
Cannabidiol-Loaded Nanostructured Lipid Carriers (NLCs) for Dermal Delivery: Enhancement of Photostability, Cell Viability, and Anti-Inflammatory Activity


 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of CBD-Loaded NLCs
2.3. Particle Sizes and Size Distributions
2.4. Zeta Potential
2.5. Particle Morphology
2.6. Drug Encapsulation Efficiency (E.E.)
2.7. Thermal Analysis
2.8. X-ray Diffraction (XRD)
2.9. Photostabilities of CBD-NLCs
2.10. In Vitro Release Study
2.11. In Vitro Skin Permeation Study
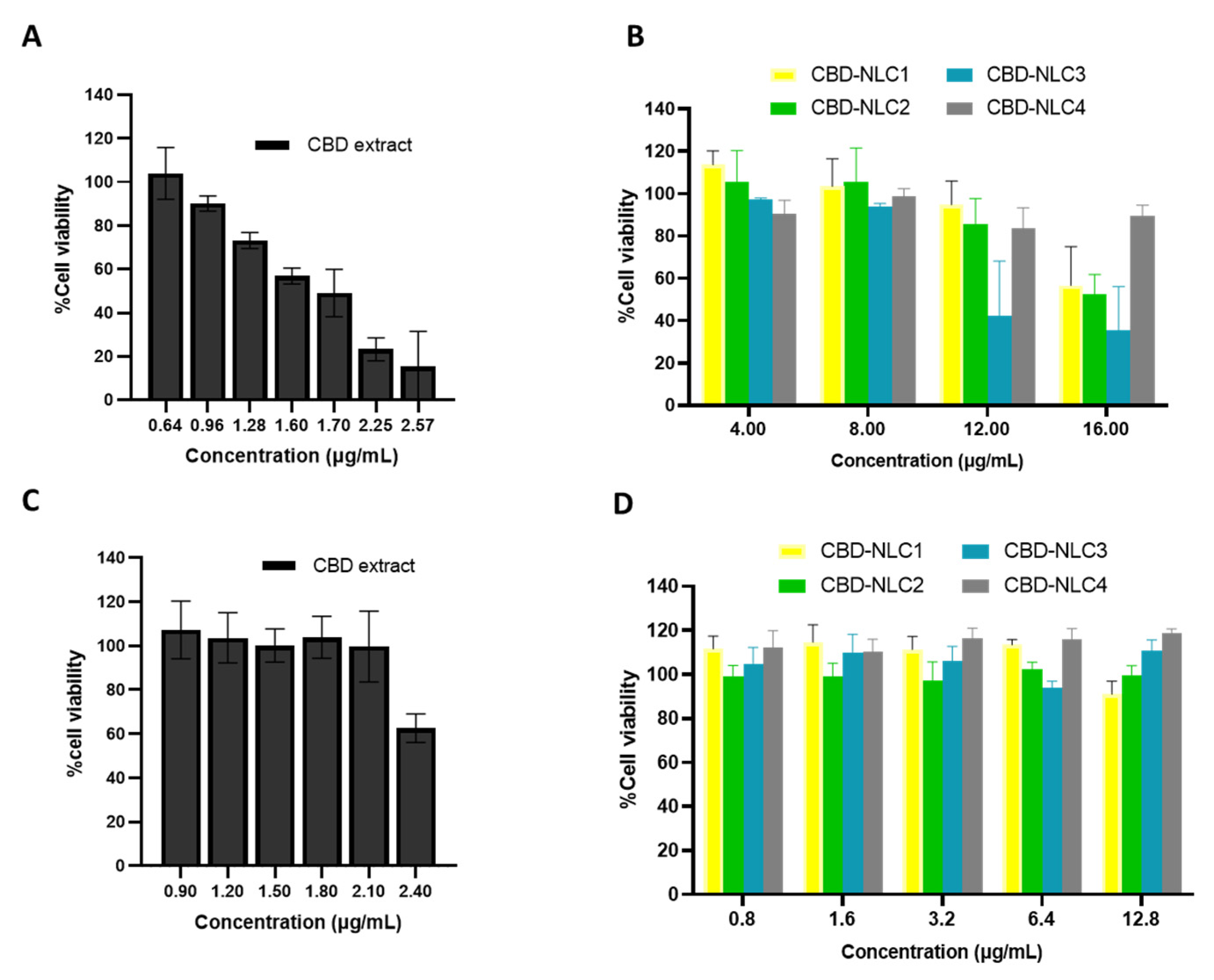
2.12. Cytotoxicity in Skin Cells
2.12.1. Cell Preparation
2.12.2. MTS Cell Proliferation Assay
2.13. Anti-Inflammatory Study
2.13.1. Cell Culture Preparation
2.13.2. Cell Viability
2.13.3. Measurement of IL-6 Production
2.14. Statistical Analysis
3. Results and Discussions
3.1. Particle Size and Size Distribution Analysis
3.2. Zeta Potential
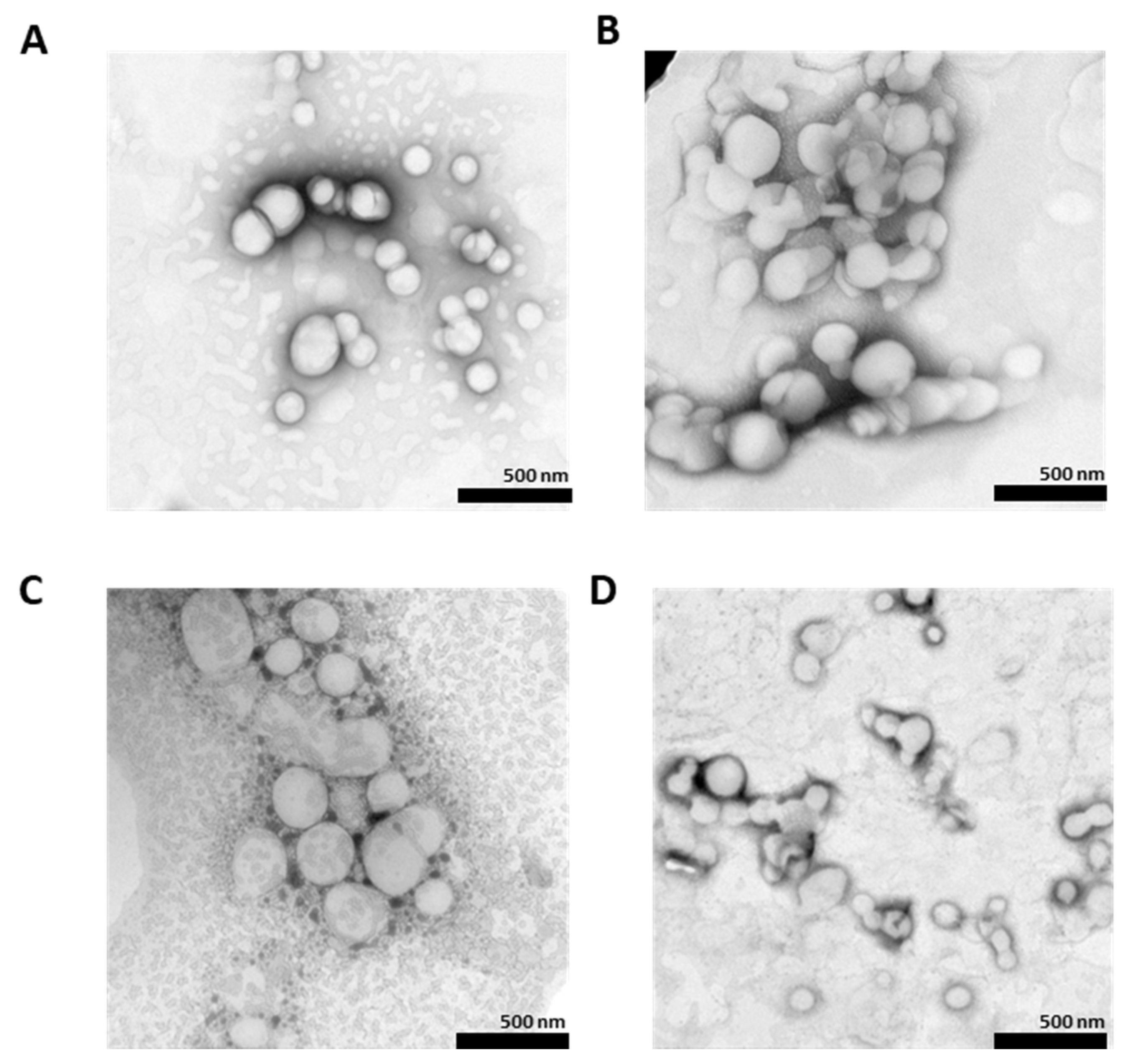
3.3. Particle Morphology
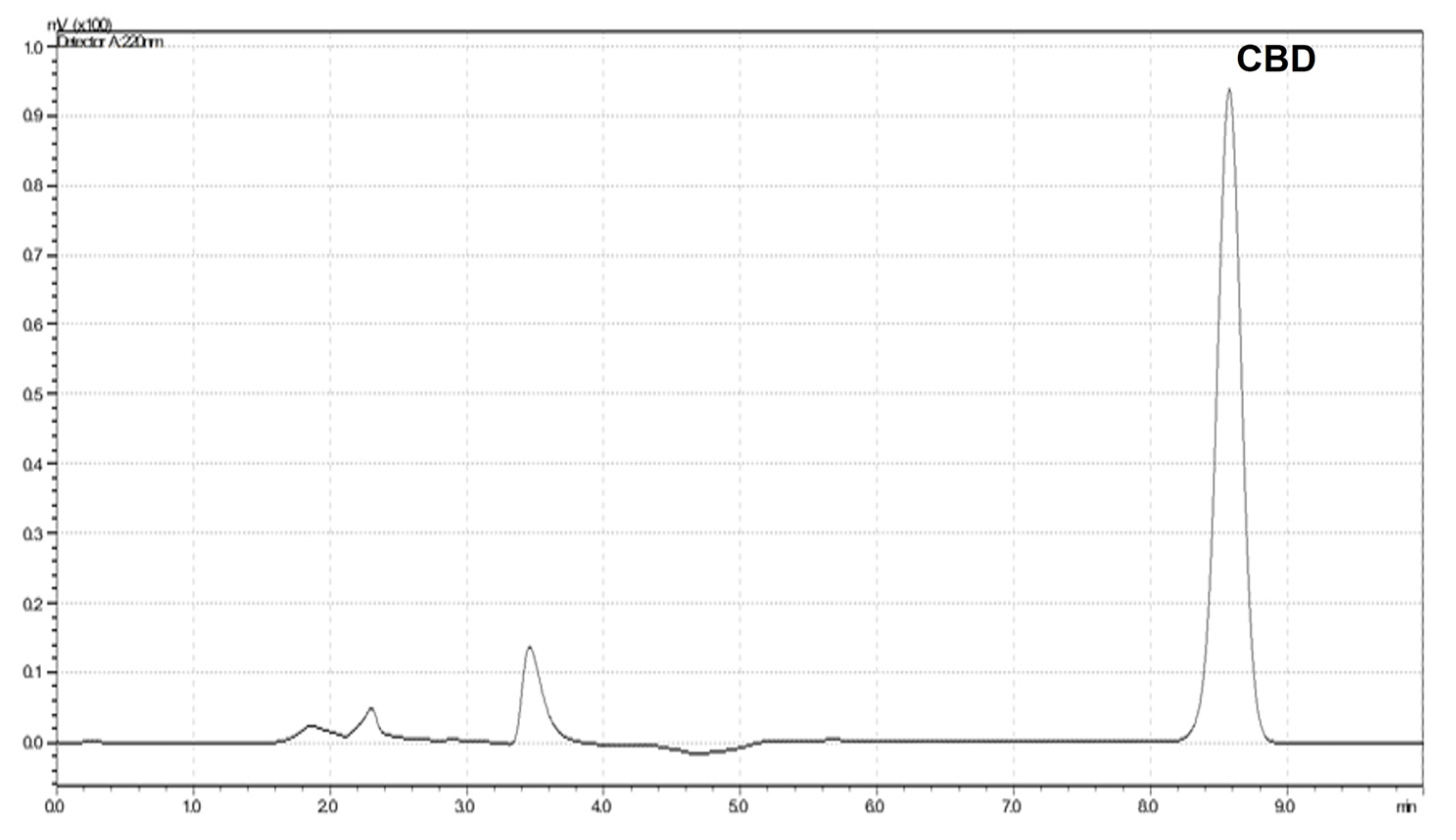
3.4. Encapsulation Efficiency
3.5. Thermal Analysis
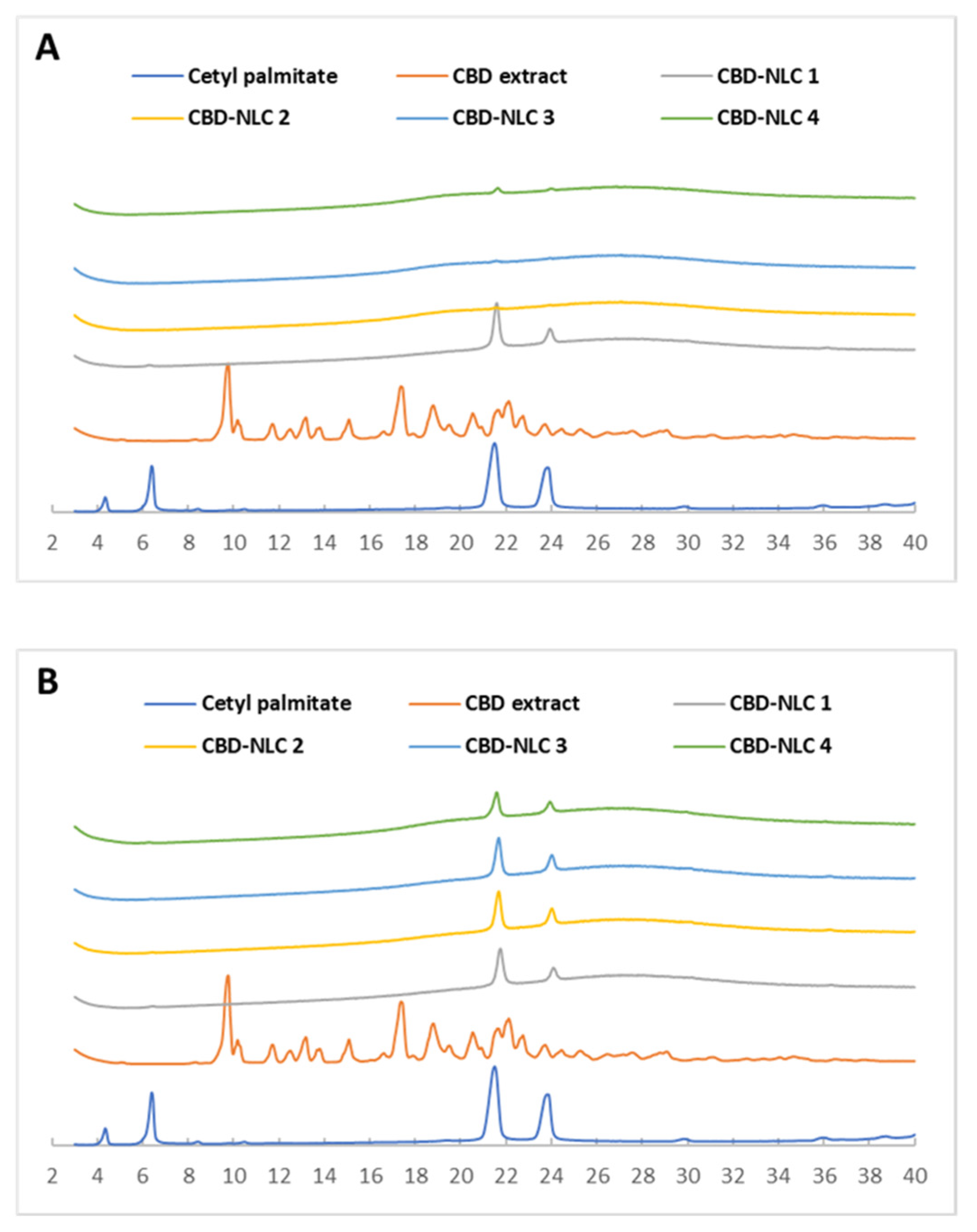
3.6. X-ray Diffraction
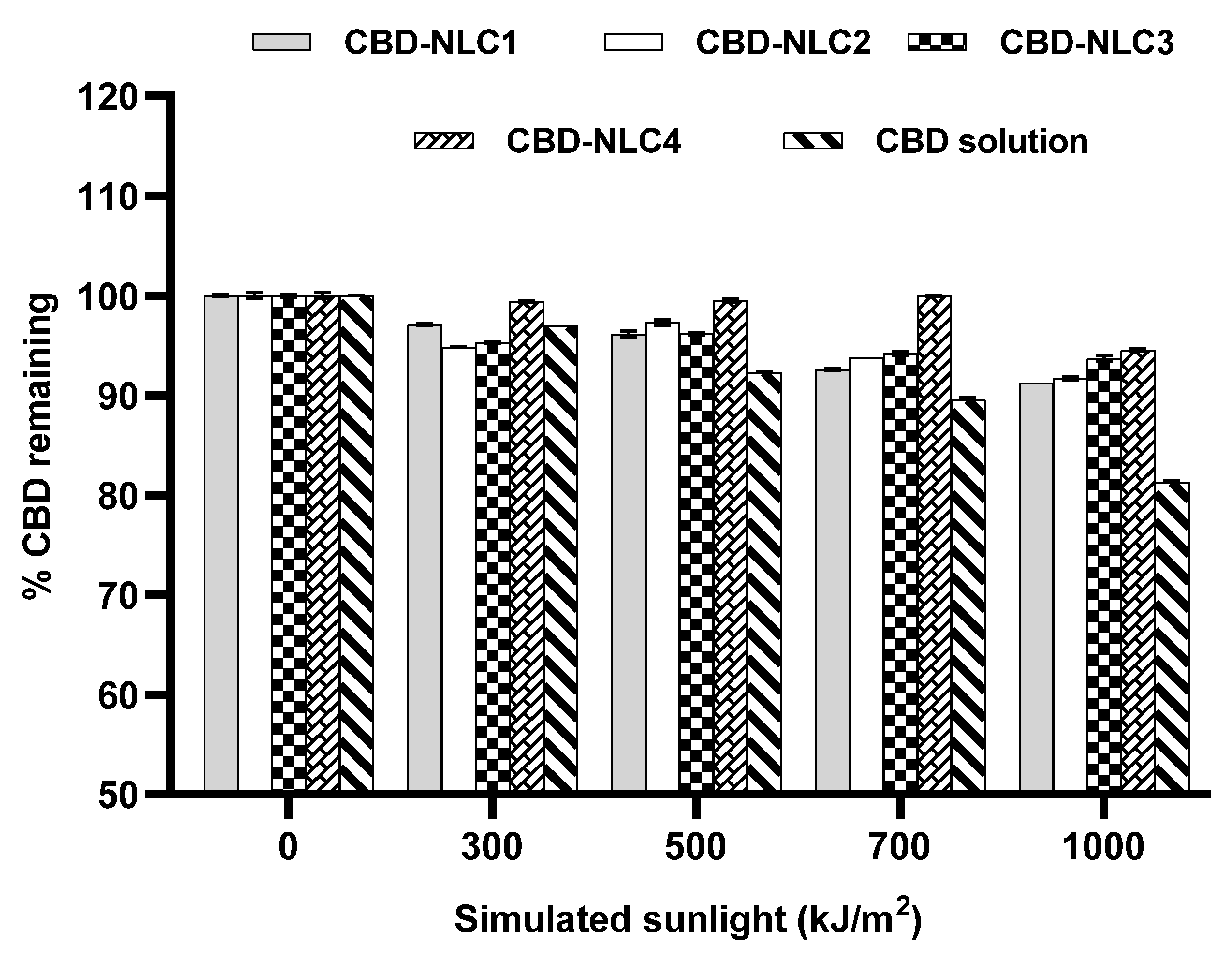
3.7. Photostability of CBD-NLCs
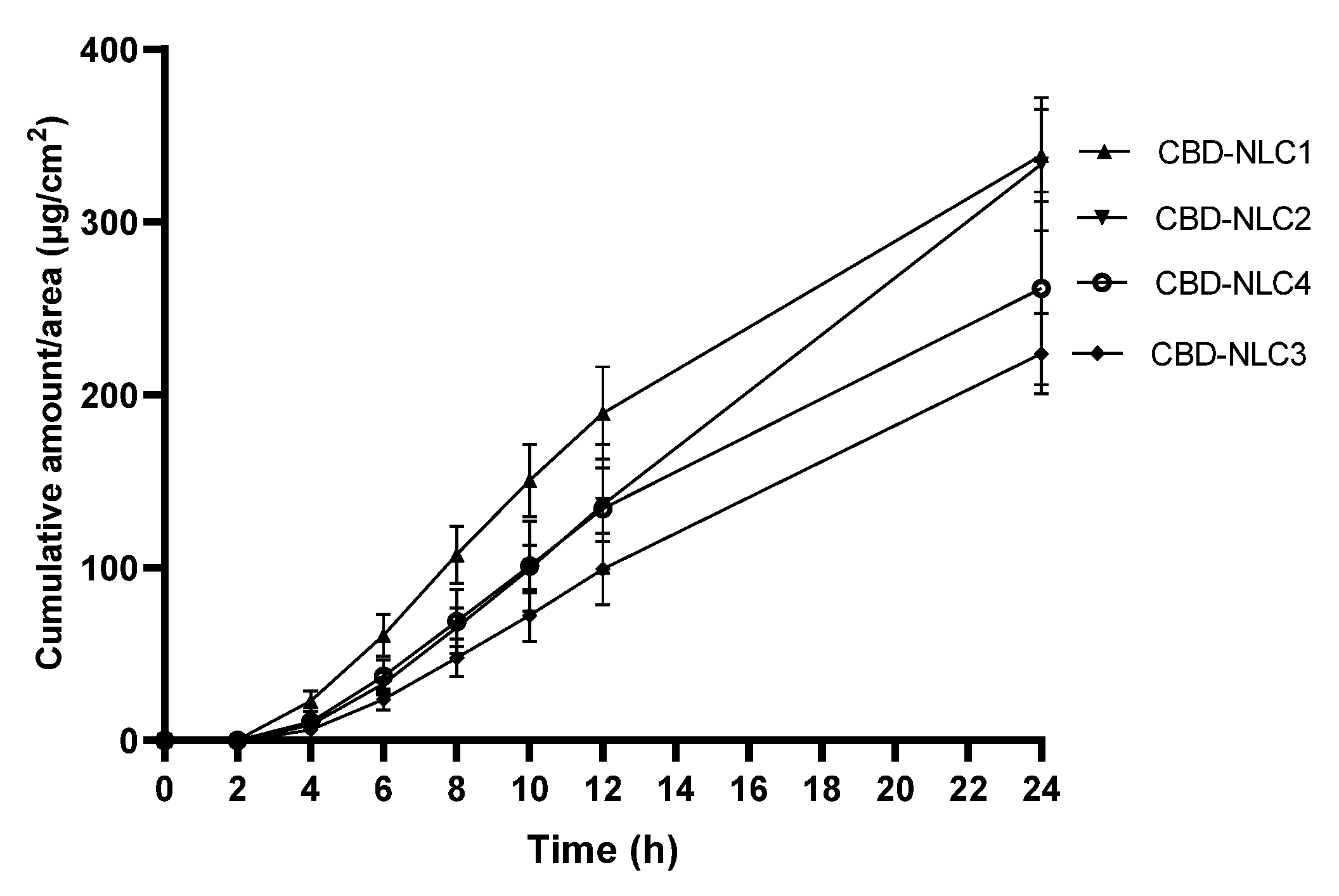
3.8. In Vitro Release
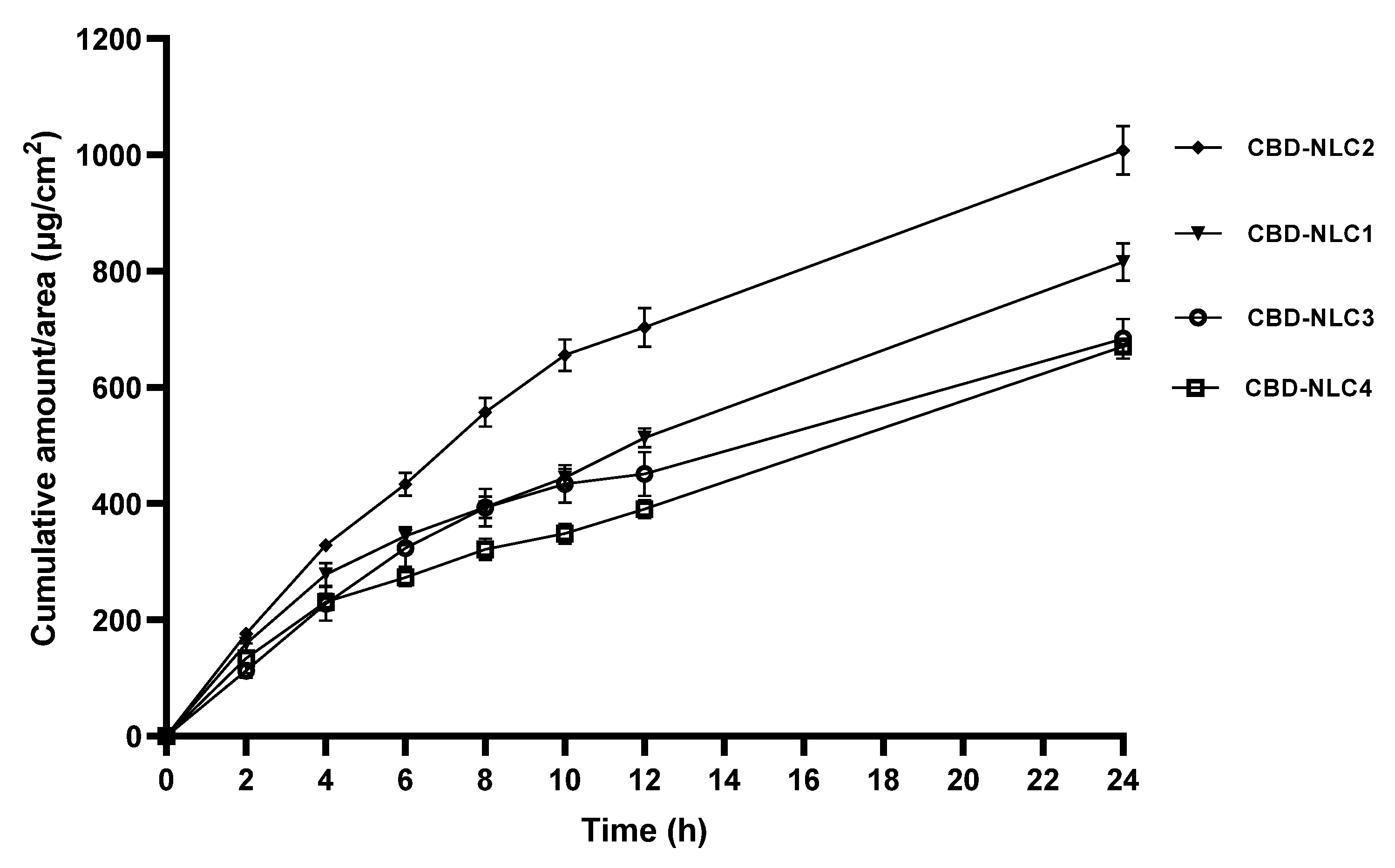
3.9. In Vitro Skin Permeation
3.10. Cytotoxicity on Skin Cells
3.11. Anti-Inflammatory Effect in RAW264.7 Cell Macrophages
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sharkawy, A.; Silva, A.M.; Rodrigues, F.; Barreiro, F.; Rodrigues, A. Pickering emulsions stabilized with chitosan/collagen peptides nanoparticles as green topical delivery vehicles for cannabidiol (CBD). Colloids Surf. A Physicochem. Eng. Asp. 2021, 631, 127677. [Google Scholar] [CrossRef]
- Szachowicz-Petelska, B.; Luczaj, W.; Wronski, A.; Jastrzab, A.; Dobrzynska, I. The Differential Effect of Cannabidiol on the Composition and Physicochemical Properties of Keratinocyte and Fibroblast Membranes from Psoriatic Patients and Healthy People. Membranes 2021, 11, 111. [Google Scholar] [CrossRef] [PubMed]
- Chelliah, M.P.; Zinn, Z.; Khuu, P.; Teng, J.M.C. Self-initiated use of topical cannabidiol oil for epidermolysis bullosa. Pediatr. Dermatol. 2018, 35, e224–e227. [Google Scholar] [CrossRef]
- Palmieri, B.; Laurino, C.; Vadalà, M. A therapeutic effect of cbd-enriched ointment in inflammatory skin diseases and cutaneous scars. Clin. Ter. 2019, 170, e93–e99. [Google Scholar] [CrossRef] [PubMed]
