1. Introduction
The discovery of RNA inference (RNAi) mechanisms has introduced investigational and therapeutic applications for exogenous short-interfering RNA (siRNA) to temporarily downregulate the expression of the targeted protein [
1,
2]. Therefore, significant efforts have been made to develop siRNA delivery systems that could bring this approach to clinical use [
3,
4]. However, there are several challenges impeding the clinical translation of siRNA therapeutics, which include poor serum stability, short half-life (due to rapid degradation and renal elimination), negligible cellular internalization, and endosomal entrapment [
5], highlighting the need for a suitable delivery vehicle to facilitate the internalization of siRNA and make it available to the RNAi machinery to elicit target mRNA degradation.
Cell-penetrating peptides (CPPs) provide promising carriers for the intracellular delivery of nucleic acids [
6]. CPPs are effective in delivering a wide range of therapeutic molecules, including small molecules, nucleic acids, and proteins with limited toxicity, and are currently further explored in drug delivery studies [
6,
7,
8,
9]. A net cationic charge is a common physicochemical property of most CPPs and is known to facilitate membrane internalization [
10]. Additionally, the abundance of cationic residues such as arginine (Arg, R) makes CPPs a suitable carrier for anionic cargos such as nucleic acids. A well-defined amphipathic structure is another essential physicochemical property identified for many CPPs [
11]. Previous reports have demonstrated the pivotal role of tryptophan (Trp, W) residues in establishing the effective hydrophobic interaction with the lipid bilayer [
12].
The potential of various CPPs (TAT, transportan, penetratin, and oligoarginines) to facilitate siRNA internalization was examined via covalent conjugation approaches [
13,
14]. These strategies have been less successful, most likely due to the resulting chemical modification to siRNA, which could hinder the biological action of the internalized oligonucleotide. Over the past decade, we have reported several Arg-Trp-based cyclic/hybrid peptides having remarkable cell-penetrating properties [
8,
15,
16]. We have thoroughly investigated the effect of the number and relative positions of cationic and hydrophobic residues and noticed that peptides composed of Arg and Trp residues enhanced internalization [
15]. After crossing the initial membrane barriers, siRNA complexes need to escape from the endosomal pathway to efficiently function in the cytoplasm. Noticeably, our studies have shown an endocytosis-independent mechanism of uptake for cyclic peptides incorporating tryptophan and arginine residues [
15]. In this study, the uptake of fluorescein-labeled [W
5R
4K] in leukemia cells (CCRF-CEM) did not significantly change in the presence of endocytosis inhibitors, suggesting a direct translocation mechanism for the uptake.
Various CPPs, including CADY, RICK (Retro-Inverso- CADY-K), TP10, MPG, R9, R9-hLF, PF14, TAT, and LAH4, have been reported for siRNA delivery. Among the CPPs, Retro-Inverso-CADY-K (RICK) and modified TAT peptides were identified to form well-defined self-assembled structures [
17,
18]. Recently, many reports have demonstrated the self-assembly ability of amphiphilic peptides, which further highlights their suitability for siRNA delivery [
19]. Panigrahi et al. observed nanostructure formation with arginine–tryptophan-based cyclic peptide with glycine as a spacer and reported the efficient intracellular delivery of GAPDH siRNA and significant RNAi efficiency [
20].
Modified linear peptides (MLPs) represent a class of amphiphilic molecules with an exceptional ability to self-assemble into functional nanostructures, such as micelles and nanovesicles [
21]. Therefore, lipidation of amphiphilic peptides is a potential synthetic approach to designing efficient molecular transporters [
22,
23]. Cholesterol is another well-studied lipid moiety enabling efficient cellular and tissue delivery. The ability of cholesterol to spontaneously intercalate into the lipid membranes upon co-incubation highlights its potential as a critical structural component of the delivery system [
24]. Many reports, including one from our group, have demonstrated the key role of cholesterol as a formulation component [
16]. Recently, one cholesteryl peptide, Chol-H
3K
2, was reported to exhibit a comparable cellular uptake and RNAi effect to PEI or Lipofectamine 2000 without showing any significant cytotoxicity [
25]. In another work, Qiu et al. have shown that attachment of PEG with KL4 peptide improves siRNA transfection efficiency in lung epithelial cells [
26].
We have also reported siRNA delivery via a wide range of specifically designed peptides, including hybrid cyclic peptides (that incorporate arginine (R) residues in a ring and tryptophan (W) residues in a hydrophobic chain) and cyclic peptides with a disulfide bridge [
7,
8]. These sequences are designed to resolve the issue of toxicity and poor pharmacokinetic/pharmacodynamic profiles observed with polyethyleneimines (PEI) and poly-L-lysine. However, toxicity could still be a limiting factor in higher concentrations. In this study, building on our previous efforts, where we designed MLPs as a siRNA delivery system, we introduce a novel strategy using peptides containing Trp and Arg residues separated with a β-alanine spacer combined with either lipophilic moieties or PEG chains. The modification of CPPs is required to act as a delivery vector for siRNA to minimize the degradation and mediate the efficient cellular uptake. It is indeed challenging to design efficient short-peptide-based siRNA transporters. We hypothesized that MLPs composed of amphiphilic peptides and optimum fatty acyl chain, or PEG, could function as an effective siRNA carrier by complementing each structural component’s self-assembly and membrane internalization properties. Based on our previous studies, we used four Arg and Trp residues separated by two β-Ala spacers to constitute the peptide region. Oleic acid or cholesterol was used for peptide lipidation.
2. Materials and Methods
The resins for solid-phase peptide synthesis and the Fmoc-protected amino acid residues were purchased from AAPPTec (Louisville, KY, USA). Crude peptides were purified using a reverse-phase high-performance liquid chromatography (RP-HPLC) system obtained from Shimadzu (Canby, OR, USA). The preparative C-18 column was used (Waters Bridge, BEH130, 10 µm, 110 Å, 21.2 × 250 mm). Acetonitrile and purified water containing 0.1% TFA (v/v) were used as solvents. The purification was performed at a flow rate of 8 mL/min, and detection was set at 214 nm. The identity of the molecular structure of peptides was determined by a MALDI-TOF (matrix-assisted laser desorption/ionization-time-of-flight) mass spectrometer (Bruker Inc., Fremont, CA, USA). The matrix for MALDI-TOF mass spectrometry was α-cyano-4-hydroxycinnamic acid.
Fetal bovine serum (FBS), Dulbecco’s Modified Eagle’s Medium (DMEM), and other cell culture supplies were obtained from Life Technologies (Grand Island, NY, USA). Western blot accessories were obtained from Bio-Rad laboratory (Hercules, CA, USA). Re-agents for preparing samples for confocal microscopy were obtained from Vector Laboratories (Burlingame, CA, USA). Monoclonal antibodies to identify GAPDH (Mouse IgG1, Catalogue No. 97166) and STAT3 (Mouse IgG2a, Catalogue No. 9139) were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). Secondary antibody (HRP conjugated polyclonal goat IgG detecting Mouse IgGs) was supplied by R&D Systems (Catalogue No. HAF007). siRNA labeled with Alexa fluor 488 (Catalogue No. 1027292), negative control (scrambled) siRNA (Catalogue No. AM4635), and STAT3-targeting siRNA (Catalogue No. SI02662338) were obtained from Life Technologies (Grand Island, NY, USA). Other chemical reagents and solvents were purchased from Millipore-Sigma (Milwaukee, WI, USA) and used without further purification. More details regarding the materials used in the present study have been previously described [
7].
