
Retinitis Pigmentosa: Novel Therapeutic Targets and Drug Development




Abstract
1. Introduction
2. Overview of Retinitis Pigmentosa
2.1. Clinical Manifestations
2.2. Classification
2.3. Management and Prognosis
3. Conventional Treatments and Limitations
3.1. Dietary Supplements (Vitamin A, DHA, Lutein) [11]
3.2. Cystoid Macular Edema (CME) Treatment
3.3. Protection from Sunlight
3.4. Visual Aids
3.5. Surgical Intervention
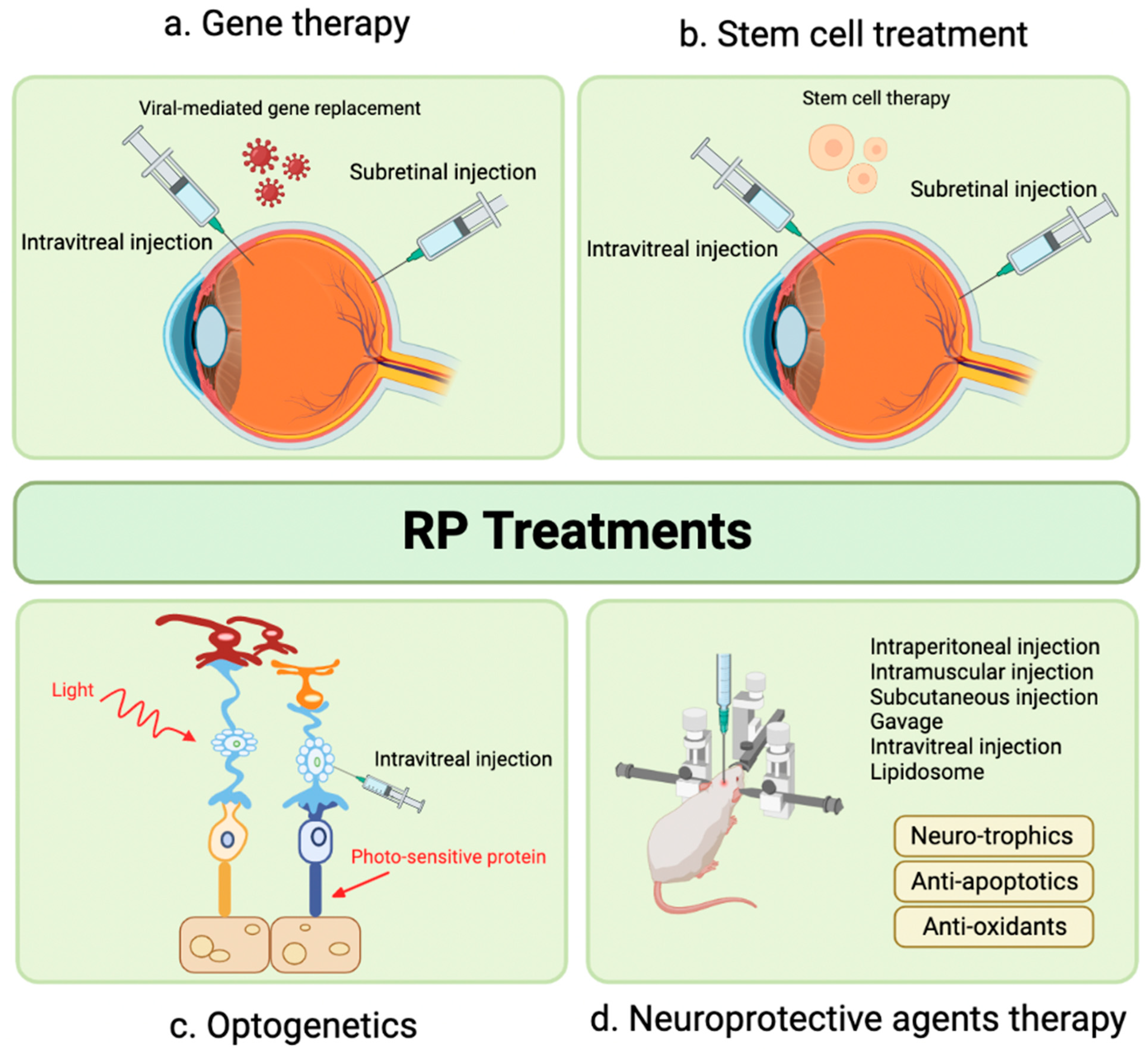
4. Recent Therapeutic Advances
5. Gene Therapy
5.1. Overview of Gene Therapy Methods
5.1.1. Viral Vectors
5.1.2. CRISPR-Cas Gene Editing System
5.1.3. RNA Replacement
5.2. Targeting Retinitis Pigmentosa Pathogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inheritance Mode | Genes | Preclinical Phase Studies |
|---|---|---|
| Autosomal Dominant Retinitis Pigmentosa | RHO | CRISPR-cas9-induced Rho mutation in Xenopus laevis tadpoles inhibits Müller glial cell proliferation [70]. T17M rhodopsin expression induces the upregulation of IL-1β, IL-6 and NFκB and the IB1A microglia markers in C57BL6 mice [71]. Proline substitution with leucine (P347L) in rhodopsin gene in transgenic rat strain induces rapid destruction of the outer nuclear layer, by CHOP and BiP activation [72]. Subretinal injection of fused zinc-finger to the KRAB domain in combination to a RHO cDNA driven by the GNAT1 promotor in pigs restores Rho gene expression and retinal function [73]. CRISPR-cas9 gene therapy in S334ter-3 rats increases visual acuity and retinal preservation [74]. AAV-miR-2014 delivery in RHO-P247S transgenic mice enhances retinal function [75] |
| Nrl | CRISPR-Cas9 engineered NRL-deficient ESC retinal organoids exhibit an abnormal number of photoreceptors expressing S-Opsin [76]. AAV-delivered CRISPR-cas9 Nrl gene knockdown in RHO-P347S Rd10 mice prevents retinal degeneration [77]. | |
| Nr2e3 | AAV-delivered CRISPR-cas9 Nr2e3 gene knockdown in Rd10 mice prevents retinal degeneration [78]. | |
| PRPF | AAV-associated CRISPR-cas9 Prpf31 gene augmentation in mice restores retinal function [79]. | |
| RP1 | Dual Cas9/sgRNA successfully reduces RP1 expression in edited Hap1-EF1a-RP1 cells [80]. | |
| Autosomal Recessive Retinitis Pigmentosa | RHO | HEK293 and HT1080 human cells lines expressing E249ter or W161ter mutants exhibit lower rhodopsin mRNA levels [81]. |
| RPGR | AAV-delivered CRISPR-cas9 restores RPGRORF15 reading frame in rd9 mice [82]. AAV-delivered CRISPR-cas9 restores photoreceptor preservation in Rpgr−/yCas9+/WT mice [83]. | |
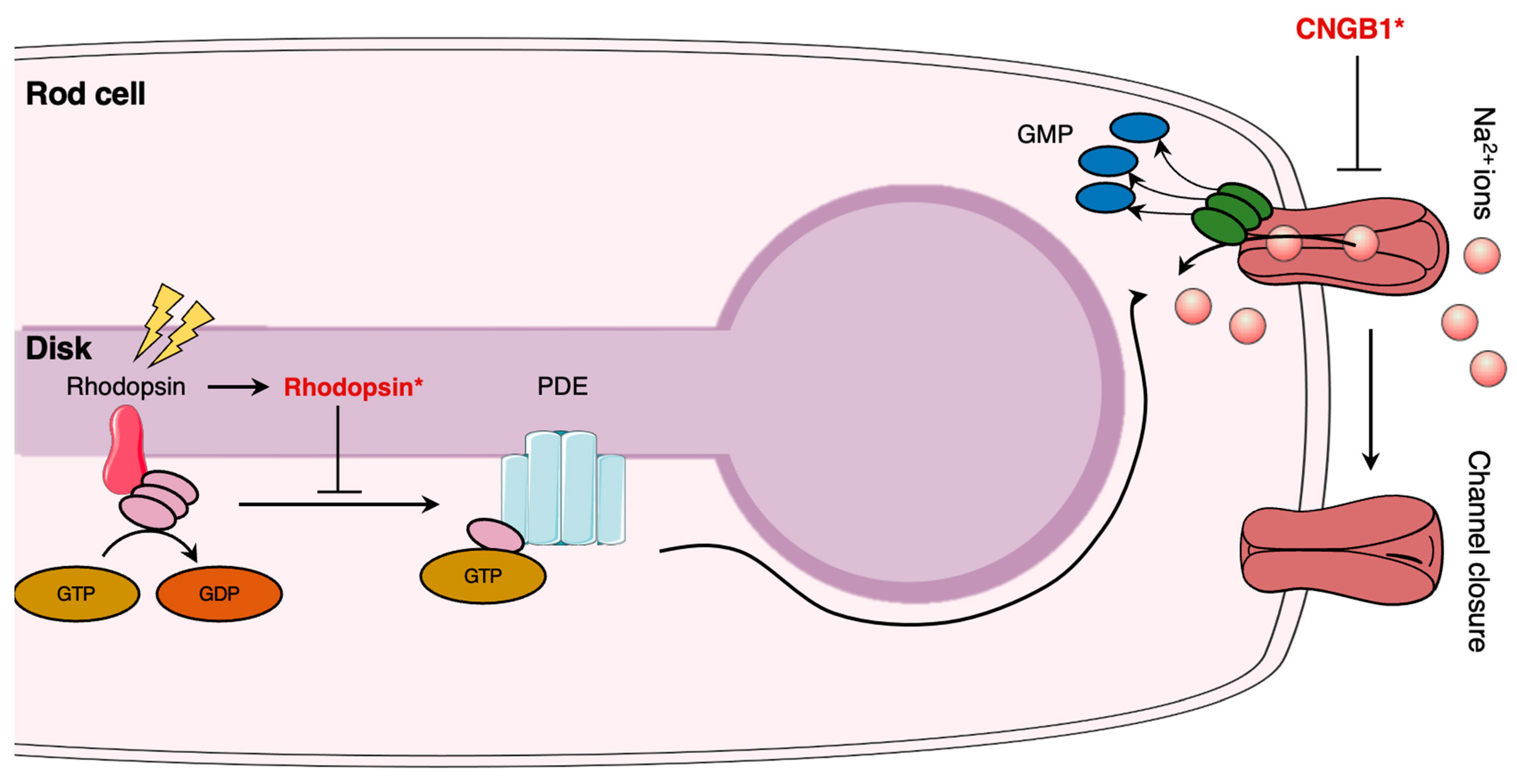
| CNGB1 | CNGB1 expression augmentation in Cngb1−/− mice using an AAV vector restores vision and delays retinal degeneration [84] (pp. 733–739). | |
| TULP1 | Single-knockout (tulp1+/−) and double-knockout zebrafish models for the expression of Tulp1 significantly alters ciliogenesis through the downregulation of tektin2 [85]. AAV-mediated Tulp1 gene expression editing restores Tulp1 mRNA and protein levels [86]. | |
| FAM161A | Fam161a knockout mice display retinal degeneration phenotype as well as enhanced molecular generative markers [87]. Homozygous Fam161a p.Arg512∗ have altered visual acuity due to complete loss of the outer nuclear layer and photoreceptor cell death [88]. |
5.2.1. Physiological Signaling Pathways of the Retina
5.2.2. Phototransduction
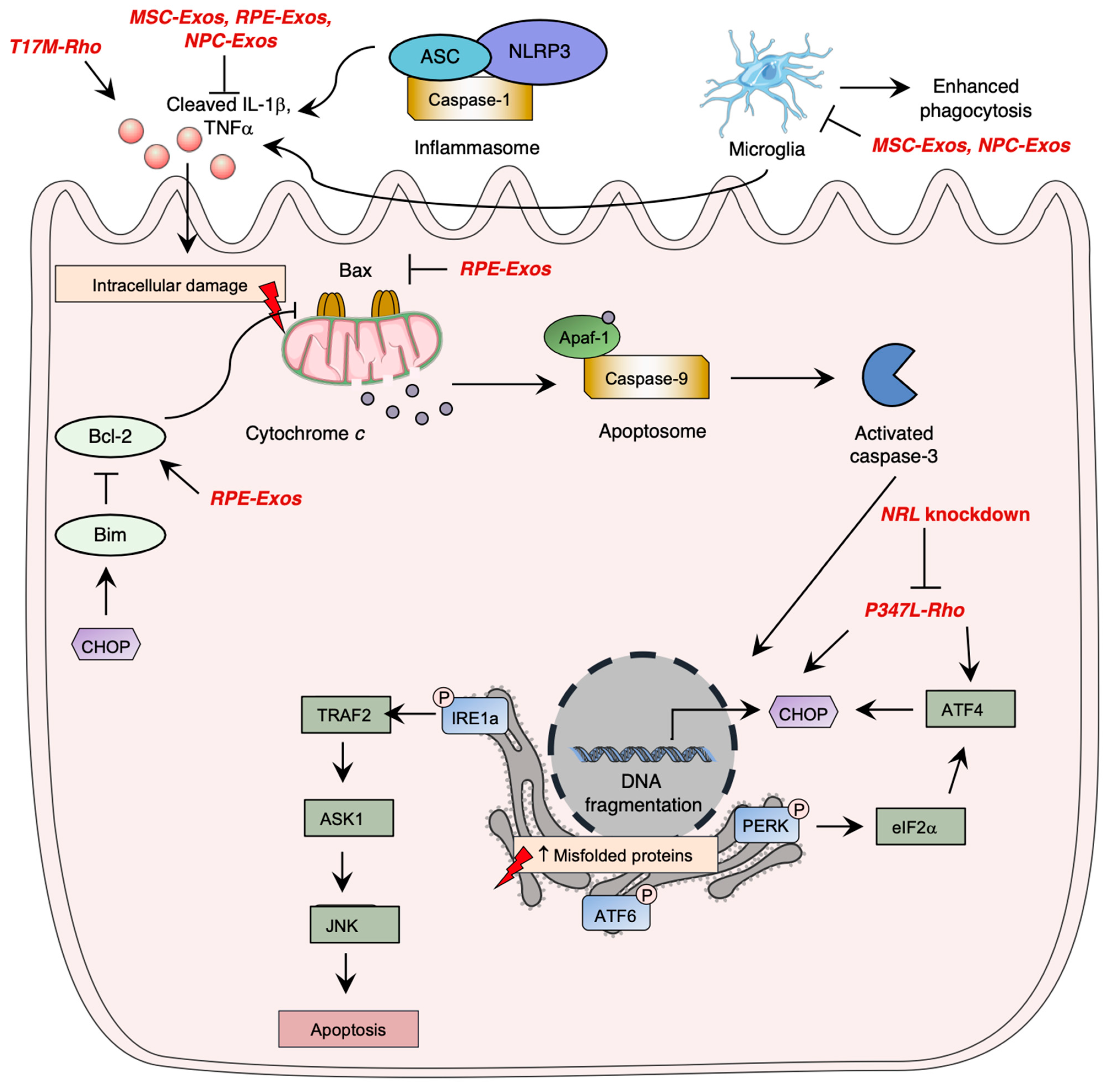
5.2.3. Photoreceptor Cell Death
5.3. Autosomal Dominant-Linked Mutations
5.3.1. Rhodopsin-Induced Autosomal Dominant Retinitis Pigmentosa
5.3.2. Non-Rhodopsin-Related Autosomal Dominant Mutations
5.4. Autosomal Recessive-Linked Mutations
5.4.1. Rhodopsin-Induced Autosomal Recessive Retinitis Pigmentosa
5.4.2. Non-Rhodopsin-Related Autosomal Recessive Mutations
5.5. Identification of Gene Targets Involved in Retinitis Pigmentosa for Novel Gene Therapy Treatments
6. Stem Cell Therapy
6.1. Various Models and Sources
6.1.1. Embryonic and Pluripotent Stem Cells
6.1.2. Bone Marrow Stem Cells Therapies
6.1.3. Therapies Based on Stem Cell-Derived RPE
6.1.4. Therapies Based on Retinal Progenitor Cells
7. Novel Therapeutic Targets in Preclinical Phase: Optogenetics
7.1. Microbial-Derived Opsins
7.2. Animal-Derived Opsins
8. Novel Therapeutic Targets in Preclinical Phase: Neuroprotective Agents
8.1. Neuroprotective Pathways
8.2. Neurotropic Factors
8.3. Anti-Apoptotic Agents
8.4. Antioxidant Agents
9. Novel Therapeutic Targets in Preclinical Phase: Exosomes
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cross, N.; van Steen, C.; Zegaoui, Y.; Satherley, A.; Angelillo, L. Retinitis Pigmentosa: Burden of Disease and Current Unmet Needs. Clin. Ophthalmol. 2022, 16, 1993–2010. [Google Scholar] [CrossRef]
- Retinitis Pigmentosa: Study Guide. Available online: https://www.aao.org/Assets/bcc226cb-7ac7-443c-897e-9b20afdfacfb/637153836910470000/r38u-pdf?inline=1&fbclid=IwAR2oP_lD1pWSAROpIfa9poqhZzeSf29_PvZztBZMSQfWKNqTbv1b6t0BXJU (accessed on 15 December 2022).
- O’Neal, T.B.; Luther, E.E. Retinitis Pigmentosa. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis Pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef] [PubMed]
- Kimberling, W.J.; Hildebrand, M.S.; Shearer, A.E.; Jensen, M.L.; Halder, J.A.; Trzupek, K.; Cohn, E.S.; Weleber, R.G.; Stone, E.M.; Smith, R.J.H. Frequency of Usher Syndrome in Two Pediatric Populations: Implications for Genetic Screening of Deaf and Hard of Hearing Children. Genet. Med. Off. J. Am. Coll. Med. Genet. 2010, 12, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Weihbrecht, K. Bardet-Biedl Syndrome. In Genetics and Genomics of Eye Disease; Elsevier: Amsterdam, The Netherlands, 2020; pp. 117–136. ISBN 978-0-12-816222-4. [Google Scholar]
- Von Sallmann, L.; Gelderman, A.H.; Laster, L. Ocular Histopathologic Changes in a Case of A-Beta-Lipoproteinemia (Bassen-Kornzweig Syndrome). Doc. Ophthalmol. Adv. Ophthalmol. 1969, 26, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.A.; Berson, E.L. Treatable Forms of Retinitis Pigmentosa Associated with Systemic Neurological Disorders. Int. Ophthalmol. Clin. 2001, 41, 103–110. [Google Scholar] [CrossRef]
- Gibberd, F.B.; Billimoria, J.D.; Goldman, J.M.; Clemens, M.E.; Evans, R.; Whitelaw, M.N.; Retsas, S.; Sherratt, R.M. Heredopathia Atactica Polyneuritiformis: Refsum’s Disease. Acta Neurol. Scand. 1985, 72, 1–17. [Google Scholar] [CrossRef]
- Berson, E.L.; Sandberg, M.A.; Rosner, B.; Birch, D.G.; Hanson, A.H. Natural Course of Retinitis Pigmentosa over a Three-Year Interval. Am. J. Ophthalmol. 1985, 99, 240–251. [Google Scholar] [CrossRef]
- Wang, A.L.; Knight, D.K.; Vu, T.-T.T.; Mehta, M.C. Retinitis Pigmentosa: Review of Current Treatment. Int. Ophthalmol. Clin. 2019, 59, 263–280. [Google Scholar] [CrossRef]
- Rayapudi, S.; Schwartz, S.G.; Wang, X.; Chavis, P. Vitamin A and Fish Oils for Retinitis Pigmentosa. Cochrane Libr. Cochrane Rev. 2013, 2013, CD008428. [Google Scholar] [CrossRef]
