
Insightful Improvement in the Design of Potent Uropathogenic E. coli FimH Antagonists

 ,
,  ,
,  ,
,  ,
,  , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
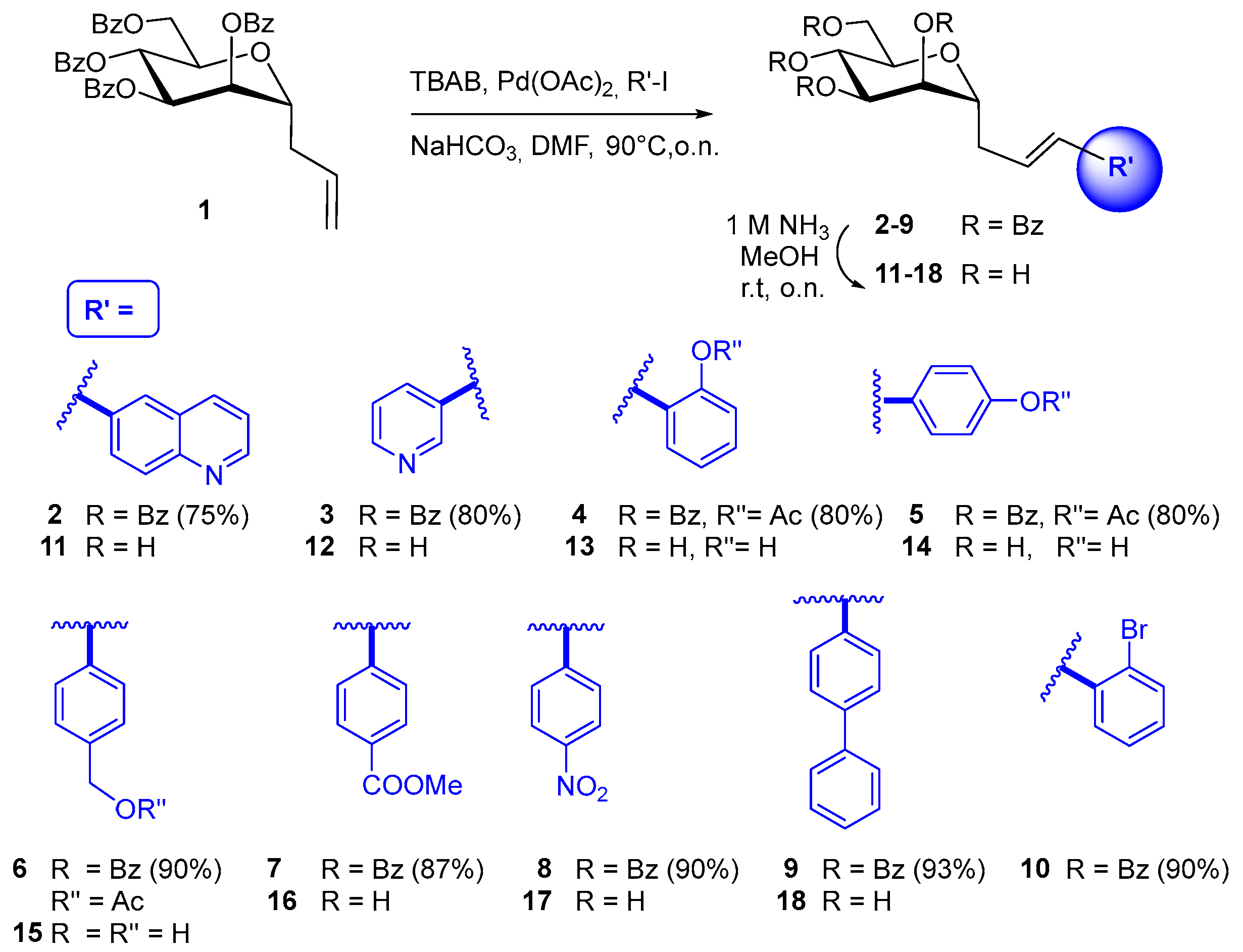
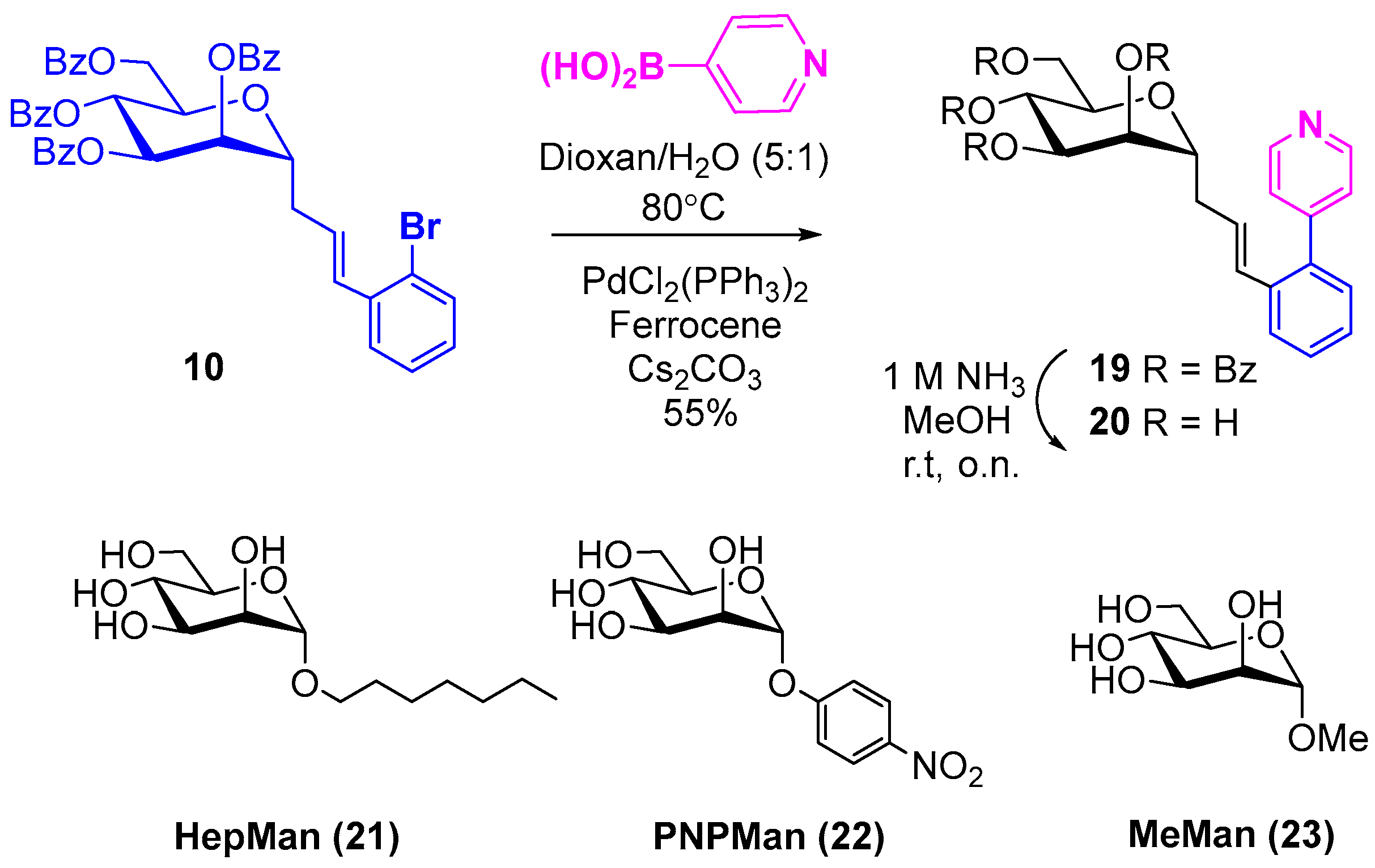
2.1. Synthesis and Structural Characterization
2.2. Synthesis of a Key Ortho-Substituted Biphenyl Derivative
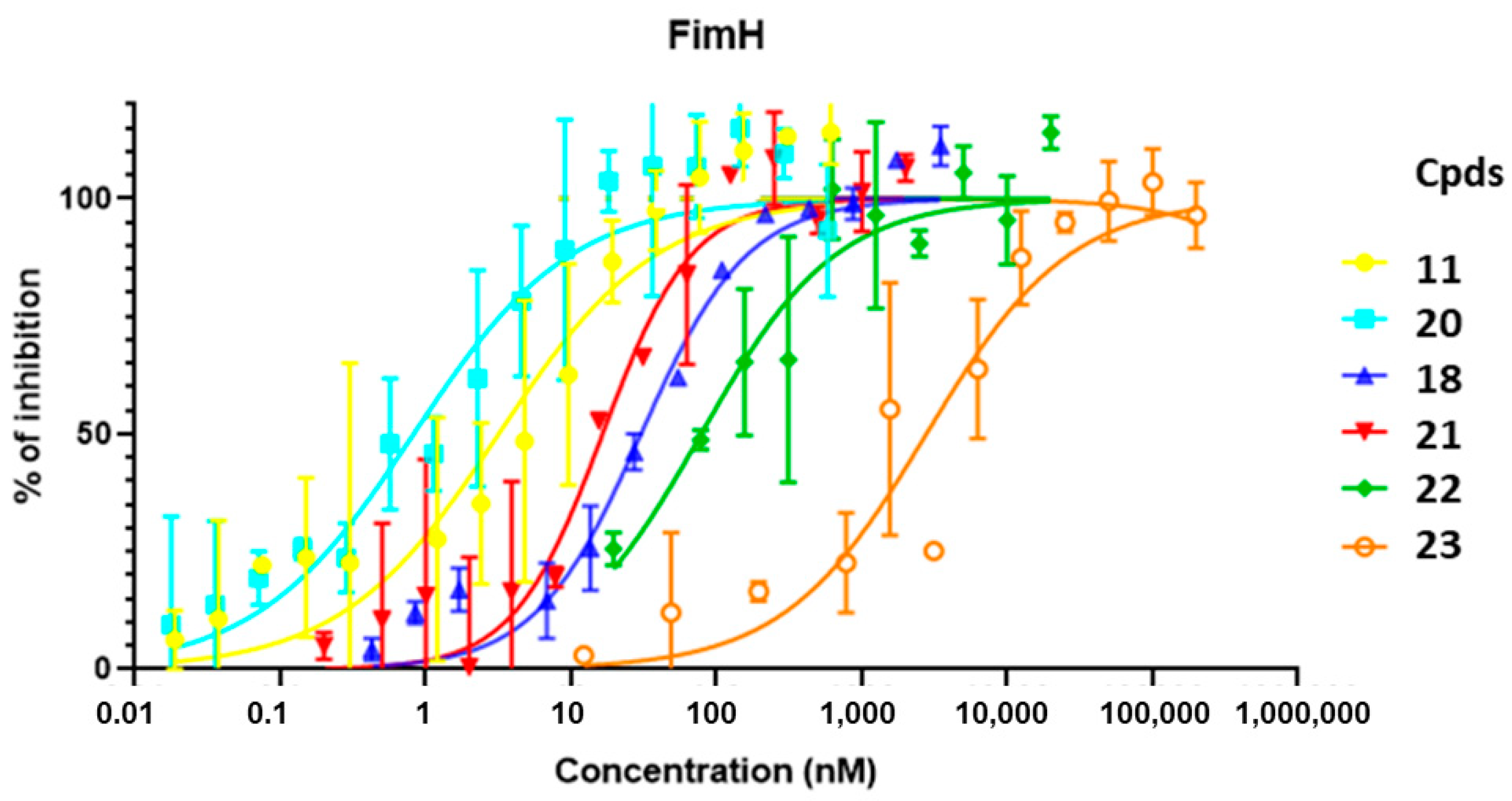
2.3. Affinity Evaluation of Mannosides through FimH LEctPROFILE Kit
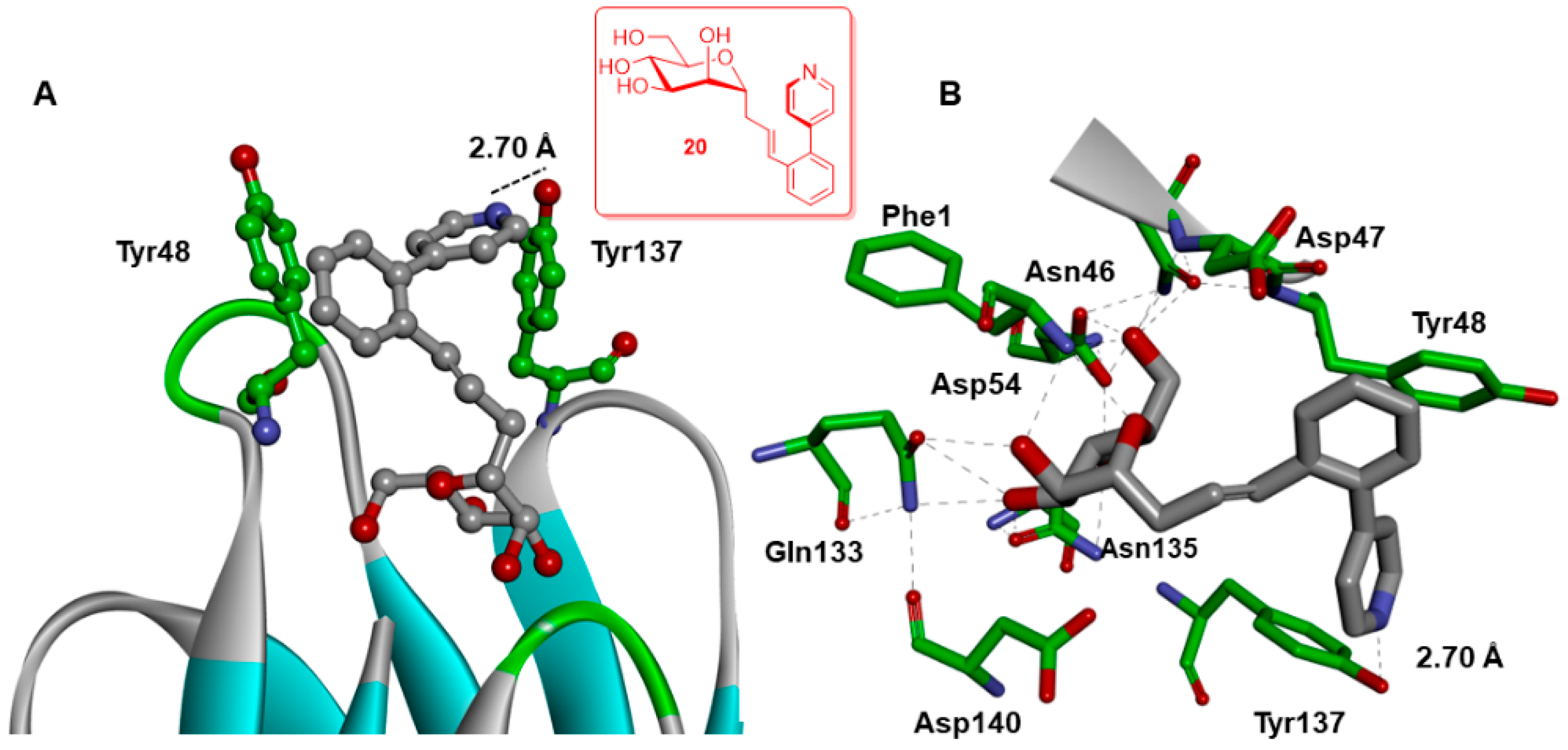
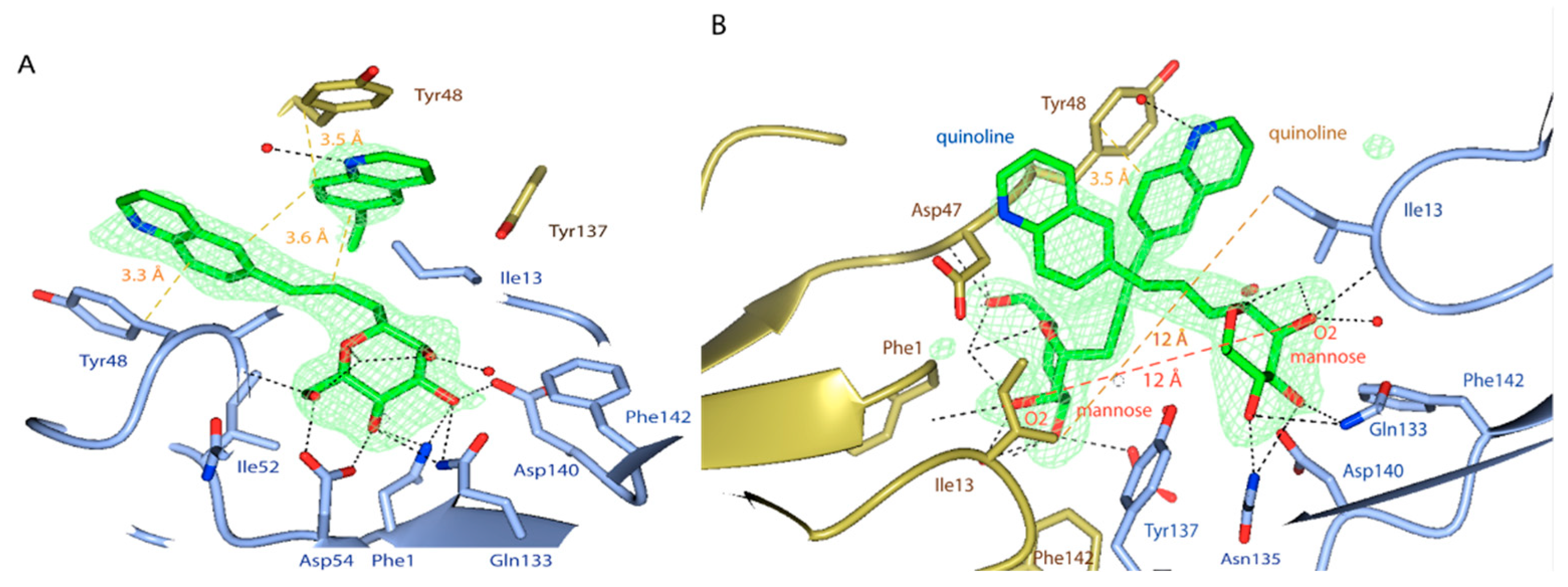
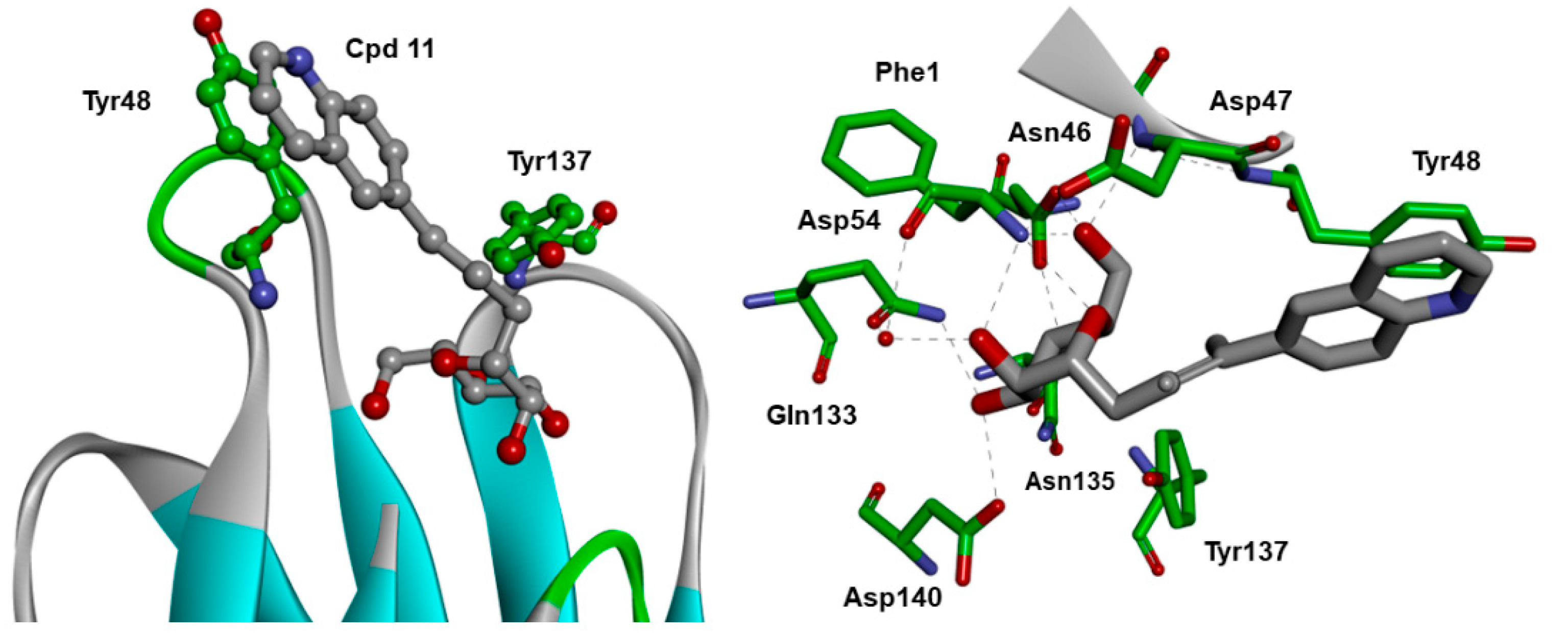
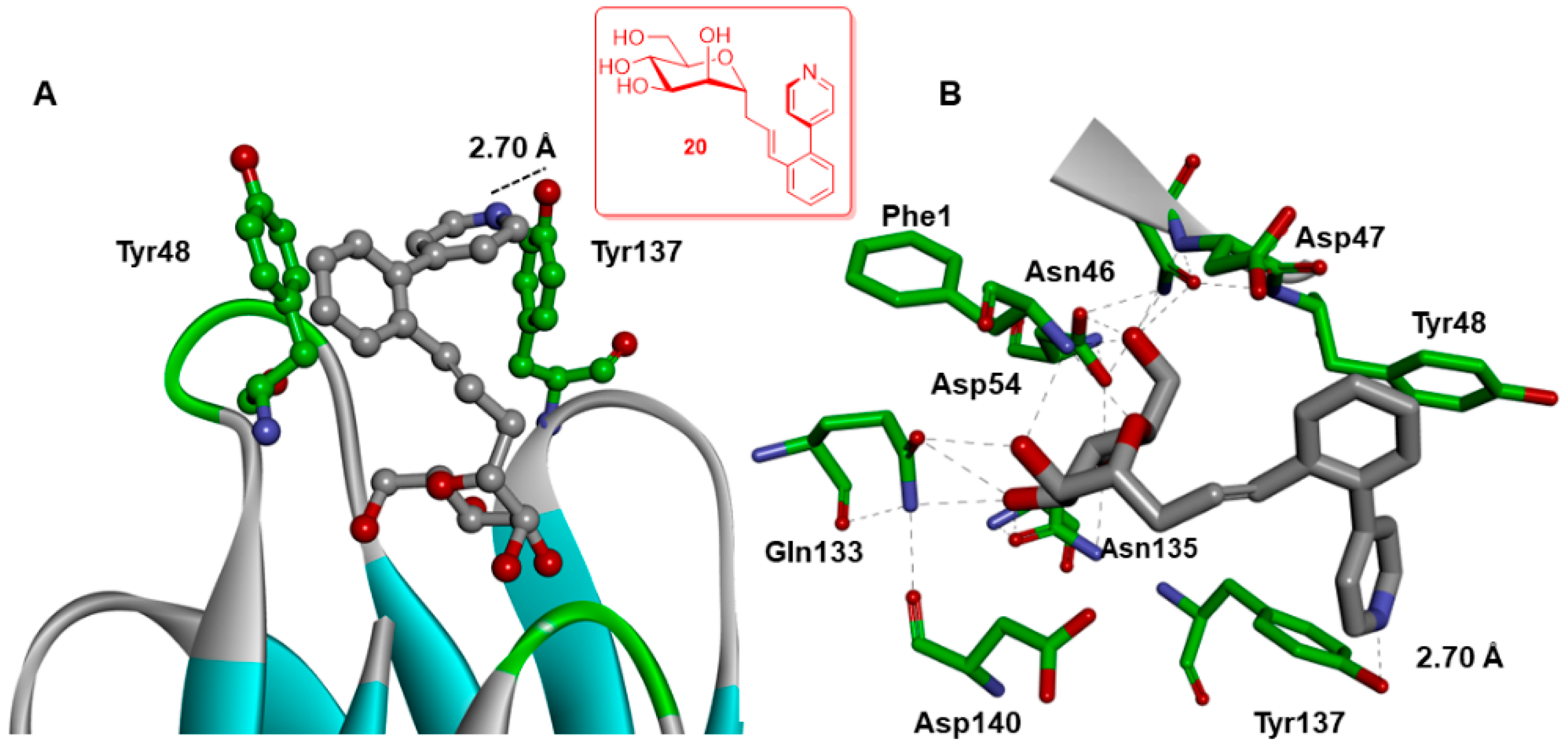
2.4. X-ray and Molecular Dynamic Simulations
Further Insights into the Design and Binding of Compound 20
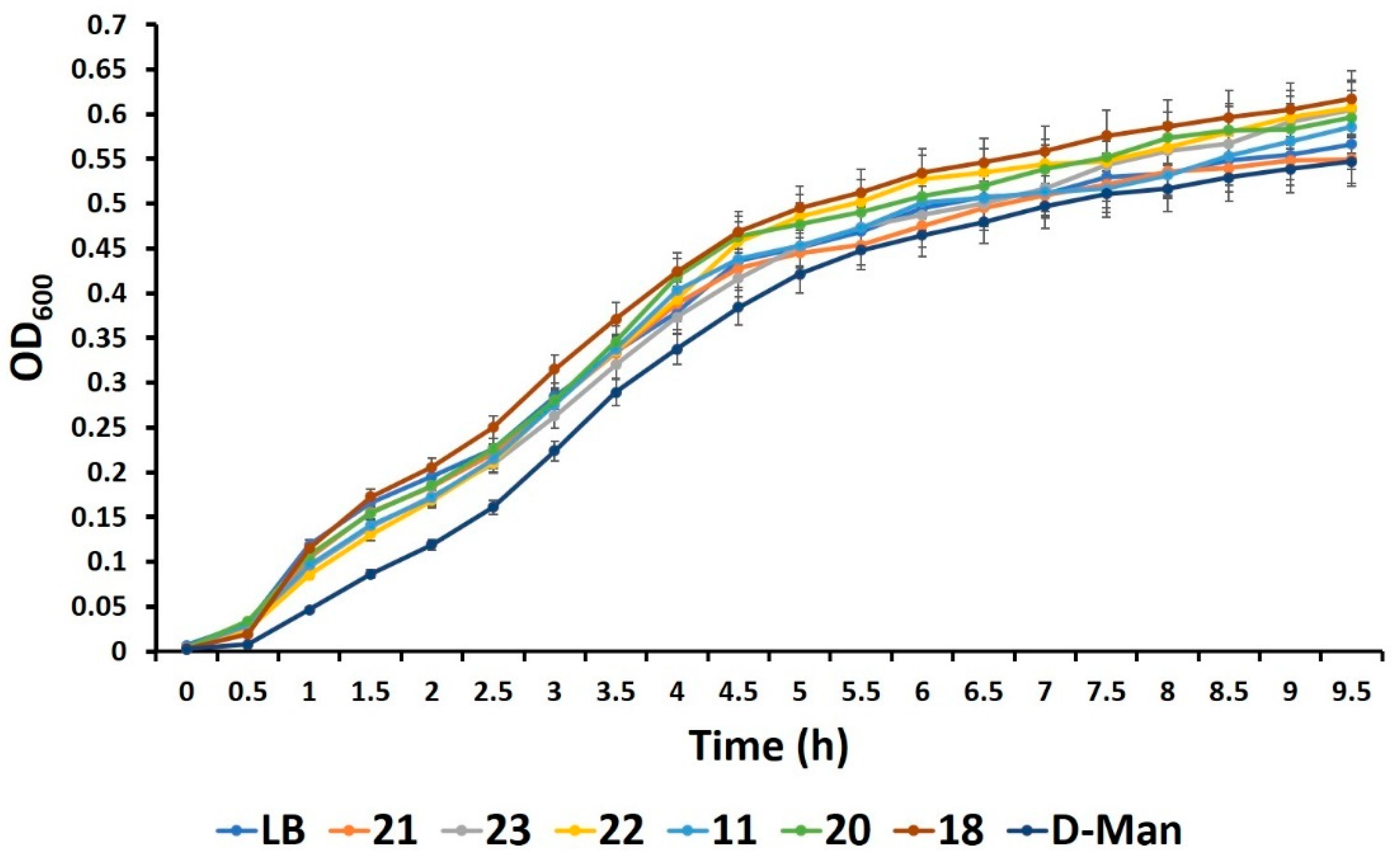
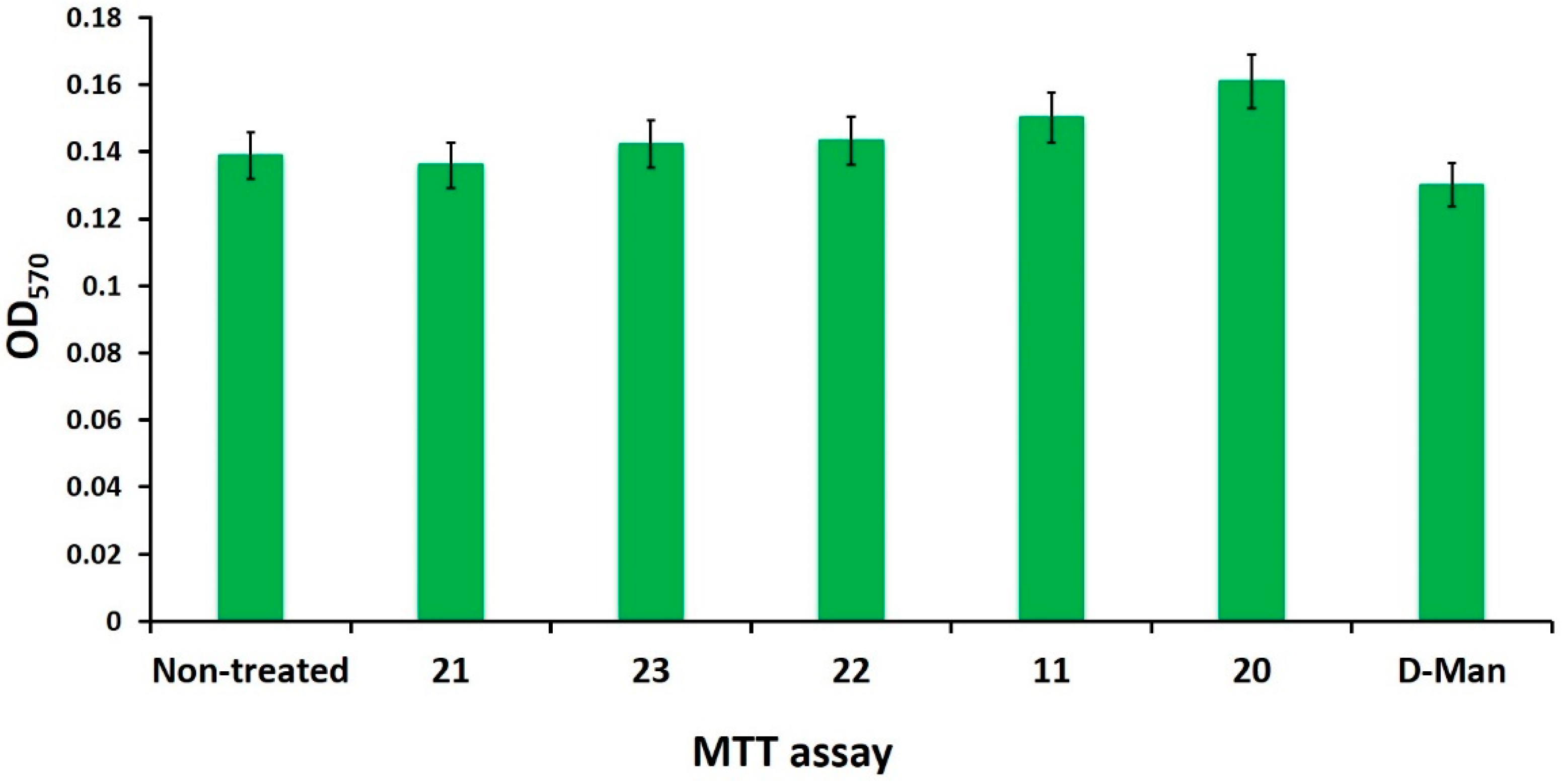
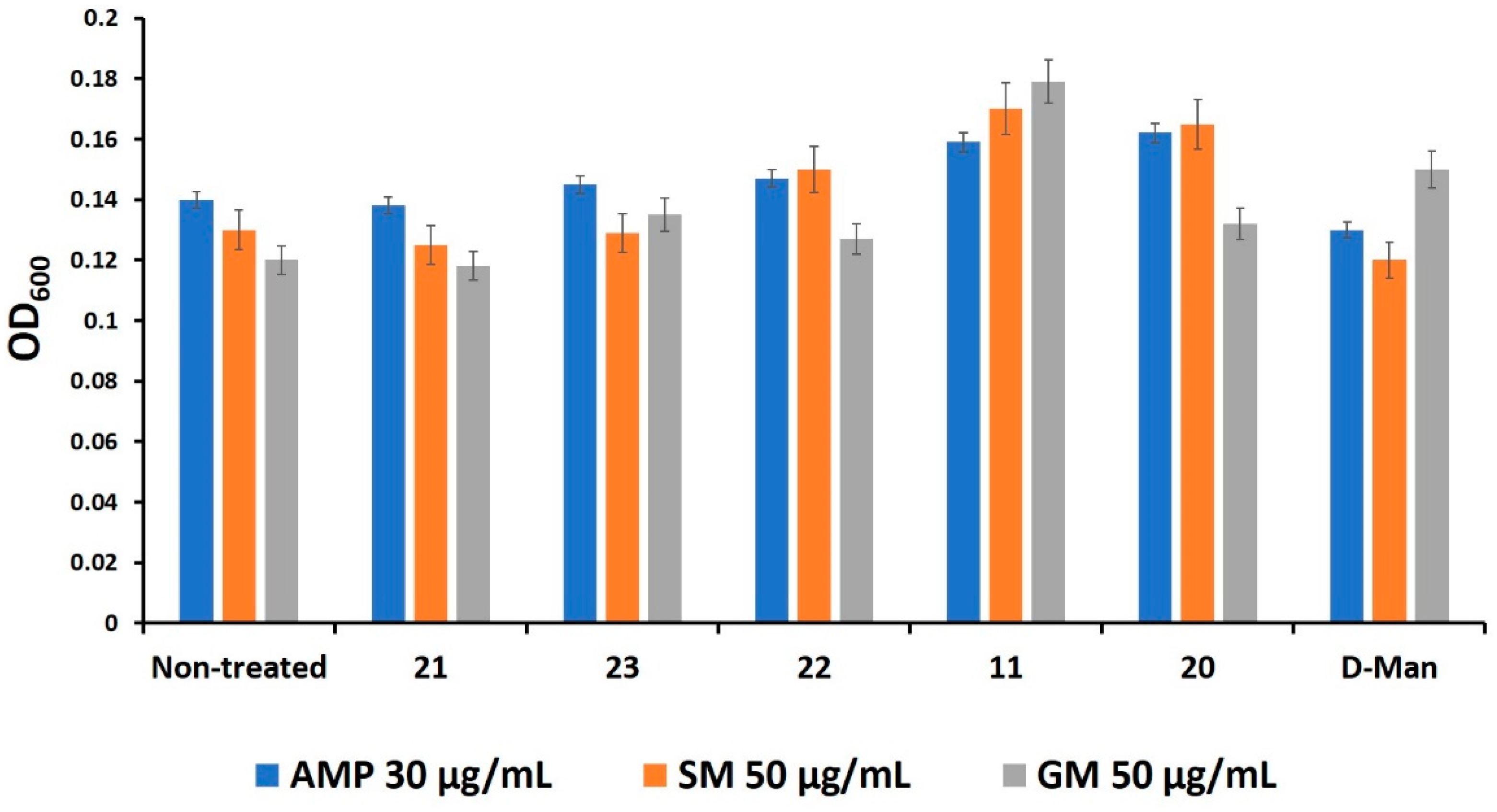
2.5. Mannosides Do Not Affect Bacterial Growth, Cell Viability, and Antibiotic Activities
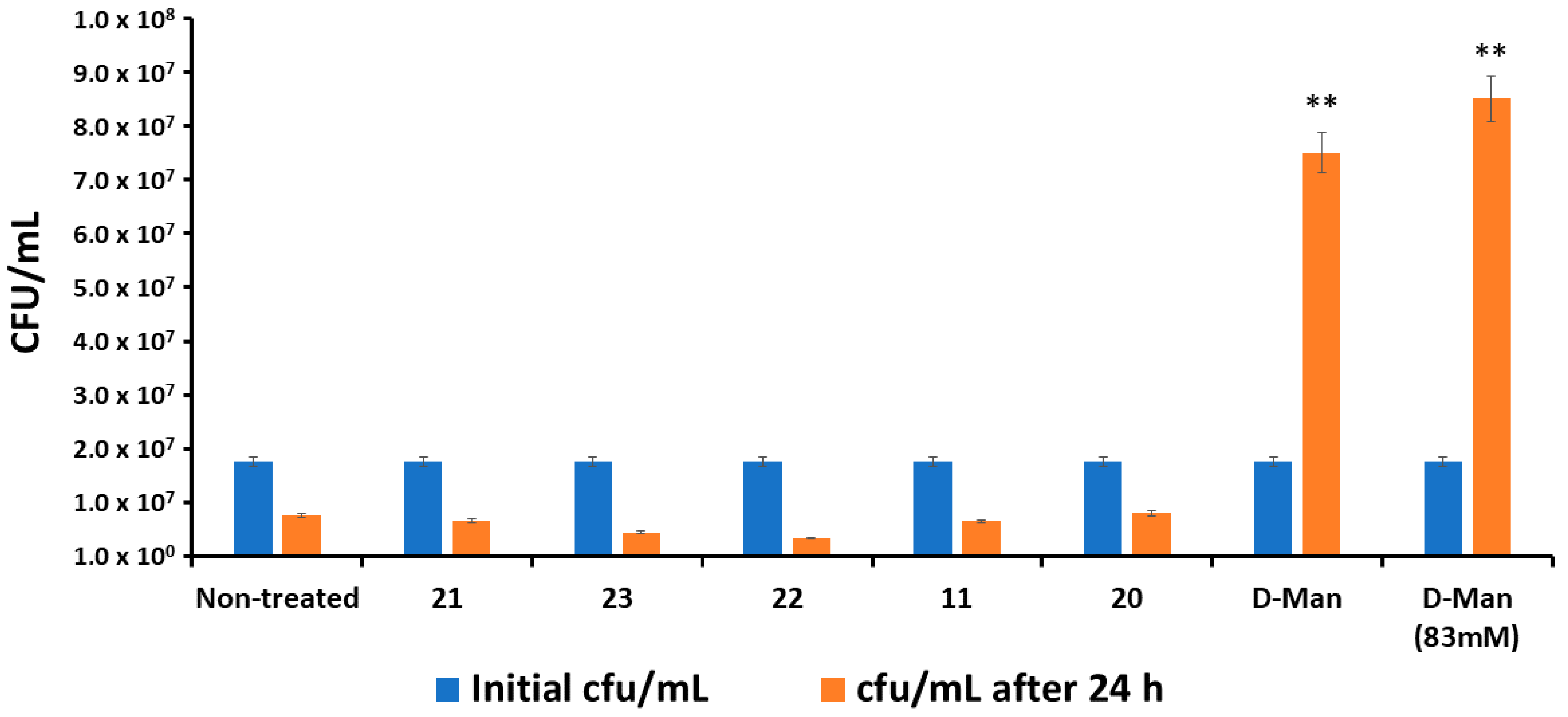
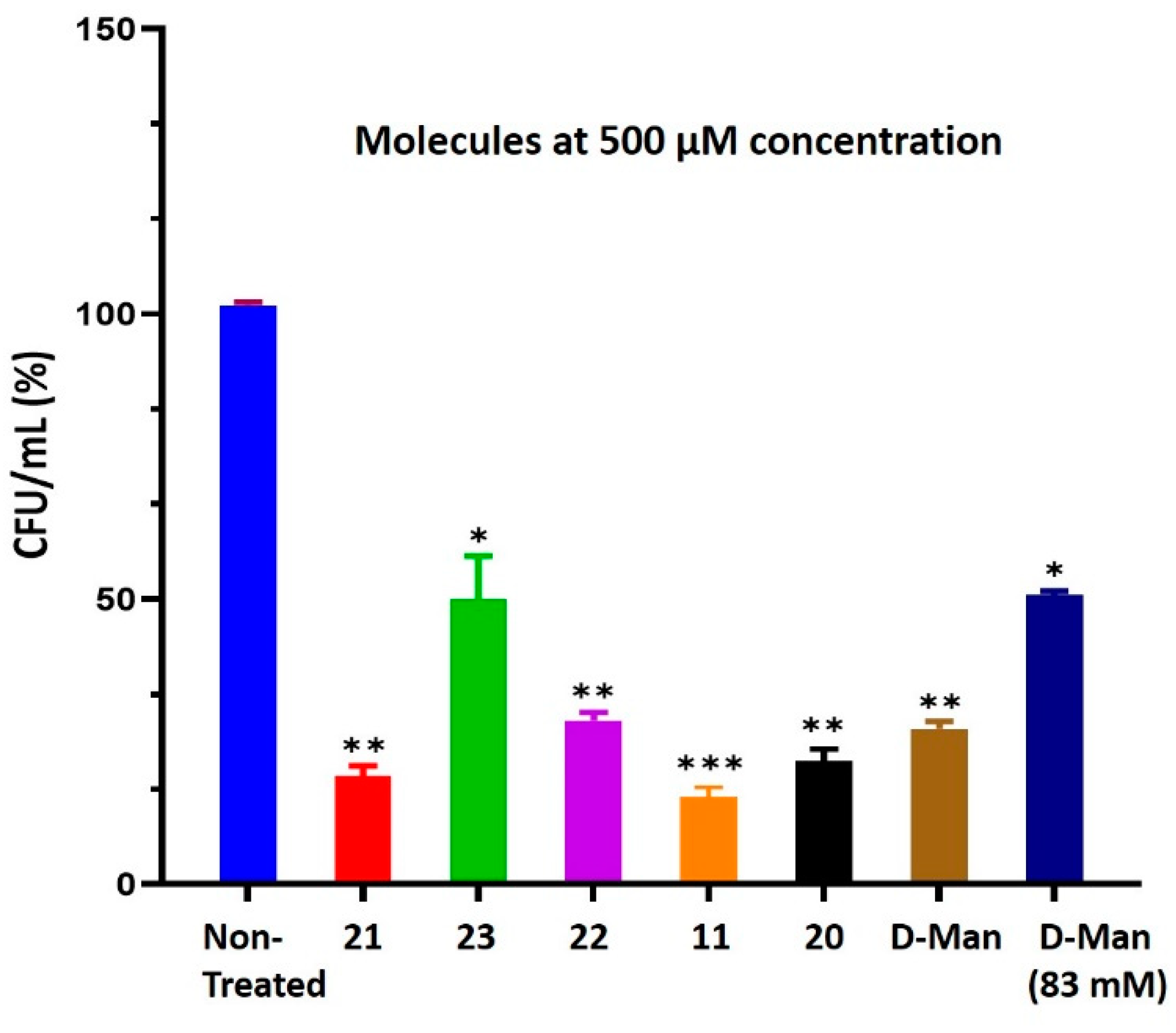
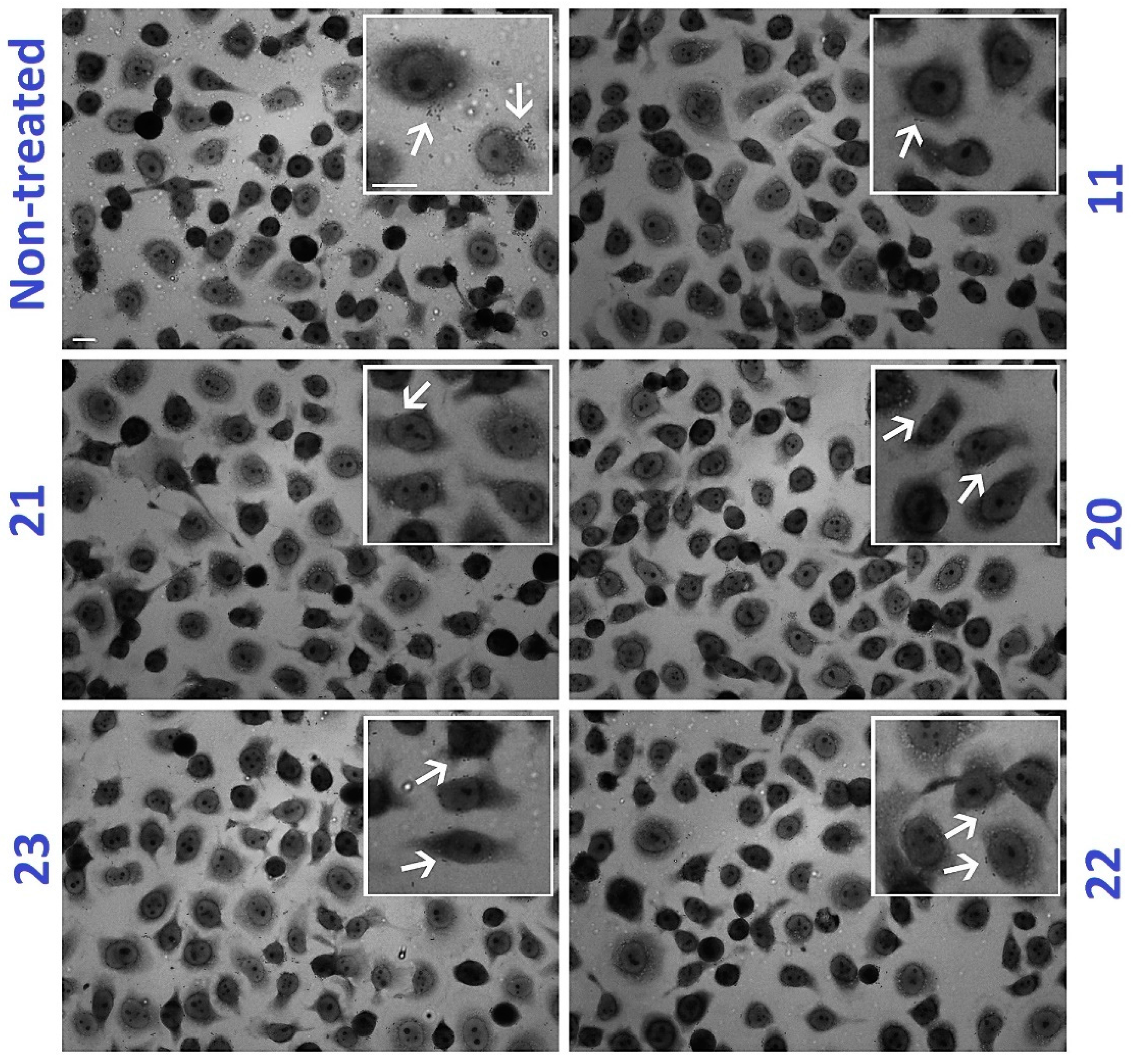
2.6. C-Mannoside Antagonists Are Effective in Decreasing Bacterial Adhesion to Human Bladder Epithelial Cells
3. Materials and Methods
3.1. General Information
3.2. Synthetic Methods and Analytic Data of Compounds
3.2.1. General procedure for Heck Coupling of Protected C-Mannopyranosides 2–10
3.2.2. General Procedure for De-O-Benzoylation
3.2.3. Synthesis of Compounds 19 and 20 by Suzuki Reaction
3.3. Bioactivity Assay
3.3.1. Expression and Purification of FimH
3.3.2. Co-Crystallization of Antagonist 11 with FimH
3.3.3. Affinity Evaluation of Mannosides through FimH LEctPROFILE Kit
3.3.4. Bacterial Strains and Cell Line
3.3.5. Effect of Mannosides on Bacterial Growth and Metabolism
3.3.6. Cell Viability and Toxicity Assay
3.3.7. Antibiotic-Mannoside Interference Assay
3.3.8. Bacterial Adhesion Assay
3.3.9. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Spaulding, C.N.; Klein, R.D.; Ruer, S.; Kau, A.L.; Schreiber, H.L.; Cusumano, Z.T.; Dodson, K.W.; Pinkner, J.S.; Fremont, D.H.; Janetka, J.W.; et al. Selective depletion of uropathogenic E. coli from the gut by a FimH antagonist. Nature 2017, 546, 528–532. [Google Scholar] [CrossRef]
- Terlizzi, M.E.; Gribaudo, G.; Maffei, M.E. Uropathogenic Escherichia coli (UPEC) infections: Virulence factors, bladder responses, antibiotic, and non-antibiotic antimicrobial strategies. Front. Microbiol. 2017, 8, 1566. [Google Scholar] [CrossRef]