2.1. Synthesis of the Conjugated Peptides
2.1.1. Synthesis of Modified Linear Peptides (MLPs)
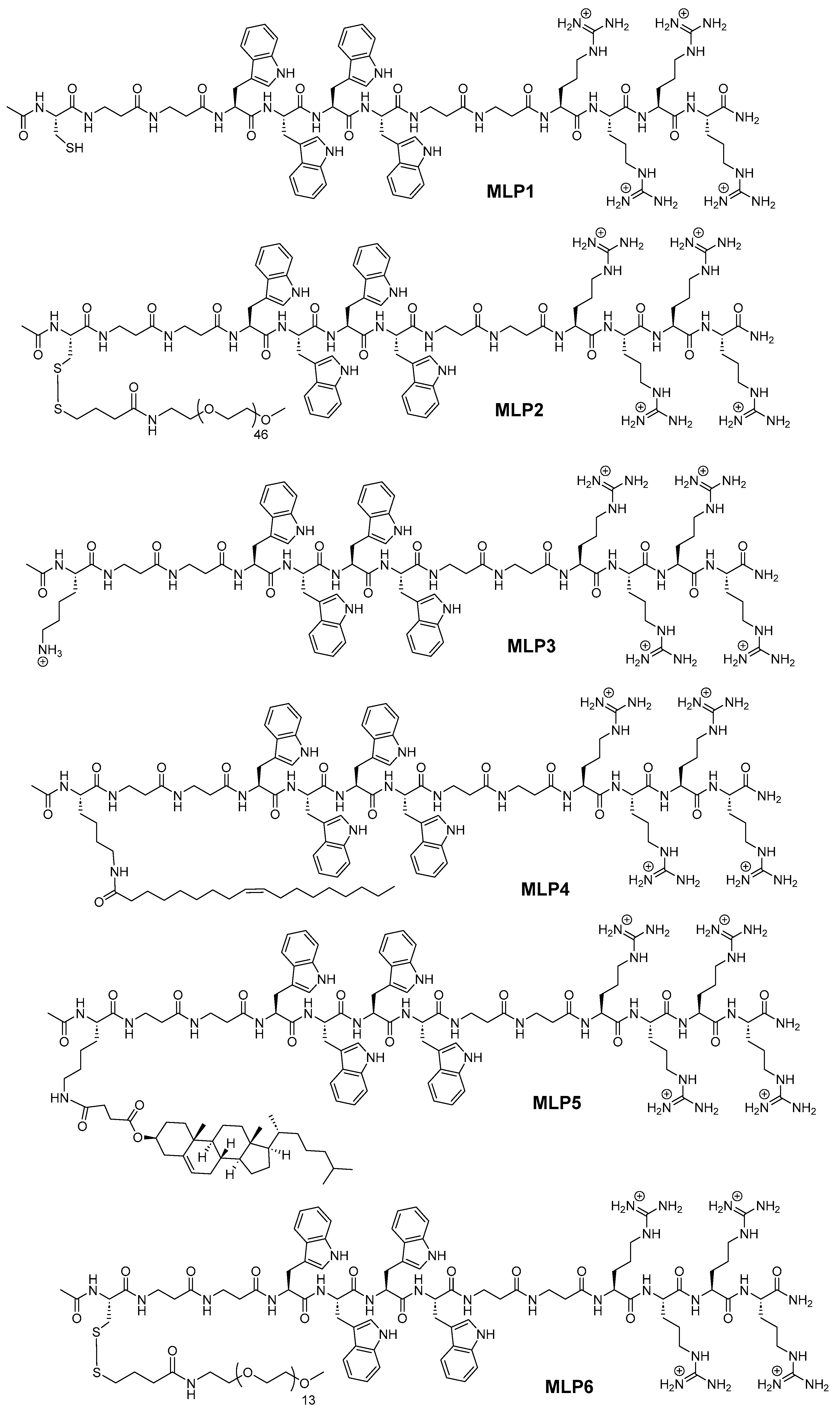
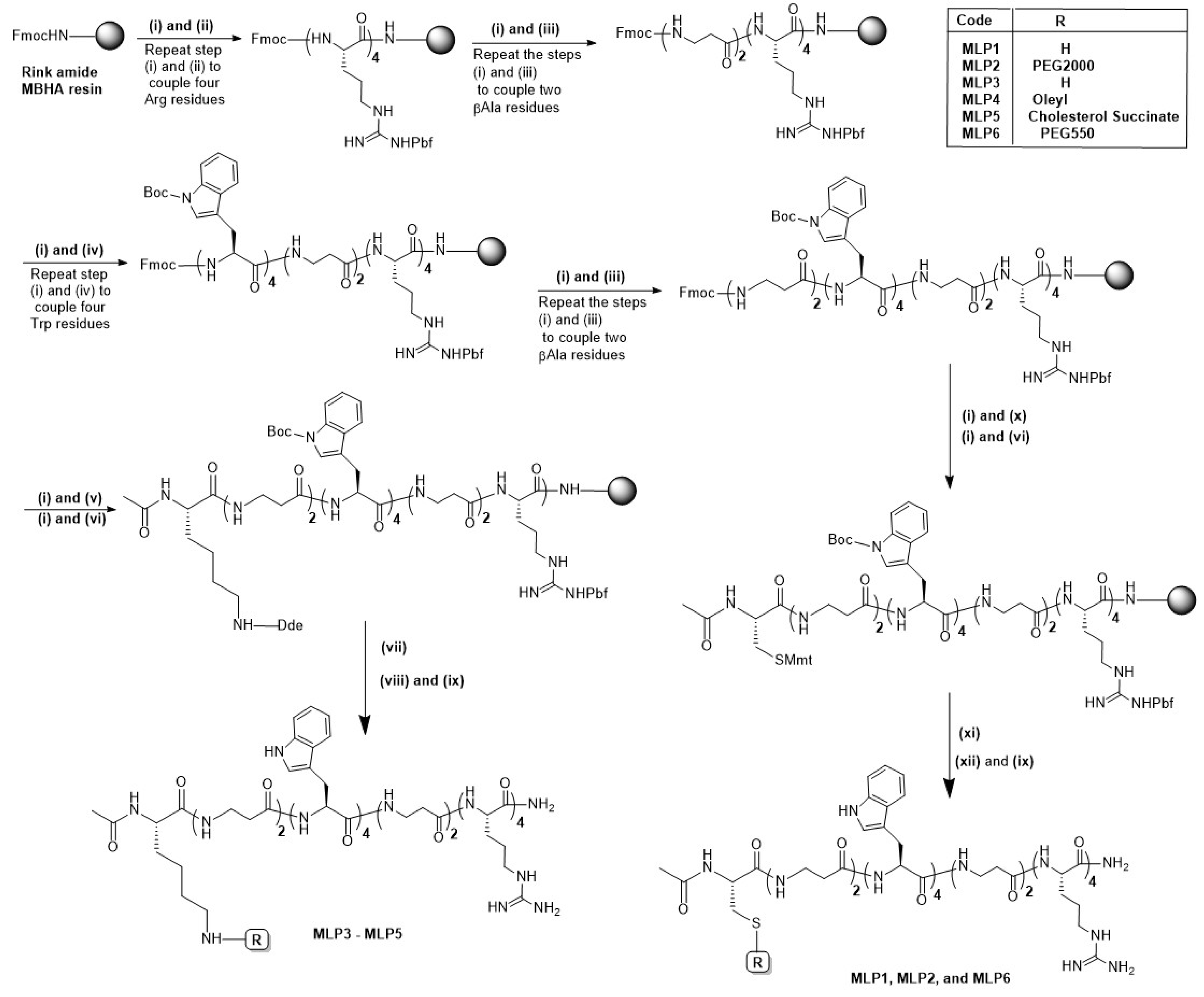
MLPs were manually synthesized on Rink amide MBHA resin (substitution 0.46 mmol/g) using the standard Fmoc/tBu solid-phase synthesis protocol. The structures of the synthesized peptides are represented in
Figure 1, and their sequences are shown in
Table 1. Briefly, the resin was allowed to swell in dry DMF for 30 min. Fmoc deprotection was conducted by treating the swelled resin with 20% piperidine in DMF (
v/
v) for 20 min. Couplings of natural (Fmoc-L-Arg(Pbf)-OH and Fmoc-L-Trp(Boc)-OH) and unnatural (Fmoc-L-βAla-OH) amino acids were carried out by using Fmoc-L-amino acid (3 equiv.), 1-hydroxy benzotriazole (HOBt, 3 equiv.), and
N,
N′-diisopropylcarbiimide (DIC, 3 equiv.), and the reaction mixture was allowed to shake at room temperature for 2–3 h. The coupling of each amino acid was confirmed by a negative Kaiser test. After each coupling, Fmoc-protecting groups were removed by treatment with 20% piperidine in DMF (
v/
v). After completing the desired peptide sequence, the
N-terminus was acetylated by treating the peptidyl resin with a mixture of acetic anhydride pyridine (2:1) in DMF. For side-chain conjugation, Fmoc-L-Lys(Dde)-OH was installed at the
N-terminal of the peptides. Selective removal of the Dde-protecting group was achieved by treating the peptidyl resin with a solution of hydrazine hydrate in DMF (2%,
v/
v). Oleic acid or cholesteryl hemi-succinate were conjugated on the Lys side chain in the presence of HOBt (3 equiv.) and DIC (3 equiv.), and the reaction mixture was allowed to shake at room temperature for 2–3 h. After each coupling and deprotection step, the peptidyl resin was thoroughly washed with DCM and DMF. After completing the desired peptide sequence, the complete cleavage was accomplished in the presence of TFA/water/triisopropyl silane (TIS) (95:2.5:2.5 (
v/
v/
v)), and the crude peptide was precipitated with ice-cooled diethyl ether. After multiple ether washes, the crude peptide was redissolved in acetonitrile/water (1:1
v/
v with 0.5% TFA), and the solution was lyophilized to obtain the crude peptide. The crude peptides were purified using RP-HPLC. The purity of all the peptides was confirmed by analytical HPLC, and mass was analyzed using the MALDI-TOF mass spectrometer. Purity and MALDI mass data of all the peptides are provided in the
Supplementary Materials.
