2.1. Chemistry
Kanamycin A was a commercially available product of Abcr GmbH (Karlsruhe, Germany). All other solvents and reagents were commercially available products from Aldrich (Saint Louis, MO, USA) and Merck (Darmstadt, Germany). DMF (dried over CaCl2, then over P2O5) and pyridine (dried over KOH, then over CaCl2) were distilled before use.
The course of reactions, purification and extraction procedures, and the identities of the obtained compounds, were monitored by TLC and HPLC methods. TLC was performed on plates with 60F254 silica gel (Merck, Darmstadt, Germany). Kanamycin and its derivatives were visualized on chromatograms in iodine vapor or by a solution of ninhydrin in ethanol or 6N sulfuric acid solution followed by heating. UV-absorbing derivatives were also detected in UV light.
The preparative isolation and purification of the compounds were performed on silica gel columns (Kieselgel G60, 0.040–0.063 mm, Merck, Darmstadt, Germany). For neutralization, DOWEX 50 WX2 (100–200 mesh) ion-exchange resin (SERVA, Heidelberg, Germany) was used. All solutions were dried over sodium sulfate and evaporated in a vacuum at a temperature below 40 °C.
Analytical HPLC was performed on an LC-20AD chromatograph (Shimadzu, Kyoto, Japan) using a UV detector and a Kromasil-100-C18, 4.6250 mm column (Phenemenex, Torrance, CA, USA) at a flow rate of 1 mL/min in systems:
System (A1): A (0.01M H3PO4, pH 2.6) and B (MeCN), linear acetonitrile concentration gradient from 20→80% acetonitrile in 30 min, then from 80→20% in 3 min;
System (A2): A (HCOONH4 0.2%, pH 4.5) and B (MeCN), linear acetonitrile concentration gradient from 40→90% for 30 min, then from 90→40% for 3 min;
System (A3): A (0.01M H3PO4, pH 2.6) and B (MeCN), linear acetonitrile concentration gradient from 80→95% for 30 min, then from 95→80% for 3 min;
System (A4): A (HCOONH4 0.2%, pH 4.5) and B (MeCN), linear acetonitrile concentration gradient from 20→90% for 30 min, then from 90→95% for 5 min, then from 95→95% for 5 min, then from 95→20% for 5 min;
System (A5): A (HCOONH4 0.2%, pH 8.4) and B (MeCN), linear acetonitrile concentration gradient from 40→90% for 30 min, then from 90→40% for 3 min;
System (A6): A (HCOONH4 0.2%, pH 8.4) and B (MeCN), linear acetonitrile concentration gradient from 10→60% for 30 min, then from 60→10% for 3 min.
The purities of the intermediates and final compounds were at least 90% by HPLC (for UV-absorbing compounds).
Elemental analysis was performed on the automated PerkinElmer 2400 CHN microanalyzer. High-resolution mass spectra (HR MS) were obtained by electrospray ionization (ESI) on a micrOTOF-Q II instrument (Bruker Daltonics GmbH, Bremen, Germany). The 1H, 13C and 15N NMR spectra were recorded in DMSO-d6 at 25 °C on a Bruker AV III 500 spectrometer (Bruker Biospin AG, Fällanden, Switzerland) at 500.2 MHz, 125.8 MHz and 50.7 MHz for 1H, 13C and 15N, respectively. Signal assignment in NMR spectra was performed using two-dimensional COSY, TOCSY, NOESY, HSQC, HMBC and 1H-15N edHSQC experiments. Standard pulse sequences were used. The 1H and 13C NMR spectra were referenced using either residual DMSO solvent signals (2.50 ppm for DMSO-d6 for 1H spectra and 49.5 ppm for DMSO-d6 for 13C spectra) or signals of internal DSS reference for D2O solutions. The 1H-15N edHSQC spectrum was externally referenced against CH3NO2 for 15N.