- Berson, E.L. Retinitis Pigmentosa. The Friedenwald Lecture. Investig. Ophthalmol. Vis. Sci. 1993, 34, 1659–1676. [Google Scholar]
- Berson, E.L.; Rosner, B.; Sandberg, M.A.; Weigel-DiFranco, C.; Moser, A.; Brockhurst, R.J.; Hayes, K.C.; Johnson, C.A.; Anderson, E.J.; Gaudio, A.R.; et al. Further Evaluation of Docosahexaenoic Acid in Patients with Retinitis Pigmentosa Receiving Vitamin A Treatment: Subgroup Analyses. Arch. Ophthalmol. 2004, 122, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Berson, E.L.; Rosner, B.; Sandberg, M.A.; Weigel-DiFranco, C.; Brockhurst, R.J.; Hayes, K.C.; Johnson, E.J.; Anderson, E.J.; Johnson, C.A.; Gaudio, A.R.; et al. Clinical Trial of Lutein in Patients with Retinitis Pigmentosa Receiving Vitamin A. Arch. Ophthalmol. 2010, 128, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.M. The Vitamin A Spectrum: From Deficiency to Toxicity. Am. J. Clin. Nutr. 2000, 71, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Pierce, E.A.; Bujakowska, K.M.; Place, E.; Navarro-Gomez, D.; Maher, M.; Weigel-DFranco, C.; Comander, J. The Effect Of Vitamin A On Progression Of Retinitis Pigmentosa Is Not Determined By The Underlying Genetic Cause Of Disease. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2011. [Google Scholar]
- Hajali, M.; Fishman, G.A.; Anderson, R.J. The Prevalence of Cystoid Macular Oedema in Retinitis Pigmentosa Patients Determined by Optical Coherence Tomography. Br. J. Ophthalmol. 2008, 92, 1065–1068. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, H.; Karacorlu, M.; Karacorlu, S. Intravitreal Triamcinolone Acetonide for Treatment of Cystoid Macular Oedema in Patients with Retinitis Pigmentosa. Acta Ophthalmol. Scand. 2005, 83, 248–251. [Google Scholar] [CrossRef]
- Bakthavatchalam, M.; Lai, F.H.P.; Rong, S.S.; Ng, D.S.; Brelen, M.E. Treatment of Cystoid Macular Edema Secondary to Retinitis Pigmentosa: A Systematic Review. Surv. Ophthalmol. 2018, 63, 329–339. [Google Scholar] [CrossRef]
- Gallenga, C.E.; Lonardi, M.; Pacetti, S.; Violanti, S.S.; Tassinari, P.; Di Virgilio, F.; Tognon, M.; Perri, P. Molecular Mechanisms Related to Oxidative Stress in Retinitis Pigmentosa. Antioxidants 2021, 10, 848. [Google Scholar] [CrossRef]
- Naash, M.L.; Peachey, N.S.; Li, Z.Y.; Gryczan, C.C.; Goto, Y.; Blanks, J.; Milam, A.H.; Ripps, H. Light-Induced Acceleration of Photoreceptor Degeneration in Transgenic Mice Expressing Mutant Rhodopsin. Investig. Ophthalmol. Vis. Sci. 1996, 37, 775–782. [Google Scholar]
- Kutsyr, O.; Sánchez-Sáez, X.; Martínez-Gil, N.; de Juan, E.; Lax, P.; Maneu, V.; Cuenca, N. Gradual Increase in Environmental Light Intensity Induces Oxidative Stress and Inflammation and Accelerates Retinal Neurodegeneration. Investig. Ophthalmol. Vis. Sci. 2020, 61, 1. [Google Scholar] [CrossRef]
- Berson, E.L.; Rabin, A.R.; Mehaffey, L. Advances in Night Vision Technology. A Pocketscope for Patients with Retinitis Pigmentosa. Arch. Ophthalmol. 1973, 90, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Hartong, D.T.; Jorritsma, F.F.; Neve, J.J.; Melis-Dankers, B.J.M.; Kooijman, A.C. Improved Mobility and Independence of Night-Blind People Using Night-Vision Goggles. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1725–1731. [Google Scholar] [CrossRef] [PubMed]
- Mancil, R.M.; Mancil, G.L.; King, E.; Legault, C.; Munday, J.; Alfieri, S.; Nowakowski, R.; Blasch, B.B. Improving Nighttime Mobility in Persons with Night Blindness Caused by Retinitis Pigmentosa: A Comparison of Two Low-Vision Mobility Devices. J. Rehabil. Res. Dev. 2005, 42, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Suzuki, E.; Kuramata, T.; Kozaki, T.; Koyama, T.; Kato, Y.; Murakami, Y.; Enaida, H.; Ishibashi, T. Development and Evaluation of a Visual Aid Using See-through Display for Patients with Retinitis Pigmentosa. Jpn. J. Ophthalmol. 2015, 59, 43–47. [Google Scholar] [CrossRef]
- Humayun, M.S.; Dorn, J.D.; da Cruz, L.; Dagnelie, G.; Sahel, J.-A.; Stanga, P.E.; Cideciyan, A.V.; Duncan, J.L.; Eliott, D.; Filley, E.; et al. Interim Results from the International Trial of Second Sight’s Visual Prosthesis. Ophthalmology 2012, 119, 779–788. [Google Scholar] [CrossRef]
- da Cruz, L.; Dorn, J.D.; Humayun, M.S.; Dagnelie, G.; Handa, J.; Barale, P.-O.; Sahel, J.-A.; Stanga, P.E.; Hafezi, F.; Safran, A.B.; et al. Five-Year Safety and Performance Results from the Argus II Retinal Prosthesis System Clinical Trial. Ophthalmology 2016, 123, 2248–2254. [Google Scholar] [CrossRef]
- Arevalo, J.F.; Al Rashaed, S.; Alhamad, T.A.; Al Kahtani, E.; Al-Dhibi, H.A.; for the KKESH Collaborative Retina Study Group; Mura, M.; Al Kahtani, E.; Nowilaty, S.; Al Rashaed, S.; et al. Argus II Retinal Prosthesis for Retinitis Pigmentosa in the Middle East: The 2015 Pan-American Association of Ophthalmology Gradle Lecture. Int. J. Retin. Vitr. 2021, 7, 65. [Google Scholar] [CrossRef]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and Safety of Voretigene Neparvovec (AAV2-HRPE65v2) in Patients with RPE65-Mediated Inherited Retinal Dystrophy: A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Maguire, A.M.; Russell, S.; Wellman, J.A.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; Marshall, K.A.; et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-Rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology 2019, 126, 1273–1285. [Google Scholar] [CrossRef]
- Maguire, A.M.; Russell, S.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Drack, A.V.; Simonelli, F.; Leroy, B.P.; Reape, K.Z.; High, K.A.; et al. Durability of Voretigene Neparvovec for Biallelic RPE65-Mediated Inherited Retinal Disease: Phase 3 Results at 3 and 4 Years. Ophthalmology 2021, 128, 1460–1468. [Google Scholar] [CrossRef]
- Dhurandhar, D.; Sahoo, N.; Mariappan, I.; Narayanan, R. Gene Therapy in Retinal Diseases: A Review. Indian J. Ophthalmol. 2021, 69, 2257–2265. [Google Scholar] [CrossRef] [PubMed]
- Ong, T.; Pennesi, M.E.; Birch, D.G.; Lam, B.L.; Tsang, S.H. Adeno-Associated Viral Gene Therapy for Inherited Retinal Disease. Pharm. Res. 2019, 36, 34. [Google Scholar] [CrossRef] [PubMed]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral Vector Platforms within the Gene Therapy Landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Berns, K.I.; Giraud, C. Biology of Adeno-Associated Virus. In Adeno-Associated Virus (AAV) Vectors in Gene Therapy; Berns, K.I., Giraud, C., Eds.; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 1996; Volume 218, pp. 1–23. ISBN 978-3-642-80209-6. [Google Scholar]
- Trapani, I.; Puppo, A.; Auricchio, A. Vector Platforms for Gene Therapy of Inherited Retinopathies. Prog. Retin. Eye Res. 2014, 43, 108–128. [Google Scholar] [CrossRef]