2.1.2. Synthesis of PEGylated MLPs
Synthesis of the pegylated peptides was conducted using solid-phase chemistry, as depicted in the synthesis scheme (
Scheme 1). The desired peptide sequence was assembled on Rink amide resin using the reaction condition mentioned in the previous section. Orthogonal-protected Cys residue (Fmoc-Cys(Mmt)-OH) was coupled on the
N-terminus to PEGylate the peptide by exploiting the side-chain thiol functionality.
N-terminal acetylation of the resulting peptide sequence was conducted by treating the peptidyl resin with a mixture of acetic anhydride and pyridine (2:1) in DMF. The methoxytrityl group was selectively removed by repeatedly treating the peptidyl resin with 2% TFA in DCM for 10 min. On-resin PEGylation was conducted using preactivated thiol derivatives of PEG (mPEG-3-(2-pyridyldithio) propionic acid, MW 550 and 2000). For the PEGylation reaction, preactivated PEG derivative (2 equiv.) was dissolved in DMF and added to the peptidyl resin. The reaction mixture was kept for shaking for 12–14 h at room temperature. The PEGylated peptides were cleaved from the resin using the cleavage cocktail composed of TFA/water/TIS (95:2.5:2.5 (
v/
v/
v)) and purified by preparative RP-HPLC. The masses of the purified fractions were confirmed by Q-TOF-LC-MS as a multiple-charged peak with a characteristic fragmentation pattern of PEG units (+44 dalton).
Table 1 shows the sequences of the synthesized peptides along with the mass data. Mass spectra of all the synthesized peptides (MLP1–MLP6) are provided in the
Supplementary Materials.
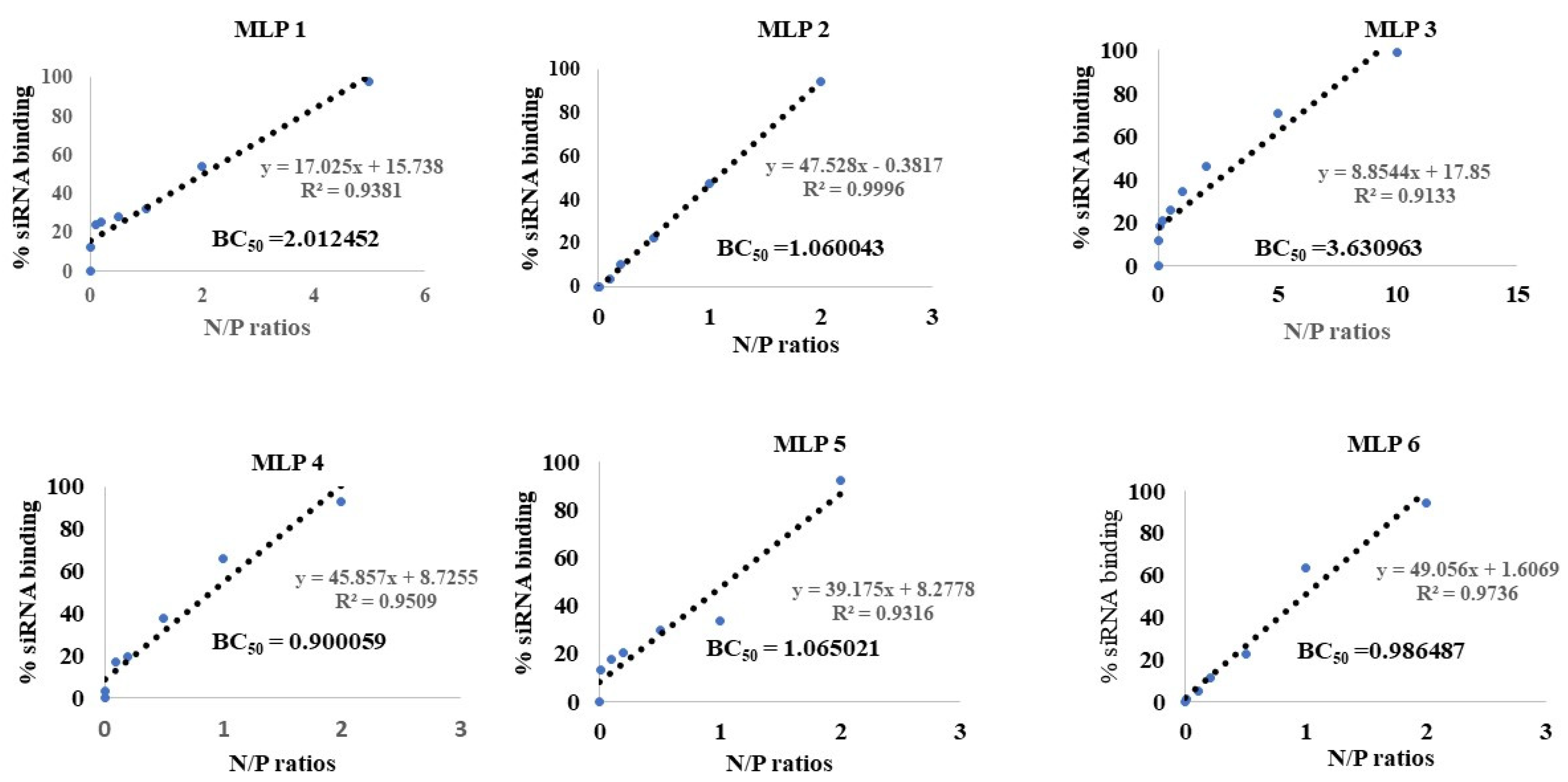
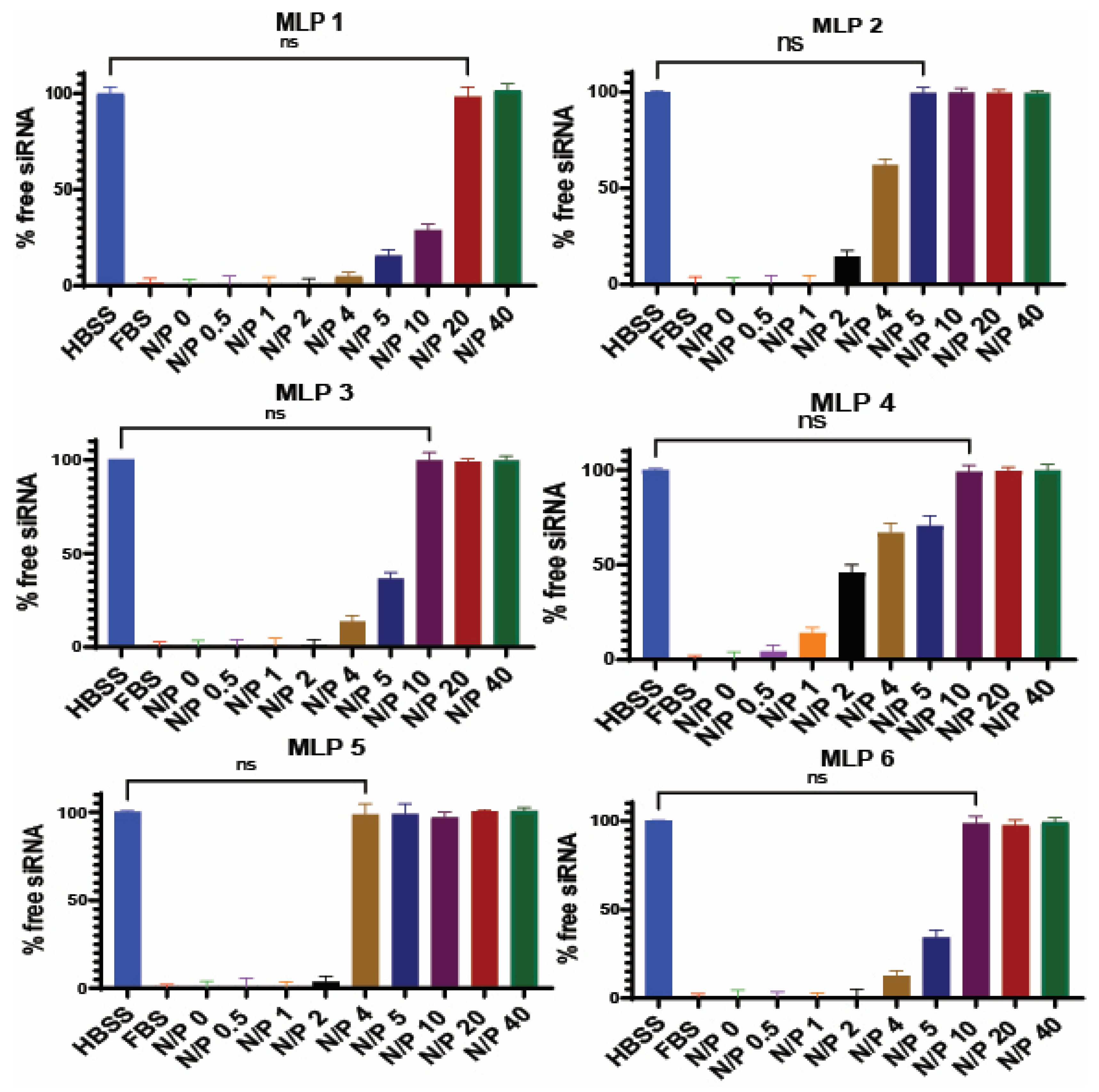
2.2. Binding Affinity
As previously reported, the binding affinity of selected peptides to siRNA was investigated using the SYBR green exclusion assay [
16]. The MLPs and scrambled siRNA were mixed in normal saline (in triplicates) with different N/P ratios ranging from 0.05 to 40. The mixture was incubated at room temperature for 30 min to ensure complete complexation of peptides and siRNA. After the incubation time, the complexes were transferred to a black 96-well