
Transferability of Published Population Pharmacokinetic Models for Apixaban and Rivaroxaban to Subjects with Obesity Treated for Venous Thromboembolism: A Systematic Review and External Evaluations
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Review of Published PPK Models
2.2. Independent External Validation Data Set
2.3. External Predictive Performance Evaluation of Apixaban and Rivaroxaban PPK Models
2.4. Prediction-Based Diagnostics
2.5. Simulation-Based Diagnostics
3. Results
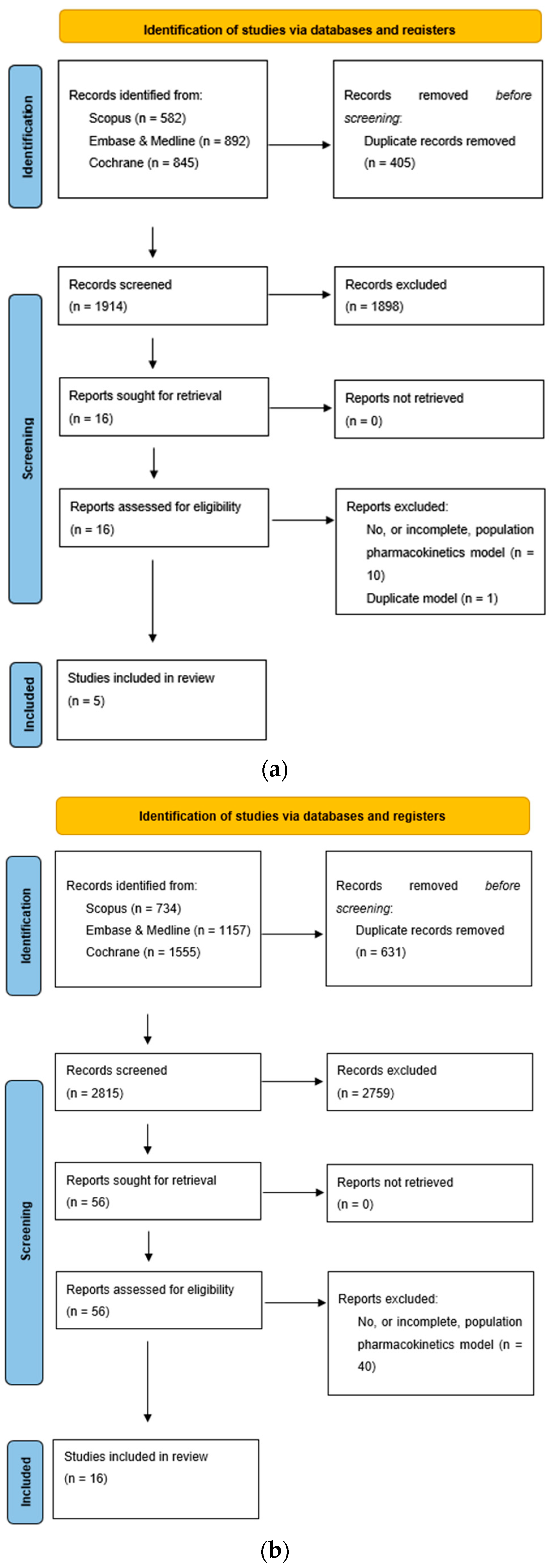
3.1. Review of Published PPK Studies
3.1.1. Apixaban
3.1.2. Rivaroxaban
3.2. External Validation Dataset Cohort
3.3. External Predictability Evaluation
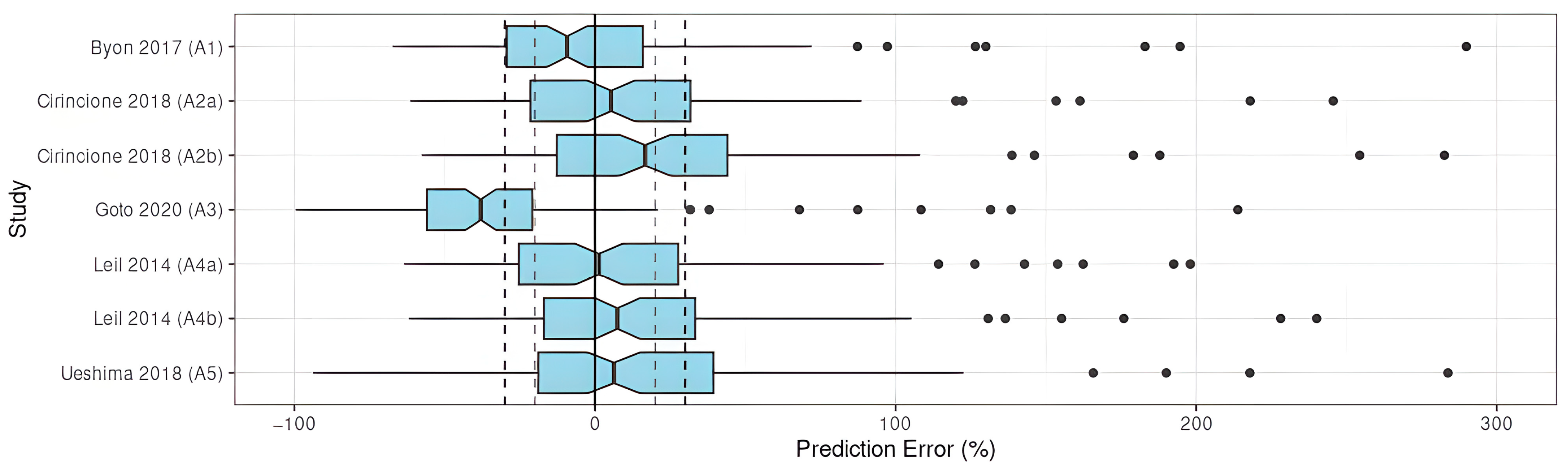
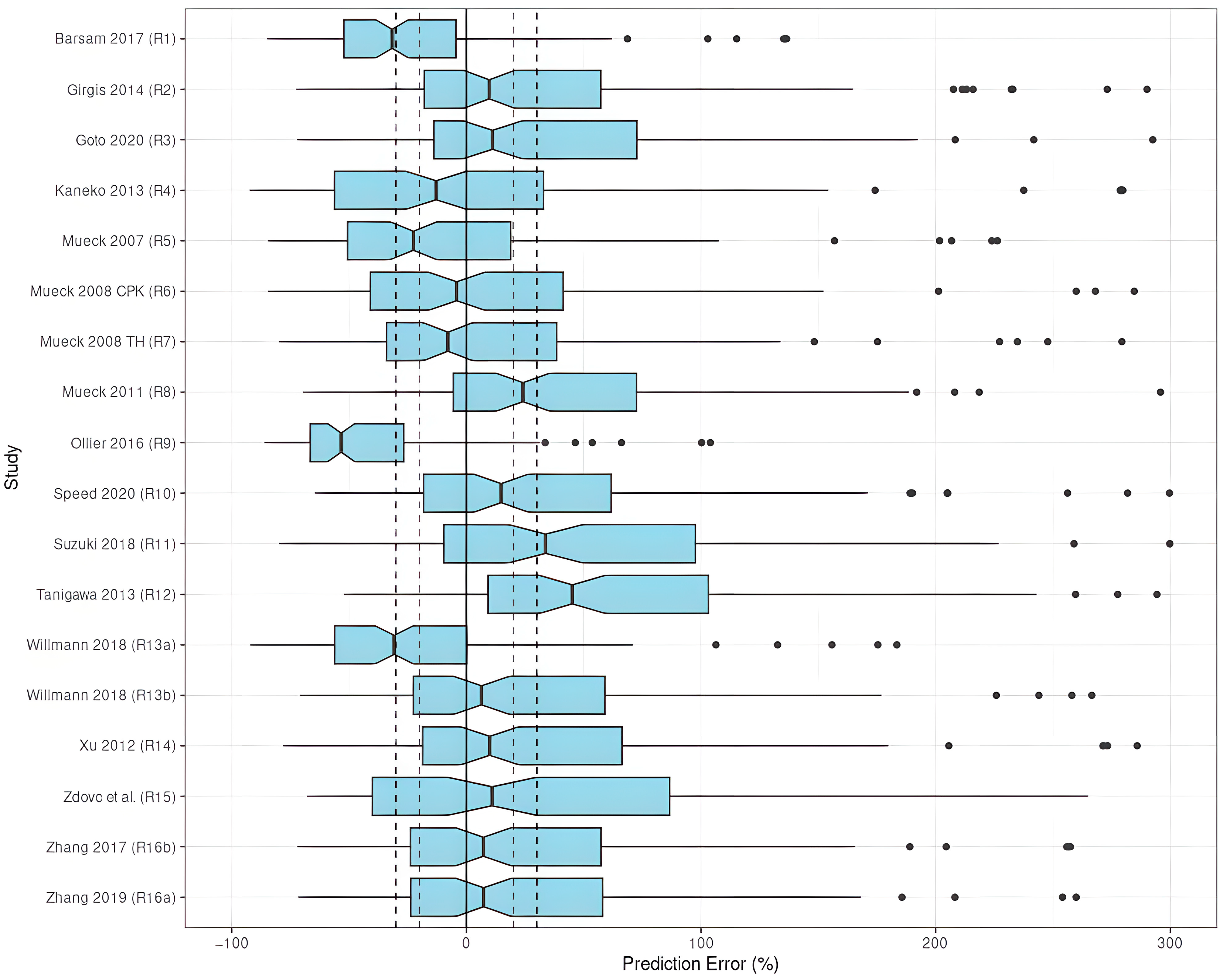
3.3.1. Prediction-Based Diagnostics
Apixaban
Rivaroxaban
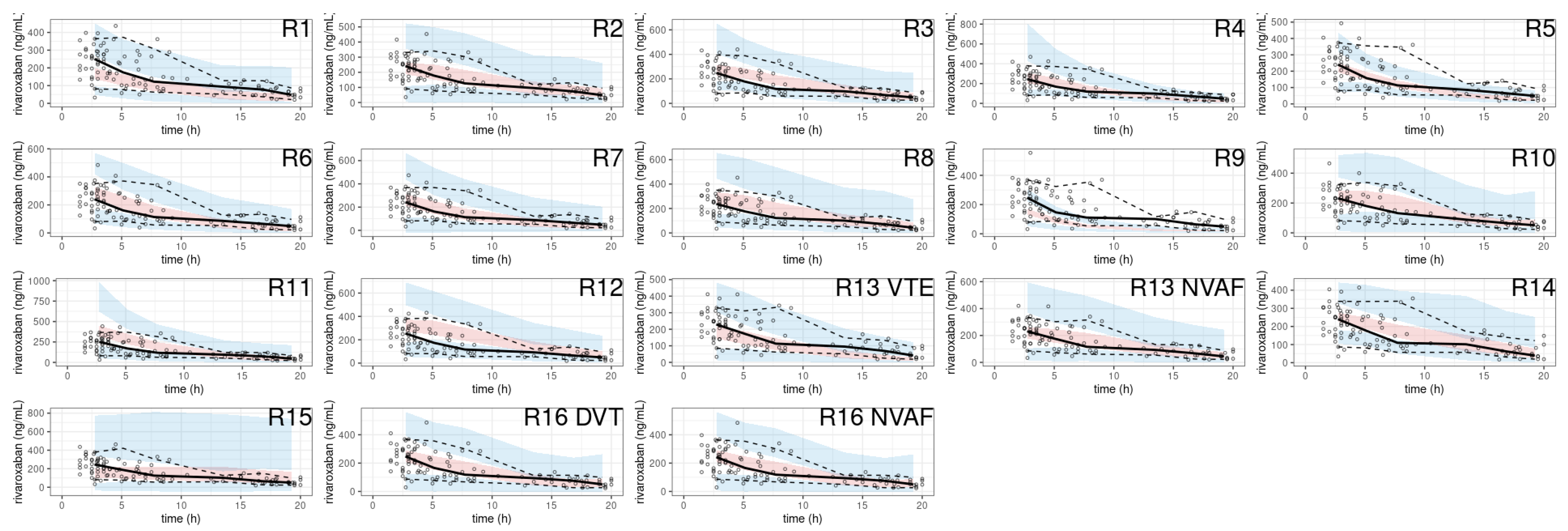
3.3.2. Simulation-Based Diagnostics
Apixaban
Rivaroxaban
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gallus, S.; Lugo, A.; Murisic, B.; Bosetti, C.; Boffetta, P.; La Vecchia, C. Overweight and Obesity in 16 European Countries. Eur. J. Nutr. 2015, 54, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.M.; Fryar, C.D.; Carroll, M.D.; Freedman, D.S.; Ogden, C.L. Trends in Obesity and Severe Obesity Prevalence in US Youth and Adults by Sex and Age, 2007–2008 to 2015–2016. JAMA 2018, 319, 1723–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, S.J.; Kompaniyets, L.; Freedman, D.S.; Kraus, E.M.; Porter, R.; Blanck, H.M.; Goodman, A.B. Longitudinal Trends in Body Mass Index Before and During the COVID-19 Pandemic Among Persons Aged 2–19 Years—United States, 2018–2020. Morb. Mortal. Wkly. Rep. 2021, 70, 1278–1283. [Google Scholar] [CrossRef] [PubMed]
- Ihaddadene, R.; Carrier, M. The Use of Anticoagulants for the Treatment and Prevention of Venous Thromboembolism in Obese Patients: Implications for Safety. Expert Opin. Drug Saf. 2016, 15, 65–74. [Google Scholar] [CrossRef]
- Kearon, C.; Ageno, W.; Cannegieter, S.C.; Cosmi, B.; Geersing, G.-J.; Kyrle, P.A. The Subcommittees on Control of Anticoagulation, and Predictive and Diagnostic Variables in Thrombotic Disease Categorization of Patients as Having Provoked or Unprovoked Venous Thromboembolism: Guidance from the SSC of ISTH. J. Thromb. Haemost. 2016, 14, 1480–1483. [Google Scholar] [CrossRef]
- van Es, N.; Coppens, M.; Schulman, S.; Middeldorp, S.; Buller, H.R. Direct Oral Anticoagulants Compared with Vitamin K Antagonists for Acute Venous Thromboembolism: Evidence from Phase 3 Trials. Blood 2014, 124, 1968–1975. [Google Scholar] [CrossRef]
- Byon, W.; Garonzik, S.; Boyd, R.A.; Frost, C.E. Apixaban: A Clinical Pharmacokinetic and Pharmacodynamic Review. Clin. Pharmacokinet. 2019, 58, 1265–1279. [Google Scholar] [CrossRef] [Green Version]
- Willmann, S.; Zhang, L.; Frede, M.; Kubitza, D.; Mueck, W.; Schmidt, S.; Solms, A.; Yan, X.; Garmann, D. Integrated Population Pharmacokinetic Analysis of Rivaroxaban Across Multiple Patient Populations. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 309–320. [Google Scholar] [CrossRef]
- Jain, R.; Chung, S.M.; Jain, L.; Khurana, M.; Lau, S.W.J.; Lee, J.E.; Vaidyanathan, J.; Zadezensky, I.; Choe, S.; Sahajwalla, C.G. Implications of Obesity for Drug Therapy: Limitations and Challenges. Clin. Pharmacol. Ther. 2011, 90, 77–89. [Google Scholar] [CrossRef]