- Chen, H.; Xiang, Z.Q.; Li, Y.; Kurupati, R.K.; Jia, B.; Bian, A.; Zhou, D.M.; Hutnick, N.; Yuan, S.; Gray, C.; et al. Adenovirus-Based Vaccines: Comparison of Vectors from Three Species of Adenoviridae. J. Virol. 2010, 84, 10522–10532. [Google Scholar] [CrossRef]
- Kotin, R.M.; Menninger, J.C.; Ward, D.C.; Berns, K.I. Mapping and Direct Visualization of a Region-Specific Viral DNA Integration Site on Chromosome 19q13-Qter. Genomics 1991, 10, 831–834. [Google Scholar] [CrossRef]
- Qu, Y.; Liu, Y.; Noor, A.; Tran, J.; Li, R. Characteristics and Advantages of Adeno-Associated Virus Vector-Mediated Gene Therapy for Neurodegenerative Diseases. Neural Regen. Res. 2019, 14, 931. [Google Scholar] [CrossRef]
- Trapani, I.; Tornabene, P.; Auricchio, A. Large Gene Delivery to the Retina with AAV Vectors: Are We There Yet? Gene Ther. 2021, 28, 220–222. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Zhao, J.; Duan, D.; Lai, Y. Design of AAV Vectors for Delivery of Large or Multiple Transgenes. In Adeno-Associated Virus Vectors; Castle, M.J., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 1950, pp. 19–33. ISBN 978-1-4939-9138-9. [Google Scholar]
- Tornabene, P.; Trapani, I. Can Adeno-Associated Viral Vectors Deliver Effectively Large Genes? Hum. Gene Ther. 2020, 31, 47–56. [Google Scholar] [CrossRef]
- Carvalho, L.S.; Turunen, H.T.; Wassmer, S.J.; Luna-Velez, M.V.; Xiao, R.; Bennett, J.; Vandenberghe, L.H. Evaluating Efficiencies of Dual AAV Approaches for Retinal Targeting. Front. Neurosci. 2017, 11, 503. [Google Scholar] [CrossRef]
- Maddalena, A.; Tornabene, P.; Tiberi, P.; Minopoli, R.; Manfredi, A.; Mutarelli, M.; Rossi, S.; Simonelli, F.; Naggert, J.K.; Cacchiarelli, D.; et al. Triple Vectors Expand AAV Transfer Capacity in the Retina. Mol. Ther. 2018, 26, 524–541. [Google Scholar] [CrossRef] [PubMed]
- McClements, M.E.; Barnard, A.R.; Singh, M.S.; Charbel Issa, P.; Jiang, Z.; Radu, R.A.; MacLaren, R.E. An AAV Dual Vector Strategy Ameliorates the Stargardt Phenotype in Adult Abca4−/− Mice. Hum. Gene Ther. 2019, 30, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Pavlou, M.; Schön, C.; Occelli, L.M.; Rossi, A.; Meumann, N.; Boyd, R.F.; Bartoe, J.T.; Siedlecki, J.; Gerhardt, M.J.; Babutzka, S.; et al. Novel AAV Capsids for Intravitreal Gene Therapy of Photoreceptor Disorders. EMBO Mol. Med. 2021, 13, e13392. [Google Scholar] [CrossRef]
- Kevany, B.; Suh, S.; Lu, J.; Padegimas, L.; Palczewski, K.; Miller, T. Novel AAV Capsids Demonstrate Strong Retinal Expression in Non-Human Primates After Intravitreal Administration. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2900. [Google Scholar]
- Katada, Y.; Kobayashi, K.; Tsubota, K.; Kurihara, T. Evaluation of AAV-DJ Vector for Retinal Gene Therapy. PeerJ 2019, 7, e6317. [Google Scholar] [CrossRef]
- Leonova, E.I.; Gainetdinov, R.R. CRISPR/Cas9 Technology in Translational Biomedicine|Cell Physiol Biochem. Cell. Physiol. Biochem. 2020, 54, 354–370. [Google Scholar]
- Doudna, J.A.; Charpentier, E. The New Frontier of Genome Engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Peddle, C.F.; MacLaren, R.E. The Application of CRISPR/Cas9 for the Treatment of Retinal Diseases. Yale J. Biol. Med. 2017, 90, 533–541. [Google Scholar]
- Ahmad, I. CRISPR/Cas9-A Promising Therapeutic Tool to Cure Blindness: Current Scenario and Future Prospects. Int. J. Mol. Sci. 2022, 23, 11482. [Google Scholar] [CrossRef]
- Reshetnikov, V.V.; Chirinskaite, A.V.; Sopova, J.V.; Ivanov, R.A.; Leonova, E.I. Cas-Based Systems for RNA Editing in Gene Therapy of Monogenic Diseases: In Vitro and in Vivo Application and Translational Potential. Front. Cell Dev. Biol. 2022, 10, 903812. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, Y.; Xu, J.; Wang, X.; Luo, S.; Mao, B.; Zhou, Q.; Li, W. Metagenomic Discovery of Novel CRISPR-Cas13 Systems. Cell Discov. 2022, 8, 107. [Google Scholar] [CrossRef] [PubMed]
- Botto, C.; Dalkara, D.; El-Amraoui, A. Progress in Gene Editing Tools and Their Potential for Correcting Mutations Underlying Hearing and Vision Loss. Front. Genome Ed. 2021, 3, 737632. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, X.; Zhou, J.; Yang, C.; Wang, G.; Tan, Y.; Wu, Y.; Zhang, S.; Yi, K.; Kang, C. The CRISPR-Cas13a Gene-Editing System Induces Collateral Cleavage of RNA in Glioma Cells. Adv. Sci. 2019, 6, 1901299. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, J.; Guo, X.; Li, Z.; Cao, L.; Liu, S.; Guo, Y.; Wang, G.; Luo, Y.; Zhang, Z.; et al. The Collateral Activity of RfxCas13d Can Induce Lethality in a RfxCas13d Knock-in Mouse Model. Genome Biol. 2023, 24, 20. [Google Scholar] [CrossRef]
- Ai, Y.; Liang, D.; Wilusz, J.E. CRISPR/Cas13 Effectors Have Differing Extents of off-Target Effects That Limit Their Utility in Eukaryotic Cells. Nucleic Acids Res. 2022, 50, e65. [Google Scholar] [CrossRef]
- Wu, Q.-W.; Kapfhammer, J.P. The Bacterial Enzyme Cas13 Interferes with Neurite Outgrowth from Cultured Cortical Neurons. Toxins 2021, 13, 262. [Google Scholar] [CrossRef]
- Fry, L.E.; McClements, M.E.; MacLaren, R.E. Comparison of CRISPR-Cas13 RNA Editing Tools for Inherited Retinal Disease. Investig. Ophthalmol. Vis. Sci. 2022, 63, 3845. [Google Scholar]
- Gemayel, M.C.; Bhatwadekar, A.D.; Ciulla, T. RNA Therapeutics for Retinal Diseases. Expert Opin. Biol. Ther. 2021, 21, 603–613. [Google Scholar] [CrossRef]
- Hannon, G.J. RNA Interference. Nature 2002, 418, 244–251. [Google Scholar] [CrossRef]
- Orlans, H.O.; McClements, M.E.; Barnard, A.R.; Martinez-Fernandez de la Camara, C.; MacLaren, R.E. Mirtron-Mediated RNA Knockdown/Replacement Therapy for the Treatment of Dominant Retinitis Pigmentosa. Nat. Commun. 2021, 12, 4934. [Google Scholar] [CrossRef]
- Bajan, S.; Hutvagner, G. RNA-Based Therapeutics: From Antisense Oligonucleotides to MiRNAs. Cells 2020, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Knott, S.R.V.; Maceli, A.; Erard, N.; Chang, K.; Marran, K.; Zhou, X.; Gordon, A.; Demerdash, O.E.; Wagenblast, E.; Kim, S.; et al. A Computational Algorithm to Predict ShRNA Potency. Mol. Cell 2014, 56, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, S.; Li, P.; Yao, K. Retinitis Pigmentosa: Progress in Molecular Pathology and Biotherapeutical Strategies. Int. J. Mol. Sci. 2022, 23, 4883. [Google Scholar] [CrossRef] [PubMed]
- Cross, N.; van Steen, C.; Zegaoui, Y.; Satherley, A.; Angelillo, L. Current and Future Treatment of Retinitis Pigmentosa. Clin. Ophthalmol. 2022, 16, 2909–2921. [Google Scholar] [CrossRef] [PubMed]
- Parain, K.; Lourdel, S.; Donval, A.; Chesneau, A.; Borday, C.; Bronchain, O.; Locker, M.; Perron, M. CRISPR/Cas9-Mediated Models of Retinitis Pigmentosa Reveal Differential Proliferative Response of Müller Cells between Xenopus Laevis and Xenopus Tropicalis. Cells 2022, 11, 807. [Google Scholar] [CrossRef]