- Abe, C.M.; Salvador, A.; Falsetti, I.N.; Blanco, E.; Blanco, M. Uropathogenic Escherichia coli ( UPEC ) strains may carry virulence properties of diarrhoeagenic E. coli. FEMS Immunol. Med. Microbiol. 2008, 52, 397–406. [Google Scholar] [CrossRef]
- Mousavifar, L.; Touaibia, M.; Roy, R. Development of Mannopyranoside Therapeutics against Adherent-Invasive Escherichia coli Infections. Acc. Chem. Res. 2018, 51, 2937–2948. [Google Scholar] [CrossRef]
- Scribano, D.; Sarshar, M.; Prezioso, C.; Lucarelli, M.; Angeloni, A.; Zagaglia, C.; Palamara, A.T.; Ambrosi, C. D-Mannose Treatment neither Affects Uropathogenic Escherichia coli Properties nor Induces Stable FimH Modifications. Molecules. 2020, 25, 316. [Google Scholar] [CrossRef]
- Sarshar, M.; Behzadi, P.; Ambrosi, C.; Zagaglia, C. FimH and Anti-Adhesive Therapeutics: A Disarming Strategy Against Uropathogens. Antibiotics. 2020, 9, 397. [Google Scholar] [CrossRef]
- Palmela, C.; Chevarin, C.; Xu, Z.; Torres, J.; Sevrin, G.; Hirten, R.; Barnich, N.; Ng, S.C.; Colombel, J.F. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut 2018, 67, 574–587. [Google Scholar] [CrossRef]
- Wagenlehner, F.; Lorenz, H.; Ewald, O.; Gerke, P. Why D -Mannose May Be as Efficient as Antibiotics in the Treatment of Acute Uncomplicated Lower Urinary Tract Infections—Preliminary Considerations and Conclusions from a Non-Interventional Study. Antibiotics 2022, 11, 314. [Google Scholar] [CrossRef]
- Hudson, R.E.; Job, K.M.; Sayre, C.L.; Krepkova, L.V.; Sherwin, C.M.; Enioutina, E.Y. Examination of Complementary Medicine for Treating Urinary Tract Infections Among Pregnant Women and Children. Front. Pharmacol. 2022, 13, 1541. [Google Scholar] [CrossRef]
- Ohman, L.; Magnusson, K.E.; Stendahl, O. Effect OF Monosaccharides and Ethyleneglycol on the Interaction Between Escherichia coli Bacteria and Octyl-Sepharose. Acta Path. Microbiol. Immunol. Scand. Sect. B 1985, 93, 133–138. [Google Scholar]
- Feenstra, T.; Thøgersen, M.S.; Wieser, E.; Peschel, A.; Ball, M.J.; Brandes, R.; Satchell, S.C.; Stockner, T.; Aarestrup, F.M.; Rees, A.J.; et al. Adhesion of Escherichia coli under flow conditions reveals potential novel effects of FimH mutations. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Scaglione, F.; Musazzi, U.M.; Minghetti, P. Considerations on D-mannose Mechanism of Action and Consequent Classification of Marketed Healthcare Products. Front. Pharmacol. 2021, 12, 636377. [Google Scholar] [CrossRef] [PubMed]