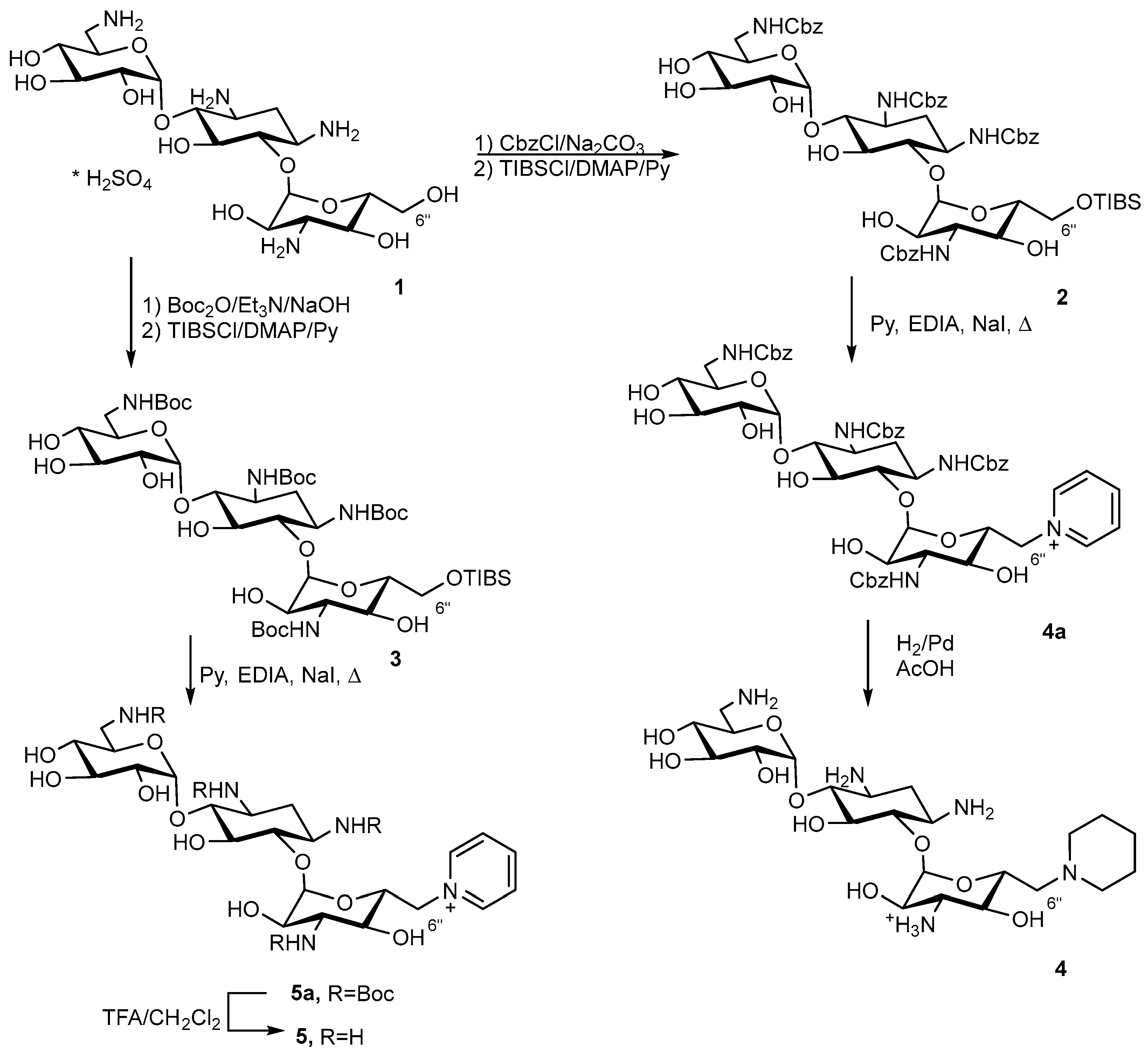
1,3,6′,3″-Tetra-N-Cbz-6″-O-(2,4,6-triisopropylbenzenesulfonyl)kanamycin A (2)
1st step
The solution of benzylchloroformate (2.40 mL, 16.7 mmol) in acetone (10 mL) was added dropwise at 0 °C to the mixture kanamycin A monosulfate (2.0 g, 3.43 mmol) and saturated water solution of Na2CO3 (26.7 mL). The reaction mixture was stirred at 0 °C for 2 h, then at rt for 8 h. The resulting precipitate was filtered off and then suspended in 1M HCl (70 mL) and stirred for 30 min. The precipitate was filtered off, washed with H2O and dried in a vacuum over P2O5. The target 1,3,6′,3″-tetra-N-Cbz-kanamycin A was obtained as a white solid (3.44 g, 98% yield) and used without the additional purification at the next stage.
HRMS (ESI): calculated for [C50H61N4O19]+ = 1021.3930 [C50H64N5O19]+ = 1038.4196 [C50H60N4O19Na]+ = 1043.3749, found [M+H]+ = 1021.4185 [M+NH4]+ = 1038.4467 [M+Na]+ = 1043.4044, Rf (CHCl3:MeOH 10:1) = 0.5, Rt (system A2) = 13.41 min.
1H NMR (δ, ppm, J/Hz): 1.46–3.56 (m, 7H, cHex), 3.07–4.93 (m, 7H, CH1′-6′), 3.31–4.99 (m, 7H, CH1″-6″), 3.67 (d, 2H, 3J = 6.1 Hz, -OH-CHCH2″,CH4″-), 4.23 (t, 1H, 3J = 5.6 Hz, -OH-CH2 CH6″-), 4.85–5.18 (m, 8H, -OCH2), 5.24 (bs, 1H, -OH-CH cHex5), 5.34 (d, 1H, 3J = 5.6 Hz, -OH-CHCH2′-CH4′-), 6.84 (s, 1H, -NH-CH2 CH6′-), 7.01 (s, 1H, -NH-CH2 CH3″-), 7.17 (s, 1H, -NH-CHcHex3-), 7.21–7.42 (m, 20H, 4 × Ph), 7.43 (s, 1H, -NH-CHcHex1-).
13C NMR (δ, ppm, J/Hz): 34.4, 41.7, 49.7, 50.1, 56.5, 60.2, 65.0, 65.3, 67.2, 70.1, 70.6, 70.7, 72.3, 72.7, 72.9, 74.1, 80.4, 84.3, 97.3, 101.1, 127.7, 128.3, 136.8–137.4, 155.6, 155.9, 156.5, 156.6.
2nd step
The 2,4,6-Triisopropylbenzenesulfonylchloride (1.28 g, 4.23 mmol) was added to the solution of 1,3,6′,3″-tetra-N-Cbz-kanamycin A (1.0 g, 0.98 mmol) and 4-(N,N-dimethylamino)pyridine (517 mg, 4.23 mmol) in dry pyridine (15 mL). The reaction mixture was stirred 6 h at room temperature (rt), then an additional amount of 2,4,6-triisopropylbenzenesulfonylchloride (1.28 g, 4.23 mmol) was added and the reaction mixture was stirred at rt for 20 h. An aqueous solution of HCl (1N, 70 mL) and water (100 mL) was added to the reaction mixture. The product was extracted with ethyl acetate (3 × 60 mL) and organic fractions were combined, dried over Na2SO4, filtered off and evaporated to dryness. The target compound was purified by the column chromatography on silica gel. The elution was carried out by CHCl3 (200 mL), followed by the mixture CHCl3-CH3OH (100:3). Fractions which contained the target compound were combined and evaporated to dryness, resulting in 2 (1.12 g, 89% yield) as a light-yellow solid.
HRMS (ESI): calculated for [C65H86N5O21S]+ = 1304.5536, found [M+NH4]+ = 1304.5655, Rf (CHCl3:MeOH 100:15) = 0.57, Rt (system A3) = 8.31 min.
