
Production, Characterization, and Assessment of Permanently Cationic and Ionizable Lipid Nanoparticles for Use in the Delivery of Self-Amplifying RNA Vaccines


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of saRNAs Encoding Reporter Proteins
2.2. Transfection of HEK 293T Cells with saRNAs and Detection of Reporter Proteins
2.3. Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
2.4. Lipid Nanoparticle Formulation of saRNAs
2.5. saRNA Quantification and Encapsulation Efficiency Assessment
2.6. Cell Viability Assay
2.7. Statistical Analysis
3. Results
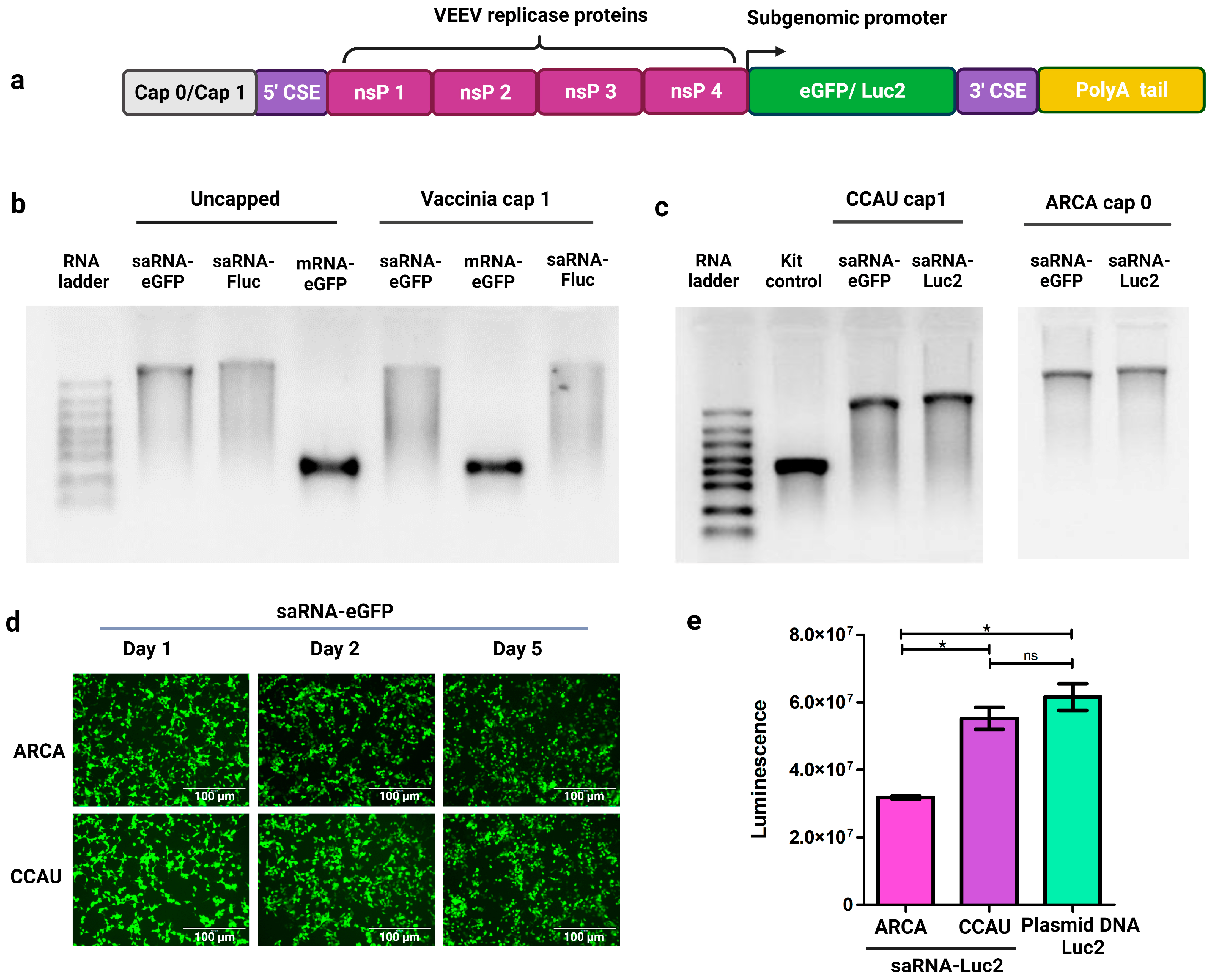
3.1. IVT of saRNAs Encoding Reporter Proteins
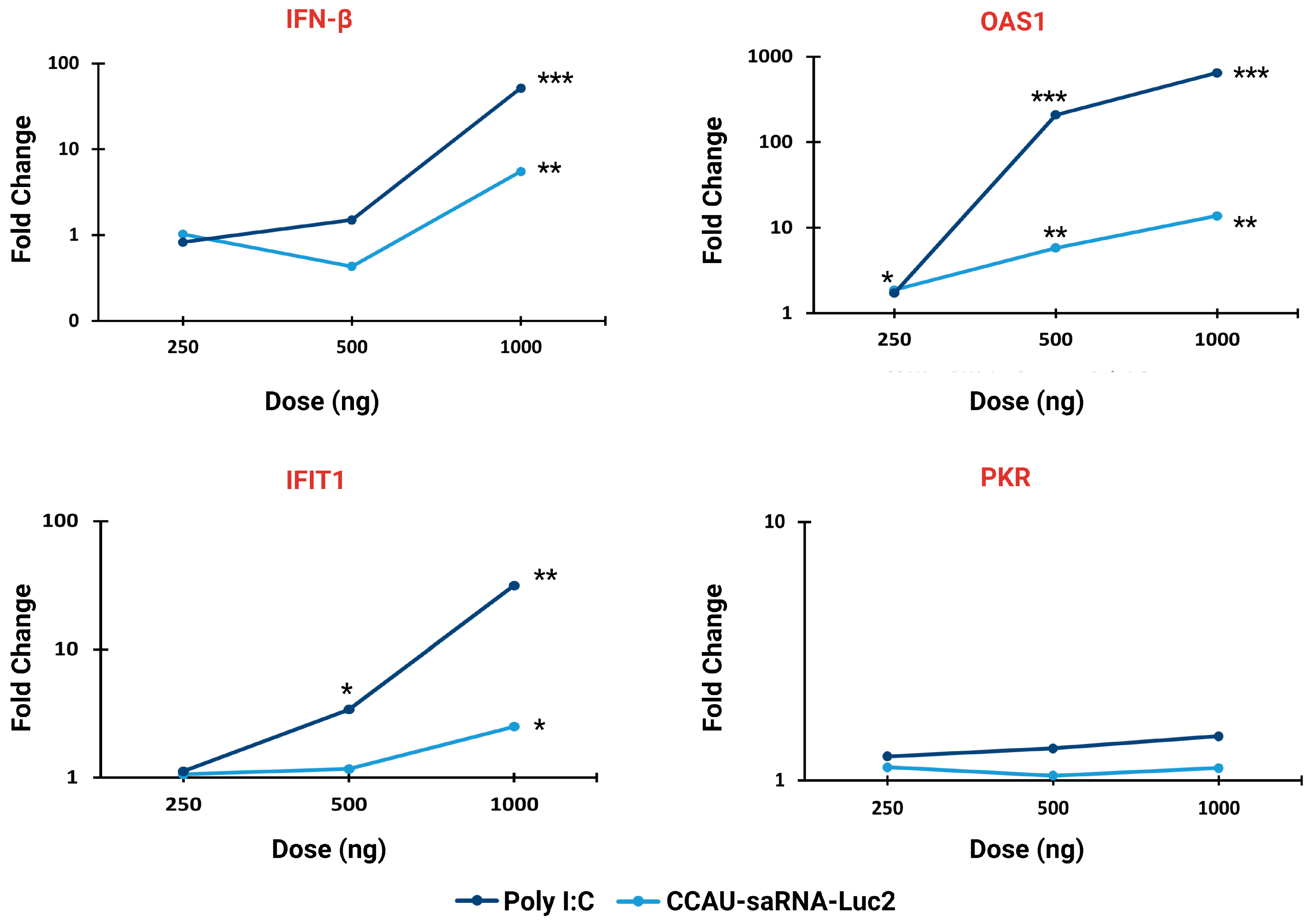
3.2. Innate Immune Response to saRNAs
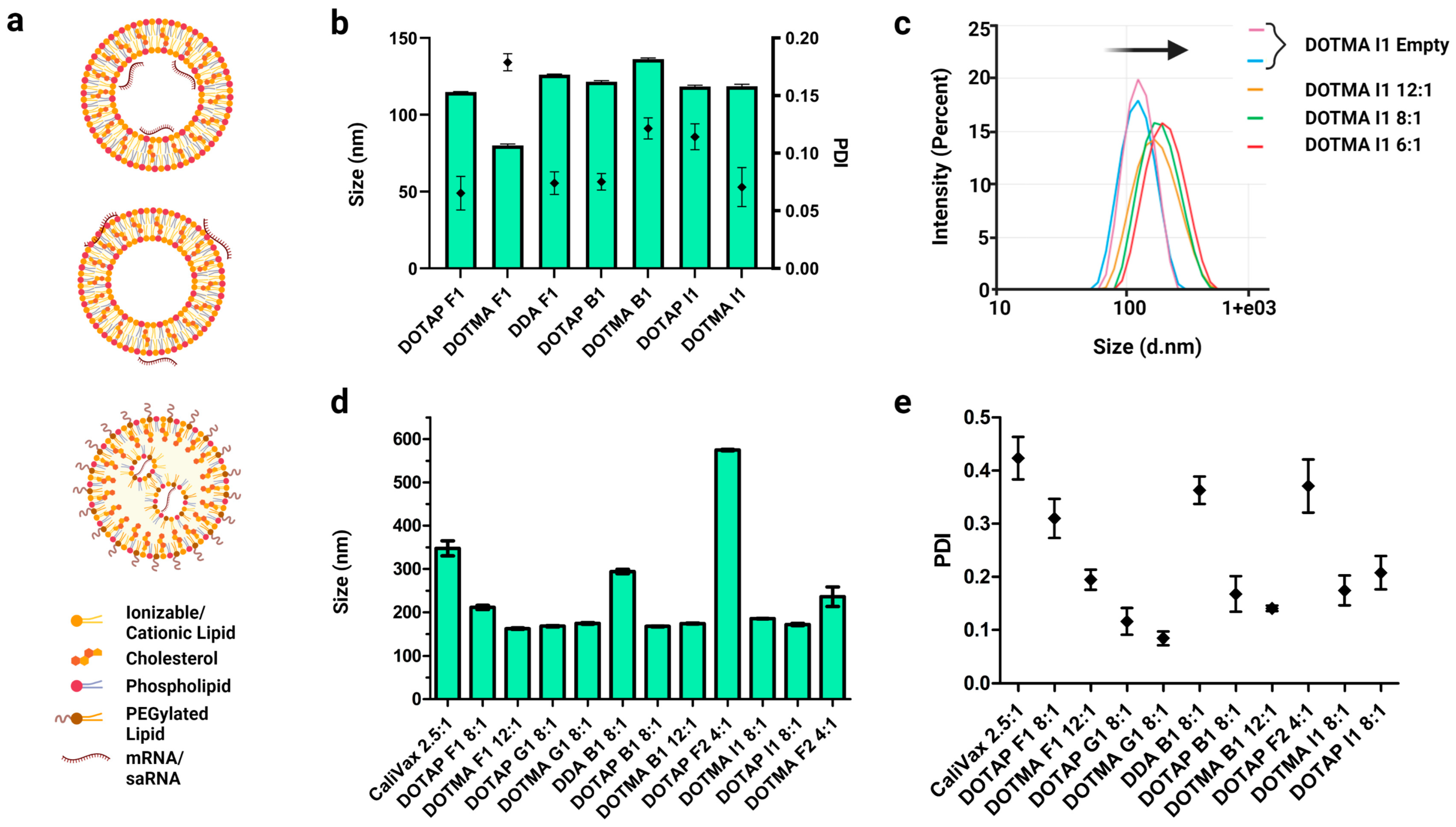
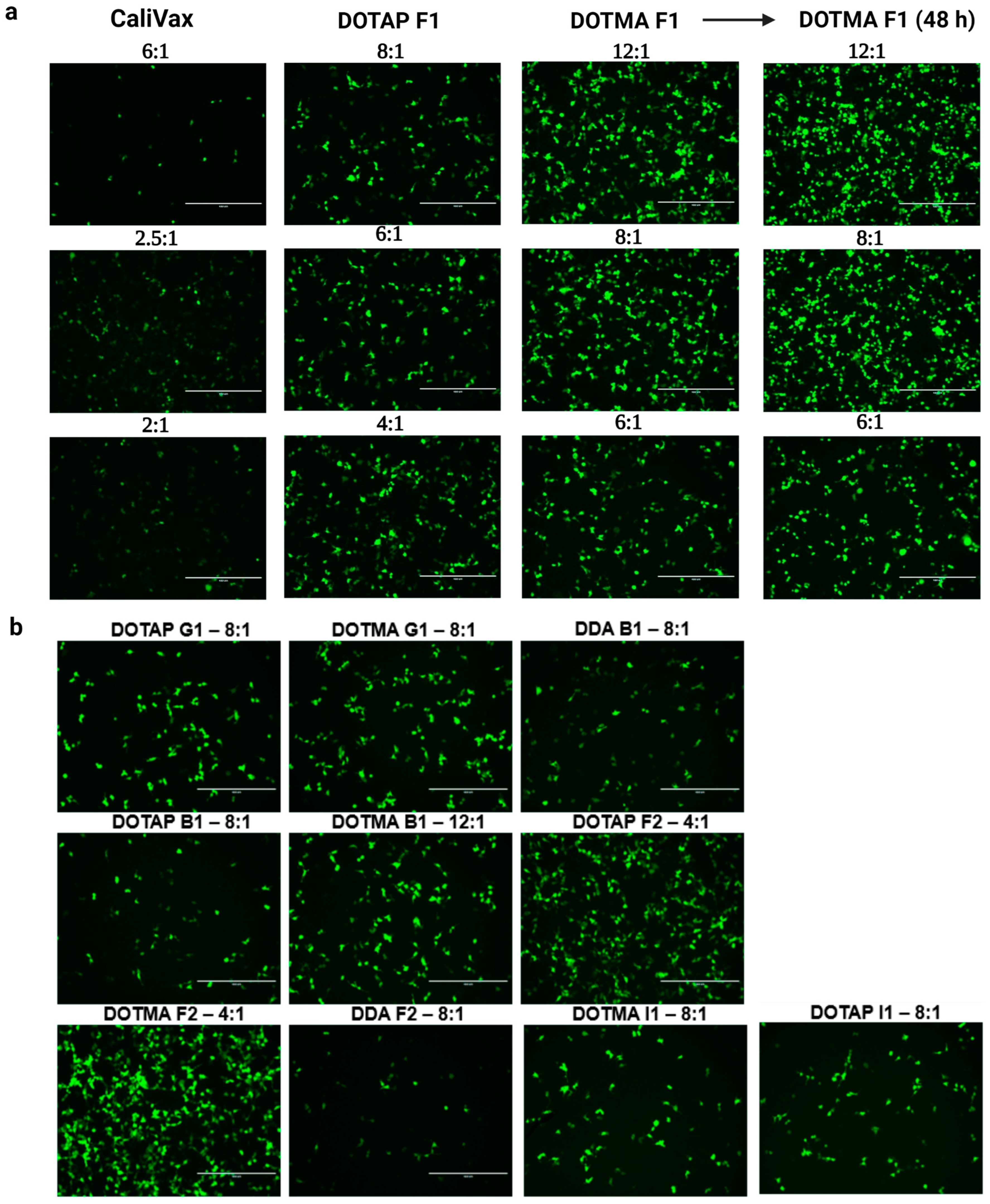
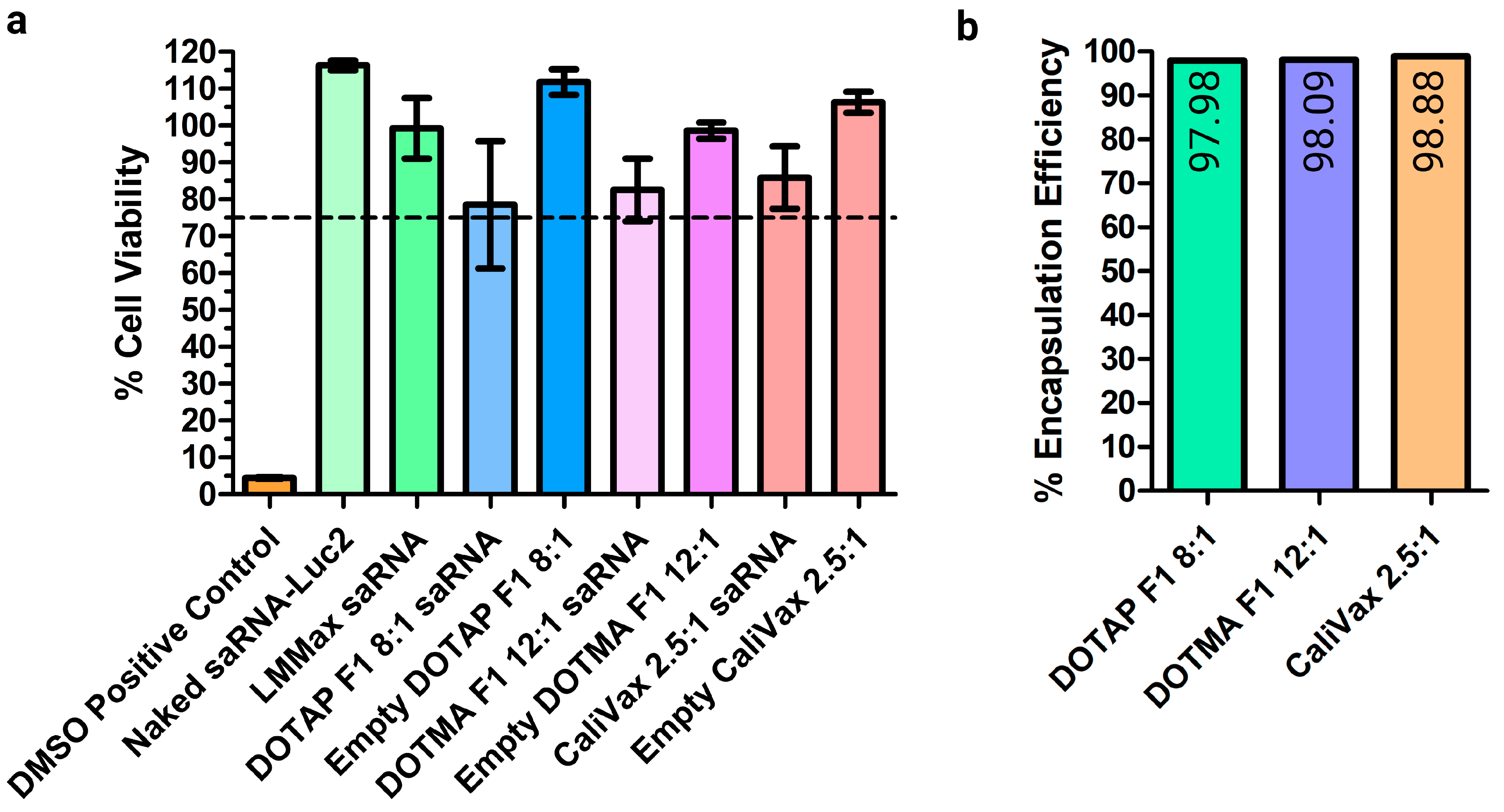
3.3. External Formulation of saRNA-cLNPs Using Lipid Film Hydration
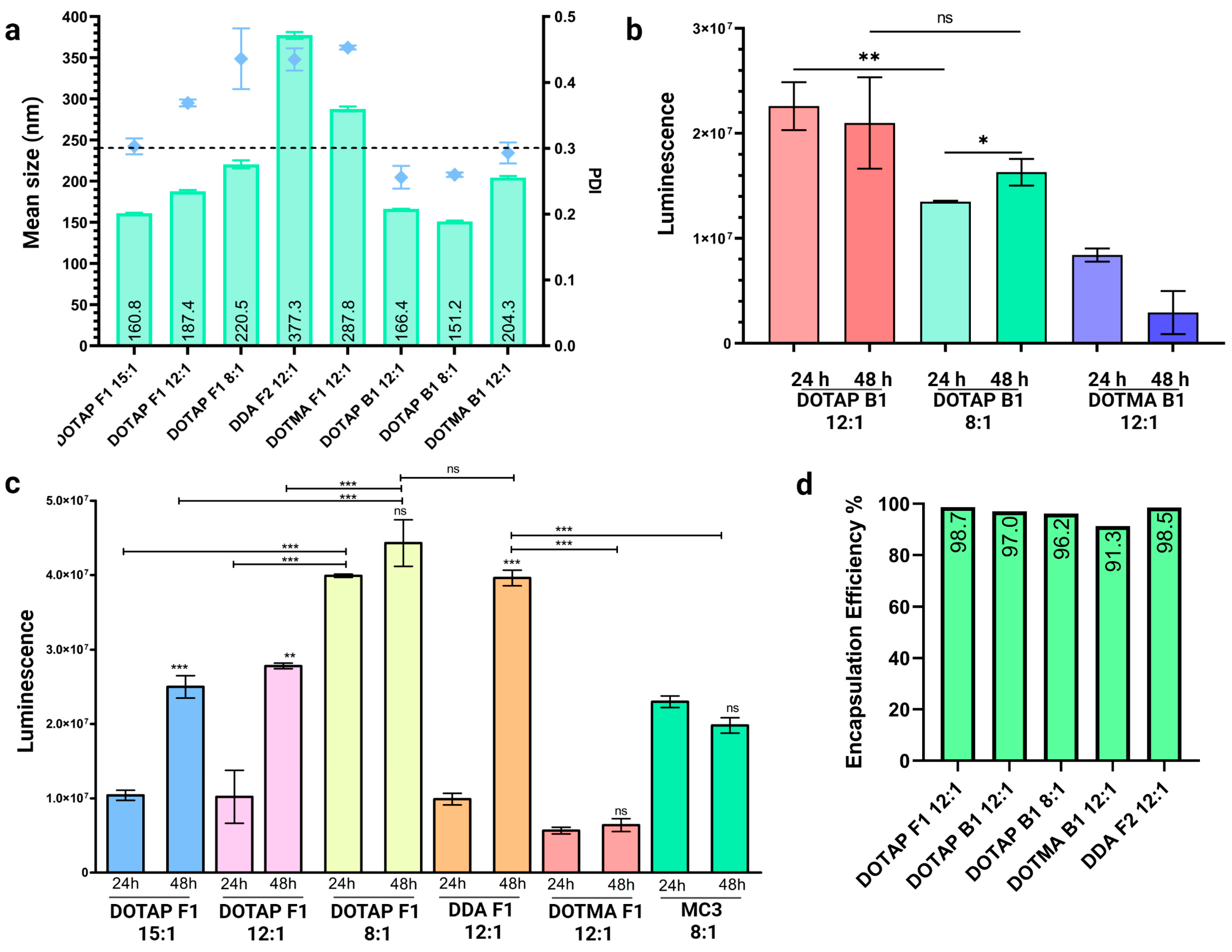
3.4. Optimization of Externally Formulated saRNA-cLNPs for In Vitro Expression