- Hatton, N.E.; Baumann, C.G.; Fascione, M.A. Developments in Mannose-Based Treatments for Uropathogenic Escherichia coli -Induced Urinary Tract Infections. Chembiochem 2021, 22, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Mousavifar, L.; Roy, R. Recent development in the design of small ‘ drug-like ’ and nanoscale glycomimetics against Escherichia coli infections. Drug Discov. Today. 2021, 26, 2124–2137. [Google Scholar] [CrossRef]
- Mydock-McGrane, L.K.; Cusumano, Z.T.; Janetka, J.W. Mannose-derived FimH antagonists: A promising anti-virulence therapeutic strategy for urinary tract infections and Crohn’s disease. Expert Opin. Ther. Pat. 2016, 26, 175–197. [Google Scholar] [CrossRef]
- Pang, L.; Kleeb, S.; Lemme, K.; Rabbani, S.; Scharenberg, M.; Zalewski, A.; Schädler, F.; Schwardt, O.; Ernst, B. FimH Antagonists: Structure-Activity and Structure-Property Relationships for Biphenyl α-D-Mannopyranosides. ChemMedChem 2012, 7, 1404–1422. [Google Scholar] [CrossRef]
- Sivignon, A.; Yan, X.; Dorta, D.A.; Bonnet, R.; Bouckaert, J.; Fleury, E.; Bernard, J.; Gouin, S.G.; Darfeuille-Michaud, A.; Barnich, N. Development of heptylmannoside-based glycoconjugate antiadhesive compounds against adherent-invasive Escherichia coli bacteria associated with crohn’s disease. MBio 2015, 6, e01298-15. [Google Scholar] [CrossRef]
- Bouckaert, J.; Mackenzie, J.; de Paz, J.L.; Chipwaza, B.; Choudhury, D.; Zavialov, A.; Mannerstedt, K.; Anderson, J.; Piérard, D.; Wyns, L.; et al. The affinity of the FimH fimbrial adhesin is receptor-driven and quasi-independent of Escherichia coli pathotypes. Mol. Microbiol. 2006, 61, 1556–1568. [Google Scholar] [CrossRef]
- Touaibia, M.; Wellens, A.; Tze, C.S.; Wang, Q.; Sirois, S.; Bouckaert, J.; Roy, R. Mannosylated G(0) dendrimers with nanomolar affinities to Escherichia coli FimH. ChemMedChem 2007, 2, 1190–1201. [Google Scholar] [CrossRef]
- Bernardi, A.; Jiménez-Barbero, J.; Casnati, A.; De Castro, C.; Darbre, T.; Fieschi, F.; Finne, J.; Funken, H.; Jaeger, K.E.; Lahmann, M.; et al. Multivalent glycoconjugates as anti-pathogenic agents. Chem. Soc. Rev. 2013, 42, 4709–4727. [Google Scholar] [CrossRef]
- Lindhorst, T.K.; Dubber, M.; Krallmann-Wenzel, U.; Ehlers, S. Cluster Mannosides as Inhibitors of Type 1 Fimbriae-Mediated Adhesion of Escherichia coli: Pentaerythritol Derivatives as Scaffolds. Eur. J. Org. Chem. 2000, 2000, 2027–2034. [Google Scholar] [CrossRef]
- Twibanire, J.A.K.; Paul, N.K.; Grindley, T.B. Synthesis of novel types of polyester glycodendrimers as potential inhibitors of urinary tract infections. New J. Chem. 2015, 39, 4115–4127. [Google Scholar] [CrossRef]