1H NMR (δ, ppm, J/Hz): 1.19–1.21 (m, 18H, 6×-CH3), 1.41–3.55 (m, 7H, cHex), 3.06–4.89 (m, 7H, CH1′-6′), 3.29–4.97 (m, 7H, CH1″-6″), 3.67 (d, 2H, 3J = 6.1 Hz, -OH-CHCH2″,CH4″-), 3.99–4.06 (m, 3H, -CH(CH3)3), 4.86–5.11 (m, 8H, -OCH2), 5.24 (bs, 1H, -OH-CHcHex5), 5.34 (d, 1H, 3J = 5.6 Hz, -OH-CHCH2′-CH4′-), 6.83 (s, 1H, -NH-CH2 CH6′-), 7.06 (s, 1H, -NH-CH2 CH3″-), 7.18 (s, 1H, -NH-CHcHex3-), 7.25–7.39 (m, 20H, 4 × Ph), 7.34 (s, 1H, -NH-CHcHex1-).
13C NMR (δ, ppm, J/Hz): 23.2, 24.37, 24.45, 29.0, 34.5, 41.6, 49.6, 50.0, 56.6, 65.0, 65.3, 66.4, 67.5, 69.6, 69.7, 70.4, 70.6, 72.2, 72.6, 74.2, 80.0, 84.6, 97.4, 101.2, 123.7, 127.6, 128.2, 137.0, 137.1, 137.2, 150.2, 153.6, 155.5, 155.8, 156.5, 156.6.
1,3, 6′,3″-Tetra-N-Boc-6″-O-(2,4,6-triisopropylbenzeneosulfonyl)kanamycin A (3)
1st step
The solution of di-tert-butyldicarbonate (1.5 g, 6.86 mmol) and Et3N (0.5 mL, 3.43 mmol) in DMSO (12 mL) was added to a suspension of kanamycin A monosulfate (500 mg, 0.86 mmol) and NaOH (34 mg, 0.858 mmol) in H2O (2 mL). The reaction mixture was stirred at rt for 12 h, then NH4OH (conc, 5 mL) was added. The obtained solid was filtered over celite, and a cake was washed with H2O (2 × 20 mL) and ethyl acetate (20 mL). The target compound was eluted from celite with methanol (50 mL) and the obtained solution was evaporated to dryness, resulting in 1,3, 6′,3″-tetra-N-Boc-kanamycin A (0.47 g, 62% yield) as white solid which was used without the additional purification at the next stage.
HRMS (ESI): calculated for [C38H68N4O19Na]+ = 907.4376, found [M+Na]+ = 907.4533, Rf (MeOH:NH3 7:10) = 0.36.
1H NMR (δ, ppm, J/Hz): 1.36 (s, 9H, 3×-CH3), 1.37 (s, 9H, 3×-CH3), 1.38 (s, 18H, 6×-CH3) 1.38–3.42 (m, 7H, cHex), 3.04–4.88 (m, 7H, CH1′-6′), 2.9–4.89 (m, 7H, CH1″-6″), 3.69 (d, 2H, 3J = 6.1 Hz, -OH-CHCH2″,CH4″-), 4.21 (t, 1H, 3J = 5.6 Hz, -OH-CH2 CH6‴-), 5.25 (bs, 1H, -OH-CH cHex5), 5.35 (d, 1H, 3J = 5.6 Hz, -OH-CHCH2′-CH4′-), 6.35 (s, 1H, -NH-CH2CH6′-), 6.5 (s, 1H, -NH-CH2 CH3″-), 6.58 (s, 1H, -NH-CHcHex3-), 6.96 (s, 1H, -NH-CHcHex1-).
13C NMR (δ, ppm, J/Hz): 28.1, 28.2, 28.3, 34.7, 41.4, 49.1, 50.0, 55.9, 60.3, 67.5, 70.1, 70.3, 70.5, 72.1, 72.7, 72.9, 75.0, 77.8, 80.4, 83.9, 97.8, 101.1, 154.9, 155.3, 156.1, 156.3.
2nd step
The 2,4,6-Triisopropylbenzenesulfonylchloride (1.74 g, 5.74 mmol) was added to the mixture of 1,3,6′,3″-tetra-N-Boc-kanamycin A (1.18 g, 1.33 mmol) and 4-(N,N-dimethylanimo)pyridine (702 mg, 5.74 mmol) in dry pyridine (30 mL). The reaction mixture was stirred at rt for 6 h, then an additional portion of 2,4,6-triisopropylbenzenesulfonylchloride (1.74 g, 5.74 mmol) was added and the reaction mixture was stirred at rt for 20 h. An aqueous solution of HCl (1N, 70 mL) and H2O (100 mL) was added to the reaction mixture. The target compound was extracted with ethyl acetate (3 × 60 mL) and the organic fraction was dried over Na2SO4, filtered off and concentrated in a vacuum. The product was purified by column chromatography in silica gel and the column was eluted by CHCl3 (150 mL), followed by the mixture of CHCl3-CH3OH (100:2). Fractions which contained the target compound were combined and evaporated to dryness, resulting in 3 (0.59 g, 39% yield) as a light-yellow solid.
HRMS (ESI): calculated for [C53H91N4O21S]+ = 1151.5896, found [M+H]+ = 1151.5847, Rf (CHCl3:MeOH 10:1) = 0.31, Rt (system A1) = 9.44 min.
6″-(Piperidin-1-yl)-6″-deoxykanamycin A acetate (4)
1st step
Compound 2 (200 mg, 0.156 mmol) was dissolved in dry pyridine (5 mL) and NaI (2 mg, 0.016 mmol) and iPr2EtN (0.1 mL, 0.77 mmol) were added to the reaction mixture. The reaction mixture was refluxed for 1 h, then cooled to rt and diluted with H2O (50 mL). The product was extracted with ethyl acetate (3 × 30 mL) and the organic fractions were combined, dried over Na2SO4 and evaporated to dryness. The residue was purified by flash-chromatography on silica gel. The elution was carried out by CHCl3 (50 mL), followed by the mixture CHCl3-CH3OH-NH4OH (10:3:1). Fractions which contained the target compound were combined and evaporated to dryness, resulting in 1,3,6′,3″-tetra-N-Cbz-6″-(pyridin-1-ium)-6″-deoxykanamycin A (167 mg, 98% yield) as a white solid.
HRMS (ESI): calculated for [C55H64N5O18]+ = 1082.4246, found [M]+ = 1082.4249, Rf (CHCl3:MeOH 100:15) = 0.17, Rt (system A2) = 11.09 min.