plate. SYBR green dye solution for this assay was made by diluting 1 part of SYBR green dye with 10,000 parts of purified water. Then, 200 µL of the freshly made SYBR green dye dilution was added to each siRNA complex in the black 96-well plate. The plate was covered with aluminum foil before reading in a micro-plate reader that detected the fluorescent signal (485 nm excitation and 527 nm emission). SYBR green only binds to free siRNA, which significantly enhances the fluorescence signal. By creating a standard curve, the intensity of the fluorescent signal was translated to the concentration of free siRNA, and an indication of the percentage of siRNA bond to the protein. The percentage of the siRNA complexed with the peptide was determined using the following equation:
% siRNA bound to the peptide was plotted against a range of nitrogen to phosphate (N/P) ratios used in the experiment, and BR50 (the N/P ratio of the complex required to bind 50% of siRNA) was determined using a sigmoidal model. The negatively charged nucleic acids interact and form complexes with positively charged carriers via interionic interactions. Therefore, the N/P ratio can be a deciding factor, not only in the percentage of siRNA bond to the carrier, but also in how the complexes interact with the cell.
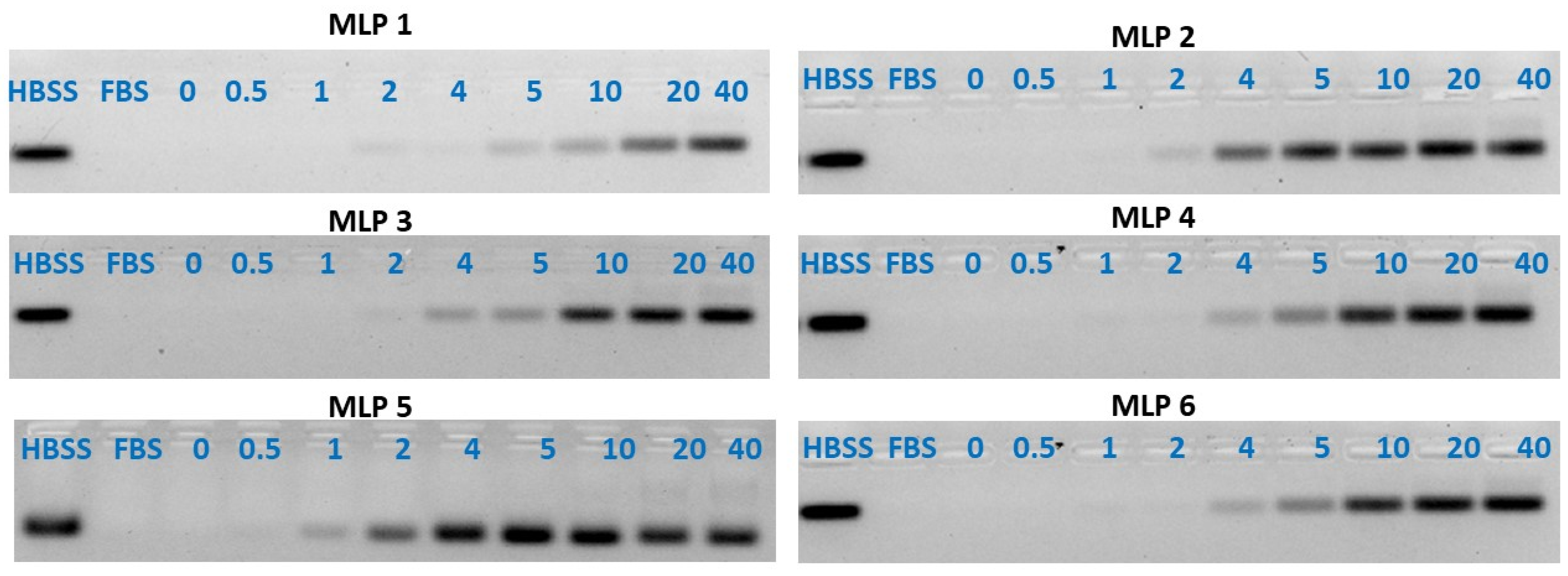
In addition to the SYBR green dye exclusion assay described above, the binding affinity of the peptides to siRNA was also determined using the gel retardation/gel shifting assay. Briefly, the MLPs-siRNA complexes were prepared at different N to P ratios ranging from 0 to 60. The N/P ratio of zero represents free siRNA. The tubes containing complexes were incubated at room temperature for 30 min for complete complex formation. After this incubation time, the complexes were mixed with gel-loading dye. Each of the samples was loaded into the wells of 1% agarose gel with (1 μg/mL) ethidium bromide, and 400 Amperes and 70 Volts were used to run the gel for 20 min. After the run, the gels were visualized under a Bio-Rad imager. The intensity of the bands was quantified by Image Lab software.