- Almant, M.; Moreau, V.; Kovensky, J.; Bouckaert, J.; Gouin, S.G. Clustering of Escherichia coli type-1 fimbrial adhesins by using multimeric heptyl α- D -mannoside probes with a carbohydrate core. Chem.-A Eur. J. 2011, 17, 10029–10038. [Google Scholar] [CrossRef]
- Bouckaert, J.; Li, Z.; Xavier, C.; Almant, M.; Caveliers, V.; Lahoutte, T.; Weeks, S.D.; Kovensky, J.; Gouin, S.G. Heptyl a-D-Mannosides Grafted on a b-Cyclodextrin Core to Interfere with Escherichia coli Adhesion: An In Vivo Multivalent Effect. Chem. Eur. J. 2013, 19, 7847–7855. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Sivignon, A.; Yamakawa, N.; Crepet, A.; Travelet, C.; Borsali, R.; Dumych, T.; Li, Z.; Bilyy, R.; Deniaud, D.; et al. Glycopolymers as Antiadhesives of E. coli Strains Inducing In fl ammatory Bowel Diseases. Bio Mac. 2015, 16, 1827–1836. [Google Scholar] [CrossRef]
- Schwardt, O.; Rabbani, S.; Hartmann, M.; Abgottspon, D.; Wittwer, M.; Kleeb, S.; Zalewski, A.; Smieško, M.; Cutting, B.; Ernst, B. Design, synthesis and biological evaluation of mannosyl triazoles as FimH antagonists. Bioorg. Med. Chem 2011, 19, 6454–6473. [Google Scholar] [CrossRef] [PubMed]
- Tomašić, T.; Rabbani, S.; Gobec, M.; Raščan, I.M.; Podlipnik, Č.; Ernst, B.; Anderluh, M. Branched a-D-mannopyranosides: A new class of potent FimH antagonists. Med. Chem. Commun. 2014, 5, 1247–1253. [Google Scholar] [CrossRef]
- Vetterli, S.U.; Moehle, K.; Robinson, J.A. Synthesis and antimicrobial activity against Pseudomonas aeruginosa of macrocyclic b -hairpin peptidomimetic antibiotics containing N -methylated amino acids. Bioorg. Med. Chem. 2016, 24, 6332–6339. [Google Scholar] [CrossRef]
- Han, Z.; Pinkner, J.S.; Ford, B.; Obermann, R.; Nolan, W.; Wildman, S.A.; Hobbs, D.; Ellenberger, T.; Cusumano, C.K.; Hultgren, S.J.; et al. Structure-based drug design and optimization of mannoside bacterial fimH antagonists. J. Med. Chem. 2010, 53, 4779–4792. [Google Scholar] [CrossRef]
- Mousavifar, L.; Vergoten, G.; Charron, G.; Roy, R. Comparative Study of Aryl O-, C-, and S-Mannopyranosides as potential Adhesion Inhibitors toward Uropathogenic E. coli FimH. Molecules. 2019, 24, 3566. [Google Scholar] [CrossRef]
- Krammer, E.M.; De Ruyck, J.; Roos, G.; Bouckaert, J.; Lensink, M.F. Targeting dynamical binding processes in the design of non-antibiotic anti-adhesives by molecular simulation—The example of FimH. Molecules 2018, 23, 1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez Dorta, D.; Sivignon, A.; Chalopin, T.; Dumych, T.I.; Roos, G.; Bilyy, R.O.; Deniaud, D.; Krammer, E.M.; De Ruyck, J.; Lensink, M.F.; et al. The Antiadhesive Strategy in Crohn’s Disease: Orally Active Mannosides to Decolonize Pathogenic Escherichia coli from the Gut. ChemBioChem 2016, 17, 936–952. [Google Scholar] [CrossRef] [PubMed]