1H NMR (δ, ppm, J/Hz): 1.43–3.46 (m, 7H, cHex), 3.1–4.56 (m, 7H, CH1′-6′), 3.15–4.95 (m, 7H, CH1″-6″), 4.86–5.11 (m, 8H, -OCH2), 6.78 (t, 1H, 3J = 4.9 Hz, -NH-CH2 CH6′-), 7.1 (s, 1H, -NH-CH2 CH3″-), 7.14 (s, 1H, -NH-CHcHex3-), 7.25–7.39 (m, 20H, 4×Ph), 7.39 (s, 1H, -NH-CHcHex1-) 8.12 (s, 2H, Py3′), 8.58 (s, 1H, Py4′), 8.81 (s, 2H, Py2′).
13C NMR (δ, ppm, J/Hz): 33.7, 41.2, 49.1, 50.3, 55.9, 61.9, 65.0, 65.3, 68.9, 69.1, 70.2, 70.3, 71.5, 72.2, 73.9, 79.1, 84.7, 98.1, 101.0, 127.1, 127.6, 128.2, 145.0, 145.4, 155.5, 155.8, 156.5, 156.6.
2nd step
1,3,6′,3″-Tetra-N-Cbz-6″-( pyridin-1-ium)-6″-deoxykanamycin A (54 mg, 0,05 mmol) was dissolved in CH3OH (2 mL), then Pd/C (5%, 85 mg) was added and the reaction mixture was acidified to pH 3 by the addition of CH3COOH. The reaction mixture was vigorously stirred at rt at H2 flow (1 atm) for 3 h. The catalyst was filtered off via celite; the cake was washed with CH3OH (5 mL) and H2O (5 mL), and the combined filtrate was concentrated in a vacuum. The target compound was precipitated by the addition of acetone (10 mL), filtered off and dried in a vacuum over P2O5, resulting in compound 4 (25 mg, 58% yield) as a white solid.
Anal. calculated for [C23H44N5O10×5AcOH×5H2O]: C, 42.12; H, 7.93; N, 7.44, O 42.51. Found: C, 42.10; H, 7.90; N, 7.43.
HRMS (ESI): calculated for [C
23H
45N
5O
10]
+ = 552.3245, found [M+H]
+ = 552.3216, R
f (NH
4OH:iPrOH 10:7) = 0.31, T
mp = 201 °C (decomp.). The assignment of the signals in the
1H and
13C NMR spectra is presented in the
Table S1.
6″-(Pyridin-1-ium)-6″-deoxykanamycin A trifluoroacetate (5)
1st step
Compound 3 (60 mg, 0.052 mmol) was dissolved in dry pyridine (2 mL), then NaI (1 mg, 0.005 mmol) and iPr2EtN (34 mg, 0.26 mmol) were added to the reaction mixture. The obtained solution was refluxed for 1 h, then cooled to room temperature and diluted with H2O (40 mL). The product was extracted with ethyl acetate (3 × 15 mL) and the organic fractions were combined, dried over Na2SO4, filtered off and evaporated to dryness in a vacuum. The product was purified by flash chromatography on silica gel and the elution was carried by the mixture CHCl3-CH3OH (10:1, 25 mL) followed by the mixture CHCl3-CH3OH-NH4OH (10:5:3). Fractions which contained the target compound were combined and evaporated to dryness in a vacuum, resulting in 1,3,6′,3″-tetra-N-Boc-6″-(pyridin-1-ium)-6″-deoxykanamycin A (35 mg, 71% yield) as a white solid.
HRMS (ESI): calculated for [C43H72N5O18]+ = 946.4872, found [M]+ = 946.4979, Rf (CHCl3:MeOH 100:15) = 0.15.
2nd step
Trifluoroacetic acid (0.2 mL, 2.752 mmol) was added dropwise to the solution of 1,3,6′,3″-tetra-N-Boc-6″-(pyridin-1-ium)-6″-deoxykanamycin A (30 mg, 0.03 mmol) in CH2Cl2 (1 mL). The reaction mixture was stirred at rt for 4 h and then evaporated to dryness in a vacuum. The addition of Et2O (10 mL) to the residue resulted in the formation of the precipitate, which was filtered off, washed with Et2O (50 mL) and dried in a vacuum over P2O5, resulting in the target compound 5 (23 mg, 63% yield) obtained as a white solid.
HRMS (ESI): calculated for [C23H40N5O10]+ = 546.2775, found [M]+ = 546.2752, Rf (NH4OH:iPrOH 10:7) = 0.3, Tmp = 205 °C (decomp.).
The assignment of the signals in the
1H and
13C spectra is presented in
Table S1.