2.3. Cell Culture
This study used the triple-negative breast cancer cell line MDA-MB-231 (ATCC® HTB-26), and ovarian cancer cells SK-OV-3 (ATCC® HTB 77). The cells were grown in DMEM medium containing 10% v/v fetal bovine serum and 1% v/v penicillin (100 U/mL) and streptomycin (100 µg/mL). The incubator for the growth of cells was set at 37 °C with 5% CO2 flow. The cells were observed daily under a microscope to determine their growth and health. The experiments were performed when the T75 plate reached 80% confluency.
2.4. Cellular Internalization
The cellular uptake of siRNA was determined using Fluorescent-Assisted Cell Sorter (FACS) flow cytometry (BD-FACSVerse; BD Biosciences, San Jose, CA, USA). For this purpose, Alexa fluor 488-labeled siRNA was used in making the peptide-siRNA complexes at different N to P ratios. The cellular uptake experiment was conducted on the breast cancer cell line MDA-MB-231. The detailed protocol is provided in our recently published manuscript [
7]. Briefly, cells were seeded in each well of the 24-well plate (approximately 200,000 cells per well) and were allowed to settle in the incubator at 37 °C for 24 h. The peptide and siRNA complexes were made at N to P ratios of 20 and 40, where the final concentration of siRNA in each complex was 36 nM, and were allowed to incubate for 30 min at room temperature. Cells seeded in the 24-well plate were treated with the freshly formed siRNA-peptide complexes and were placed in an incubator for another 24 h to allow the siRNA uptake by the cells. After 24 h of incubation, the media was drained from each well, and the cells were washed three times with HBSS. Following rigorous washing, the cells were trypsinized with colorless trypsin and were quickly visualized under the microscope for their detachment from the surface of the well. The trypsinized cells were fixed in a 3.7% formaldehyde solution, and these suspended cells were investigated via FACS flow cytometry to determine the internalization of Alexa-fluor 488-labeled siRNA. Non-treated cells served as a negative control. The FACS machine was calibrated with a signal gate using non-treated cells, where the fluorescent signal was set at ~1% of the population. The percentage of cells with a fluorescent signal and the mean fluorescence of the cell population were analyzed for each sample.
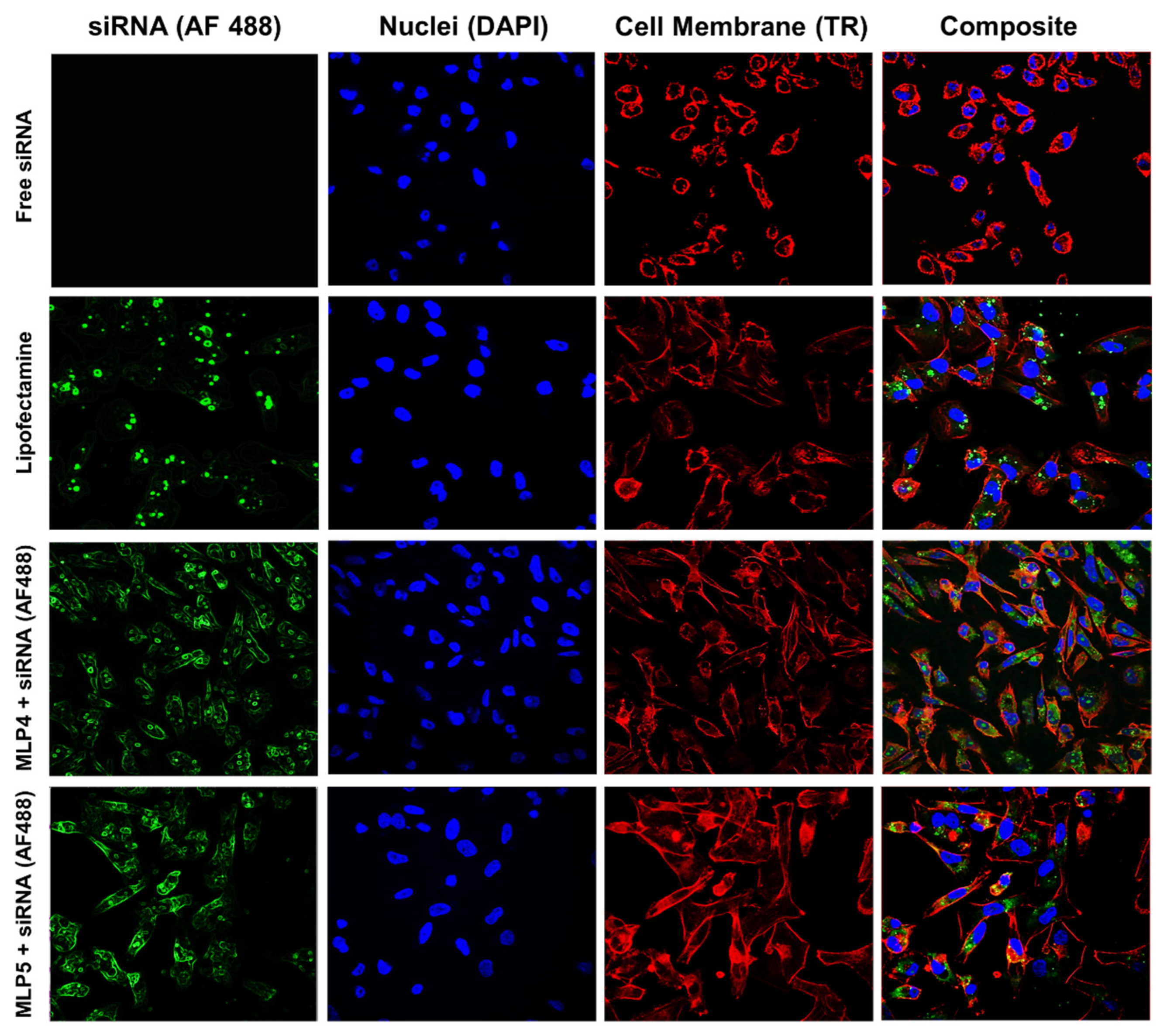
In order to visualize and locate the internalized siRNA, samples for confocal microscopy were prepared using MDA-MB-231 cells, adopting the protocol described previously by our lab [
7]. Briefly, sterile 6-well plates were used, and sterile coverslips were placed at the bottom of each well. MDA-MB-231 cells suspended in 2 mL of media were seeded in each well, and the plates were incubated in an incubator at standard growth conditions. After incubation, the cells were exposed to MLPs-fluorescent-labeled siRNA complexes at a N/P ratio of 40 using the same method described above for the FACS assay. The treated cells were again incubated for 24 h to allow for the internalization of siRNA. After overnight incubation, the media was drained from each well, and the cells were rigorously washed with HBSS three times, followed by fixation with 3.7% formaldehyde in the HBSS solution. Following fixation, the cells were treated with Texas Red for 1 h at room temperature to stain the cell membrane. After 1 h of staining with Texas Red, the cells were stained with DAPI overnight at room temperature in a dark environment to stain the nuclei. Following successful nuclei staining, the coverslips containing fixed stained cells were transferred to slides for viewing under a high-definition confocal microscope (Nikon A1 R).