- Rana, T.; Shinde, V.M.; Starr, C.R.; Kruglov, A.A.; Boitet, E.R.; Kotla, P.; Zolotukhin, S.; Gross, A.K.; Gorbatyuk, M.S. An Activated Unfolded Protein Response Promotes Retinal Degeneration and Triggers an Inflammatory Response in the Mouse Retina. Cell Death Dis. 2014, 5, e1578. [Google Scholar] [CrossRef]
- Inoue, C.; Takeuchi, T.; Shiota, A.; Kondo, M.; Nshizawa, Y. A Rat Model for Retinitis Pigmentosa with Rapid Retinal Degeneration Enables Drug Evaluation in Vivo. Biol. Proced. Online 2021, 23, 11. [Google Scholar] [CrossRef]
- Marrocco, E.; Maritato, R.; Botta, S.; Esposito, M.; Surace, E.M. Challenging Safety and Efficacy of Retinal Gene Therapies by Retinogenesis. Int. J. Mol. Sci. 2021, 22, 5767. [Google Scholar] [CrossRef]
- Bakondi, B.; Lv, W.; Lu, B.; Jones, M.K.; Tsai, Y.; Kim, K.J.; Levy, R.; Akhtar, A.A.; Breunig, J.J.; Svendsen, C.N.; et al. In Vivo CRISPR/Cas9 Gene Editing Corrects Retinal Dystrophy in the S334ter-3 Rat Model of Autosomal Dominant Retinitis Pigmentosa. Mol. Ther. 2016, 24, 556–563. [Google Scholar] [CrossRef]
- Karali, M.; Guadagnino, I.; Marrocco, E.; De Cegli, R.; Carissimo, A.; Pizzo, M.; Casarosa, S.; Conte, I.; Surace, E.M.; Banfi, S. AAV-MiR-204 Protects from Retinal Degeneration by Attenuation of Microglia Activation and Photoreceptor Cell Death. Mol. Ther.-Nucleic Acids 2020, 19, 144–156. [Google Scholar] [CrossRef]
- Cuevas, E.; Holder, D.L.; Alshehri, A.H.; Tréguier, J.; Lakowski, J.; Sowden, J.C. NRL−/− Gene Edited Human Embryonic Stem Cells Generate Rod-Deficient Retinal Organoids Enriched in S-Cone-like Photoreceptors. Stem Cells 2021, 39, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Mookherjee, S.; Chaitankar, V.; Hiriyanna, S.; Kim, J.-W.; Brooks, M.; Ataeijannati, Y.; Sun, X.; Dong, L.; Li, T.; et al. Nrl Knockdown by AAV-Delivered CRISPR/Cas9 Prevents Retinal Degeneration in Mice. Nat. Commun. 2017, 8, 14716. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Ming, C.; Fu, X.; Duan, Y.; Hoang, D.A.; Rutgard, J.; Zhang, R.; Wang, W.; Hou, R.; Zhang, D.; et al. Gene and Mutation Independent Therapy via CRISPR-Cas9 Mediated Cellular Reprogramming in Rod Photoreceptors. Cell Res. 2017, 27, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Vats, A.; Sahel, J.-A.; Chen, Y.; Byrne, L.C. Gene Augmentation Prevents Retinal Degeneration in a CRISPR/Cas9-Based Mouse Model of PRPF31 Retinitis Pigmentosa. Nat. Commun. 2022, 13, 7695. [Google Scholar] [CrossRef] [PubMed]
- Collin, C.; Liu, Q. CRISPR/Cas9-Based Genome Editing Approaches for RP1 Associated Autosomal Dominant Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2021, 62, 1486. [Google Scholar]
- Roman-Sanchez, R.; Wensel, T.G.; Wilson, J.H. Nonsense Mutations in the Rhodopsin Gene That Give Rise to Mild Phenotypes Trigger MRNA Degradation in Human Cells by Nonsense-Mediated Decay. Exp. Eye Res. 2016, 145, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Gumerson, J.D.; Alsufyani, A.; Yu, W.; Lei, J.; Sun, X.; Dong, L.; Wu, Z.; Li, T. Restoration of RPGR Expression in Vivo Using CRISPR/Cas9 Gene Editing. Gene Ther. 2022, 29, 81–93. [Google Scholar] [CrossRef]
- Hu, S.; Du, J.; Chen, N.; Jia, R.; Zhang, J.; Liu, X.; Yang, L. In Vivo CRISPR/Cas9-Mediated Genome Editing Mitigates Photoreceptor Degeneration in a Mouse Model of X-Linked Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2020, 61, 31. [Google Scholar] [CrossRef]
- Michalakis, S.; Koch, S.; Sothilingam, V.; Garrido, M.G.; Tanimoto, N.; Schulze, E.; Becirovic, E.; Koch, F.; Seide, C.; Beck, S.C.; et al. Gene Therapy Restores Vision and Delays Degeneration in the CNGB1−/− Mouse Model of Retinitis Pigmentosa. In Retinal Degenerative Diseases; Ash, J.D., Grimm, C., Hollyfield, J.G., Anderson, R.E., LaVail, M.M., Bowes Rickman, C., Eds.; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2014; Volume 801, pp. 733–739. ISBN 978-1-4614-3208-1. [Google Scholar]
- Jia, D.; Gao, P.; Lv, Y.; Huang, Y.; Reilly, J.; Sun, K.; Han, Y.; Hu, H.; Chen, X.; Zhang, Z.; et al. Tulp1 Deficiency Causes Early-Onset Retinal Degeneration through Affecting Ciliogenesis and Activating Ferroptosis in Zebrafish. Cell Death Dis. 2022, 13, 962. [Google Scholar] [CrossRef]
- Palfi, A.; Yesmambetov, A.; Millington-Ward, S.; Shortall, C.; Humphries, P.; Kenna, P.F.; Chadderton, N.; Farrar, G.J. AAV-Delivered Tulp1 Supplementation Therapy Targeting Photoreceptors Provides Minimal Benefit in Tulp1−/− Retinas. Front. Neurosci. 2020, 14, 891. [Google Scholar] [CrossRef]
- Beryozkin, A.; Matsevich, C.; Obolensky, A.; Kostic, C.; Arsenijevic, Y.; Wolfrum, U.; Banin, E.; Sharon, D. A New Mouse Model for Retinal Degeneration Due to Fam161a Deficiency. Sci. Rep. 2021, 11, 2030. [Google Scholar] [CrossRef] [PubMed]
- Matsevich, C.; Gopalakrishnan, P.; Obolensky, A.; Banin, E.; Sharon, D.; Beryozkin, A. Retinal Structure and Function in a Knock-in Mouse Model for the FAM161A-p.Arg523∗ Human Nonsense Pathogenic Variant. Ophthalmol. Sci. 2023, 3, 100229. [Google Scholar] [CrossRef] [PubMed]
- García Bohórquez, B.; Aller, E.; Rodríguez Muñoz, A.; Jaijo, T.; García García, G.; Millán, J.M. Updating the Genetic Landscape of Inherited Retinal Dystrophies. Front. Cell Dev. Biol. 2021, 9, 645600. [Google Scholar] [CrossRef] [PubMed]
- Nash, B.M.; Wright, D.C.; Grigg, J.R.; Bennetts, B.; Jamieson, R.V. Retinal Dystrophies, Genomic Applications in Diagnosis and Prospects for Therapy. Transl. Pediatr. 2015, 4, 139–163. [Google Scholar] [CrossRef]
- Perea-Romero, I.; Gordo, G.; Iancu, I.F.; Del Pozo-Valero, M.; Almoguera, B.; Blanco-Kelly, F.; Carreño, E.; Jimenez-Rolando, B.; Lopez-Rodriguez, R.; Lorda-Sanchez, I.; et al. Genetic Landscape of 6089 Inherited Retinal Dystrophies Affected Cases in Spain and Their Therapeutic and Extended Epidemiological Implications. Sci. Rep. 2021, 11, 1526. [Google Scholar] [CrossRef] [PubMed]
- Parmeggiani, F.S.; Sorrentino, F.; Ponzin, D.; Barbaro, V.; Ferrari, S.; Di Iorio, E. Retinitis Pigmentosa: Genes and Disease Mechanisms. Curr. Genom. 2011, 12, 238–249. [Google Scholar] [CrossRef]
- Palczewski, K. Chemistry and Biology of Vision. J. Biol. Chem. 2012, 287, 1612–1619. [Google Scholar] [CrossRef]
- Lenahan, C.; Sanghavi, R.; Huang, L.; Zhang, J.H. Rhodopsin: A Potential Biomarker for Neurodegenerative Diseases. Front. Neurosci. 2020, 14, 326. [Google Scholar] [CrossRef]
- Kandori, H.; Shichida, Y.; Yoshizawa, T. Photoisomerization in Rhodopsin. Biochemistry 2001, 66, 1197–1209. [Google Scholar] [CrossRef]
- Palczewski, K. G Protein-Coupled Receptor Rhodopsin. Annu. Rev. Biochem. 2006, 75, 743–767. [Google Scholar] [CrossRef]
- Kramer, R.H.; Molokanova, E. Modulation of Cyclic-Nucleotide-Gated Channels and Regulation of Vertebrate Phototransduction. J. Exp. Biol. 2001, 204, 2921–2931. [Google Scholar] [CrossRef] [PubMed]
- BIO254: Phototransduction—OpenWetWare. Available online: https://openwetware.org/mediawiki/index.php?title=BIO254:Phototransduction&oldid=85424 (accessed on 12 February 2023).