- Morrish, G.A.; Pai, M.P.; Green, B. The Effects of Obesity on Drug Pharmacokinetics in Humans. Expert Opin. Drug Metab. Toxicol. 2011, 7, 697–706. [Google Scholar] [CrossRef]
- Smit, C.; De Hoogd, S.; Brüggemann, R.J.M.; Knibbe, C.A.J. Obesity and Drug Pharmacology: A Review of the Influence of Obesity on Pharmacokinetic and Pharmacodynamic Parameters. Expert Opin. Drug Metab. Toxicol. 2018, 14, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Han, P.Y.; Duffull, S.B.; Kirkpatrick, C.M.J.; Green, B. Dosing in Obesity: A Simple Solution to a Big Problem. Clin. Pharmacol. Ther. 2007, 82, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.; Beyer-Westendorf, J.; Davidson, B.L.; Huisman, M.V.; Sandset, P.M.; Moll, S. Use of the Direct Oral Anticoagulants in Obese Patients: Guidance from the SSC of the ISTH. J. Thromb. Haemost. 2016, 14, 1308–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, K.A.; Beyer-Westendorf, J.; Davidson, B.L.; Huisman, M.V.; Sandset, P.M.; Moll, S. Use of Direct Oral Anticoagulants in Patients with Obesity for Treatment and Prevention of Venous Thromboembolism: Updated Communication from the ISTH SSC Subcommittee on Control of Anticoagulation. J. Thromb. Haemost. 2021, 19, 1874–1882. [Google Scholar] [CrossRef]
- Brum, L. Population Pharmacokinetics; FDA Guidance; Food and Drug Administration: Rockville, MD, USA, 2022; p. 27. [Google Scholar]
- Ballerie, A.; Nguyen Van, R.; Lacut, K.; Galinat, H.; Rousseau, C.; Pontis, A.; Nédelec-Gac, F.; Lescoat, A.; Belhomme, N.; Guéret, P.; et al. Apixaban and Rivaroxaban in Obese Patients Treated for Venous Thromboembolism: Drug Levels and Clinical Outcomes. Thromb. Res. 2021, 208, 39–44. [Google Scholar] [CrossRef]
- Beal, S.L. Ways to Fit a PK Model with Some Data Below the Quantification Limit. J. Pharmacokinet. Pharmacodyn. 2001, 28, 481–504. [Google Scholar] [CrossRef]
- Hanafin, P.O.; Nation, R.L.; Scheetz, M.H.; Zavascki, A.P.; Sandri, A.M.; Kwa, A.L.; Cherng, B.P.Z.; Kubin, C.J.; Yin, M.T.; Wang, J.; et al. Assessing the Predictive Performance of Population Pharmacokinetic Models for Intravenous Polymyxin B in Critically Ill Patients. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 1525–1537. [Google Scholar] [CrossRef]
- Zhang, H.; Sheng, C.; Liu, L.; Luo, B.; Fu, Q.; Zhao, Q.; Li, J.; Liu, Y.; Deng, R.; Jiao, Z.; et al. Systematic External Evaluation of Published Population Pharmacokinetic Models of Mycophenolate Mofetil in Adult Kidney Transplant Recipients Co-administered with Tacrolimus. Br. J. Clin. Pharmacol. 2019, 85, 746–761. [Google Scholar] [CrossRef]
- Bergstrand, M.; Hooker, A.C.; Wallin, J.E.; Karlsson, M.O. Prediction-Corrected Visual Predictive Checks for Diagnosing Nonlinear Mixed-Effects Models. AAPS J. 2011, 13, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Comets, E.; Brendel, K.; Mentré, F. Computing Normalised Prediction Distribution Errors to Evaluate Nonlinear Mixed-Effect Models: The Npde Add-on Package for R. Comput. Methods Programs Biomed. 2008, 90, 154–166. [Google Scholar] [CrossRef] [Green Version]