2.5. Serum Stability
The capability of MLPs to protect siRNA against enzymatic degradation in serum was determined using the gel retardation assay described previously [
7]. Samples of MLPs-siRNA complexes were made at room temperature with N to P ratios ranging from 0 to 60. The N to P ratio 0 only represents the siRNA. In each of these samples, 25%
v/
v fetal bovine serum was added, and the resultant mixture was incubated at 37 °C for 24 h. After incubation, the MLPs-siRNA complexes were dissociated using a heparin competition assay. Heparin was used to dissociate the peptide/siRNA complex as it is anionic in nature and binds with the cationic carrier. The samples were treated with dithiothreitol (DTT, 1 M), followed by treatment with heparin:EDTA (2:3) solution. The concentration of heparin used in this assay was 5% (
w/
v), whereas ethylenediaminetetraacetic acid (EDTA) was 0.5 mM. After dissociation of the complexes, the samples were loaded in 1% agarose gel wells containing 1 μg/mL of ethidium bromide. The gel was run for 25 min at 70 V. The gels were finally visualized by ultraviolet illumination using a Bio-Rad imager and the intensity of the bands was quantified using Image Lab software.
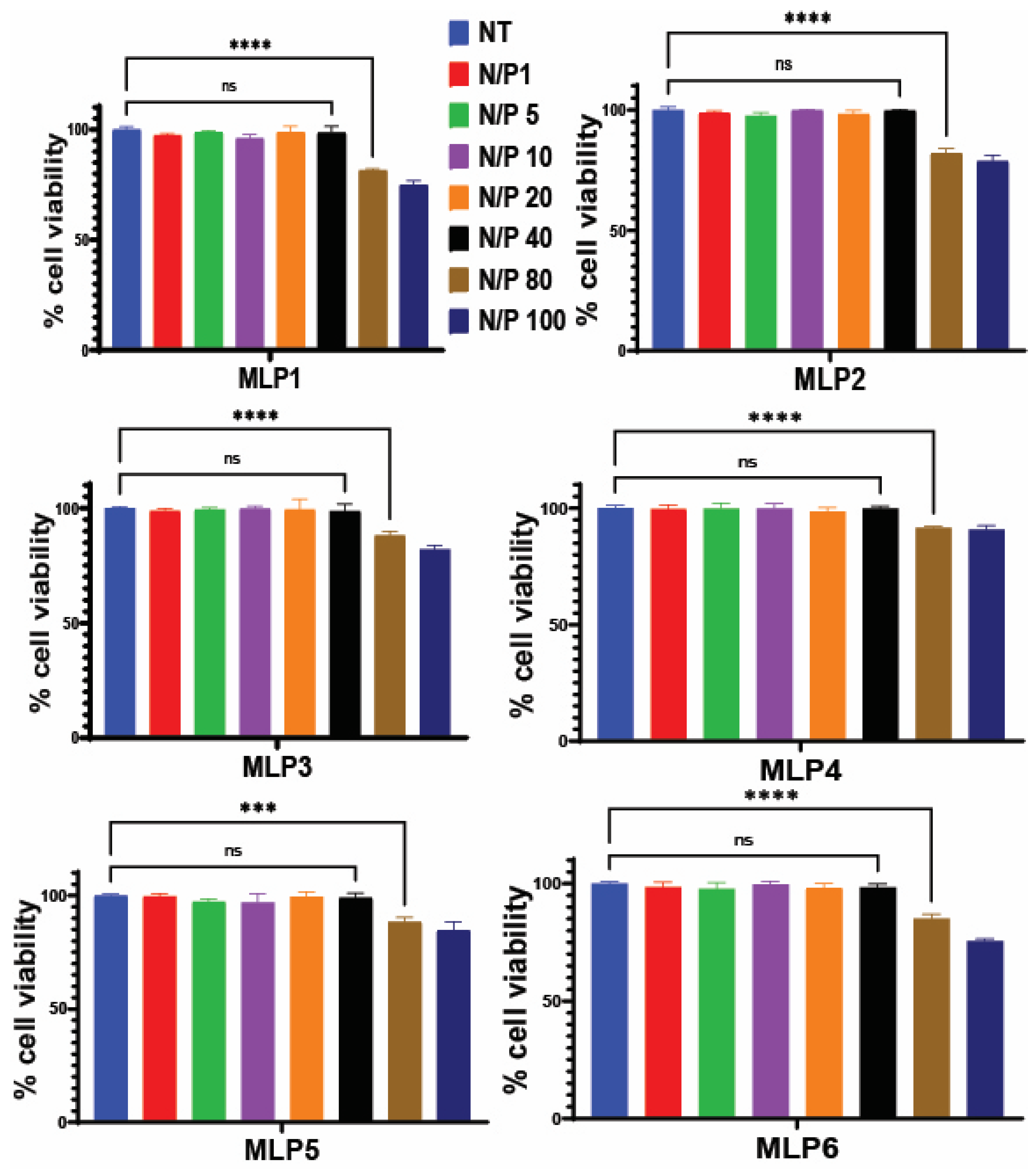
2.6. Cytotoxicity of Peptide and Peptide/siRNA Complex
The toxicity of the MLPs and MLP-siRNA complexes was determined in MDA-MB-231 cells using the MTS assay, as previously described [