- Newton, F.; Megaw, R. Mechanisms of Photoreceptor Death in Retinitis Pigmentosa. Genes 2020, 11, 1120. [Google Scholar] [CrossRef]
- Choudhury, S.; Bhootada, Y.; Gorbatyuk, O.; Gorbatyuk, M. Caspase-7 Ablation Modulates UPR, Reprograms TRAF2-JNK Apoptosis and Protects T17M Rhodopsin Mice from Severe Retinal Degeneration. Cell Death Dis. 2013, 4, e528. [Google Scholar] [CrossRef] [PubMed]
- Comitato, A.; Sanges, D.; Rossi, A.; Humphries, M.M.; Marigo, V. Activation of Bax in Three Models of Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3555. [Google Scholar] [CrossRef] [PubMed]
- Kulbay, M.; Paimboeuf, A.; Ozdemir, D.; Bernier, J. Review of Cancer Cell Resistance Mechanisms to Apoptosis and Actual Targeted Therapies. J. Cell. Biochem. 2022, 123, 1736–1761. [Google Scholar] [CrossRef]
- Zhao, L.; Hou, C.; Yan, N. Neuroinflammation in Retinitis Pigmentosa: Therapies Targeting the Innate Immune System. Front. Immunol. 2022, 13, 1059947. [Google Scholar] [CrossRef] [PubMed]
- Read, A.; Schröder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef]
- Junjappa, R.P.; Patil, P.; Bhattarai, K.R.; Kim, H.-R.; Chae, H.-J. IRE1α Implications in Endoplasmic Reticulum Stress-Mediated Development and Pathogenesis of Autoimmune Diseases. Front. Immunol. 2018, 9, 1289. [Google Scholar] [CrossRef]
- Teske, B.F.; Wek, S.A.; Bunpo, P.; Cundiff, J.K.; McClintick, J.N.; Anthony, T.G.; Wek, R.C. The EIF2 Kinase PERK and the Integrated Stress Response Facilitate Activation of ATF6 during Endoplasmic Reticulum Stress. Mol. Biol. Cell 2011, 22, 4390–4405. [Google Scholar] [CrossRef]
- Hillary, R.F.; FitzGerald, U. A Lifetime of Stress: ATF6 in Development and Homeostasis. J. Biomed. Sci. 2018, 25, 48. [Google Scholar] [CrossRef]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the Unfolded Protein Response Regulator GRP78/BiP in Development, Cancer, and Neurological Disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A Regulated Inflammatory Mode of Cell Death. J. Neuroinflammation 2018, 15, 199. [Google Scholar] [CrossRef] [PubMed]
- Daiger, S.P.; Bowne, S.J.; Sullivan, L.S. Genes and Mutations Causing Autosomal Dominant Retinitis Pigmentosa. Cold Spring Harb. Perspect. Med. 2014, 5, a017129. [Google Scholar] [CrossRef] [PubMed]
- Massengill, M.T.; Lewin, A.S. Gene Therapy for Rhodopsin-Associated Autosomal Dominant Retinitis Pigmentosa. Int. Ophthalmol. Clin. 2021, 61, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Snyder, W.K.; Olsson, J.E.; Dryja, T.P. Transgenic Mice Carrying the Dominant Rhodopsin Mutation P347S: Evidence for Defective Vectorial Transport of Rhodopsin to the Outer Segments. Proc. Natl. Acad. Sci. USA 1996, 93, 14176–14181. [Google Scholar] [CrossRef] [PubMed]
- Kraft, T.W.; Allen, D.; Petters, R.M.; Hao, Y.; Peng, Y.-W.; Wong, F. Altered Light Responses of Single Rod Photoreceptors in Transgenic Pigs Expressing P347L or P347S Rhodopsin. Mol. Vis. 2005, 11, 1246–1256. [Google Scholar] [PubMed]
- Mussolino, C.; Sanges, D.; Marrocco, E.; Bonetti, C.; Di Vicino, U.; Marigo, V.; Auricchio, A.; Meroni, G.; Surace, E.M. Zinc-Finger-Based Transcriptional Repression of Rhodopsin in a Model of Dominant Retinitis Pigmentosa. EMBO Mol. Med. 2011, 3, 118–128. [Google Scholar] [CrossRef]
- Chadderton, N.; Millington-Ward, S.; Palfi, A.; O’Reilly, M.; Tuohy, G.; Humphries, M.M.; Li, T.; Humphries, P.; Kenna, P.F.; Farrar, G.J. Improved Retinal Function in a Mouse Model of Dominant Retinitis Pigmentosa Following AAV-Delivered Gene Therapy. Mol. Ther. 2009, 17, 593–599. [Google Scholar] [CrossRef]
- Palfi, A.; Millington-Ward, S.; Chadderton, N.; O’Reilly, M.; Goldmann, T.; Humphries, M.M.; Li, T.; Wolfrum, U.; Humphries, P.; Kenna, P.F.; et al. Adeno-Associated Virus-Mediated Rhodopsin Replacement Provides Therapeutic Benefit in Mice with a Targeted Disruption of the Rhodopsin Gene. Hum. Gene Ther. 2010, 21, 311–323. [Google Scholar] [CrossRef]
- Millington-Ward, S.; Chadderton, N.; O’Reilly, M.; Palfi, A.; Goldmann, T.; Kilty, C.; Humphries, M.; Wolfrum, U.; Bennett, J.; Humphries, P.; et al. Suppression and Replacement Gene Therapy for Autosomal Dominant Disease in a Murine Model of Dominant Retinitis Pigmentosa. Mol. Ther. 2011, 19, 642–649. [Google Scholar] [CrossRef]
- Moore, S.M.; Skowronska-Krawczyk, D.; Chao, D.L. Targeting of the NRL Pathway as a Therapeutic Strategy to Treat Retinitis Pigmentosa. J. Clin. Med. 2020, 9, 2224. [Google Scholar] [CrossRef] [PubMed]
- Conte, I.; Hadfield, K.D.; Barbato, S.; Carrella, S.; Pizzo, M.; Bhat, R.S.; Carissimo, A.; Karali, M.; Porter, L.F.; Urquhart, J.; et al. MiR-204 Is Responsible for Inherited Retinal Dystrophy Associated with Ocular Coloboma. Proc. Natl. Acad. Sci. USA 2015, 112, E3236–E3245. [Google Scholar] [CrossRef] [PubMed]
- Piano, I.; D’Antongiovanni, V.; Novelli, E.; Biagioni, M.; Dei Cas, M.; Paroni, R.C.; Ghidoni, R.; Strettoi, E.; Gargini, C. Myriocin Effect on Tvrm4 Retina, an Autosomal Dominant Pattern of Retinitis Pigmentosa. Front. Neurosci. 2020, 14, 372. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Xiong, S.; Xia, X. Retinitis Pigmentosa-Associated Rhodopsin Mutant T17M Induces Endoplasmic Reticulum (ER) Stress and Sensitizes Cells to ER Stress-Induced Cell Death. Mol. Med. Rep. 2014, 9, 1737–1742. [Google Scholar] [CrossRef]
- Bhootada, Y.; Gully, C.; Gorbatyuk, M.S. Controlling PERK-ATF4-CHOP Branch of the UPR Is the Key to Reverse Retinal Degeneration of T17M Retina. Investig. Ophthalmol. Vis. Sci. 2015, 56, 5409. [Google Scholar]
- Yang, C.; Georgiou, M.; Atkinson, R.; Collin, J.; Al-Aama, J.; Nagaraja-Grellscheid, S.; Johnson, C.; Ali, R.; Armstrong, L.; Mozaffari-Jovin, S.; et al. Pre-MRNA Processing Factors and Retinitis Pigmentosa: RNA Splicing and Beyond. Front. Cell Dev. Biol. 2021, 9, 700276. [Google Scholar] [CrossRef]
- Bowne, S. Mutations in the RP1 Gene Causing Autosomal Dominant Retinitis Pigmentosa. Hum. Mol. Genet. 1999, 8, 2121–2128. [Google Scholar] [CrossRef]
- Liu, Q.; Zhou, J.; Daiger, S.P.; Farber, D.B.; Heckenlively, J.R.; Smith, J.E.; Sullivan, L.S.; Zuo, J.; Milam, A.H.; Pierce, E.A. Identification and Subcellular Localization of the RP1 Protein in Human and Mouse Photoreceptors. Investig. Ophthalmol. Vis. Sci. 2002, 43, 22–32. [Google Scholar]
- Liu, Q.; Saveliev, A.; Hu, J.; Cao, H.; Auricchio, A.; Pierce, E.A. The Degeneration of Photoreceptor Cells in the Rp1 Form of Retinitis Pigmentosa Is Caused by a Dominant Mechanism. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2285. [Google Scholar]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Li, W.; McCulley, C.; Reeves, P.J.; Cheetham, M.E. The Molecular and Cellular Basis of Rhodopsin Retinitis Pigmentosa Reveals Potential Strategies for Therapy. Prog. Retin. Eye Res. 2018, 62, 1–23. [Google Scholar] [CrossRef]
- Saqib, M.A.N.; Nikopoulos, K.; Ullah, E.; Sher Khan, F.; Iqbal, J.; Bibi, R.; Jarral, A.; Sajid, S.; Nishiguchi, K.M.; Venturini, G.; et al. Homozygosity Mapping Reveals Novel and Known Mutations in Pakistani Families with Inherited Retinal Dystrophies. Sci. Rep. 2015, 5, 9965. [Google Scholar] [CrossRef] [PubMed]
- Van Schil, K.; Karlstetter, M.; Aslanidis, A.; Dannhausen, K.; Azam, M.; Qamar, R.; Leroy, B.P.; Depasse, F.; Langmann, T.; De Baere, E. Autosomal Recessive Retinitis Pigmentosa with Homozygous Rhodopsin Mutation E150K and Non-Coding Cis-Regulatory Variants in CRX-Binding Regions of SAMD7. Sci. Rep. 2016, 6, 21307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Kolesnikov, A.V.; Jastrzebska, B.; Mustafi, D.; Sawada, O.; Maeda, T.; Genoud, C.; Engel, A.; Kefalov, V.J.; Palczewski, K. Autosomal Recessive Retinitis Pigmentosa E150K Opsin Mice Exhibit Photoreceptor Disorganization. J. Clin. Investig. 2013, 123, 121–137. [Google Scholar] [CrossRef] [PubMed]
- Collin, R.W.J.; van den Born, L.I.; Klevering, B.J.; de Castro-Miró, M.; Littink, K.W.; Arimadyo, K.; Azam, M.; Yazar, V.; Zonneveld, M.N.; Paun, C.C.; et al. High-Resolution Homozygosity Mapping Is a Powerful Tool to Detect Novel Mutations Causative of Autosomal Recessive RP in the Dutch Population. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2227. [Google Scholar] [CrossRef]
- Nassisi, M.; Smirnov, V.M.; Solis Hernandez, C.; Mohand-Saïd, S.; Condroyer, C.; Antonio, A.; Kühlewein, L.; Kempf, M.; Kohl, S.; Wissinger, B.; et al. CNGB1-related Rod-cone Dystrophy: A Mutation Review and Update. Hum. Mutat. 2021, 42, 641–666. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Cideciyan, A.V.; Huang, W.C.; Sumaroka, A.; Roman, A.J.; Schwartz, S.B.; Luo, X.; Sheplock, R.; Dauber, J.M.; Swider, M.; et al. TULP1 Mutations Causing Early-Onset Retinal Degeneration: Preserved but Insensitive Macular Cones. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5354. [Google Scholar] [CrossRef]
- Chhetri, J.; Jacobson, G.; Gueven, N. Zebrafish—On the Move towards Ophthalmological Research. Eye 2014, 28, 367–380. [Google Scholar] [CrossRef]
- Martinez-Fernandez De La Camara, C.; Nanda, A.; Salvetti, A.P.; Fischer, M.D.; MacLaren, R.E. Gene Therapy for the Treatment of X-Linked Retinitis Pigmentosa. Expert Opin. Orphan Drugs 2018, 6, 167–177. [Google Scholar] [CrossRef]
- Koyanagi, Y.; Akiyama, M.; Nishiguchi, K.M.; Momozawa, Y.; Kamatani, Y.; Takata, S.; Inai, C.; Iwasaki, Y.; Kumano, M.; Murakami, Y.; et al. Genetic Characteristics of Retinitis Pigmentosa in 1204 Japanese Patients. J. Med. Genet. 2019, 56, 662–670. [Google Scholar] [CrossRef]
- González-del Pozo, M.; Fernández-Suárez, E.; Martín-Sánchez, M.; Bravo-Gil, N.; Méndez-Vidal, C.; Rodríguez-de la Rúa, E.; Borrego, S.; Antiñolo, G. Unmasking Retinitis Pigmentosa Complex Cases by a Whole Genome Sequencing Algorithm Based on Open-Access Tools: Hidden Recessive Inheritance and Potential Oligogenic Variants. J. Transl. Med. 2020, 18, 73. [Google Scholar] [CrossRef]