- Sheriff, T.; Lin, M.J.; Dubin, D.; Khorasani, H. The potential role of cannabinoids in dermatology. J. Dermatolog. Treat. 2020, 31, 839–845. [Google Scholar] [CrossRef] [PubMed]
- di Giacomo, V.; Recinella, L.; Chiavaroli, A.; Orlando, G.; Cataldi, A.; Rapino, M.; Di Valerio, V.; Politi, M.; Antolini, M.D.; Acquaviva, A.; et al. Metabolomic Profile and Antioxidant/Anti-Inflammatory Effects of Industrial Hemp Water Extract in Fibroblasts, Keratinocytes and Isolated Mouse Skin Specimens. Antioxidants 2021, 10, 44. [Google Scholar] [CrossRef]
- Baswan, S.M.; Klosner, A.E.; Glynn, K.; Rajgopal, A.; Malik, K.; Yim, S.; Stern, N. Therapeutic Potential of Cannabidiol (CBD) for Skin Health and Disorders. Clin. Cosmet. Investig. Dermatol. 2020, 13, 927–942. [Google Scholar] [CrossRef]
- Lodzki, M.; Godin, B.; Rakou, L.; Mechoulam, R.; Gallily, R.; Touitou, E. Cannabidiol-transdermal delivery and anti-inflammatory effect in a murine model. J. Control. Release 2003, 93, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Junaid, M.S.A.; Tijani, A.O.; Puri, A.; Banga, A.K. In vitro percutaneous absorption studies of cannabidiol using human skin: Exploring the effect of drug concentration, chemical enhancers, and essential oils. Int. J. Pharm. 2022, 616, 121540. [Google Scholar] [CrossRef] [PubMed]
- Stinchcomb, A.L.; Valiveti, S.; Hammell, D.C.; Ramsey, D.R. Human skin permeation of Delta8-tetrahydrocannabinol, cannabidiol and cannabinol. J. Pharm. Pharmacol. 2004, 56, 291–297. [Google Scholar] [CrossRef]
- Kosovic, E.; Sykora, D.; Kuchar, M. Stability Study of Cannabidiol in the Form of Solid Powder and Sunflower Oil Solution. Pharmaceutics 2021, 13, 412. [Google Scholar] [CrossRef] [PubMed]
- Mazzetti, C.; Ferri, E.; Pozzi, M.; Labra, M. Quantification of the content of cannabinol in commercially available e-liquids and studies on their thermal and photo-stability. Sci. Rep. 2020, 10, 3697. [Google Scholar] [CrossRef]
- Grifoni, L.; Vanti, G.; Donato, R.; Sacco, C.; Bilia, A.R. Promising Nanocarriers to Enhance Solubility and Bioavailability of Cannabidiol for a Plethora of Therapeutic Opportunities. Molecules 2022, 27, 6070. [Google Scholar] [CrossRef] [PubMed]
- Gupta, T.; Singh, J.; Kaur, S.; Sandhu, S.; Singh, G.; Kaur, I.P. Enhancing Bioavailability and Stability of Curcumin Using Solid Lipid Nanoparticles (CLEN): A Covenant for Its Effectiveness. Front. Bioeng. Biotechnol. 2020, 8, 879. [Google Scholar] [CrossRef]