- Byon, W.; Sweeney, K.; Frost, C.; Boyd, R. Population Pharmacokinetics, Pharmacodynamics, and Exploratory Exposure-Response Analyses of Apixaban in Subjects Treated for Venous Thromboembolism: Subjects Treated for Venous Thromboembolism. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Cirincione, B.; Kowalski, K.; Nielsen, J.; Roy, A.; Thanneer, N.; Byon, W.; Boyd, R.; Wang, X.; Leil, T.; LaCreta, F.; et al. Population Pharmacokinetics of Apixaban in Subjects with Nonvalvular Atrial Fibrillation. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 728–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, E.; Horinaka, S.; Ishimitsu, T.; Kato, T. Factor Xa Inhibitors in Clinical Practice: Comparison of Pharmacokinetic Profiles. Drug Metab. Pharmacokinet. 2020, 35, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Leil, T.A.; Frost, C.; Wang, X.; Pfister, M.; LaCreta, F. Model-Based Exposure–Response Analysis of Apixaban to Quantify Bleeding Risk in Special Populations of Subjects Undergoing Orthopedic Surgery. CPT Pharmacomet. Syst. Pharmacol. 2014, 3, 136. [Google Scholar] [CrossRef]
- Ueshima, S.; Hira, D.; Kimura, Y.; Fujii, R.; Tomitsuka, C.; Yamane, T.; Tabuchi, Y.; Ozawa, T.; Itoh, H.; Ohno, S.; et al. Population Pharmacokinetics and Pharmacogenomics of Apixaban in Japanese Adult Patients with Atrial Fibrillation. Br. J. Clin. Pharmacol. 2018, 84, 1301–1312. [Google Scholar] [CrossRef] [Green Version]
- Barsam, S.J.; Patel, J.P.; Roberts, L.N.; Kavarthapu, V.; Patel, R.K.; Green, B.; Arya, R. The Impact of Body Weight on Rivaroxaban Pharmacokinetics. Res. Pract. Thromb. Haemost. 2017, 1, 180–187. [Google Scholar] [CrossRef]
- Girgis, I.G.; Patel, M.R.; Peters, G.R.; Moore, K.T.; Mahaffey, K.W.; Nessel, C.C.; Halperin, J.L.; Califf, R.M.; Fox, K.A.A.; Becker, R.C. Population Pharmacokinetics and Pharmacodynamics of Rivaroxaban in Patients with Non-Valvular Atrial Fibrillation: Results from ROCKET AF. J. Clin. Pharmacol. 2014, 54, 917–927. [Google Scholar] [CrossRef]
- Kaneko, M.; Tanigawa, T.; Hashizume, K.; Kajikawa, M.; Tajiri, M.; Mueck, W. Confirmation of Model-Based Dose Selection for a Japanese Phase III Study of Rivaroxaban in Non-Valvular Atrial Fibrillation Patients. Drug Metab. Pharmacokinet. 2013, 28, 321–331. [Google Scholar] [CrossRef]
- Mueck, W.; Becka, M.; Kubitza, D.; Voith, B.; Zuehlsdorf, M. Population Model of the Pharmacokinetics and Pharmacodynamics of Rivaroxaban--an Oral, Direct Factor Xa Inhibitor--in Healthy Subjects. Int. J. Clin. Pharmacol. Ther. 2007, 45, 335–344. [Google Scholar] [CrossRef]
- Mueck, W.; Eriksson, B.I.; Bauer, K.A.; Borris, L.; Dahl, O.E.; Fisher, W.D.; Gent, M.; Haas, S.; Huisman, M.V.; Kakkar, A.K.; et al. Population Pharmacokinetics and Pharmacodynamics of Rivaroxaban—an Oral, Direct Factor Xa Inhibitor—in Patients Undergoing Major Orthopaedic Surgery: Clin. Pharmacokinet. 2008, 47, 203–216. [Google Scholar] [CrossRef]
- Mueck, W.; Borris, L.C.; Dahl, O.E.; Haas, S.; Huisman, M.V.; Kakkar, A.K.; Kälebo, P.; Muelhofer, E.; Misselwitz, F.; Eriksson, B.I. Population Pharmacokinetics and Pharmacodynamics of Once and Twice-Daily Rivaroxaban for the Prevention of Venous Thromboembolism in Patients Undergoing Total Hip Replacement. Thromb. Haemost. 2008, 100, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Mueck, W.; Lensing, A.W.A.; Agnelli, G.; Decousus, H.; Prandoni, P.; Misselwitz, F. Rivaroxaban: Population Pharmacokinetic Analyses in Patients Treated for Acute Deep-Vein Thrombosis and Exposure Simulations in Patients with Atrial Fibrillation Treated for Stroke Prevention. Clin. Pharmacokinet. 