7]. The peptides were tested at concentrations ranging from 1 µM to 100 µM, whereas the MLP-siRNA complexes were tested at different N/P ratios ranging from 1 to 80. Briefly, cells were seeded in 96-well plates (approximately 10,000 cells suspended in 100 µL) and were allowed to settle in the incubator for 24 h. After 24 h, the cells were visualized for their adherence and health under a microscope and were exposed to different concentrations of peptides as well as different N to P ratios of MLP-siRNA complexes and were incubated at 37 °C, 5% CO
2, for 48 h. After incubation, 20 µL of the MTS reagent was added into each well using a multichannel pipette. After 2 h of incubation at 37 °C, the absorbance of each well was determined using the microplate reader (SpectraMax M5, Molecular Devices, San Jose, CA, USA), and the wavelength chosen for absorbance was 490 nm. The cell viability was measured using the following equation:
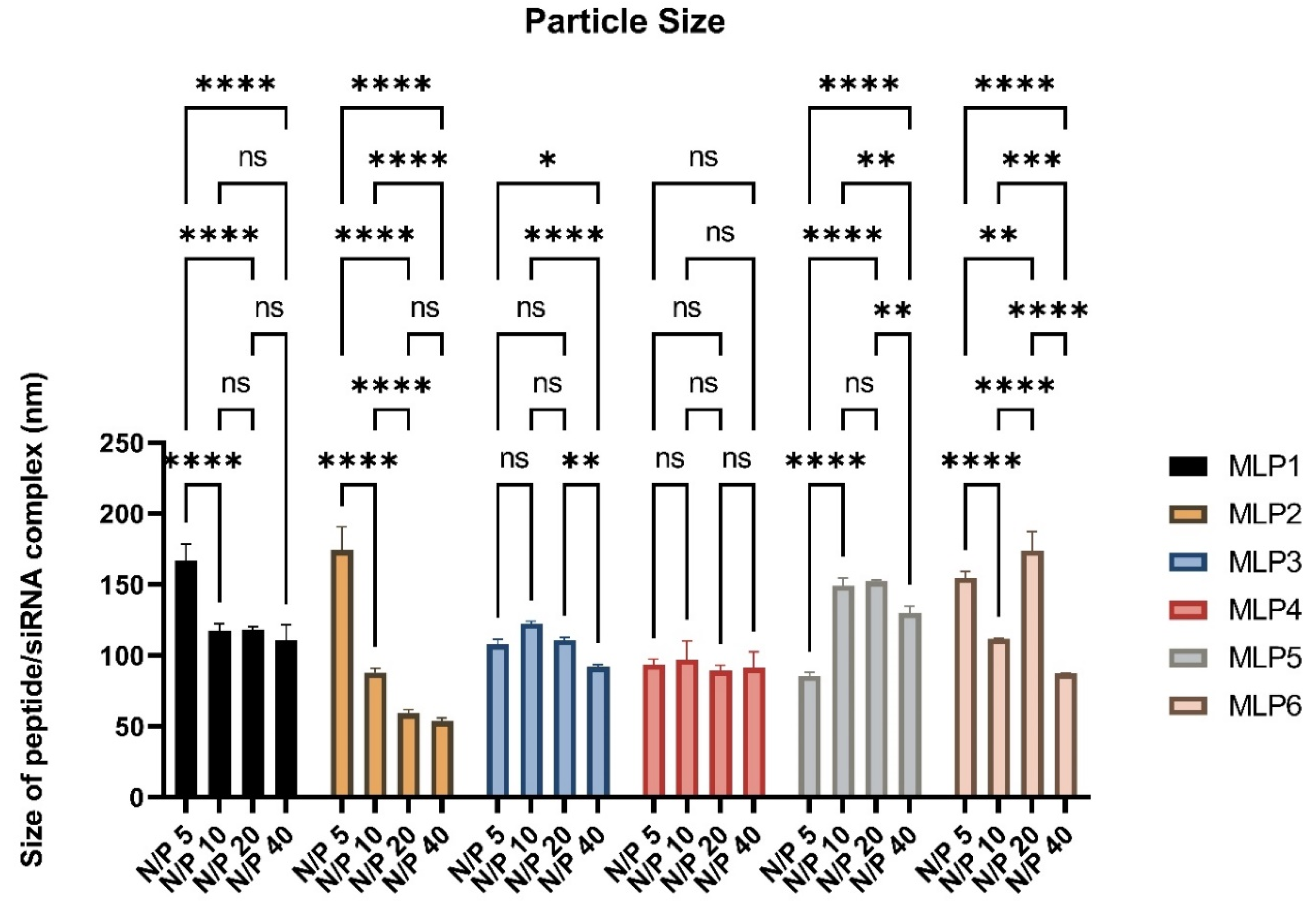
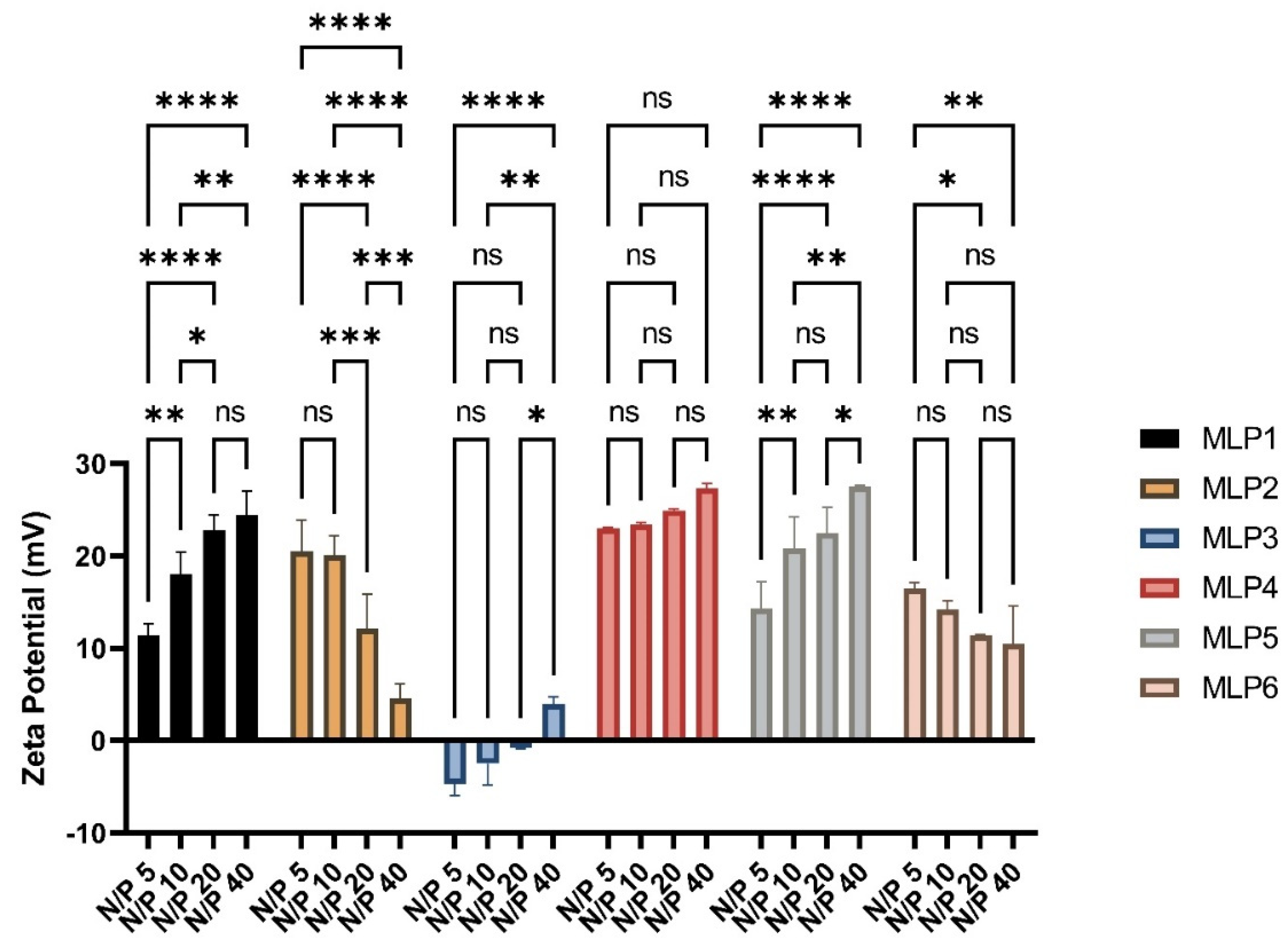
2.7. Size and ζ-Potential of Peptide/siRNA Complex
MLPs-siRNA complexes were prepared using scrambled siRNA at N/P ratios of 5, 10, 20, and 40, and were analyzed for hydrodynamic diameter and surface charge by a Nano ZS Zetasizer (Malvern, Westborough, MA, USA) at room temperature. The disposable cuvettes were used for the determination of particle size, whereas disposable folded capillary cells were used for zeta potential measurements. All measurements were performed three times using the protocol adopted from our previous study [
7]. The ζ-potential was measured at 40 V using the Smoluchowski approximation.