- Coimbra, M.; Isacchi, B.; van Bloois, L.; Torano, J.S.; Ket, A.; Wu, X.; Broere, F.; Metselaar, J.M.; Rijcken, C.J.F.; Storm, G.; et al. Improving solubility and chemical stability of natural compounds for medicinal use by incorporation into liposomes. Int. J. Pharm. 2011, 416, 433–442. [Google Scholar] [CrossRef]
- Abdulbaqi, I.M.; Darwis, Y.; Khan, N.A.; Assi, R.A.; Khan, A.A. Ethosomal nanocarriers: The impact of constituents and formulation techniques on ethosomal properties, in vivo studies, and clinical trials. Int. J. Nanomed. 2016, 11, 2279–2304. [Google Scholar] [CrossRef] [PubMed]
- Soliman, G.M.; Zhang, Y.L.; Merle, G.; Cerruti, M.; Barralet, J. Hydrocaffeic acid-chitosan nanoparticles with enhanced stability, mucoadhesion and permeation properties. Eur. J. Pharm. Biopharm. 2014, 88, 1026–1037. [Google Scholar] [CrossRef]
- Kim, M.H.; Jeon, Y.E.; Kang, S.; Lee, J.Y.; Lee, K.W.; Kim, K.T.; Kim, D.D. Lipid Nanoparticles for Enhancing the Physicochemical Stability and Topical Skin Delivery of Orobol. Pharmaceutics 2020, 12, 845. [Google Scholar] [CrossRef]
- Souto, E.B.; Fangueiro, J.F.; Fernandes, A.R.; Cano, A.; Sanchez-Lopez, E.; Garcia, M.L.; Severino, P.; Paganelli, M.O.; Chaud, M.V.; Silva, A.M. Physicochemical and biopharmaceutical aspects influencing skin permeation and role of SLN and NLC for skin drug delivery. Heliyon 2022, 8, e08938. [Google Scholar] [CrossRef]
- Garcês, A.; Amaral, M.H.; Sousa Lobo, J.M.; Silva, A.C. Formulations based on solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) for cutaneous use: A review. Eur. J. Pharm. Sci. 2018, 112, 159–167. [Google Scholar] [CrossRef]
- Pardeike, J.; Hommoss, A.; Müller, R.H. Lipid nanoparticles (SLN, NLC) in cosmetic and pharmaceutical dermal products. Int. J. Pharm. 2009, 366, 170–184. [Google Scholar] [CrossRef]
- Weber, S.; Zimmer, A.; Pardeike, J. Solid Lipid Nanoparticles (SLN) and Nanostructured Lipid Carriers (NLC) for pulmonary application: A review of the state of the art. Eur. J. Pharm. Biopharm. 2014, 86, 7–22. [Google Scholar] [CrossRef]
- Matarazzo, A.P.; Elisei, L.M.S.; Carvalho, F.C.; Bonfilio, R.; Ruela, A.L.M.; Galdino, G.; Pereira, G.R. Mucoadhesive nanostructured lipid carriers as a cannabidiol nasal delivery system for the treatment of neuropathic pain. Eur. J. Pharm. Sci. 2021, 159, 105698. [Google Scholar] [CrossRef] [PubMed]