2011, 50, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Ollier, E.; Hodin, S.; Lanoiselée, J.; Escal, J.; Accassat, S.; De Magalhaes, E.; Basset, T.; Bertoletti, L.; Mismetti, P.; Delavenne, X. Effect of Activated Charcoal on Rivaroxaban Complex Absorption. Clin. Pharmacokinet. 2017, 56, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Speed, V.; Green, B.; Roberts, L.N.; Woolcombe, S.; Bartoli-Abdou, J.; Barsam, S.; Byrne, R.; Gee, E.; Czuprynska, J.; Brown, A.; et al. Fixed Dose Rivaroxaban Can Be Used in Extremes of Bodyweight: A Population Pharmacokinetic Analysis. J. Thromb. Haemost. 2020, 18, 2296–2307. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Yamashita, T.; Kasai, H.; Otsuka, T.; Sagara, K. An Analysis on Distribution and Inter-Relationships of Biomarkers under Rivaroxaban in Japanese Patients with Non-Valvular Atrial Fibrillation (CVI ARO 1). Drug Metab. Pharmacokinet. 2018, 33, 188–193. [Google Scholar] [CrossRef]
- Tanigawa, T.; Kaneko, M.; Hashizume, K.; Kajikawa, M.; Ueda, H.; Tajiri, M.; Paolini, J.F.; Mueck, W. Model-Based Dose Selection for Phase III Rivaroxaban Study in Japanese Patients with Non-Valvular Atrial Fibrillation. Drug Metab. Pharmacokinet. 2013, 28, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.S.; Moore, K.; Burton, P.; Stuyckens, K.; Mueck, W.; Rossenu, S.; Plotnikov, A.; Gibson, M.; Vermeulen, A. Population Pharmacokinetics and Pharmacodynamics of Rivaroxaban in Patients with Acute Coronary Syndromes: Pharmacokinetics and Pharmacodynamics of Rivaroxaban. Br. J. Clin. Pharmacol. 2012, 74, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Zdovc, J.; Petre, M.; Pišlar, M.; Repnik, K.; Mrhar, A.; Vogrin, M.; Potočnik, U.; Grabnar, I. Downregulation of ABCB1 Gene in Patients with Total Hip or Knee Arthroplasty Influences Pharmacokinetics of Rivaroxaban: A Population Pharmacokinetic-Pharmacodynamic Study. Eur. J. Clin. Pharmacol. 2019, 75, 817–824. [Google Scholar] [CrossRef]
- Zhang, L.; Peters, G.; Haskell, L.; Patel, P.; Nandy, P.; Moore, K.T. A Cross-Study Analysis Evaluating the Effects of Food on the Pharmacokinetics of Rivaroxaban in Clinical Studies. J. Clin. Pharmacol. 2017, 57, 1607–1615. [Google Scholar] [CrossRef] [Green Version]
- Tishkoff, S.A.; Kidd, K.K. Implications of Biogeography of Human Populations for “race” and Medicine. Nat. Genet. 2004, 36, S21–S27. [Google Scholar] [CrossRef]
- Olafuyi, O.; Parekh, N.; Wright, J.; Koenig, J. Inter-Ethnic Differences in Pharmacokinetics—Is There More That Unites than Divides? Pharmacol. Res. Perspect. 2021, 9, e00890. [Google Scholar] [CrossRef]
- Gosselin, R.C.; Adcock, D.M.; Bates, S.M.; Douxfils, J.; Favaloro, E.J.; Gouin-Thibault, I.; Guillermo, C.; Kawai, Y.; Lindhoff-Last, E.; Kitchen, S. International Council for Standardization in Haematology (ICSH) Recommendations for Laboratory Measurement of Direct Oral Anticoagulants. Thromb. Haemost. 2018, 118, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Leven, C.; Coste, A.; Mané, C. Free and Open-Source Posologyr Software for Bayesian Dose Individualization: An Extensive Validation on Simulated Data. Pharmaceutics 2022, 14, 442. [Google Scholar] [CrossRef]