3.5. Internal Formulation of saRNA-cLNPs by a Modified Solvent Injection Method
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chaudhary, N.; Weissman, D.; Whitehead, K.A. mRNA vaccines for infectious diseases: Principles, delivery and clinical translation. Nat. Rev. Drug Discov. 2021, 20, 817–838. [Google Scholar] [CrossRef] [PubMed]
- Bloom, K.; van den Berg, F.; Arbuthnot, P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 2021, 28, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Kairuz, D.; Samudh, N.; Ely, A.; Arbuthnot, P.; Bloom, K. Advancing mRNA technologies for therapies and vaccines: An African context. Front. Immunol. 2022, 13, 1018961. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.B.; Lambert, L.; Kinnear, E.; Busse, D.; Erbar, S.; Reuter, K.C.; Wicke, L.; Perkovic, M.; Beissert, T.; Haas, H.; et al. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol. Ther. 2018, 26, 446–455. [Google Scholar] [CrossRef] [Green Version]
- Beissert, T.; Koste, L.; Perkovic, M.; Walzer, K.C.; Erbar, S.; Selmi, A.; Diken, M.; Kreiter, S.; Türeci, Ö.; Sahin, U. Improvement of In Vivo Expression of Genes Delivered by Self-Amplifying RNA Using Vaccinia Virus Immune Evasion Proteins. Hum. Gene Ther. 2017, 28, 1138–1146. [Google Scholar] [CrossRef] [Green Version]
- Probst, J.; Brechtel, S.; Scheel, B.; Hoerr, I.; Jung, G.; Rammensee, H.-G.; Pascolo, S. Characterization of the ribonuclease activity on the skin surface. Genet. Vaccines Ther. 2006, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Zeng, C.; Zhang, C.; Walker, P.G.; Dong, Y. Formulation and Delivery Technologies for mRNA Vaccines. In mRNA Vaccines; Yu, D., Petsch, B., Eds.; Current Topics in Microbiology and Immunology; Springer International Publishing: Cham, Switzerland, 2020; Volume 440, pp. 71–110. ISBN 978-3-031-18069-9. [Google Scholar]
- Blakney, A.K.; Ip, S.; Geall, A.J. An Update on Self-Amplifying mRNA Vaccine Development. Vaccines 2021, 9, 97. [Google Scholar] [CrossRef]
- Crawford, R.; Dogdas, B.; Keough, E.; Haas, R.M.; Wepukhulu, W.; Krotzer, S.; Burke, P.A.; Sepp-Lorenzino, L.; Bagchi, A.; Howell, B.J. Analysis of lipid nanoparticles by Cryo-EM for characterizing siRNA delivery vehicles. Int. J. Pharm. 