- Teeranachaideekul, V.; Morakul, B.; Boonme, P.; Pornputtapitak, W.; Junyaprasert, V. Effect of Lipid and Oil Compositions on Physicochemical Properties and Photoprotection of Octyl Methoxycinnamate-loaded Nanostructured Lipid Carriers (NLC). J. Oleo Sci. 2020, 69, 1627–1639. [Google Scholar] [CrossRef]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. 2010, 67, 217–223. [Google Scholar]
- Arce, F.J.; Asano, N.; See, G.L.; Itakura, S.; Todo, H.; Sugibayashi, K. Usefulness of Artificial Membrane, Strat-M((R)), in the Assessment of Drug Permeation from Complex Vehicles in Finite Dose Conditions. Pharmaceutics 2020, 12, 173. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Henning, S.M.; Heber, D. Limitations of MTT and MTS-based assays for measurement of antiproliferative activity of green tea polyphenols. PLoS ONE 2010, 5, e10202. [Google Scholar] [CrossRef] [PubMed]
- Riebeling, C.; Piret, J.P.; Trouiller, B.; Nelissen, I.; Saout, C.; Toussaint, O.; Haase, A. A guide to nanosafety testing: Considerations on cytotoxicity testing in different cell models. NanoImpact 2018, 10, 1–10. [Google Scholar] [CrossRef]
- Teeranachaideekul, V.; Parichatikanond, W.; Junyaprasert, V.B.; Morakul, B. Pumpkin Seed Oil-Loaded Niosomes for Topical Application: 5alpha-Reductase Inhibitory, Anti-Inflammatory, and In Vivo Anti-Hair Loss Effects. Pharmaceuticals 2022, 15, 930. [Google Scholar] [CrossRef]
- Takechi-Haraya, Y.; Ohgita, T.; Demizu, Y.; Saito, H.; Izutsu, K.I.; Sakai-Kato, K. Current Status and Challenges of Analytical Methods for Evaluation of Size and Surface Modification of Nanoparticle-Based Drug Formulations. AAPS PharmSciTech 2022, 23, 150. [Google Scholar] [CrossRef]
- Andalib, S.; Varshosaz, J.; Hassanzadeh, F.; Sadeghi, H. Optimization of LDL targeted nanostructured lipid carriers of 5-FU by a full factorial design. Adv. Biomed. Res. 2012, 1, 45. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Blanco, J.; Sebastian, V.; Benoit, J.P.; Torres-Suarez, A.I. Lipid nanocapsules decorated and loaded with cannabidiol as targeted prolonged release carriers for glioma therapy: In vitro screening of critical parameters. Eur. J. Pharm. Biopharm. 2019, 134, 126–137. [Google Scholar] [CrossRef]
- Vanti, G.; Grifoni, L.; Bergonzi, M.C.; Antiga, E.; Montefusco, F.; Caproni, M.; Bilia, A.R. Development and optimisation of biopharmaceutical properties of a new microemulgel of cannabidiol for locally-acting dermatological delivery. Int. J. Pharm. 2021, 607, 121036. [Google Scholar] [CrossRef]