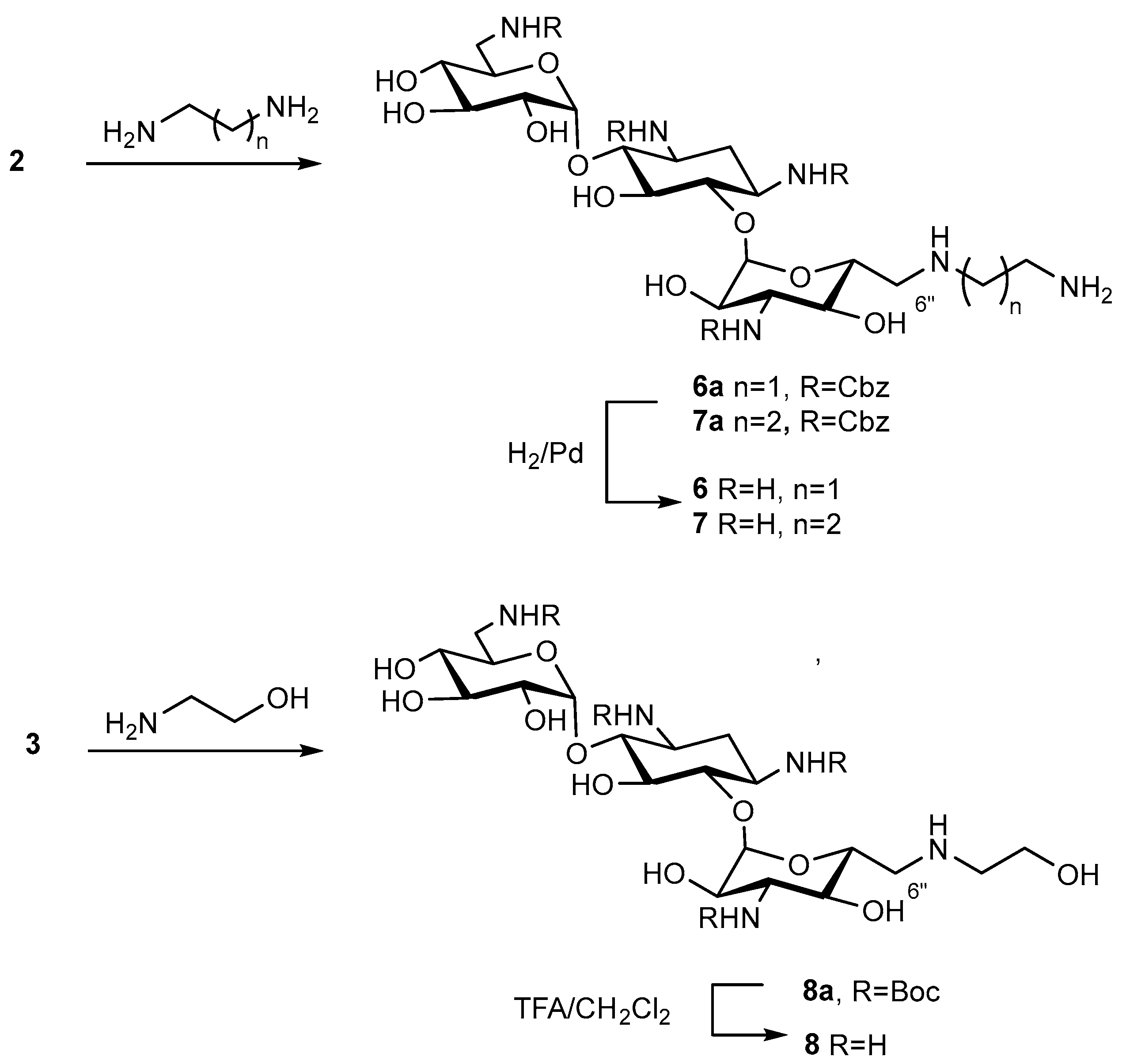
6″-(2-Aminoethylamino)-6″-deoxykanamycin A acetate (6)
1st step
6″-(2-Aminoethylamino)-1,3,6′,3″-tetra-N-Cbz-6″-deoxykanamycin A (6a)
A mixture of compound 2 (805 mg, 0.626 mmol) and ethylenediamine (3.5 mL) was stirred at rt for 24 h, then the reaction mixture was diluted with H2O (150 mL). The forming precipitate was filtered off, washed with H2O and dried in a vacuum at 45 °C and then in a vacuum over P2O5. The target 6″-(2-aminoethylamino)-1,3,6′,3″-tetra-N-Cbz-6″-deoxykanamycin A (6a) (612 mg, 92%) was obtained as a light-yellow solid and used without additional purification on the next stage.
HRMS (ESI): calculated for [C52H67N6O18]+ = 1063.4511 [C52H66N6O18Na]+ = 1085.1150 [C52H66N6O18K]+ = 1101.4071, found [M+H]+ = 1063.4506 [M+Na]+ = 1085.1326 [M+K]+ = 1101.4065, Rf (CHCl3:MeOH:HCOOH 13:5:0.1) = 0.45, Rt (System A4) = 17.5 min.
2nd step
6″-(2-Aminoethylamino)-1,3,6′,3″-tetra-N-Cbz-6″-deoxykanamycin A (6a) (61 mg, 0.057 mmol) was dissolved in CH3OH (2 mL) and 5% Pd/C (95 mg) was added to the reaction mixture. The CH3COOH was added to the reaction mixture until pH 3 was reached, and the mixture was vigorously stirred at H2 flow (1 atm.) for 4 h. The catalyst was filtered off via a celite layer; the cake was washed with CH3OH (5 mL) and H2O (5 mL), and the combined filtrate was concentrated in a vacuum. The target compound was precipitated by the addition of acetone (10 mL), filtered off and dried in a vacuum over P2O5, resulting in the target compound 6 (32.8 mg, 69%) as a white solid.
Anal. calculated for [C20H42N6O10×5AcOH×3H2O]: C, 40.91; H, 7.78; N, 9.54, O 41.77. Found: C, 40.91; H, 7.63; N, 9.50.
HRMS (ESI): calculated for [C20H43N6O10]+ = 527.3040, found [M+H]+ = 527.3032, Rf (NH4OH:iPrOH 10:7) = 0.32, Tmp = 181 °C (decomp.).
The assignment of the signals in the
1H and
13C spectra is presented in
Table S1.
6″-(3-Aminopropyl-1-amino)-6″-deoxykanamycin A acetate (7)
1st step
6″-(3-Aminopropyl-1-amino)-1,3,6′,3″-tetra-N-Cbz-6″-deoxykanamycin A (7a)
A mixture of compound 2 (200 mg, 0.156 mmol) and propan-1,3-diamine (3.5 mL) was stirred at rt for 24 h, then the reaction mixture was diluted with H2O (100 mL). The forming precipitate was filtered off, washed with H2O and dried in a vacuum at 45 °C and then in a vacuum over P2O5 at rt. The target 6″-(3-aminopropyl-1-amino)-1,3,6′,3″-tetra-N-Cbz-6″-deoxykanamycin A (7a) (136 mg, 81%) was obtained as a white solid and used without additional purification on the next stage.
HRMS (ESI): calculated for [C53H69N6O18]+ = 1077.4668, found [M+H]+ = 1077.4667, Rf (CHCl3:MeOH:HCOOH 13:5:0.1) = 0.42, Rt (System A4) = 16.5 min.