- Birtel, J.; Gliem, M.; Mangold, E.; Müller, P.L.; Holz, F.G.; Neuhaus, C.; Lenzner, S.; Zahnleiter, D.; Betz, C.; Eisenberger, T.; et al. Next-Generation Sequencing Identifies Unexpected Genotype-Phenotype Correlations in Patients with Retinitis Pigmentosa. PLoS ONE 2018, 13, e0207958. [Google Scholar] [CrossRef] [PubMed]
- Millo, T.; Rivera, A.; Obolensky, A.; Marks-Ohana, D.; Xu, M.; Li, Y.; Wilhelm, E.; Gopalakrishnan, P.; Gross, M.; Rosin, B.; et al. Identification of Autosomal Recessive Novel Genes and Retinal Phenotypes in Members of the Solute Carrier (SLC) Superfamily. Genet. Med. 2022, 24, 1523–1535. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.-B.; Huang, X.-F.; Lv, J.-N.; Xiang, L.; Li, D.-Q.; Chen, J.; Huang, C.; Wu, J.; Lu, F.; Qu, J. SLC7A14 Linked to Autosomal Recessive Retinitis Pigmentosa. Nat. Commun. 2014, 5, 3517. [Google Scholar] [CrossRef] [PubMed]
- Branham, K.; Matsui, H.; Biswas, P.; Guru, A.A.; Hicks, M.; Suk, J.J.; Li, H.; Jakubosky, D.; Long, T.; Telenti, A.; et al. Establishing the Involvement of the Novel Gene AGBL5 in Retinitis Pigmentosa by Whole Genome Sequencing. Physiol. Genom. 2016, 48, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Jindal, N.; Banik, A.; Prabhakar, S.; Vaiphie, K.; Anand, A. Alteration of Neurotrophic Factors After Transplantation of Bone Marrow Derived Lin-ve Stem Cell in NMDA-Induced Mouse Model of Retinal Degeneration. J. Cell. Biochem. 2017, 118, 1699–1711. [Google Scholar] [CrossRef]
- Kashani, A.H.; Uang, J.; Mert, M.; Rahhal, F.; Chan, C.; Avery, R.L.; Dugel, P.; Chen, S.; Lebkowski, J.; Clegg, D.O.; et al. Surgical Method for Implantation of a Biosynthetic Retinal Pigment Epithelium Monolayer for Geographic Atrophy: Experience from a Phase 1/2a Study. Ophthalmol. Retin. 2020, 4, 264–273. [Google Scholar] [CrossRef]
- Mandai, M.; Fujii, M.; Hashiguchi, T.; Sunagawa, G.A.; Ito, S.; Sun, J.; Kaneko, J.; Sho, J.; Yamada, C.; Takahashi, M. IPSC-Derived Retina Transplants Improve Vision in Rd1 End-Stage Retinal-Degeneration Mice. Stem Cell Rep. 2017, 8, 69–83. [Google Scholar] [CrossRef]
- Salas, A.; Duarri, A.; Fontrodona, L.; Ramírez, D.M.; Badia, A.; Isla-Magrané, H.; Ferreira-de-Souza, B.; Zapata, M.Á.; Raya, Á.; Veiga, A.; et al. Cell Therapy with HiPSC-Derived RPE Cells and RPCs Prevents Visual Function Loss in a Rat Model of Retinal Degeneration. Mol. Ther.-Methods Clin. Dev. 2021, 20, 688–702. [Google Scholar] [CrossRef]
- Surendran, H.; Nandakumar, S.; Reddy, K.V.B.; Stoddard, J.; Mohan, K.V.; Upadhyay, P.K.; McGill, T.J.; Pal, R. Transplantation of Retinal Pigment Epithelium and Photoreceptors Generated Concomitantly via Small Molecule-Mediated Differentiation Rescues Visual Function in Rodent Models of Retinal Degeneration. Stem Cell Res. Ther. 2021, 12, 70. [Google Scholar] [CrossRef]
- Lin, B.; McLelland, B.T.; Aramant, R.B.; Thomas, B.B.; Nistor, G.; Keirstead, H.S.; Seiler, M.J. Retina Organoid Transplants Develop Photoreceptors and Improve Visual Function in RCS Rats With RPE Dysfunction. Investig. Ophthalmol. Vis. Sci. 2020, 61, 34. [Google Scholar] [CrossRef]
- Duarri, A.; Rodríguez-Bocanegra, E.; Martínez-Navarrete, G.; Biarnés, M.; García, M.; Ferraro, L.L.; Kuebler, B.; Aran, B.; Izquierdo, E.; Aguilera-Xiol, E.; et al. Transplantation of Human Induced Pluripotent Stem Cell-Derived Retinal Pigment Epithelium in a Swine Model of Geographic Atrophy. Int. J. Mol. Sci. 2021, 22, 10497. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.B.; Lin, B.; Martinez-Camarillo, J.C.; Zhu, D.; McLelland, B.T.; Nistor, G.; Keirstead, H.S.; Humayun, M.S.; Seiler, M.J. Co-Grafts of Human Embryonic Stem Cell Derived Retina Organoids and Retinal Pigment Epithelium for Retinal Reconstruction in Immunodeficient Retinal Degenerate Royal College of Surgeons Rats. Front. Neurosci. 2021, 15, 752958. [Google Scholar] [CrossRef] [PubMed]
- Ferroni, L.; Gardin, C.; Tocco, I.; Epis, R.; Casadei, A.; Vindigni, V.; Mucci, G.; Zavan, B. Potential for Neural Differentiation of Mesenchymal Stem Cells. Adv. Biochem. Eng. Biotechnol. 2013, 129, 89–115. [Google Scholar] [CrossRef]
- Bassi, Ê.J.; de Almeida, D.C.; Moraes-Vieira, P.M.M.; Câmara, N.O.S. Exploring the Role of Soluble Factors Associated with Immune Regulatory Properties of Mesenchymal Stem Cells. Stem Cell Rev. Rep. 2012, 8, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Soleymaninejadian, E.; Pramanik, K.; Samadian, E. Immunomodulatory Properties of Mesenchymal Stem Cells: Cytokines and Factors. Am. J. Reprod. Immunol. 2012, 67, 1–8. [Google Scholar] [CrossRef]
- Ding, S.L.S.; Kumar, S.; Mok, P.L. Cellular Reparative Mechanisms of Mesenchymal Stem Cells for Retinal Diseases. Int. J. Mol. Sci. 2017, 18, 1406. [Google Scholar] [CrossRef]
- Brown, C.; Agosta, P.; McKee, C.; Walker, K.; Mazzella, M.; Alamri, A.; Svinarich, D.; Chaudhry, G.R. Human Primitive Mesenchymal Stem Cell-Derived Retinal Progenitor Cells Improved Neuroprotection, Neurogenesis, and Vision in Rd12 Mouse Model of Retinitis Pigmentosa. Stem Cell Res. Ther. 2022, 13, 148. [Google Scholar] [CrossRef]
- Kolomeyer, A.M.; Zarbin, M.A. Trophic Factors in the Pathogenesis and Therapy for Retinal Degenerative Diseases. Surv. Ophthalmol. 2014, 59, 134–165. [Google Scholar] [CrossRef]
- Talcott, K.E.; Ratnam, K.; Sundquist, S.M.; Lucero, A.S.; Lujan, B.J.; Tao, W.; Porco, T.C.; Roorda, A.; Duncan, J.L. Longitudinal Study of Cone Photoreceptors during Retinal Degeneration and in Response to Ciliary Neurotrophic Factor Treatment. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2219. [Google Scholar] [CrossRef]
- Moisseiev, E.; Smit-McBride, Z.; Oltjen, S.; Zhang, P.; Zawadzki, R.J.; Motta, M.; Murphy, C.J.; Cary, W.; Annett, G.; Nolta, J.A.; et al. Intravitreal Administration of Human Bone Marrow CD34+ Stem Cells in a Murine Model of Retinal Degeneration. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4125. [Google Scholar] [CrossRef]
- Soleimannejad, M.; Ebrahimi-Barough, S.; Nadri, S.; Riazi-Esfahani, M.; Soleimani, M.; Tavangar, S.M.; Ai, J. Retina Tissue Engineering by Conjunctiva Mesenchymal Stem Cells Encapsulated in Fibrin Gel: Hypotheses on Novel Approach to Retinal Diseases Treatment. Med. Hypotheses 2017, 101, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, P.; Zhao, G.; He, S.; Xu, D.; Jiang, W.; Peng, Q.; Li, Z.; Xie, Z.; Zhang, H.; et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles Protect Retina in a Mouse Model of Retinitis Pigmentosa by Anti-Inflammation through MiR-146a-Nr4a3 Axis. Stem Cell Res. Ther. 2022, 13, 394. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Shen, H.; Yuan, S.; Lu, G.; Zhang, Y.; Wang, H.; Zhao, Y.; Sun, X.; Liu, Q. Combined Transplantation With Human Mesenchymal Stem Cells Improves Retinal Rescue Effect of Human Fetal RPE Cells in Retinal Degeneration Mouse Model. Investig. Ophthalmol. Vis. Sci. 2020, 61, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.-K.; Tosi, J.; Kasanuki, J.M.; Chou, C.L.; Kong, J.; Parmalee, N.; Wert, K.J.; Allikmets, R.; Lai, C.-C.; Chien, C.-L.; et al. Transplantation of Reprogrammed Embryonic Stem Cells Improves Visual Function in a Mouse Model for Retinitis Pigmentosa. Transplantation 2010, 89, 911–919. [Google Scholar] [CrossRef]
- Klassen, H.J.; Ng, T.F.; Kurimoto, Y.; Kirov, I.; Shatos, M.; Coffey, P.; Young, M.J. Multipotent Retinal Progenitors Express Developmental Markers, Differentiate into Retinal Neurons, and Preserve Light-Mediated Behavior. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4167. [Google Scholar] [CrossRef]
- Qiu, G.; Seiler, M.J.; Thomas, B.B.; Wu, K.; Radosevich, M.; Sadda, S.R. Revisiting Nestin Expression in Retinal Progenitor Cells in Vitro and after Transplantation in Vivo. Exp. Eye Res. 2007, 84, 1047–1059. [Google Scholar] [CrossRef]
- MacLaren, R.E.; Pearson, R.A.; MacNeil, A.; Douglas, R.H.; Salt, T.E.; Akimoto, M.; Swaroop, A.; Sowden, J.C.; Ali, R.R. Retinal Repair by Transplantation of Photoreceptor Precursors. Nature 2006, 444, 203–207. [Google Scholar] [CrossRef]
- He, X.-Y.; Zhao, C.-J.; Xu, H.; Chen, K.; Bian, B.-S.-J.; Gong, Y.; Weng, C.-H.; Zeng, Y.-X.; Fu, Y.; Liu, Y.; et al. Synaptic Repair and Vision Restoration in Advanced Degenerating Eyes by Transplantation of Retinal Progenitor Cells. Stem Cell Rep. 2021, 16, 1805–1817. [Google Scholar] [CrossRef]
- Stern, J.H.; Tian, Y.; Funderburgh, J.; Pellegrini, G.; Zhang, K.; Goldberg, J.L.; Ali, R.R.; Young, M.; Xie, Y.; Temple, S. Regenerating Eye Tissues to Preserve and Restore Vision. Cell Stem Cell 2018, 22, 834–849. [Google Scholar] [CrossRef]
- Ellingford, J.M.; Barton, S.; Bhaskar, S.; O’Sullivan, J.; Williams, S.G.; Lamb, J.A.; Panda, B.; Sergouniotis, P.I.; Gillespie, R.L.; Daiger, S.P.; et al. Molecular Findings from 537 Individuals with Inherited Retinal Disease. J. Med. Genet. 2016, 53, 761–767. [Google Scholar] [CrossRef]
- Bi, A.; Cui, J.; Ma, Y.-P.; Olshevskaya, E.; Pu, M.; Dizhoor, A.M.; Pan, Z.-H. Ectopic Expression of a Microbial-Type Rhodopsin Restores Visual Responses in Mice with Photoreceptor Degeneration. Neuron 2006, 50, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Tomita, H.; Sugano, E.; Fukazawa, Y.; Isago, H.; Sugiyama, Y.; Hiroi, T.; Ishizuka, T.; Mushiake, H.; Kato, M.; Hirabayashi, M.; et al. Visual Properties of Transgenic Rats Harboring the Channelrhodopsin-2 Gene Regulated by the Thy-1.2 Promoter. PLoS ONE 2009, 4, e7679. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.-J.; Sahel, J.-A.; Duebel, J.; Herlitze, S.; Dalkara, D. Opsins for Vision Restoration. Biochem. Biophys. Res. Commun. 2020, 527, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Vierock, J.; Yizhar, O.; Fenno, L.E.; Tsunoda, S.; Kianianmomeni, A.; Prigge, M.; Berndt, A.; Cushman, J.; Polle, J.; et al. The Microbial Opsin Family of Optogenetic Tools. Cell 2011, 147, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Deisseroth, K. Optogenetics: 10 Years of Microbial Opsins in Neuroscience. Nat. Neurosci. 2015, 18, 1213–1225. [Google Scholar] [CrossRef]