- Banerjee, A.; Binder, J.; Salama, R.; Trant, J.F. Synthesis, characterization and stress-testing of a robust quillaja saponin stabilized oil-in-water phytocannabinoid nanoemulsion. J. Cannabis. Res. 2021, 3, 43. [Google Scholar] [CrossRef] [PubMed]
- Francke, N.M.; Schneider, F.; Baumann, K.; Bunjes, H. Formulation of Cannabidiol in Colloidal Lipid Carriers. Molecules 2021, 26, 1469. [Google Scholar] [CrossRef] [PubMed]
- Souto, E.B.; Baldim, I.; Oliveira, W.P.; Rao, R.; Yadav, N.; Gama, F.M.; Mahant, S. SLN and NLC for topical, dermal, and transdermal drug delivery. Expert. Opin. Drug. Deliv. 2020, 17, 357–377. [Google Scholar] [CrossRef]
- Kovacevic, A.; Savic, S.; Vuleta, G.; Müller, R.H.; Keck, C.M. Polyhydroxy surfactants for the formulation of lipid nanoparticles (SLN and NLC): Effects on size, physical stability and particle matrix structure. Int. J. Pharm. 2011, 406, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Kovačević, A.B.; Müller, R.H.; Savić, S.D.; Vuleta, G.M.; Keck, C.M. Solid lipid nanoparticles (SLN) stabilized with polyhydroxy surfactants: Preparation, characterization and physical stability investigation. Colloids Surf. A Physicochem. Eng. Asp. 2014, 444, 15–25. [Google Scholar] [CrossRef]
- Zarrintaj, P.; Ramsey, J.D.; Samadi, A.; Atoufi, Z.; Yazdi, M.K.; Ganjali, M.R.; Amirabad, L.M.; Zangene, E.; Farokhi, M.; Formela, K.; et al. Poloxamer: A versatile tri-block copolymer for biomedical applications. Acta Biomater. 2020, 110, 37–67. [Google Scholar] [CrossRef]
- Gundogdu, E.; Demir, E.S.; Ekinci, M.; Ozgenc, E.; Ilem-Ozdemir, D.; Senyigit, Z.; Gonzalez-Alvarez, I.; Bermejo, M. An Innovative Formulation Based on Nanostructured Lipid Carriers for Imatinib Delivery: Pre-Formulation, Cellular Uptake and Cytotoxicity Studies. Nanomaterials 2022, 12, 250. [Google Scholar] [CrossRef]
- Chantaburanan, T.; Teeranachaideekul, V.; Chantasart, D.; Jintapattanakit, A.; Junyaprasert, V.B. Effect of binary solid lipid matrix of wax and triglyceride on lipid crystallinity, drug-lipid interaction and drug release of ibuprofen-loaded solid lipid nanoparticles (SLN) for dermal delivery. J. Colloid. Interface. Sci. 2017, 504, 247–256. [Google Scholar] [CrossRef]
- Sharif Makhmal Zadeh, B.; Niro, H.; Rahim, F.; Esfahani, G. Ocular Delivery System for Propranolol Hydrochloride Based on Nanostructured Lipid Carrier. Sci. Pharm. 2018, 86, 16. [Google Scholar] [CrossRef] [PubMed]