2nd step
6″-(3-Aminopropyl-1-amino)-1,3,6′,3″-tetra-N-Cbz-6″-deoxykanamycin A (7a) (138 mg, 0.129 mmol) was dissolved in CH3OH (2.5 mL) and 5% Pd/C (180 mg) was added to the reaction mixture. The CH3COOH was added to the reaction mixture until pH 3 was reached, and the mixture was vigorously stirred at H2 flow (1 atm.) for 4 h. The catalyst was filtered off via a celite layer; the cake was washed with CH3OH (5 mL) and H2O (5 mL), and the combined filtrate was concentrated in a vacuum. The target compound was precipitated by the addition of acetone (10 mL), filtered off and dried in a vacuum over P2O5, resulting in the target compound 7 (103 mg, 95%) as a white solid.
Anal. calculated for [C21H44N6O10×5AcOH×1H2O]: C, 43.35; H, 7.75; N, 9.78, O 39.12. Found: C, 43.35; H, 7.75; N, 9.78.
HRMS (ESI): calculated for [C21H45N6O10]+ = 541.3197, found [M+H]+ = 541.3122, Rf (NH4OH:iPrOH 1:1) = 0.15, Tmp = 176 °C (decomp).
The assignment of the signals in the
1H and
13C spectra is presented in
Table S1.
6″-(2-Hydroxyethyl-1-amino)-6″-deoxykanamycin A trifluoroacetate (8)
1st step
1,3,6′,3″-Tetra-N-Boc-6″-(2-hydroxyethyl-1-amino)-6″-deoxykanamycin A (8a)
A mixture of compound 3 (100 mg, 0.087 mmol) and 2-aminoethanol (1.5 mL) was stirred at rt for 24 h, then the reaction mixture was diluted with H2O (100 mL). The forming precipitate was filtered off, washed with H2O and dried in a vacuum at 45 °C and then in a vacuum over P2O5 at rt. The target 1,3,6′,3″-tetra-N-Boc-6″-(2-hydroxyethyl-1-amino)-6″-deoxykanamycin A (8a) (35 mg, 45%) was obtained as a white solid and used without additional purification on the next stage.
HRMS (ESI): calculated for [C40H74N5O19]+ = 928.4978, found [M+H]+ = 928.4988, Rf (CHCl3:MeOH:HCOOH 7:1:0.3) = 0.28.
1H NMR (δ, ppm, J/Hz): 1.36 (s, 9H, 3×-CH3), 1.37 (s, 9H, 3×-CH3), 1.38 (s, 18H, 6×-CH3), 1.47–3.47 (m, 7H, cHex), 2.90–4.91 (m, 7H, -CH1″-6″), 3.0 (s, 2H, -NH-CH2-), 3.1–4.92 (m, 7H, -CH1′-6′), 3.63 (s, 2H, -CH2-OH), 6.35 (bt, 1H, 3J = 4.5 Hz, -NH-CH2 CH6′-), 6.57 (d, 1H, 3J = 9.3 Hz, -NH-CH2 CH3″-), 6.64 (bd, 1H, 3J = 7.0 Hz, -NH-CHcHex3-), 6.97 (bs, 1H, -NH-CHcHex1-).
13C NMR (δ, ppm, J/Hz): 28.1–28.3, 34.5, 41.3, 48.5, 48.9, 49.8, 50.4, 55.6, 56.8, 68.4, 69.5, 69.9, 70.0, 70.5, 72.6, 75.4, 77.4–77.9, 78.0, 80.2, 84.3, 98.6, 101.1, 154.9–156.2.
2nd step
Trifluoroacetic acid (0.2 mL, 2.752 mmol) was added dropwise to a solution of 1,3,6′,3″-tetra-N-Boc-6″-(2-hydroxyethyl-1-amino)-6″-deoxykanamycin A (8a) (30 mg, 0.032 mmol) in CH2Cl2 (2 mL). The reaction mixture was stirred at rt for 24 h and then evaporated to dryness in a vacuum. The addition of acetone (10 mL) to the residue resulted in the formation of a white solid which was filtered off, washed with acetone (3 × 10 mL) and dried in a vacuum over P2O5. Finally, 6″-(2-Hydroxyethylamino)-6″-deoxykanamycin A (8) (10 mg, 27%) was obtained as a white solid in the form of trifluoroacetate.
Anal. calculated for [C20H41N6O10×5TFA×1H2O]: C, 32.35; H, 4.34; N, 7.55, F 25.59, O 30.17. Found: C, 32.35; H, 4.60; N, 7.50.
HRMS (ESI): calculated for [C20H42N5O11]+ = 528.2881, found [M+H]+ = 528.2870, Rf (NH4OH:iPrOH 1:1) = 0.32, Tmp = 186 °C (decomp.).
The assignment of the signals in the
1H and
13C spectra is presented in
Table S1.