- Gradinaru, V.; Zhang, F.; Ramakrishnan, C.; Mattis, J.; Prakash, R.; Diester, I.; Goshen, I.; Thompson, K.R.; Deisseroth, K. Molecular and Cellular Approaches for Diversifying and Extending Optogenetics. Cell 2010, 141, 154–165. [Google Scholar] [CrossRef]
- Bedbrook, C.N.; Yang, K.K.; Rice, A.J.; Gradinaru, V.; Arnold, F.H. Machine Learning to Design Integral Membrane Channelrhodopsins for Efficient Eukaryotic Expression and Plasma Membrane Localization. PLoS Comput. Biol. 2017, 13, e1005786. [Google Scholar] [CrossRef]
- Garita-Hernandez, M.; Guibbal, L.; Toualbi, L.; Routet, F.; Chaffiol, A.; Winckler, C.; Harinquet, M.; Robert, C.; Fouquet, S.; Bellow, S.; et al. Optogenetic Light Sensors in Human Retinal Organoids. Front. Neurosci. 2018, 12, 789. [Google Scholar] [CrossRef]
- Gaub, B.M.; Berry, M.H.; Holt, A.E.; Reiner, A.; Kienzler, M.A.; Dolgova, N.; Nikonov, S.; Aguirre, G.D.; Beltran, W.A.; Flannery, J.G.; et al. Restoration of Visual Function by Expression of a Light-Gated Mammalian Ion Channel in Retinal Ganglion Cells or ON-Bipolar Cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5574–E5583. [Google Scholar] [CrossRef]
- Ameline, B.; Tshilenge, K.-T.; Weber, M.; Biget, M.; Libeau, L.; Caplette, R.; Mendes-Madeira, A.; Provost, N.; Guihal, C.; Picaud, S.; et al. Long-Term Expression of Melanopsin and Channelrhodopsin Causes No Gross Alterations in the Dystrophic Dog Retina. Gene Ther. 2017, 24, 735–741. [Google Scholar] [CrossRef]
- Nikonov, S.; Aravand, P.; Lyubarsky, A.; Nikonov, R.; Luo, A.J.; Wei, Z.; Maguire, A.M.; Phelps, N.T.; Shpylchak, I.; Willett, K.; et al. Restoration of Vision and Retinal Responses After Adeno-Associated Virus–Mediated Optogenetic Therapy in Blind Dogs. Transl. Vis. Sci. Technol. 2022, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Chaffiol, A.; Caplette, R.; Jaillard, C.; Brazhnikova, E.; Desrosiers, M.; Dubus, E.; Duhamel, L.; Macé, E.; Marre, O.; Benoit, P.; et al. A New Promoter Allows Optogenetic Vision Restoration with Enhanced Sensitivity in Macaque Retina. Mol. Ther. 2017, 25, 2546–2560. [Google Scholar] [CrossRef] [PubMed]
- Ganjawala, T.H.; Lu, Q.; Fenner, M.D.; Abrams, G.W.; Pan, Z.-H. Improved CoChR Variants Restore Visual Acuity and Contrast Sensitivity in a Mouse Model of Blindness under Ambient Light Conditions. Mol. Ther. 2019, 27, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Tabata, K.; Sugano, E.; Hatakeyama, A.; Watanabe, Y.; Suzuki, T.; Ozaki, T.; Fukuda, T.; Tomita, H. Phototoxicities Caused by Continuous Light Exposure Were Not Induced in Retinal Ganglion Cells Transduced by an Optogenetic Gene. Int. J. Mol. Sci. 2021, 22, 6732. [Google Scholar] [CrossRef]
- Watanabe, Y.; Sugano, E.; Tabata, K.; Hatakeyama, A.; Sakajiri, T.; Fukuda, T.; Ozaki, T.; Suzuki, T.; Sayama, T.; Tomita, H. Development of an Optogenetic Gene Sensitive to Daylight and Its Implications in Vision Restoration. Npj Regen. Med. 2021, 6, 64. [Google Scholar] [CrossRef]
- Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein-Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef]
- Katada, Y.; Yoshida, K.; Serizawa, N.; Kobayashi, K.; Neghisi, K.; Okano, H.; Kandori, H.; Tsubota, K.; Kurihara, T. High-Sensitivity Vision Restoration via Ectopic Expression of Chimeric Rhodopsin in Mice. bioRxiv 2020. [CrossRef]
- Buch, P.; MacLaren, R.; Ali, R. Neuroprotective Gene Therapy for the Treatment of Inherited Retinal Degeneration. Curr. Gene Ther. 2007, 7, 434–445. [Google Scholar] [CrossRef]
- Delplace, V.; Ortin-Martinez, A.; Tsai, E.L.S.; Amin, A.N.; Wallace, V.; Shoichet, M.S. Controlled Release Strategy Designed for Intravitreal Protein Delivery to the Retina. J. Control. Release 2019, 293, 10–20. [Google Scholar] [CrossRef]
- Gupta, V.; You, Y.; Gupta, V.; Klistorner, A.; Graham, S. TrkB Receptor Signalling: Implications in Neurodegenerative, Psychiatric and Proliferative Disorders. Int. J. Mol. Sci. 2013, 14, 10122–10142. [Google Scholar] [CrossRef]
- Pardue, M.T.; Allen, R.S. Neuroprotective Strategies for Retinal Disease. Prog. Retin. Eye Res. 2018, 65, 50–76. [Google Scholar] [CrossRef] [PubMed]
- Wen, R.; Tao, W.; Li, Y.; Sieving, P.A. CNTF and Retina. Prog. Retin. Eye Res. 2012, 31, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, M.; Alizadeh, E.; Saei Arezoumand, K.; Fallahi Motlagh, B.; Zarghami, N. Ciliary Neurotrophic Factor (CNTF) Delivery to Retina: An Overview of Current Research Advancements. Artif. Cells Nanomed. Biotechnol. 2017, 46, 1694–1707. [Google Scholar] [CrossRef]
- Chew, E.Y.; Clemons, T.E.; Jaffe, G.J.; Johnson, C.A.; Farsiu, S.; Lad, E.M.; Guymer, R.; Rosenfeld, P.; Hubschman, J.-P.; Constable, I.; et al. Effect of Ciliary Neurotrophic Factor on Retinal Neurodegeneration in Patients with Macular Telangiectasia Type 2. Ophthalmology 2019, 126, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Bielmeier, C.B.; Roth, S.; Schmitt, S.I.; Boneva, S.K.; Schlecht, A.; Vallon, M.; Tamm, E.R.; Ergün, S.; Neueder, A.; Braunger, B.M. Transcriptional Profiling Identifies Upregulation of Neuroprotective Pathways in Retinitis Pigmentosa. Int. J. Mol. Sci. 2021, 22, 6307. [Google Scholar] [CrossRef] [PubMed]
- Bramall, A.N.; Szego, M.J.; Pacione, L.R.; Chang, I.; Diez, E.; D’Orleans-Juste, P.; Stewart, D.J.; Hauswirth, W.W.; Yanagisawa, M.; McInnes, R.R. Endothelin-2-Mediated Protection of Mutant Photoreceptors in Inherited Photoreceptor Degeneration. PLoS ONE 2013, 8, e58023. [Google Scholar] [CrossRef]
- Froger, N.; Matonti, F.; Roubeix, C.; Forster, V.; Ivkovic, I.; Brunel, N.; Baudouin, C.; Sahel, J.-A.; Picaud, S. VEGF Is an Autocrine/Paracrine Neuroprotective Factor for Injured Retinal Ganglion Neurons. Sci. Rep. 2020, 10, 12409. [Google Scholar] [CrossRef]
- Tao, W.; Wen, R.; Goddard, M.B.; Sherman, S.D.; O’Rourke, P.J.; Stabila, P.F.; Bell, W.J.; Dean, B.J.; Kauper, K.A.; Budz, V.A.; et al. Encapsulated Cell-Based Delivery of CNTF Reduces Photoreceptor Degeneration in Animal Models of Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3292–3298. [Google Scholar]
- Yang, J.-Y.; Lu, B.; Feng, Q.; Alfaro, J.S.; Chen, P.-H.; Loscalzo, J.; Wei, W.-B.; Zhang, Y.-Y.; Lu, S.-J.; Wang, S. Retinal Protection by Sustained Nanoparticle Delivery of Oncostatin M and Ciliary Neurotrophic Factor Into Rodent Models of Retinal Degeneration. Transl. Vis. Sci. Technol. 2021, 10, 6. [Google Scholar] [CrossRef]
- Fernández-Sánchez, L.; Lax, P.; Pinilla, I.; Martín-Nieto, J.; Cuenca, N. Tauroursodeoxycholic Acid Prevents Retinal Degeneration in Transgenic P23H Rats. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4998. [Google Scholar] [CrossRef]
- Olivares-González, L.; Velasco, S.; Campillo, I.; Rodrigo, R. Retinal Inflammation, Cell Death and Inherited Retinal Dystrophies. Int. J. Mol. Sci. 2021, 22, 2096. [Google Scholar] [CrossRef] [PubMed]
- Omura, T.; Asari, M.; Yamamoto, J.; Oka, K.; Hoshina, C.; Maseda, C.; Awaya, T.; Tasaki, Y.; Shiono, H.; Yonezawa, A.; et al. Sodium Tauroursodeoxycholate Prevents Paraquat-Induced Cell Death by Suppressing Endoplasmic Reticulum Stress Responses in Human Lung Epithelial A549 Cells. Biochem. Biophys. Res. Commun. 2013, 432, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Baehr, W.; Fu, Y. Chemical Chaperone TUDCA Preserves Cone Photoreceptors in a Mouse Model of Leber Congenital Amaurosis. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3349. [Google Scholar] [CrossRef]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664. [Google Scholar] [CrossRef]
- Eltony, S.A.; Mohaseb, H.S.; Ahmed, A.A.; Sayed, M.M. Can Metformin Modulate the Retinal Degenerative Changes in a Rat Model of Retinitis Pigmentosa? Tissue Cell 2022, 76, 101786. [Google Scholar] [CrossRef] [PubMed]
- Kilicarslan, I.; Zanetti, L.; Novelli, E.; Schwarzer, C.; Strettoi, E.; Koschak, A. Knockout of CaV1.3 L-Type Calcium Channels in a Mouse Model of Retinitis Pigmentosa. Sci. Rep. 2021, 11, 15146. [Google Scholar] [CrossRef]
- Aloe, L.; Rocco, M.L.; Bianchi, P.; Manni, L. Nerve Growth Factor: From the Early Discoveries to the Potential Clinical Use. J. Transl. Med. 2012, 10, 239. [Google Scholar] [CrossRef]
- Li, B.; Ning, B.; Yang, F.; Guo, C. Nerve Growth Factor Promotes Retinal Neurovascular Unit Repair: A Review. Curr. Eye Res. 2022, 47, 1095–1105. [Google Scholar] [CrossRef]
- Wiatrak, B.; Kubis-Kubiak, A.; Piwowar, A.; Barg, E. PC12 Cell Line: Cell Types, Coating of Culture Vessels, Differentiation and Other Culture Conditions. Cells 2020, 9, 958. [Google Scholar] [CrossRef]
- Rocco, M.L.; Balzamino, B.O.; Petrocchi Passeri, P.; Micera, A.; Aloe, L. Effect of Purified Murine NGF on Isolated Photoreceptors of a Rodent Developing Retinitis Pigmentosa. PLoS ONE 2015, 10, e0124810. [Google Scholar] [CrossRef]
- Rocco, M.L.; Calzà, L.; Aloe, L. NGF and Retinitis Pigmentosa: Structural and Molecular Studies. In Recent Advances in NGF and Related Molecules; Calzà, L., Aloe, L., Giardino, L., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2021; Volume 1331, pp. 255–263. ISBN 978-3-030-74045-0. [Google Scholar]
- Cocchiaro, P.; Di Donato, V.; Rubbini, D.; Mastropasqua, R.; Allegretti, M.; Mantelli, F.; Aramini, A.; Brandolini, L. Intravitreal Administration of RhNGF Enhances Regenerative Processes in a Zebrafish Model of Retinal Degeneration. Front. Pharmacol. 2022, 13, 822359. [Google Scholar] [CrossRef]
- Bielmeier, C.B.; Schmitt, S.I.; Kleefeldt, N.; Boneva, S.K.; Schlecht, A.; Vallon, M.; Tamm, E.R.; Hillenkamp, J.; Ergün, S.; Neueder, A.; et al. Deficiency in Retinal TGFβ Signaling Aggravates Neurodegeneration by Modulating Pro-Apoptotic and MAP Kinase Pathways. Int. J. Mol. Sci. 2022, 23, 2626. [Google Scholar] [CrossRef]