- Unnisa, A.; Chettupalli, A.K.; Al Hagbani, T.; Khalid, M.; Jandrajupalli, S.B.; Chandolu, S.; Hussain, T. Development of Dapagliflozin Solid Lipid Nanoparticles as a Novel Carrier for Oral Delivery: Statistical Design, Optimization, In-Vitro and In-Vivo Characterization, and Evaluation. Pharmaceuticals 2022, 15, 568. [Google Scholar] [CrossRef] [PubMed]
- Haq, A.; Goodyear, B.; Ameen, D.; Joshi, V.; Michniak-Kohn, B. Strat-M(R) synthetic membrane: Permeability comparison to human cadaver skin. Int. J. Pharm. 2018, 547, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Haq, A.; Dorrani, M.; Goodyear, B.; Joshi, V.; Michniak-Kohn, B. Membrane properties for permeability testing: Skin versus synthetic membranes. Int. J. Pharm. 2018, 539, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Kadhum, W.R.; Kanai, S.; Todo, H.; Oshizaka, T.; Sugibayashi, K. Prediction of skin permeation by chemical compounds using the artificial membrane, Strat-M. Eur. J. Pharm. Sci. 2015, 67, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Sangiovanni, E.; Fumagalli, M.; Pacchetti, B.; Piazza, S.; Magnavacca, A.; Khalilpour, S.; Melzi, G.; Martinelli, G.; Dell’Agli, M. Cannabis sativa L. extract and cannabidiol inhibit in vitro mediators of skin inflammation and wound injury. Phytother. Res. 2019, 33, 2083–2093. [Google Scholar] [CrossRef]
- Li, Y.; Hao, D.; Wei, D.; Xiao, Y.; Liu, L.; Li, X.; Wang, L.; Gan, Y.; Yan, W.; Ke, B.; et al. Photoprotective Effects of Cannabidiol against Ultraviolet-B-Induced DNA Damage and Autophagy in Human Keratinocyte Cells and Mouse Skin Tissue. Molecules 2022, 27, 6740. [Google Scholar] [CrossRef]
- Gegotek, A.; Atalay, S.; Domingues, P.; Skrzydlewska, E. The Differences in the Proteome Profile of Cannabidiol-Treated Skin Fibroblasts following UVA or UVB Irradiation in 2D and 3D Cell Cultures. Cells 2019, 8, 995. [Google Scholar] [CrossRef]
- Josiah, A.J.; Pillai, S.K.; Cordier, W.; Nell, M.; Twilley, D.; Lall, N.; Ray, S.S. Cannabidiol-Mediated Green Synthesis, Characterization, and Cytotoxicity of Metal Nanoparticles in Human Keratinocyte Cells. ACS Omega 2021, 6, 29078–29090. [Google Scholar] [CrossRef]
- Petrosino, S.; Verde, R.; Vaia, M.; Allara, M.; Iuvone, T.; Di Marzo, V. Anti-inflammatory Properties of Cannabidiol, a Nonpsychotropic Cannabinoid, in Experimental Allergic Contact Dermatitis. J. Pharmacol. Exp. Ther. 2018, 365, 652–663. [Google Scholar] [CrossRef]