2011, 403, 237–244. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Darjuan, M.M.; Mercer, J.E.; Chen, S.; van der Meel, R.; Thewalt, J.L.; Tam, Y.Y.C.; Cullis, P.R. On the Formation and Morphology of Lipid Nanoparticles Containing Ionizable Cationic Lipids and siRNA. ACS Nano 2018, 12, 4787–4795. [Google Scholar] [CrossRef] [Green Version]
- Melo, M.; Porter, E.; Zhang, Y.; Silva, M.; Li, N.; Dobosh, B.; Liguori, A.; Skog, P.; Landais, E.; Menis, S.; et al. Immunogenicity of RNA Replicons Encoding HIV Env Immunogens Designed for Self-Assembly into Nanoparticles. Mol. Ther. 2019, 27, 2080–2090. [Google Scholar] [CrossRef]
- Blakney, A.K.; McKay, P.F.; Yus, B.I.; Aldon, Y.; Shattock, R.J. Inside out: Optimization of lipid nanoparticle formulations for exterior complexation and in vivo delivery of saRNA. Gene Ther. 2019, 26, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Blakney, A.K.; McKay, P.F.; Ibarzo Yus, B.; Hunter, J.E.; Dex, E.A.; Shattock, R.J. The Skin You Are In: Design-of-Experiments