- Eastlake, K.; Lamb, W.D.B.; Luis, J.; Khaw, P.T.; Jayaram, H.; Limb, G.A. Prospects for the Application of Müller Glia and Their Derivatives in Retinal Regenerative Therapies. Prog. Retin. Eye Res. 2021, 85, 100970. [Google Scholar] [CrossRef]
- Karademir, D.; Todorova, V.; Ebner, L.J.A.; Samardzija, M.; Grimm, C. Single-Cell RNA Sequencing of the Retina in a Model of Retinitis Pigmentosa Reveals Early Responses to Degeneration in Rods and Cones. BMC Biol. 2022, 20, 86. [Google Scholar] [CrossRef]
- Rasmussen, M.; Zhou, J.; Schwede, F.; Ekström, P. Enhanced CGMP Interactor Rap Guanine Exchange Factor 4 (EPAC2) Expression and Activity in Degenerating Photoreceptors: A Neuroprotective Response? Int. J. Mol. Sci. 2022, 23, 4619. [Google Scholar] [CrossRef]
- Ozawa, Y.; Toda, E.; Homma, K.; Osada, H.; Nagai, N.; Tsubota, K.; Okano, H. Effects of Epigenetic Modification of PGC-1α by a Chemical Chaperon on Mitochondria Biogenesis and Visual Function in Retinitis Pigmentosa. Cells 2022, 11, 1497. [Google Scholar] [CrossRef]
- Campochiaro, P.A.; Mir, T.A. The Mechanism of Cone Cell Death in Retinitis Pigmentosa. Prog. Retin. Eye Res. 2018, 62, 24–37. [Google Scholar] [CrossRef]
- Komeima, K.; Rogers, B.S.; Lu, L.; Campochiaro, P.A. Antioxidants Reduce Cone Cell Death in a Model of Retinitis Pigmentosa. Proc. Natl. Acad. Sci. USA 2006, 103, 11300–11305. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Usui, S.; Zafar, A.; Oveson, B.C.; Jo, Y.-J.; Lu, L.; Masoudi, S.; Campochiaro, P.A. N-Acetylcysteine Promotes Long-Term Survival of Cones in a Model of Retinitis Pigmentosa. J. Cell. Physiol. 2011, 226, 1843–1849. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Ikeda, Y.; Notomi, S.; Ishikawa, K.; Murakami, Y.; Hisatomi, T.; Enaida, H.; Ishibashi, T. Laboratory Evidence of Sustained Chronic Inflammatory Reaction in Retinitis Pigmentosa. Ophthalmology 2013, 120, e5–e12. [Google Scholar] [CrossRef] [PubMed]
- Johansson, I.; Monsen, V.T.; Pettersen, K.; Mildenberger, J.; Misund, K.; Kaarniranta, K.; Schønberg, S.; Bjørkøy, G. The Marine N-3 PUFA DHA Evokes Cytoprotection against Oxidative Stress and Protein Misfolding by Inducing Autophagy and NFE2L2 in Human Retinal Pigment Epithelial Cells. Autophagy 2015, 11, 1636–1651. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.S.; Calandria, J.M.; Gordon, W.C.; Jun, B.; Zhou, Y.; Gelfman, C.M.; Li, S.; Jin, M.; Knott, E.J.; Chang, B.; et al. Adiponectin Receptor 1 Conserves Docosahexaenoic Acid and Promotes Photoreceptor Cell Survival. Nat. Commun. 2015, 6, 6228. [Google Scholar] [CrossRef] [PubMed]
- Morshedian, A.; Kaylor, J.J.; Ng, S.Y.; Tsan, A.; Frederiksen, R.; Xu, T.; Yuan, L.; Sampath, A.P.; Radu, R.A.; Fain, G.L.; et al. Light-Driven Regeneration of Cone Visual Pigments through a Mechanism Involving RGR Opsin in Müller Glial Cells. Neuron 2019, 102, 1172–1183.e5. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, T.; Nakazawa, M.; Kudo, T.; Hirano, S.; Suzuki, K.; Ishiguro, S. Protection of Cone Photoreceptor M-Opsin Degradation with 9-Cis-β-Carotene-Rich Alga Dunaliella Bardawil in Rpe65(−/−) Mouse Retinal Explant Culture. Curr. Eye Res. 2014, 39, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Pawlyk, B.S.; Li, T.; Scimeca, M.S.; Sandberg, M.A.; Berson, E.L. Absence of Photoreceptor Rescue with D-Cis-Diltiazem in the Rd Mouse. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1912–1915. [Google Scholar]
- Yang, J.; Weimer, R.M.; Kallop, D.; Olsen, O.; Wu, Z.; Renier, N.; Uryu, K.; Tessier-Lavigne, M. Regulation of Axon Degeneration after Injury and in Development by the Endogenous Calpain Inhibitor Calpastatin. Neuron 2013, 80, 1175–1189. [Google Scholar] [CrossRef]
- Yamamoto, S.; Sugawara, T.; Murakami, A.; Nakazawa, M.; Nao-I, N.; Machida, S.; Wada, Y.; Mashima, Y.; Myake, Y. Topical Isopropyl Unoprostone for Retinitis Pigmentosa: Microperimetric Results of the Phase 2 Clinical Study. Ophthalmol. Ther. 2012, 1, 5. [Google Scholar] [CrossRef]
- Sahaboglu, A.; Vidal-Gil, L.; Sancho-Pelluz, J. Release of Retinal Extracellular Vesicles in a Model of Retinitis Pigmentosa. In Retinal Degenerative Diseases; Bowes Rickman, C., Grimm, C., Anderson, R.E., Ash, J.D., LaVail, M.M., Hollyfield, J.G., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2019; Volume 1185, pp. 431–436. ISBN 978-3-030-27377-4. [Google Scholar]
- Knickelbein, J.E.; Liu, B.; Arakelyan, A.; Zicari, S.; Hannes, S.; Chen, P.; Li, Z.; Grivel, J.-C.; Chaigne-Delalande, B.; Sen, H.N.; et al. Modulation of Immune Responses by Extracellular Vesicles From Retinal Pigment Epithelium. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4101. [Google Scholar] [CrossRef]
- Ma, M.; Li, B.; Zhang, M.; Zhou, L.; Yang, F.; Ma, F.; Shao, H.; Li, Q.; Li, X.; Zhang, X. Therapeutic Effects of Mesenchymal Stem Cell-Derived Exosomes on Retinal Detachment. Exp. Eye Res. 2020, 191, 107899. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, T.; Yang, Z.; Sun, X.; Huang, Z.; Deng, X.; He, C.; Liu, X. Bone Marrow Mesenchymal Stem Cells-Derived Exosomes Attenuate Neuroinflammation and Promote Survival of Photoreceptor in Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3108. [Google Scholar]
- Odagiu, L.; May, J.; Boulet, S.; Baldwin, T.A.; Labrecque, N. Role of the Orphan Nuclear Receptor NR4A Family in T-Cell Biology. Front. Endocrinol. 2020, 11, 624122. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Q.; Yang, G.; Wei, Y.; Li, M.; Du, E.; Li, H.; Song, Z.; Tao, Y. RPE-Derived Exosomes Rescue the Photoreceptors during Retina Degeneration: An Intraocular Approach to Deliver Exosomes into the Subretinal Space. Drug Deliv. 2021, 28, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.-L.; Hu, C.-B.; Ling, S.-T.; Zhao, N.; Bao, L.-H.; Zhou, F.; Xiong, Y.-C.; Chen, T.; Sui, B.-D.; Yu, X.-R.; et al. Photoreceptor Protection by Mesenchymal Stem Cell Transplantation Identifies Exosomal MiR-21 as a Therapeutic for Retinal Degeneration. Cell Death Differ. 2021, 28, 1041–1061. [Google Scholar] [CrossRef] [PubMed]
- Jenike, A.E.; Halushka, M.K. MiR-21: A Non-specific Biomarker of All Maladies. Biomark. Res. 2021, 9, 18. [Google Scholar] [CrossRef]
- Bian, B.; Zhao, C.; He, X.; Gong, Y.; Ren, C.; Ge, L.; Zeng, Y.; Li, Q.; Chen, M.; Weng, C.; et al. Exosomes Derived from Neural Progenitor Cells Preserve Photoreceptors during Retinal Degeneration by Inactivating Microglia. J. Extracell. Vesicles 2020, 9, 1748931. [Google Scholar] [CrossRef]
- Zhang, Z.; Mugisha, A.; Fransisca, S.; Liu, Q.; Xie, P.; Hu, Z. Emerging Role of Exosomes in Retinal Diseases. Front. Cell Dev. Biol. 2021, 9, 643680. [Google Scholar] [CrossRef]
- Elliott, R.O.; He, M. Unlocking the Power of Exosomes for Crossing Biological Barriers in Drug Delivery. Pharmaceutics 2021, 13, 122. [Google Scholar] [CrossRef]
- Liang, Y.; Duan, L.; Lu, J.; Xia, J. Engineering Exosomes for Targeted Drug Delivery. Theranostics 2021, 11, 3183–3195. [Google Scholar] [CrossRef]
- Hung, M.E.; Leonard, J.N. Stabilization of Exosome-Targeting Peptides via Engineered Glycosylation. J. Biol. Chem. 2015, 290, 8166–8172. [Google Scholar] [CrossRef]
- Boukouris, S.; Mathivanan, S. Exosomes in Bodily Fluids Are a Highly Stable Resource of Disease Biomarkers. Proteom. Clin. Appl. 2015, 9, 358–367. [Google Scholar] [CrossRef]
- Wassmer, S.J.; Carvalho, L.S.; György, B.; Vandenberghe, L.H.; Maguire, C.A. Exosome-Associated AAV2 Vector Mediates Robust Gene Delivery into the Murine Retina upon Intravitreal Injection. Sci. Rep. 2017, 7, 45329. [Google Scholar] [CrossRef] [PubMed]




| Exosome Source | Models | Observed Effect |
|---|---|---|
| MSC-Exos | Hyaluronic acid-induced retinal detachment in Sprague–Dawley rat | Diminished TNFα and IL-β expression Enhancement of LC3-II and LC3-I ratio Decrease of Atg5 cleavage Decrease in photoreceptor cell apoptosis [228] |
| Rd10 mice | Diminished TNFα, IL-β and IL-6 expression Enhanced photoreceptor cell survival Microglial, Müller cell and macrophage activation inhibition miR-146a upregulation [159,229] | |
| N-methyl-N-nitrosourea (MNU)-induced retinal detachment in C57BL6 mice | Decrease in photoreceptor cell apoptosis miR-21 upregulation [232]. | |
| RPE-Exos | N-methyl-N-nitrosourea (MNU)-induced retinal detachment in C57BL6 mice | Enhanced photoreceptor cell survival Diminished TNFα, IL-β and IL-6 mRNA expression Diminished Bax and caspase-3 mRNA expression Enhanced Bcl-2 mRNA expression [231]. |
| Inflammatory cytokine-induced extracellular vesicle release in ARPE-19 cell line | T cell proliferation inhibition by TNFα and IL-6 production [227]. | |
| NPC-Exos | Royal College of Surgeons rats | Decrease in photoreceptor cell apoptosis Inhibition of retinal microglia activation Diminished TNFα, IL-β and COX-2 expression [234]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, K.Y.; Kulbay, M.; Toameh, D.; Xu, A.Q.; Kalevar, A.; Tran, S.D. Retinitis Pigmentosa: Novel Therapeutic Targets and Drug Development. Pharmaceutics 2023, 15, 685. https://doi.org/10.3390/pharmaceutics15020685
Wu KY, Kulbay M, Toameh D, Xu AQ, Kalevar A, Tran SD. Retinitis Pigmentosa: Novel Therapeutic Targets and Drug Development. Pharmaceutics. 2023; 15(2):685. https://doi.org/10.3390/pharmaceutics15020685
Chicago/Turabian StyleWu, Kevin Y., Merve Kulbay, Dana Toameh, An Qi Xu, Ananda Kalevar, and Simon D. Tran. 2023. "Retinitis Pigmentosa: Novel Therapeutic Targets and Drug Development" Pharmaceutics 15, no. 2: 685. https://doi.org/10.3390/pharmaceutics15020685
APA StyleWu, K. Y., Kulbay, M., Toameh, D., Xu, A. Q., Kalevar, A., & Tran, S. D. (2023). Retinitis Pigmentosa: Novel Therapeutic Targets and Drug Development. Pharmaceutics, 15(2), 685. https://doi.org/10.3390/pharmaceutics15020685