- Muller, A.K.; Albrecht, F.; Rohrer, C.; Koeberle, A.; Werz, O.; Schlormann, W.; Glei, M.; Lorkowski, S.; Wallert, M. Olive Oil Extracts and Oleic Acid Attenuate the LPS-Induced Inflammatory Response in Murine RAW264.7 Macrophages but Induce the Release of Prostaglandin E2. Nutrients 2021, 13, 4437. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Yang, Y.; Quan, Q.; Huo, T.; Yang, S.; Ju, R.; An, Q. Comparison of the in vitro Anti-Inflammatory Effect of Cannabidiol to Dexamethasone. Clin. Cosmet. Investig. Dermatol. 2022, 15, 1959–1967. [Google Scholar] [CrossRef]
- BenSaad, L.A.; Kim, K.H.; Quah, C.C.; Kim, W.R.; Shahimi, M. Anti-inflammatory potential of ellagic acid, gallic acid and punicalagin A&B isolated from Punica granatum. BMC Complement. Altern. Med. 2017, 17, 47. [Google Scholar] [CrossRef]
- Santamarina, A.B.; Pisani, L.P.; Baker, E.J.; Marat, A.D.; Valenzuela, C.A.; Miles, E.A.; Calder, P.C. Anti-inflammatory effects of oleic acid and the anthocyanin keracyanin alone and in combination: Effects on monocyte and macrophage responses and the NF-kappaB pathway. Food Funct. 2021, 12, 7909–7922. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Go, G.W.; Kim, W. Medium Chain Triglyceride (MCT) Oil Affects the Immunophenotype via Reprogramming of Mitochondrial Respiration in Murine Macrophages. Foods 2019, 8, 553. [Google Scholar] [CrossRef]
- Janik-Hazuka, M.; Szafraniec-Szczęsny, J.; Kamiński, K.; Odrobińska, J.; Zapotoczny, S. Uptake and in vitro anticancer activity of oleic acid delivered in nanocapsules stabilized by amphiphilic derivatives of hyaluronic acid and chitosan. Int. J. Biol. Macromol. 2020, 164, 2000–2009. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compositions | CBD-NLC1 | CBD-NLC2 | CBD-NLC3 | CBD-NLC4 |
|---|---|---|---|---|
| CP | 8 | 8 | 8 | 8 |
| CBD extract | 1 | 1 | 1 | 1 |
| TG450 | 1.8 | - | - | - |
| P188 | - | 2.5 | 2.5 | 2.5 |
| Transcutol® P | 2 | 2 | - | - |
| MCT | - | - | 2 | - |
| OA | - | - | - | 2 |
| Unigerm G2 | 1 | 1 | 1 | 1 |
| Water q.s. to | 100 | 100 | 100 | 100 |
| Formulation | Particle Size (nm) | PDI | Zeta Potential (mV) |
|---|---|---|---|
| CBD-NLC1 | 165.4 ± 0.8 | 0.009 ± 0.011 | −57.0 ± 1.7 |
| CBD-NLC2 | 163.2 ± 1.2 | 0.073 ± 0.009 | −29.0 ± 0.5 |
| CBD-NLC3 | 175.5 ± 1.9 | 0.087 ± 0.023 | −31.1 ± 0.8 |
| CBD-NLC4 | 166.9 ± 1.5 | 0.061 ± 0.020 | −39.8 ± 1.0 |
| Formulation | Condition | Onset (°C) | Endset (°C) | Melting Point (°C) | %CI |
|---|---|---|---|---|---|
| CBD extract | Initial | 65.7 | 68.0 | 67.0 | - |
| CP | Initial | 44.2 | 50.3 | 48.4 | 100.00 |
| CBD-NLC1 | Initial | 35.0 | 47.5 | 43.3 | 9.6 |