Optimization of Lipid Nanoparticle Self-Amplifying RNA Formulations in Human Skin Explants. ACS Nano 2019, 13, 5920–5930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, G.; Anderluzzi, G.; Schmidt, S.T.; Woods, S.; Gallorini, S.; Brazzoli, M.; Giusti, F.; Ferlenghi, I.; Johnson, R.N.; Roberts, C.W.; et al. Delivery of self-amplifying mRNA vaccines by cationic lipid nanoparticles: The impact of cationic lipid selection. J. Control. Release 2020, 325, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Anderluzzi, G.; Lou, G.; Gallorini, S.; Brazzoli, M.; Johnson, R.; O’Hagan, D.T.; Baudner, B.C.; Perrie, Y. Investigating the Impact of Delivery System Design on the Efficacy of Self-Amplifying RNA Vaccines. Vaccines 2020, 8, 212. [Google Scholar] [CrossRef]
- Yoshioka, N.; Gros, E.; Li, H.-R.; Kumar, S.; Deacon, D.C.; Maron, C.; Muotri, A.R.; Chi, N.C.; Fu, X.-D.; Yu, B.D.; et al. Efficient Generation of Human iPSCs by a Synthetic Self-Replicative RNA. Cell Stem Cell 2013, 13, 246–254. [Google Scholar] [CrossRef] [Green Version]