- Zhang, D.; Chen, W.; Qin, W.; Du, W.; Wang, X.; Zuo, X.; Li, P. Population Pharmacokinetics and Hemorrhagic Risk Analysis of Rivaroxaban in Elderly Chinese Patients with Nonvalvular Atrial Fibrillation. J. Clin. Pharmacol. 2023, 63, 66–76. [Google Scholar] [CrossRef]
- Zhang, F.; Chen, X.; Wu, T.; Huang, N.; Li, L.; Yuan, D.; Xiang, J.; Wang, N.; Chen, W.; Zhang, J. Population Pharmacokinetics of Rivaroxaban in Chinese Patients with Non-Valvular Atrial Fibrillation: A Prospective Multicenter Study. Clin. Pharmacokinet. 2022, 61, 881–893. [Google Scholar] [CrossRef]
- Singkham, N.; Phrommintikul, A.; Pacharasupa, P.; Norasetthada, L.; Gunaparn, S.; Prasertwitayakij, N.; Wongcharoen, W.; Punyawudho, B. Population Pharmacokinetics and Dose Optimization Based on Renal Function of Rivaroxaban in Thai Patients with Non-Valvular Atrial Fibrillation. Pharmaceutics 2022, 14, 1744. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Ding, H.; Yan, M.; Jiao, Z.; Zhong, M.; Ma, C. Population Pharmacokinetic and Pharmacodynamic Analysis of Rivaroxaban in Chinese Patients with Non-Valvular Atrial Fibrillation. Acta Pharmacol. Sin. 2022, 43, 2723–2734. [Google Scholar] [CrossRef]
- Esmaeili, T.; Rezaee, M.; Abdar Esfahani, M.; Davoudian, A.; Omidfar, D.; Rezaee, S. Rivaroxaban Population Pharmacokinetic and Pharmacodynamic Modeling in Iranian Patients. J. Clin. Pharm. Ther. 2022, 47, 1284–1292. [Google Scholar] [CrossRef]
- University Hospital. Brest Pharmacokinetics and Pharmacodynamics of RivAroxaban After Bariatric Surgery and in MORBid Obesity; clinicaltrials.gov: Bethesda, MD, USA, 2022. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Reference | PK Study Reference | N Patients | Age | Weight | Daily Dose (mg) | Dosing Frequency | N PK Samples | Sampling Regimen | Assay | Intended Application of the PPK Model |
|---|---|---|---|---|---|---|---|---|---|---|
| EVD apixaban | Ballerie 2021 [16] | 69 | 55 (20–86) | 99 (79–150) | 2.5, 5 | BID | 116 | Sparse | Anti-Xa chromogenic assay LLOQ 20 ng/mL | No PPK model |
| A1 | Byon 2017 [22] | 970 | (18–89) | 167 patients > 100 kg | 2.5–50 | single dose, QD, BID | 8323 | Intensive + sparse | LC-MS/MS LLOQ 1 ng/mL | PKPD EER analysis in patients with VTE |
| A2 | Cirincione 2018 [23] | 4385 | 68 (18–94) | 81.4 (32–198.2) | 2.5–50 | single dose, QD, BID | 11,968 | Intensive + sparse | LC-MS/MS LLOQ 1 ng/mL | Explain PK heterogeneity in patients with NVAF |
| A3 | Goto 2020 [24] | 140 | 79.1 * ± 5.8 (2.5 mg BID) 70.9 * ± 7.5 (5 mg BID) | 55.7 * ± 10.6 (2.5 mg BID) 62.8 * ± 11.7 (5 mg BID) | 2.5, 5 | BID | 183 | Sparse | Anti-Xa chromogenic assay LLOQ 20 ng/mL | Compare anti-Xa DOAC PK |
| A4 | Leil 2014 [25] | 1284 | NA | NA | 2.5–50 | single dose, QD, BID | 11,252 | Intensive + sparse | LC-MS/MS LLOQ 1 ng/mL | PKPD EER analysis in patients undergoing orthopedic surgery |
| A5 | Ueshima 2018 [26] | 81 | 68 (40–85) | 65 (41–92) | 5–20 | BID | 276 | Sparse | LC-MS/MS LLOQ 2.5 ng/mL | Explain PK heterogeneity in patients with NVAF |
| EVD rivaroxaban | Ballerie 2021 [16] | 81 | 64.5 (20–85) | 102 (73.0–178) | 20 | QD | 121 | Sparse | Anti-Xa chromogenic assay LLOQ 20 ng/mL | No PPK model |
| R1 | Barsam 2017 [27] | 101 | 52 * (20–86) | 88 * ± 23.4 | 10–30 | QD, BID | 193 | Sparse | Anti-Xa chromogenic assay LLOQ 20 ng/mL | Study the impact of weight on rivaroxaban PK |