| 30 days | 41.5 | 47.5 | 46.0 | 49.6 | |
| CBD-NLC2 | Initial | 44.6 | 47.9 | 46.9 | 2.1 |
| 30 days | 43.8 | 47.8 | 46.7 | 37.6 | |
| CBD-NLC3 | Initial | 44.1 | 48.0 | 46.2 | 2.6 |
| 30 days | 44.4 | 47.9 | 46.8 | 39.6 | |
| CBD-NLC4 | Initial | 42.3 | 48.4 | 46.0 | 0.3 |
| 30 days | 43.0 | 47.0 | 45.6 | 17.7 |
| Formulation | Zero-Order | First-Order | Higuchi’s Model | Korsmeyer–Peppas |
|---|---|---|---|---|
| CBD-NLC1 | 0.9503 | 0.9630 | 0.9813 | 0.9441 |
| CBD-NLC2 | 0.9128 | 0.9326 | 0.9820 | 0.9761 |
| CBD-NLC3 | 0.9439 | 0.9186 | 0.9815 | 0.9800 |
| CBD-NLC4 | 0.9549 | 0.9640 | 0.9720 | 0.9246 |
| Formulation | Fluxss (µg/cm2/h) |
|---|---|
| CBD-NLC1 | 15.56 ± 1.22 a,b |
| CBD-NLC2 | 16.47 ± 1.81 a |
| CBD-NLC3 | 11.00 ± 1.02 b |
| CBD-NLC4 | 12.52 ± 2.71 a,b |
| Formulation | IL-6 Level (pg/mL) | %IL-6 Inhibition over Positive Control |
|---|---|---|
| Negative control | 3.4 ± 1.8 | - |
| Positive control | 573.4 ± 4.0 | 0.00 |
| CBD extract | 483.1 ± 16.3 | 15.7 ± 2.8 |
| CBD-NLC1 | 540.0 ± 8.4 | 5.8 ± 1.5 |
| CBD-NLC2 | 540.7 ± 9.0 | 5.7 ± 1.6 |
| CBD-NLC3 | 433.98 ± 38.7 | 24.3 ± 6.7 |
| CBD-NLC4 | 361.4 ± 4.2 | 37.0 ± 0.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morakul, B.; Junyaprasert, V.B.; Sakchaisri, K.; Teeranachaideekul, V. Cannabidiol-Loaded Nanostructured Lipid Carriers (NLCs) for Dermal Delivery: Enhancement of Photostability, Cell Viability, and Anti-Inflammatory Activity. Pharmaceutics 2023, 15, 537. https://doi.org/10.3390/pharmaceutics15020537
Morakul B, Junyaprasert VB, Sakchaisri K, Teeranachaideekul V. Cannabidiol-Loaded Nanostructured Lipid Carriers (NLCs) for Dermal Delivery: Enhancement of Photostability, Cell Viability, and Anti-Inflammatory Activity. Pharmaceutics. 2023; 15(2):537. https://doi.org/10.3390/pharmaceutics15020537
Chicago/Turabian StyleMorakul, Boontida, Varaporn Buraphacheep Junyaprasert, Krisada Sakchaisri, and Veerawat Teeranachaideekul. 2023. "Cannabidiol-Loaded Nanostructured Lipid Carriers (NLCs) for Dermal Delivery: Enhancement of Photostability, Cell Viability, and Anti-Inflammatory Activity" Pharmaceutics 15, no. 2: 537. https://doi.org/10.3390/pharmaceutics15020537
APA StyleMorakul, B., Junyaprasert, V. B., Sakchaisri, K., & Teeranachaideekul, V. (2023). Cannabidiol-Loaded Nanostructured Lipid Carriers (NLCs) for Dermal Delivery: Enhancement of Photostability, Cell Viability, and Anti-Inflammatory Activity. Pharmaceutics, 15(2), 537. https://doi.org/10.3390/pharmaceutics15020537