Z.; Qiu, N.; Shen, Y. Rational design of cancer nanomedicine: Nanoproperty integration and synchronization. Adv. Mater. 2017, 29, 1904156. [Google Scholar] [CrossRef]
- Thanuja, M.Y.; Anupama, C.; Ranganath, S.H. Bioengineered cellular and cell membrane-derived vehicles for actively targeted drug delivery: So near and yet so far. Adv. Drug Deliv. Rev. 2018, 132, 57–80. [Google Scholar] [CrossRef]
- Li, J.; Kataoka, K. Chemo-physical strategies to advance the in vivo functionality of targeted nanomedicine: The next generation. J. Am. Chem. Soc. 2021, 143, 538–559. [Google Scholar] [CrossRef]
- Zhu, L.; Ding, Z.; Li, X.; Wei, H.; Chen, Y. Research progress of radiolabeled Asn-Gly-Arg (NGR) peptides for imaging and therapy. Mol. Imaging 2020, 19, 1536012120934957. [Google Scholar] [CrossRef]
- Huang, N.; Cheng, S.; Zhang, X.; Tian, Q.; Pi, J.; Tang, J.; Huang, Q.; Wang, F.; Chen, J.; Xie, Z.; et al. Efficacy of NGR peptide-modified PEGylated quantum dots for crossing the blood-brain barrier and targeted fluorescence imaging of glioma and tumor vasculature. Nanomedicine 2017, 13, 83–93. [Google Scholar] [CrossRef]
- Shi, X.; Zhang, Y.; Tian, Y.; Xu, S.; Ren, E.; Bai, S.; Chen, X.; Chu, C.; Xu, Z.; Liu, G. Multi-responsive bottlebrush-like unimolecules self-assembled nano-riceball for synergistic sono-chemotherapy. Small Methods 2021, 5, e2000416. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, Y.; Yao, Q.; Fan, J.; Sun, W.; Long, S.; Shao, K.; Du, J.; Wang, J.; Peng, X. In situ imaging of aminopeptidase N activity in hepatocellular carcinoma: A migration model for tumour using an activatable two-photon NIR fluorescent probe. Chem. Sci. 2018, 10, 1619–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torkhovskaya, T.I.; Kostryukova, L.V.; Tereshkina, Y.A.; Tikhonova, E.G.; Morozevich, G.E.; Plutinskaya, A.D.; Lupatov, A.Y.; Pankratov, A.A. Chlorin e6 embedded in phospholipid nanoparticles equipped with specific peptides: Interaction with tumor cells with different aminopeptidase N expression. Biomed. Pharmacother. 2021, 134, 111154. [Google Scholar] [CrossRef]
- Xu, Y.; Zi, Y.; Lei, J.; Mo, X.; Shao, Z.; Wu, Y.; Tian, Y.; Li, D.; Mu, C. pH-Responsive nanoparticles based on cholesterol/imidazole modified oxidized-starch for targeted anticancer drug delivery. Carbohydr. Polym. 2020, 233, 115858. [Google Scholar] [CrossRef]
- Feng, L.; Dong, Z.; Tao, D.; Zhang, Y.; Liu, Z. The acidic tumor microenvironment: A target for smart cancer nano-theranostics. Natl. Sci. Rev. 2018, 5, 269–286. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Yang, J.; Liu, R.; Yi, Y.; Huang, M.; Wu, Y.; Tu, H.; Zhang, L. Efficient fabrication of reversible pH-induced carboxymethyl chitosan nanoparticles for antitumor drug delivery under weakly acidic microenvironment. Int. J. Biol. Macromol. 2019, 126, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Leonhard, M.; Moser, D.; Ma, S.; Schneider-Stickler, B. Inhibition of mixed fungal and bacterial biofilms on silicone by carboxymethyl chitosan. Colloids Surf. B Biointerfaces 2016, 148, 193–199. [Google Scholar] [CrossRef]
- Kalliola, S.; Repo, E.; Srivastava, V.; Heiskanen, J.P.; Sirviö, J.A.; Liimatainen, H.; Sillanpää, M. The pH sensitive properties of carboxymethyl chitosan nanoparticles cross-linked with calcium ions. Colloids Surf. B Biointerfaces 2017, 153, 229–236. [Google Scholar] [CrossRef]
- Yan, X.; Tong, Z.; Chen, Y.; Mo, Y.; Feng, H.; Li, P.; Qu, X.; Jin, S. Bioresponsive materials for drug delivery based on carboxymethyl chitosan/poly(γ-glutamic acid) composite microparticles. Mar. Drugs 2017, 15, 127. [Google Scholar] [CrossRef] [Green Version]