- Chalopin, T.; Dorta, D.A.; Sivignon, A.; Caudan, M.; Dumych, T.I.; Bilyy, R.O.; Deniaud, D.; Barnich, N.; Bouckaert, J.; Gouin, S.G. Second generation of thiazolylmannosides, FimH antagonists for E. coli-induced Crohn’s disease. Org. Biomol. Chem. 2016, 14, 3913–3925. [Google Scholar] [CrossRef]
- Brument, S.; Sivignon, A.; Dumych, T.I.; Moreau, N.; Roos, G.; Guérardel, Y.; Gouin, S.G. Thiazolylaminomannosides As Potent Antiadhesives of Type 1 Piliated Escherichia coli Isolated from Crohn’ s Disease Patients. J. Med. Chem. 2013, 56, 5395–5406. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Pinkner, J.S.; Ford, B.; Chorell, E.; Crowley, J.M.; Cusumano, C.K.; Campbell, S.; Henderson, J.P.; Hultgren, S.J.; Janetka, J.W. Lead optimization studies on FimH antagonists: Discovery of potent and orally bioavailable ortho-substituted biphenyl mannosides. J. Med. Chem. 2012, 55, 3945–3959. [Google Scholar] [CrossRef]
- Mydock-McGrane, L.K.; Hannan, T.J.; Janetka, J.W. Rational Design Strategies for FimH Antagonists: New Drugs on the Horizon for Urinary Tract Infection and Crohn’s Disease. Expert Opin. Drug Discov. 2017, 12, 711–731. [Google Scholar] [CrossRef]
- Huggins, D.J.; Sherman, W.; Tidor, B. Rational approaches to improving selectivity in drug design. J. Med.Chem. 2012, 55, 1424–1444. [Google Scholar] [CrossRef]
- Toma, T.; Maier, T.; Ernst, B.; Anderluh, M. Does targeting Arg98 of FimH lead to high af fi nity antagonists? Eur. J. Med. Chem. 2021, 211, 113093. [Google Scholar] [CrossRef]
- Mydock-McGrane, L.; Cusumano, Z.; Han, Z.; Binkley, J.; Kostakioti, M.; Hannan, T.; Pinkner, J.S.; Klein, R.; Kalas, V.; Crowley, J.; et al. Antivirulence C-Mannosides as Antibiotic-Sparing, Oral Therapeutics for Urinary Tract Infections. J. Med. Chem. 2016, 59, 9390–9408. [Google Scholar] [CrossRef]
- Mousavifar, L.; Vergoten, G.; Roy, R. Deciphering the conformation of C -linked α-D-mannopyranosides and their application toward the synthesis of low nanomolar E. coli FimH ligands. ARKIVOC Free. Online J. Org. Chem. 2018, 2018, 384–397. [Google Scholar] [CrossRef]
- Touaibia, M.; Krammer, E.-M.; Shiao, T.C.; Yamakawa, N.; Wang, Q.; Glinschert, A.; Papadopoulos, A.; Mousavifar, L.; Maes, E.; Oscarson, S.; et al. Sites for dynamic protein-carbohydrate interactions of O- and C-linked mannosides on the E. coli FimH adhesin. Molecules 2017, 22, 1101. [Google Scholar] [CrossRef] [Green Version]