| R2 | Girgis 2014 [28] | 161 | NA | NA | 15–20 | QD | 801 | Sparse | LC-MS/MS LLOQ 0.5 ng/mL | Confirm dose selection in patients with NVAF |
| R3 | Goto 2020 [24] | 119 | 73.1 * ± 10.0 (10 mg QD) 66.7 * ± 10.0 (15 mg QD) | 60.3 * ± 15.5 (10 mg QD) 67.3 * ± 13.8 (15 mg QD) | 10, 15 | QD | 162 | Sparse | Anti-Xa chromogenic assay LLOQ 20 ng/mL | Compare anti-Xa DOAC PK |
| R4 | Kaneko 2013 [29] | 597 | 72 (34–89) | 63.9 (35–104) | 10, 15 | QD | 1834 | Sparse | LC-MS/MS LLOQ 0.5 ng/mL | Confirm dose selection in Japanese patients with NVAF |
| R5 | Mueck 2007 [30] | 43 | 33 * (20–45) | NA | 5–60 | QD, BID | 1809 | Intensive | LC-MS/MS LLOQ 0.5 ng/mL | Describe rivaroxaban PK in healthy subjects |
| R6 | Mueck 2008 CPK [31] | 1009 | 65 (26–87) (hip study) 67 (39–92) (knee study) | 76 (45–125) (hip study) 86 (50–173) (knee study) | 5–60 | QD, BID | 7568 | Intensive + sparse | LC-MS/MS LLOQ 2.5 ng/mL | Describe rivaroxaban PK in patients undergoing major orthopaedic surgery |
| R7 | Mueck 2008 TH [32] | 758 | 66 (26–93) | 75 (45–120) | 5–20 | QD, BID | 5743 | Sparse | LC-MS/MS LLOQ 2.5 ng/mL | Compare the PKPD of QD and BID rivaroxaban in patients undergoing total hip replacement |
| R8 | Mueck 2011 [33] | 870 | 61 (18–94) | 85 * ± 17 (male) 73 * ± 16 (female) | 10–60 | QD, BID | 4634 | Sparse | LC-MS/MS LLOQ 2.5 ng/mL | Describe rivaroxaban PK in patients treated for acute DVT and simulate exposure in patients with NVAF |
| R9 | Ollier 2016 [34] | 12 | 26 (20–30) | 71 (62–88) | 40 | Single dose | 192 | Intensive | LC-MS/MS LLOQ 5 ng/mL | Study the effect of activated charcoal on rivaroxaban absorption |
| R10 | Speed 2020 [35] | 913 | 67.0 * ± 15.0 | 85.8 * ± 23.1 | 15–30 | QD, BID | 1108 | Sparse | Anti-Xa chromogenic assay LLOQ 20 ng/mL | Understand the influence of WT on rivaroxaban PK |
| R11 | Suzuki 2018 [36] | 96 | 68.0 * ± 9.5 | 69.1 ± 11.4 | 10–15 | QD | 192 | Sparse | LC-MS/MS LLOQ 1 ng/mL | Describe rivaroxaban PK in Japanese patients with NVAF |
| R12 | Tanigawa 2013 [37] | 182 | 65.6 (30–92) | 67.2 (45–103) | 5–40 | QD, BID | 842 | Sparse | LC-MS/MS LLOQ 0.5 ng/mL | Select dose for Japanese patients with NVAF |
| R13 | Willman 2018 [8] | 4918 | 60.5 * ± 11.8 | 82.5 * ± 16.9 | 5–60 | QD, BID | 22,843 | Sparse | LC-MS/MS LLOQ 0.5 ng/mL | Describe rivaroxaban PK across multiple patient populations |
| R14 | Xu 2012 [38] | 2290 | 57 (24–87) | 84 (36–181) | 5–20 | QD, BID | 6644 ** | Sparse | LC-MS/MS LLOQ 0.5 ng/mL | Describe rivaroxaban PKPD in patients with ACS |
| R15 | Zdovc 2019 [39] | 17 | 64 (49–82) | 84 (54–125) | 10 | QD | 82 | Sparse | Anti-Xa chromogenic assay LLOQ 1 ng/mL | Investigate the influence of ABCB1 polymorphism on rivaroxaban PKPD |
| R16 | Zhang 2017 [40] | 285 | 59 (31–83) (DVT study) 65 (51–81) (NVAF study) | 54.1 (40.1–72.7) (DVT study) 56.6 (42.5–73.6) (NVAF study) | 20–40 | QD | NA | Sparse | LC-MS/MS LLOQ 0.5 ng/mL | Evaluate the effect of food on rivaroxaban PK |
| Model Reference | Modeling Software | Structural Model | Relative Bioavailability | Parameter Values | Covariates | Interpatient Variability | Residual Error |
|---|---|---|---|---|---|---|---|
| A1 | NONMEM 7.2 | 2 CMT | NA | Ka (1/h) = 0.440 CL (L/h) = 4.35 Vc (L) = 32.1 Q (L/h) = 1.62 Vp (L) = 19.8 | Ka: evening dosing CL: Sex, WT, CrCL *, Race, INH Vc: WT | ωKa = 0.474 ωCL = 0.322 ωVc = 0.232 | Additive |