- Vallet-Regí, M.; Schüth, F.; Lozano, D.; Colilla, M.; Manzano, M. Engineering mesoporous silica nanoparticles for drug delivery: Where are we after two decades? Chem. Soc. Rev. 2022, 51, 5365–5451. [Google Scholar] [CrossRef]
- Ahmadi, F.; Sodagar-Taleghani, A.; Ebrahimnejad, P.; Moghaddam, S.P.H.; Ebrahimnejad, F.; Asare-Addo, K.; Nokhodchi, A. A review on the latest developments of mesoporous silica nanoparticles as a promising platform for diagnosis and treatment of cancer. Int. J. Pharm. 2022, 625, 122099. [Google Scholar] [CrossRef]
- Li, B.; Zhang, X.; Wu, Z.; Chu, T.; Yang, Z.; Xu, S.; Wu, S.; Qie, Y.; Lu, Z.; Qi, F.; et al. Reducing Postoperative Recurrence of Early-Stage Hepatocellular Carcinoma by a Wound-Targeted Nanodrug. Adv. Sci. 2022, 9, e2200477. [Google Scholar] [CrossRef]
- Cordeiro, R.; Carvalho, A.; Durães, L.; Faneca, H. Triantennary GalNAc-Functionalized Multi-Responsive Mesoporous Silica Nanoparticles for Drug Delivery Targeted at Asialoglycoprotein Receptor. Int. J. Mol. Sci. 2022, 23, 6243. [Google Scholar] [CrossRef]
- Gao, Y.; Gao, D.; Shen, J.; Wang, Q. A Review of Mesoporous Silica Nanoparticle Delivery Systems in Chemo-Based Combination Cancer Therapies. Front Chem. 2020, 8, 598722. [Google Scholar] [CrossRef]
- Zhou, S.; Zhong, Q.; Wang, Y.; Hu, P.; Zhong, W.; Huang, C.B.; Yu, Z.Q.; Ding, C.D.; Liu, H.X.; Fu, J.J. Chemically engineered mesoporous silica nanoparticles-based intelligent delivery systems for theranostic applications in multiple cancerous/non-cancerous diseases. Coord. Chem. Rev 2022, 452, 21430. [Google Scholar] [CrossRef]
- Li, Q.; Wang, W.Q.; Hu, G.W.; Cui, X.L.; Sun, D.J.; Jin, Z.; Zhao, K. Evaluation of Chitosan Derivatives Modified Mesoporous Silica Nanoparticles as Delivery Carrier. Molecules 2021, 26, 2490. [Google Scholar] [CrossRef]
- Cui, L.; Liu, W.T.; Liu, H.; Qin, Q.; Wu, S.X.; He, S.Q.; Pang, X.C.; Zhu, C.S.; Shen, P.H. pH-Triggered Charge-Reversal Mesoporous Silica Nanoparticles Stabilized by Chitosan Oligosaccharide/Carboxymethyl Chitosan Hybrids for Effective Intracellular Delivery of Doxorubicin. ACS Appl. Biol. Mater. 2019, 2, 1907–1919. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, H.; Li, Y. Stimuli-Responsive Nanomedicines for Overcoming Cancer Multidrug Resistance. Theranostics 2018, 8, 1059–1074. [Google Scholar] [CrossRef]
- Wang, X.; Wang, X.; Jin, S.; Muhammad, N.; Guo, Z. Stimuli-responsive therapeutic metallodrugs. Chem. Rev. 2019, 119, 1138–1192. [Google Scholar] [CrossRef]
- Wang, X.; Chen, Y.; Dahmani, F.Z.; Yin, L.; Zhou, J.; Yao, J. Amphiphilic carboxymethyl chitosan-quercetin conjugate with P-gp inhibitory properties for oral delivery of paclitaxel. Biomaterials 2014, 35, 7654–7665. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, M.; Liu, C.; Liu, Y.; Zhang, N. pH-sensitive self-assembled carboxymethyl chitosan-modified DNA/polyethylenimine complexes for efficient gene delivery. J. Biomed. Nanotechnol. 2014, 10, 3397–3406. [Google Scholar] [CrossRef] [PubMed]
- Mu, S.; Liu, Y.; Wang, T.; Zhang, J.; Jiang, D.; Yu, X.; Zhang, N. Unsaturated nitrogen-rich polymer poly(l-histidine) gated reversibly switchable mesoporous silica nanoparticles using “graft to” strategy for drug controlled release. Acta Biomater. 2017, 63, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Chen, Y.; Zhang, L.; Li, X.; Cai, X.; Du, Y.; Zhang, L.; Shi, J. A salt-assisted acid etching strategy for hollow mesoporous silica/organosilica for pH-responsive drug and gene co-delivery. J. Mater. Chem. B 2015, 3, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Li, W.Q.; Sun, L.P.; Xia, Y.; Hao, S.; Cheng, G.; Wang, Z.; Wan, Y.; Zhu, C.; He, H.; Zheng, S.Y. Preoccupation of Empty Carriers Decreases Endo-/Lysosome Escape and Reduces the Protein Delivery Efficiency of Mesoporous Silica Nanoparticles. ACS Appl. Mater. Interfaces 2018, 10, 5340–5347. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Tian, F.; Dahmani, F.Z.; Yang, H.; Yue, D.; He, S.; Zhou, J.; Yao, J. Curcumin-carboxymethyl chitosan (CNC) conjugate and CNC/LHR mixed polymeric micelles as new approaches to improve the oral absorption of P-gp substrate drugs. Drug Deliv. 2016, 23, 3424–3435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worthen, A.J.; Foster, L.M.; Dong, J.; Bollinger, J.A.; Peterman, A.H.; Pastora, L.E.; Bryant, S.L.; Truskett, T.M.; Bielawski, C.W.; Johnston, K.P. Synergistic formation and stabilization of oil-in-water emulsions by a weakly interacting mixture of zwitterionic surfactant and silica nanoparticles. Langmuir 2014, 30, 984–994. [Google Scholar] [CrossRef]
- Samji, H.; Yu, A.; Kuo, M.; Alavi, M.; Woods, R.; Alvarez, M.; Dore, G.J.; Tyndall, M.; Krajden, M.; Janjua, N.Z.; et al. Late hepatitis B and C diagnosis in relation to disease decompensation and hepatocellular carcinoma development. J. Hepatol. 2017, 67, 909–917. [Google Scholar] [CrossRef]
- Lyu, N.; Kong, Y.; Mu, L.; Lin, Y.; Li, J.; Liu, Y.; Zhang, Z.; Zheng, L.; Deng, H.; Li, S.; et al. Hepatic arterial infusion of oxaliplatin plus fluorouracil/leucovorin vs. sorafenib for advanced hepatocellular carcinoma. J. Hepatol. 2018, 69, 60–69. [Google Scholar] [CrossRef]
- Xiao, P.; Zhao, J.; Huang, Y.; Jin, R.; Tang, Z.; Wang, P.; Song, X.; Zhu, H.; Yang, Z.; Yu, N. A novel long-circulating DOX liposome: Formulation and pharmacokinetics studies. Pharm. Nanotechnol. 2020, 8, 391–398. [Google Scholar] [CrossRef]
- Li, S.J.; Dai, W.; Yin, Z.Z.; Gao, J.; Wu, D.T.; Kong, Y. Synthesis of oxidized pullulan coated mesoporous silica for pH-sensitive drug delivery. Eur. Polym. J. 2020, 122, 109399. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | |
|---|---|---|---|
| APTES (µL) | 20 | 40 | 60 |
| Zeta potential (mV) | −0.26 ± 0.52 | 26.40 ± 1.87 | 35.23 ± 1.18 |
| Size (nm) | 104.15 ± 12.07 | 99.45 ± 8.10 | 101.91 ± 10.83 |
| Size (nm) | PDI | Zeta Potential (mV) | EE (%) | DL (%) | |
|---|---|---|---|---|---|
| DOX/MSN-NH2 | 104.24 ± 4.32 | 0.195 ± 0.019 | 35.23 ± 1.18 | 52.52 ± 0.87 | 11.61 ± 0.17 |
| DOX/MSN-CPN | 125.01 ± 1.52 | 0.213 ± 0.015 | −20.20 ± 0.45 | — | 10.55 ± 0.15 |
| BET Surface Area (cm2/g) | BJH Adsorption Cumulative Volume of Pores (cm3/g) | BJH Desorption Average Pore Diameter (nm) | |
|---|---|---|---|
| MSN-NH2 | 1185.12 | 0.89 | 2.87 |
| DOX/MSN-CPN | 41.07 | 0.25 | — |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Mu, W.; Gao, T.; Fang, Y.; Zhang, N.; Liu, Y. CD13-Mediated Pegylated Carboxymethyl Chitosan-Capped Mesoporous Silica Nanoparticles for Enhancing the Therapeutic Efficacy of Hepatocellular Carcinoma. Pharmaceutics 2023, 15, 426. https://doi.org/10.3390/pharmaceutics15020426
Liu J, Mu W, Gao T, Fang Y, Zhang N, Liu Y. CD13-Mediated Pegylated Carboxymethyl Chitosan-Capped Mesoporous Silica Nanoparticles for Enhancing the Therapeutic Efficacy of Hepatocellular Carcinoma. Pharmaceutics. 2023; 15(2):426. https://doi.org/10.3390/pharmaceutics15020426
Chicago/Turabian StyleLiu, Jinhu, Weiwei Mu, Tong Gao, Yuxiao Fang, Na Zhang, and Yongjun Liu. 2023. "CD13-Mediated Pegylated Carboxymethyl Chitosan-Capped Mesoporous Silica Nanoparticles for Enhancing the Therapeutic Efficacy of Hepatocellular Carcinoma" Pharmaceutics 15, no. 2: 426. https://doi.org/10.3390/pharmaceutics15020426