- Samnuan, K.; Blakney, A.K.; McKay, P.F.; Shattock, R.J. Design-of-experiments in vitro transcription yield optimization of self-amplifying RNA. F1000Research 2022, 11, 333. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Atochina-Vasserman, E.N.; Lu, J.; Maurya, D.S.; Xiao, Q.; Liu, M.; Adamson, J.; Ona, N.; Reagan, E.K.; Ni, H.; et al. The Unexpected Importance of the Primary Structure of the Hydrophobic Part of One-Component Ionizable Amphiphilic Janus Dendrimers in Targeted mRNA Delivery Activity. J. Am. Chem. Soc. 2022, 144, 4746–4753. [Google Scholar] [CrossRef]
- Cagigi, A.; Loré, K. Immune Responses Induced by mRNA Vaccination in Mice, Monkeys and Humans. Vaccines 2021, 9, 61. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef]
- Hyde, J.L.; Chen, R.; Trobaugh, D.W.; Diamond, M.S.; Weaver, S.C.; Klimstra, W.B.; Wilusz, J. The 5′ and 3′ ends of alphavirus RNAs—Non-coding is not non-functional. Virus Res. 2015, 206, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blakney, A.K.; McKay, P.F.; Hu, K.; Samnuan, K.; Jain, N.; Brown, A.; Thomas, A.; Rogers, P.; Polra, K.; Sallah, H.; et