| A2 | NONMEM 7.1 | 2 CMT | I50 = −0.322 Gamma = 0.857 | Ka (1/h) = 0.473 CL (L/h) = 3.59 Vc (L) = 30.0 Q (L/h) = 1.91 Vp (L) = 27.0 | Ka: AMPM CL: CrCL *, Age, Sex, Race, INH, SUB Vc: WT, SUB | ωKa = 0.513 ωK = 0.309 ωk12 = 1.245 ωk21 = 0.490 ωVc = 0.172 | Proportional = 0.31 |
| A3 | Phoenix NLME 8.1 | 1 CMT | NA | Ka (1/h) = 0.42 CL (L/h) = 4.74 Vc (L) = 30 | CL: CrCL | ωCL = 0.266 ωVc = 0.566 | Proportional = 0.34 |
| A4 | NONMEM 6.1.1 | 2 CMT | ED50 = 32.5 Imax = 0.705 Gamma = 2.21 | Ka (1/h) = 0.188 CL (L/h) = 4.75 ** Vc (L) = 22.9 Q (L/h) = 2.60 Vp (L) = 22.2 | Ka: SUB CL: Age, Sex, Dose ***, CrCL, Vc: WT, HCT | ωKa = 0.532 ωCL = 0.375 ωVc = 0.252 ωQ = 0.491 ωVp = 0.735 correlation ωVc ωCL = 0.915 | Proportional = 0.34 Additive = 3.38 |
| A5 | NONMEM 7.3.0 | 1 CMT | NA | Ka (1/h) = 0.42 CL (L/h) = 1.53 Vc (L) = 24.7 | CL: CrCL, PGx | ωCL = 0.266 ωVc = 0.566 | Proportional = 0.34 |
| R1 | NONMEM 7.2.13 | 1 CMT | NA | Ka (1/h) = 1.21 CL (L/h) = 8.86 Vc (L) = 101 | CL: CrCL | ωCL = 0.480 ωVc = 0.600 | Proportional = 0.31 |
| R2 | NONMEM 7.10 | 1 CMT | NA | Ka (1/h) = 1.16 CL (L/h) = 6.10 Vc (L) = 79.7 | CL: Age, SCre Vc: LBM, Age | ωCL = 0.342 ωVc = 0.175 | Proportional = 0.479 |
| R3 | Phoenix NLME 8.1 | 1 CMT | F1 = 1 | Ka (1/h) = 0.617 CL (L/h) = 5.59 Vc (L) = 50.9 | CL: CrCL | ωKa = 0.540 ωCL = 0.394 ωVc = 0.583 ωF1 = 0.365 | Proportional = 0.131 |
| R4 | NONMEM 6.2.0 | 1 CMT | F1 = 1 | Ka (1/h) = 0.617 CL (L/h) = 4.73 Vc (L) = 43.8 | CL: CrCL, HCT | ωKa = 0.582 ωCL = 0.410 ωVc = 0.636 ωF1 = 0.377 correlation ωVc ωCL = 0.729 | Proportional = 0.131 |
| R5 | NONMEM 5.1.1 | 2 CMT | NA | Tlag (h) = 0.25 Ka (1/h) = 0.97 CL (L/h) = 9.17 Vc (L) = 55.3 Q (L/h) = 1.35 Vp (L) = 12.6 | Vc: Dose Vp: Dose | IOV Tlag = 0.847 ωKa = 0.497 IOV Ka = 0.794 ωCL = 0.173 ωVc = 0.300 ωVp = 0.373 | Proportional = 0.254 |
| R6 | NONMEM 5.1.1 | 1 CMT | F1 = 1 | Ka (1/h) = 1.81 CL (L/h) = 7.3 Vc (L) = 49.1 | F1: Dose | ωCL = 0.373 | Proportional = 0.371 |
| R7 | NONMEM 5.1.1 | 1 CMT | F1 = 1 | Ka (1/h) = 1.49 CL (L/h) = 7.51 Vc (L) = 58.2 | F1: Dose | ωCL = 0.369 ωVc = 0.316 | Proportional = 0.526 |
| R8 | NONMEM 5.1.1 | 1 CMT | F1 = 1 | Ka (1/h) = 1.23 CL (L/h) = 5.67 Vc (L) = 54.4 | F1: Dose CL: Age, SCr Vc: LBM, Age | ωCL = 0.384 ωVc = 0.282 | Proportional = 0.407 |
| R9 | Monolix 4.3.2 | 1 CMT | F = 0.569 | f1 = 0.748 f2 = 0.348 Tmax1 (h) = 0.274 dTmax2 (h) = 1.94 dTmax3 (h) = 11.5 CV1 = 0.495 CV2 = 0.167 CV3 = 0.651 CL (L/h)= 7.4 Vc (L) = 28.4 | Activated charcoal effect on input rate | ωF = 0.253 IOV F = 0.728 IOV f1 = 0.997 IOV correlation ωF ωf1 = −0.717 ωCV1 = 0.570 ωVc = 0.085 | Proportional = 0.194 |
| R10 | NONMEM 7.4.2 | 1 CMT | NA | Ka (1/h) = 0.707 CL (L/h) = 5.57 Vc (L) = 59.4 Lambda = −1.83 | CL: CrCl ***** Vc: LBM ****** | ωCL = 0.227 ******* | Proportional = 0.4637 |
| R11 | Phoenix NLME 1.4 | 1 CMT | NA | Ka (1/h) = 1.37 CL (L/h) = 4.40 Vc (L) = 38.2 | CL: CrCL, ALT, INH | ωKa = 0.426 ωCL = 0.204 ωVc = 0.583 | Proportional = 0.418 |
| R12 | NONMEM 5.1.1 | 1 CMT | F1 = 1 | Ka (1/h) = 0.60 CL (L/h) = 4.72 Vc (L) = 42.9 | CL: BUN | ωF1 = 0.244 ωKa = 0.680 ωCL = 0.213 | Proportional = 0.402 |
| R13 | NONMEM 7.3 | 1 CMT | Fmin = 0.590 Fmax = 1.25 D50 = 14.4 | Ka (1/h) = 0.821 CL (L/h) = 6.58 Vc (L) = 62.5 | CL: CrCL, WT, INH, SUB Vc: WT, Age, Sex | ωKa = 0.792 ωCL = 0.409 ωVc = 0.198 correlation ωCL ωVc = 0.834 | Proportional = 0.451 |