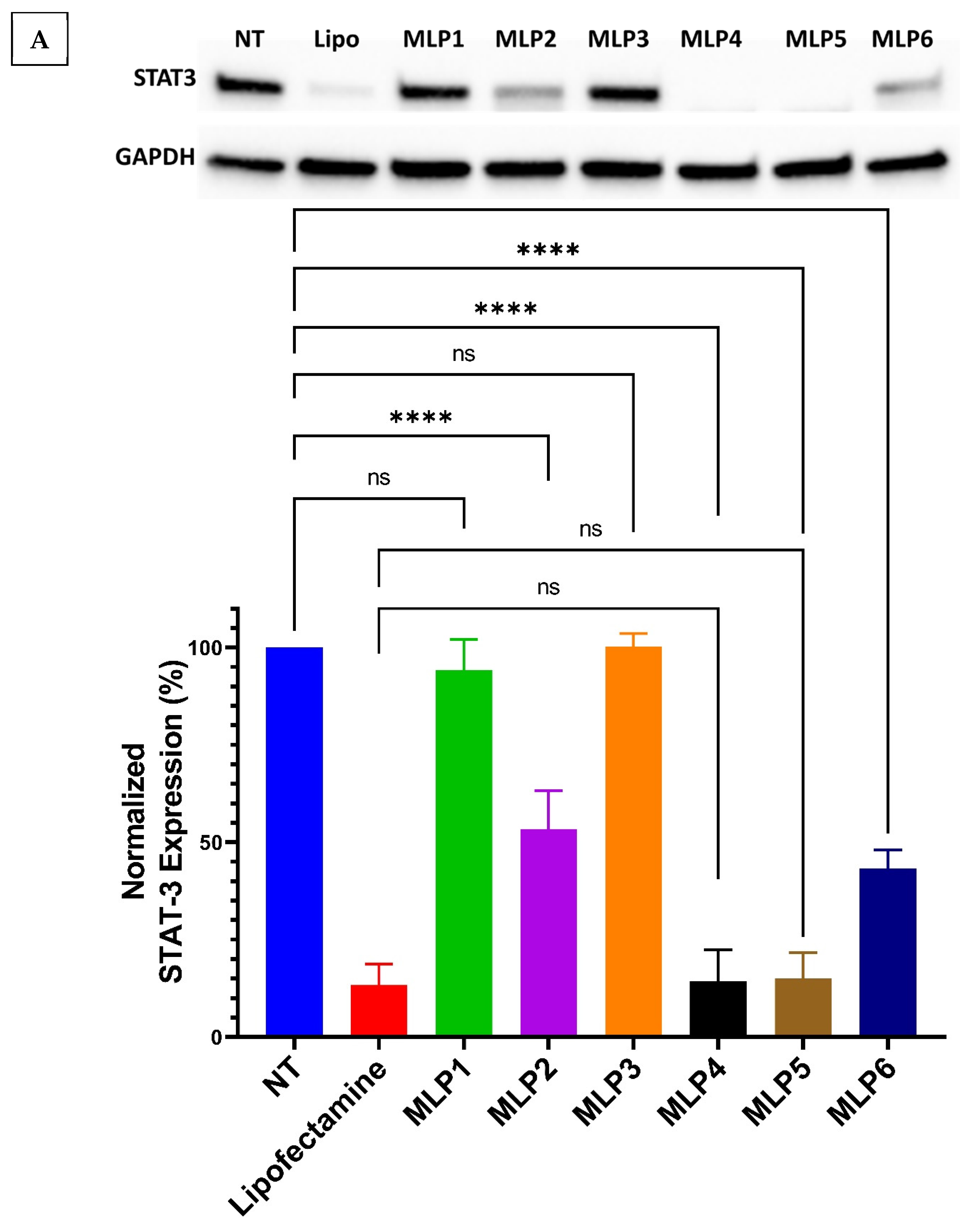
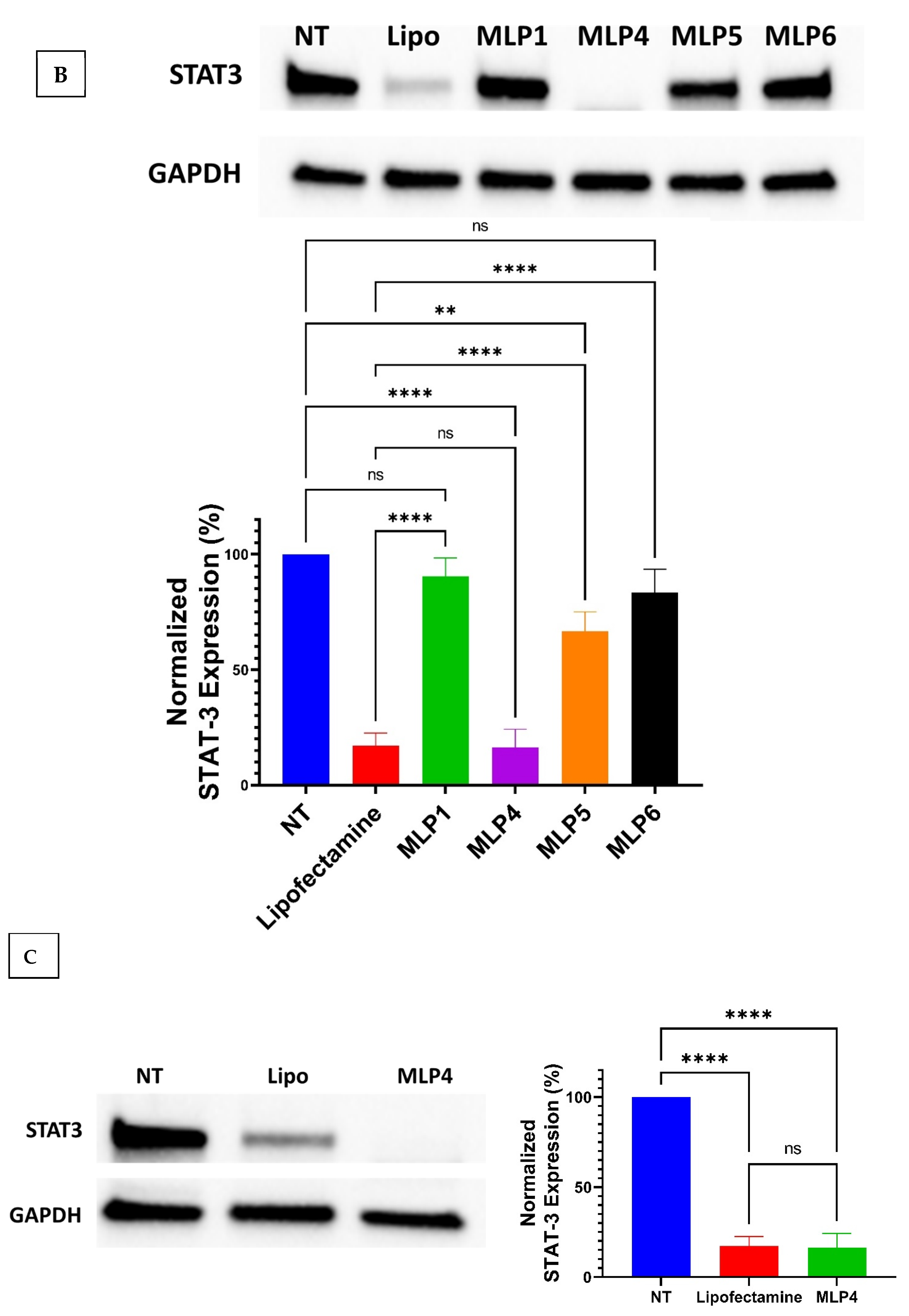
2.8. Western Blot
The Western blot assay was used to evaluate the expression of STAT-3 protein in MDA-MB-231 cells using the protocol adopted from our previous study [
7]. Briefly, the cells were grown in T25 flasks and treated with MLP-siRNA complexes at N/P ratios of 20 and 40 for 48 h. Following treatment, the cells were harvested as pellets in microtubes and were lysed using RIPA lysis buffer supplanted with a protease inhibitor. The lysate was centrifuged for 15 min at 12,000 rpm at 4 °C to allow the separation of all cell proteins. The supernatant containing all cell proteins was separated in pre-cooled microtubes and the protein was quantified using the standard Bovine Serum Albumin (BSA) assay.
Following the quantification of protein, the samples representing 15 µg of protein were mixed with gel loading dye and loaded in the wells of pre-formed gels and the electrophoresis was carried out at 200 V for 40 min. After the complete run, the gels were transferred to the Western blotting membrane using the Trans-Blot® machine. After successfully transferring gels onto the membrane, the membrane was blocked in 5% Bovine Serum Albumin in TBS-T buffer consisting of 1X tris-buffered saline and 0.1% Tween 20. The membrane blocking was performed for 1 h, followed by washing with TBS-T buffer 3 times each for 5 min. Subsequently, the membrane was incubated with the primary antibody in 1 to 1000 dilution with TBS-T buffer overnight. After overnight treatment with the primary antibody, the membrane was washed with TBS-T buffer four times each for five minutes and was incubated with the secondary antibody (HRP-linked antibody) for one hour. After secondary antibody treatment, the membrane was again washed with TBS-T buffer four times each for 5 min. The same steps were repeated for housekeeping protein (GAPDH). Finally, the membrane was visualized under the Bio-Rad Imager, and the bands were quantified using Image Lab software.
2.9. Statistical Analysis
Data obtained from the above-described experiments are shown as the mean ± SD of three independent experiments, unless stated otherwise. Ordinary one-way analyses of variance (ANOVA) with multiple comparisons using Bartlett’s test were conducted to analyze flow cytometry and Western blotting data. Two-way ANOVA was performed using Tukey’s multiple comparisons test to determine the effect of changing the N/P ratio on the particle size and zeta potential. Paired Student’s t-test was performed for comparison between the two groups. A p-value < 0.05 was considered statistically significant.


 ,
, 

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}