- Thakur, A.; Zhang, K.; Louie, J. Suzuki-Miyaura coupling of heteroaryl boronic acids and vinyl chlorides. Chem. Commun. 2012, 48, 203–205. [Google Scholar] [CrossRef]
- Suzuki, A. Recent advances in the cross-coupling reactions of organoboron derivatives with organic electrophiles, 1995–1998. J. Organomet. Chem. 1999, 576, 147–168. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. Sect. D Struct. Biol. 2019, D75, 861–877. [Google Scholar] [CrossRef]
- Miroux, B.; Walker, J.E. Over-production of Proteins in Escherichia coli: Mutant Hosts that Allow Synthesis of some Membrane Proteins and Globular Proteins at High Levels. J. Mol. Biol. 1996, 260, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Wellens, A.; Garofalo, C.; Nguyen, H.; Van Gerven, N.; Slättegard, R.; Hernalsteens, J.P.; Wyns, L.; Oscarson, S.; De Greve, H.; Hultgren, S.; et al. Intervening with urinary tract infectious using anti-adhesives based on the crystal structure of the FimH-oligomannose-3 complex. PLoS ONE 2008, 3, e2040. [Google Scholar] [CrossRef]
- Sauer, M.M.; Jakob, R.P.; Luber, T.; Canonica, F.; Ernst, B.; Unverzagt, C.; Maier, T.; Glockshuber, R. Binding of the bacterial adhesin FimH to its natural, multivalent high-mannose type glycan targets Binding of the bacterial adhesin FimH to its natural, multivalent high- mannose type glycan targets. J. Am. Chem. Soc. 2018, 141, 936–944. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald:An N*log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Andersen, H.C. Rattle: A “velocity” version of the shake algorithm for molecular dynamics calculations. J. Comput. Phys. 1983, 52, 24–34. [Google Scholar] [CrossRef]
- Tuckerman, M.; Berne, B.J.; Martyna, G.J. Reversible multiple time scale molecular dynamics. J. Chem. Phys. 1992, 97, 1990. [Google Scholar] [CrossRef] [Green Version]
- Hung, C.S.; Bouckaert, J.; Hung, D.; Pinkner, J.; Widberg, C.; DeFusco, A.; Auguste, C.G.; Strouse, R.; Langermann, S.; Waksman, G.; et al. Structure basis of tropism of Escherichia coli to the bladder during urinary tract infection. Mol. Microbiol. 2002, 44, 903–915. [Google Scholar] [CrossRef]
- Bianco, A.; Cavarischia, C.; Guiso, M. Total synthesis of anthocyanidins via Heck reaction. Nat. Prod. Res. 2006, 20, 93–97. [Google Scholar] [CrossRef]
- Lindgren, A.E.G.; Öberg, C.T.; Hillgren, J.M.; Elofsson, M. Total synthesis of the resveratrol oligomers (±)-Ampelopsin B and (±)-σ-Viniferin. Eur. J. Org. Chem. 2016, 2016, 426–429. [Google Scholar] [CrossRef]