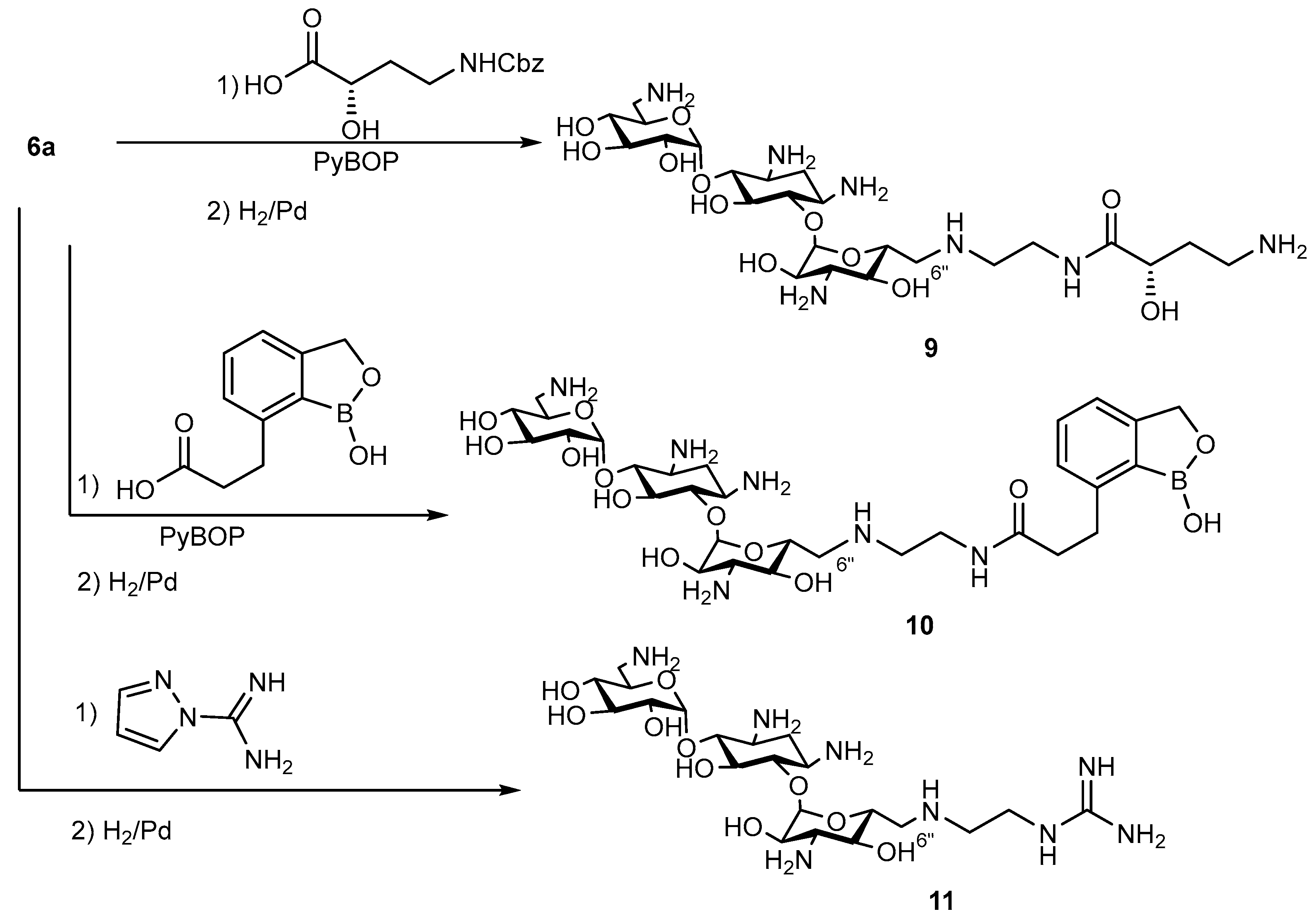
6″-((S)-7-((2-(4-Amino-2-hydroxybutanamido)ethyl)amino))-6″-deoxykanamycin A acetate (9)
1st step
Benzotriazol-1-yloxytripirrolidonophosphonium hexafluorophosphate (PyBoP) (196 mg, 0.39 mmol) was added portionwise to the solution of 6″-(2-aminoethylamino)-1,3,6′,3″-tetra-N-Cbz-6″-deoxykanamycin A (6a) (200 mg, 0.188 mmol) and (S)-4-(((benzyloxy)carbonyl)amino)-2-hydroxybutanoic acid (143 mg, 0.57 mmol) in DMSO (2 mL). The pH of the reaction mixture was kept ~8 by the addition of Et3N (~200 µL). The reaction mixture was stirred at rt for 48 h, then H2O (100 mL) was added. The forming precipitate was filtered off, washed with Et2O (50 mL), dried and purified by the column chromatography on silica gel. The elution was carried out by the mixture CHCl3:CH3OH:HCOOH (5:1:0.3). Fractions which contained the target compound were combined and evaporated to dryness, resulting in the target intermediate 6″-((S)-7-((2-(4-(benzyloxycarbonyl)amino-2-hydroxybutanamido)ethyl)amino))-1,3,6′,3″-tetra-N-Cbz-6″-deoxykanamycin A (164 mg, 67%) as a white solid.
HRMS (ESI): calculated for [C64H80N7O22]+ = 1298.53, found [M+H]+ = 1298.5398, Rf (CHCl3:MeOH:HCOOH 5:1:0.3) = 0.41, Rt (System A4) = 20.8 min.
1H NMR (δ, ppm, J/Hz): 1.51–3.56 (m, 7H, cHex), 1.57 (s, 1H, -CH1′- CH2′-), 1.83 (s, 1H, -CH1′- CH2′-), 2.65 (s, 2H, -CH2-NHCO), 2.66–5.00 (m, 7H, CH1″-6″), 3.08–4.98 (m, 7H, CH1′-6′), 3.1 (s, 2H, -CH2′- CH3′-), 3.18 (s, 2H, -NH-CH2-), 3.88 (s, 1H, -NHCO-CH1′-), 4.87–5.09 (m, 10H, -OCH2), 6.83 (t, 1H, 3J = 4.7 Hz, -NH-CH2 CH6′-), 7.02 (d, 1H, 3J = 9.0 Hz, -NH-CH2 CH3″-), 7.18 (d, 1H, 3J = 8.6 Hz, -NH-CHcHex3-), 7.22 (t, 1H, 3J = 5.5 Hz, -CH3′-NH-), 7.26–7.37 (m, 20H, 4×Ph), 7.41 (s, 1H, -NH-CHcHex1-), 7.81 (t, 1H, 3J = 5.3 Hz, -CH2-NH-CO).
13C NMR (δ, ppm, J/Hz): 34.3, 34.5, 37.1, 37.6, 41.6, 48.5, 49.6, 49.6, 50.2, 56.4, 64.9, 65.0, 65.1, 69.1, 69.4, 69.9, 70.5, 70.5, 70.7, 72.3, 72.7, 74.3, 80.4, 84.5, 97.4, 101.2, 127.5, 127.6, 128.2, 155.5, 155.9, 156.0, 156.4, 156.5, 164.5.