| R14 | NONMEM 6.1.1 | 1 CMT | F1 = 1 | Ka (1/h) = 1.24 CL (L/h) = 6.48 Vc (L) = 57.9 | F: Dose CL: Age, SCr **** Vc: LBM, Age | ωKa = 1.037 ωCL = 0.306 IOV CL = 0.316 ωVc = 0.010 | Additive = 0.352 |
| R15 | NONMEM 7.3 | 1 CMT | NA | Ka (1/h) = 0.147 CL (L/h) = 6.12 Vc (L) = 96.8 | CL: PGx | ωKa = 2.004 ωCL = 0.709 | Proportional = 0.595 |
| R16 | NONMEM 7.2 | 1 CMT | F1 = 1 | Ka (1/h) = 0.982 CL (L/h) = 6.31 Vc (L) = 70.3 | F1: SUB CL: Age, SCr **** Vc: LBM, Age | ωCL = 0.336 ωVc = 0.154 | Proportional = 0.475 |
| MDPE (%) | MDAE (%) | F20 (%) | F30 (%) | |
|---|---|---|---|---|
| A1 (Byon 2017 [22]) | −7.8 | 25.0 | 42.2 | 56.0 |
| A2 (Cirincione 2018 SUB = ACS [23]) | 17.0 | 29.0 | 37.1 | 51.7 |
| A2 (Cirincione 2018 SUB = NVAF [23]) | 5.5 | 24.8 | 41.4 | 55.2 |
| A3 (Goto 2020 [24]) | −38.0 | 39.7 | 16.4 | 31.9 |
| A4 (Leil 2014 SUB = patients [25]) | 1.7 | 27.3 | 37.1 | 52.6 |
| A4 (Leil 2014 SUB = non patients [25]) | 7.6 | 27.2 | 39.7 | 56.0 |
| A5 (Ueshima 2018 [26]) | 7.2 | 25.8 | 40.5 | 54.3 |
| R1 (Barsam 2017 [27]) | −31.5 | 39.6 | 21.5 | 34.7 |
| R2 (Girgis 2014 [28]) | 9.7 | 29.9 | 33.9 | 50.4 |
| R3 (Goto 2020 [24]) | 11.6 | 34.0 | 36.4 | 45.4 |
| R4 (Kaneko 2013 [29]) | −12.2 | 45.8 | 26.4 | 33.9 |
| R5 (Mueck 2007 [30]) | −22.2 | 41.2 | 24.0 | 39.7 |
| R6 (Mueck 2008 CPK [31]) | −3.4 | 41.7 | 25.6 | 38.8 |
| R7 (Mueck 2008 TH [32]) | −7.3 | 36.0 | 25.6 | 42.1 |
| R8 (Mueck 2011 [33]) | 24.8 | 35.4 | 34.7 | 43.8 |
| R9 (Ollier 2016 [34]) | −53.4 | 53.9 | 14.9. | 20.7 |
| R10 (Speed 2020 [35]) | 17.3 | 32.2 | 32.2 | 47.9 |
| R11 (Suzuki 2018 [36]) | 36.2 | 46.4 | 19.8 | 33.1 |
| R12 (Tanigawa 2013 [37]) | 49.0 | 49.4 | 25.6 | 35.5 |
| R13 (Willman 2018 SUB = VTE [8]) | −30.4 | 42.7 | 23.1 | 33.1 |
| R13 (Willman 2018 SUB = NVAF [8]) | 6.6 | 28.5 | 32.2 | 51.2 |
| R14 (Xu 2012 [38]) | 10.9 | 37.5 | 30.6 | 46.2 |
| R15 (Zdovc 2019 [39]) | 18.6 | 51.8 | 19.0 | 24.8 |
| R16 (Zhang 2017 SUB = DVT [40]) | 7.6 | 30.1 | 25.6 | 48.8 |
| R16 (Zhang 2017 SUB = NVAF [40]) | 8.5 | 31.0 | 25.6 | 48.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leven, C.; Ménard, P.; Gouin-Thibault, I.; Ballerie, A.; Lacut, K.; Ollier, E.; Théreaux, J. Transferability of Published Population Pharmacokinetic Models for Apixaban and Rivaroxaban to Subjects with Obesity Treated for Venous Thromboembolism: A Systematic Review and External Evaluations. Pharmaceutics 2023, 15, 665. https://doi.org/10.3390/pharmaceutics15020665
Leven C, Ménard P, Gouin-Thibault I, Ballerie A, Lacut K, Ollier E, Théreaux J. Transferability of Published Population Pharmacokinetic Models for Apixaban and Rivaroxaban to Subjects with Obesity Treated for Venous Thromboembolism: A Systematic Review and External Evaluations. Pharmaceutics. 2023; 15(2):665. https://doi.org/10.3390/pharmaceutics15020665
Chicago/Turabian StyleLeven, Cyril, Pauline Ménard, Isabelle Gouin-Thibault, Alice Ballerie, Karine Lacut, Edouard Ollier, and Jérémie Théreaux. 2023. "Transferability of Published Population Pharmacokinetic Models for Apixaban and Rivaroxaban to Subjects with Obesity Treated for Venous Thromboembolism: A Systematic Review and External Evaluations" Pharmaceutics 15, no. 2: 665. https://doi.org/10.3390/pharmaceutics15020665