- Cabrera-Afonso, M.J.; Lu, Z.P.; Kelly, C.B.; Lang, S.B.; Dykstra, R.; Gutierrez, O.; Molander, G.A. Engaging Sulfinate Salts via Ni/Photoredox Dual Catalysis Enables Facile C–SO2R Coupling María. Chem. Sci. 2018, 9, 3186–3191. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics research papers. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Joosten, R.P.; Long, F.; Murshudov, G.N.; Perrakis, A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ 2014, 1, 213–220. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Vonrhein, C.; Tickle, I.J.; Flensburg, C.; Keller, P.; Paciorek, W.; Sharff, A.; Bricogne, G. Advances in automated data and processing within autoPROC, combined with improved characterization, mitigation, and visualization of the anisotropy of diffraction limits using STARANISO. Acta Crystallogr. Sect. A 2018, 74, a360. [Google Scholar] [CrossRef]
- Cauwel, M.; Sivignon, A.; Bridot, C.; Nongbe, M.C.; Deniaud, D.; Roubinet, B.; Landemarre, L. Heptylmannose-functionalized cellulose for the binding and specific detection of pathogenic E. coli. Chem. Commun. 2019, 55, 10158–10161. [Google Scholar] [CrossRef]
- Landemarre, L.; Duverger, E. Lectin glycoprofiling of recombinant therapeutic interleukin-7. In Glycosylation Engineering of Biopharmaceuticals. Methods in Molecular Biology; Alain beck, Ed.; Humana Press of Springer Science: Totowa, NJ, USA, 2013; pp. 221–226. ISBN 9781627033268. [Google Scholar]
- Brissonnet, Y.; Compain, G.; Renoux, B.; Krammer, E.; Daligault, F.; Deniaud, D. Monitoring glycosidase activity for clustered sugar substrates, a study on b -glucuronidase. RSC Adv. 2019, 9, 40263–40267. [Google Scholar] [CrossRef] [Green Version]
- Sarshar, M.; Scribano, D.; Marazzato, M.; Ambrosi, C.; Rita, M.; Aleandri, M.; Pronio, A.; Longhi, C.; Nicoletti, M.; Zagaglia, C.; et al. Genetic diversity, phylogroup distribution and virulence gene pro fi le of pks positive Escherichia coli colonizing human intestinal polyps. Microb. Pathog. 2017, 112, 274–278. [Google Scholar] [CrossRef]
- Ambrosi, C.; Sarshar, M.; Rita, M.; Pompilio, A.; Di, G.; Strati, F.; Pronio, A.; Nicoletti, M.; Zagaglia, C.; Teresa, A.; et al. Colonic adenoma-associated Escherichia coli express speci fi c phenotypes. Microbes Infect. 2019, 21, 305–312. [Google Scholar] [CrossRef]
- Ambrosi, C.; Pompili, M.; Scribano, D.; Zagaglia, C.; Ripa, S.; Nicoletti, M. Outer Membrane Protein A ( OmpA ): A New Player in Shigella flexneri Protrusion Formation and Inter-Cellular Spreading. PLoS ONE 2012, 7, e49625. [Google Scholar] [CrossRef]