al. Polymeric and lipid nanoparticles for delivery of self-amplifying RNA vaccines. J. Control. Release 2021, 338, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Geall, A.J.; Verma, A.; Otten, G.R.; Shaw, C.A.; Hekele, A.; Banerjee, K.; Cu, Y.; Beard, C.W.; Brito, L.A.; Krucker, T.; et al. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci. USA 2012, 109, 14604–14609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blakney, A.K.; Deletic, P.; McKay, P.F.; Bouton, C.R.; Ashford, M.; Shattock, R.J.; Sabirsh, A. Effect of complexing lipids on cellular uptake and expression of messenger RNA in human skin explants. J. Control. Release 2021, 330, 1250–1261. [Google Scholar] [CrossRef]
- Chen, W.C.; May, J.P.; Li, S.-D. Immune responses of therapeutic lipid nanoparticles. Nanotechnol. Rev. 2013, 2, 201–213. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kairuz, D.; Samudh, N.; Ely, A.; Arbuthnot, P.; Bloom, K. Production, Characterization, and Assessment of Permanently Cationic and Ionizable Lipid Nanoparticles for Use in the Delivery of Self-Amplifying RNA Vaccines. Pharmaceutics 2023, 15, 1173. https://doi.org/10.3390/pharmaceutics15041173
Kairuz D, Samudh N, Ely A, Arbuthnot P, Bloom K. Production, Characterization, and Assessment of Permanently Cationic and Ionizable Lipid Nanoparticles for Use in the Delivery of Self-Amplifying RNA Vaccines. Pharmaceutics. 2023; 15(4):1173. https://doi.org/10.3390/pharmaceutics15041173
Chicago/Turabian StyleKairuz, Dylan, Nazia Samudh, Abdullah Ely, Patrick Arbuthnot, and Kristie Bloom. 2023. "Production, Characterization, and Assessment of Permanently Cationic and Ionizable Lipid Nanoparticles for Use in the Delivery of Self-Amplifying RNA Vaccines" Pharmaceutics 15, no. 4: 1173. https://doi.org/10.3390/pharmaceutics15041173
APA StyleKairuz, D., Samudh, N., Ely, A., Arbuthnot, P., & Bloom, K. (2023). Production, Characterization, and Assessment of Permanently Cationic and Ionizable Lipid Nanoparticles for Use in the Delivery of Self-Amplifying RNA Vaccines. Pharmaceutics, 15(4), 1173. https://doi.org/10.3390/pharmaceutics15041173