- Wellens, A.; Lahmann, M.; Touaibia, M.; Vaucher, J.; Oscarson, S.; Roy, R.; Remaut, H.; Bouckaert, J. The tyrosine gate as a potential entropic lever in the receptor-binding site of the bacterial adhesin FimH. Biochemistry 2012, 51, 4790–4799. [Google Scholar] [CrossRef]
- Bas, D.C.; Rogers, D.M.; Jensen, J.H. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins 2008, 73, 765–783. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Feig, M.; Brooks, C.L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- De Nunzio, C.; Bartoletti, R.; Tubaro, A.; Simonato, A. Role of D-Mannose in the Prevention of Recurrent Uncomplicated Cystitis: State of the Art and Future Perspectives. Antibiotics 2021, 10, 373. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd | Structure | IC50 (nM) | RIP a | cLogP |
|---|---|---|---|---|
| 11 | 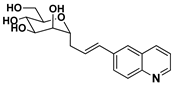 | 3.17 ± 2.3 | 887 | 1.16 |
| 18 |  | 30.28 ± 9.0 | 93 | 3.16 |
| 20 |  | 0.82 ± 0.4 | 3428 | 1.66 |
| 21 |  | 19.4 ± 5.2 | 145 | 1.44 |
| 22 |  | 74.13 ± 48.1 | 38 | 0.02 |
| 23 |  | 2810.74 ± 2546 | 1 | −1.58 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mousavifar, L.; Sarshar, M.; Bridot, C.; Scribano, D.; Ambrosi, C.; Palamara, A.T.; Vergoten, G.; Roubinet, B.; Landemarre, L.; Bouckaert, J.; et al. Insightful Improvement in the Design of Potent Uropathogenic E. coli FimH Antagonists. Pharmaceutics 2023, 15, 527. https://doi.org/10.3390/pharmaceutics15020527
Mousavifar L, Sarshar M, Bridot C, Scribano D, Ambrosi C, Palamara AT, Vergoten G, Roubinet B, Landemarre L, Bouckaert J, et al. Insightful Improvement in the Design of Potent Uropathogenic E. coli FimH Antagonists. Pharmaceutics. 2023; 15(2):527. https://doi.org/10.3390/pharmaceutics15020527
Chicago/Turabian StyleMousavifar, Leila, Meysam Sarshar, Clarisse Bridot, Daniela Scribano, Cecilia Ambrosi, Anna Teresa Palamara, Gérard Vergoten, Benoît Roubinet, Ludovic Landemarre, Julie Bouckaert, and et al. 2023. "Insightful Improvement in the Design of Potent Uropathogenic E. coli FimH Antagonists" Pharmaceutics 15, no. 2: 527. https://doi.org/10.3390/pharmaceutics15020527
APA StyleMousavifar, L., Sarshar, M., Bridot, C., Scribano, D., Ambrosi, C., Palamara, A. T., Vergoten, G., Roubinet, B., Landemarre, L., Bouckaert, J., & Roy, R. (2023). Insightful Improvement in the Design of Potent Uropathogenic E. coli FimH Antagonists. Pharmaceutics, 15(2), 527. https://doi.org/10.3390/pharmaceutics15020527