2nd step
The 6″-((S)-7-((2-(4-(Benzyloxycarbonyl)amino-2-hydroxybutanamido)ethyl)amino))-1,3,6′,3″-tetra-N-Cbz-6″-deoxykanamycin (150 mg, 0.116 mmol) was dissolved in CH3OH (3 mL), then 5% Pd/C (200 mg) was added and the reaction mixture was acidified to pH 3 by the addition of CH3COOH. The reaction mixture was vigorously stirred at rt at H2 flow (1 atm) for 2 h. The catalyst was filtered off via a celite layer; the cake was washed with CH3OH (5 mL) and H2O (5 mL), and the combined filtrate was concentrated in a vacuum. The target compound was precipitated by the addition of acetone (10 mL), filtered off and dried in a vacuum over P2O5, resulting in compound 9 in the form of acetate (97 mg, 91% yield) as a white solid.
Anal. calculated for [C24H49N7O12×5AcOH×1H2O]: C, 43.17; H, 7.57; N, 10.36, O 38.9. Found: C, 43.17; H, 7.57; N, 10.36.
HRMS (ESI): calculated for [C24H50N7O12]+ = 628.3517, found [M+H]+ = 628.3479, Rf (NH4OH:iPrOH 1:1) = 0.27, Tmp = 179 °C (decomp.).
The assignment of the signals in the
1H and
13C spectra is presented in
Table S1.
6″-(2-(3-(1-Hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-7-yl)-N-(ethyl-1-amino)propanamide)-6″-deoxykanamycin acetate (10)
1st step
1,3,6′,3″-Tetra-N-Cbz-6″-(2-(3-(1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-7-yl)-N-(ethyl-1-amino)propanamide)-6″-deoxykanamycin A
PyBoP (98 mg, 0.188 mmol) was added portionwise to the solution of 6″-(2-aminoethylamino)-1,3,6′,3″-tetra-N-Cbz-6″-deoxykanamycin A (6a) (100 mg, 0.094 mmol) and 3-(1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-7-yl)propanoic acid (58 mg, 0.282 mmol) in DMSO (2 mL). The pH of the reaction mixture was kept ~8 by the addition of Et3N (~50 µL). The reaction mixture was stirred at rt for 24 h, then Et2O (50 mL) was added. The forming precipitate was filtered off, washed with Et2O (5 × 10 mL) and dried over P2O5, and the obtained product was used without additional purification on the next stage (70 mg, 60%).
HRMS (ESI): calculated for [C62H76BN6O21]+ = 1251.5156, found [M+H]+ = 1251.5124, Rf (CHCl3:MeOH:HCOOH 11.5:3:1.5) = 0.37, Rt (System A5) = 16.0 min
2nd step
The 1,3,6′,3″-Tetra-N-Cbz-6″-(2-(3-(1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-7-yl)-N-(ethyl-1-amino)propanamide)-6″-deoxykanamycin A (60 mg, 0.048 mmol) was dissolved in CH3OH (3.5 mL), then 5% Pd/C (85 mg) was added and the reaction mixture was acidified to pH 3 by the addition of CH3COOH. The reaction mixture was vigorously stirred at rt at H2 flow (1 atm) for 2 h. The catalyst was filtered off via celite layer; the cake was washed with CH3OH (5 mL) and H2O (5 mL), and the combined filtrate was concentrated in a vacuum. The target compound was precipitated by the addition of acetone (10 mL) and was filtered off and dried in a vacuum over P2O5, resulting in compound 10 (22 mg, 45% yield) as a white solid.
HRMS (ESI): calculated for [C30H51BN6O13] = 714.3607, found [M+H]2+ = 357.1795, Rf (NH4OH:iPrOH 10:7) = 0.3, Rt (System A6) = 20.2 min, Tmp = 245 °C (decomp.).
The assignment of the signals in the
1H and
13C spectra is presented in
Table S1.
6″-(2-Guanidinoethylamino)-6″-deoxykanamycin A acetate (11)
1st step
1,3,6′,3″-Tetra-N-Cbz-6″-(2-guanidinoethylamino)-6″-deoxykanamycin A
Ethyldiisopropylamine (EDIA, 80 µL, 0.452 mmol) was added to mixture of 6″-(2-aminoethylamino)-1,3,6′,3″-tetra-N-Cbz-6″-deoxykanamycin A (6a) (160 mg, 0.151 mmol) and 1H-pyrazole-1-carboximidamide hydrochloride (33 mg, 0.25 mmol) in DMF (2 mL). The reaction mixture was stirred at rt for 24 h, then Et2O (50 mL) was added. The forming precipitate was filtered off, washed with Et2O (5 × 10 mL) and dried over P2O5, and the obtained product was used without additional purification on the next stage (125 mg, 75%).
HRMS (ESI): calculated for [C53H69N8O18]+ = 1105.4729, found [M+H]+ = 1105.4456, Rf (CHCl3:MeOH:HCOOH 5:1:0.3) = 0.4, Rt (System A4) = 16.9 min.
2nd step
The 1,3,6′,3″-Tetra-N-Cbz-6″-(2-guanidinoethylamino)-6″-deoxykanamycin A (125 mg, 0.113 mmol) was dissolved in CH3OH (4 mL), then 5% Pd/C (190 mg) was added and the reaction mixture was acidified to pH 3 by the addition of CH3COOH. The reaction mixture was vigorously stirred at rt at H2 flow (1 atm) for 4 h. The catalyst was filtered off via a celite layer; the cake was washed with CH3OH (5 mL) and H2O (5 mL), and the combined filtrate was concentrated in a vacuum. The target compound was precipitated by the addition of acetone (10 mL), filtered off and dried in a vacuum over P2O5, resulting in compound 11 (83 mg, 85% yield) as a white solid.
Anal. calculated for [C21H44N8O10×5AcOH×6H2O]: C, 38.11; H, 7.84; N, 11.47, O 42.58. Found: C, 38.11; H, 7.84; N, 11.47.
HRMS (ESI): calculated for [C21H45N8O10]+ = 569.3259, found [M+H]+ = 569.3467, Rf (NH4OH:iPrOH 1:1) = 0.28, Tmp = 202 °C (decomp.).
The assignment of the signals in the
1H and
13C spectra is presented in
Table S1.
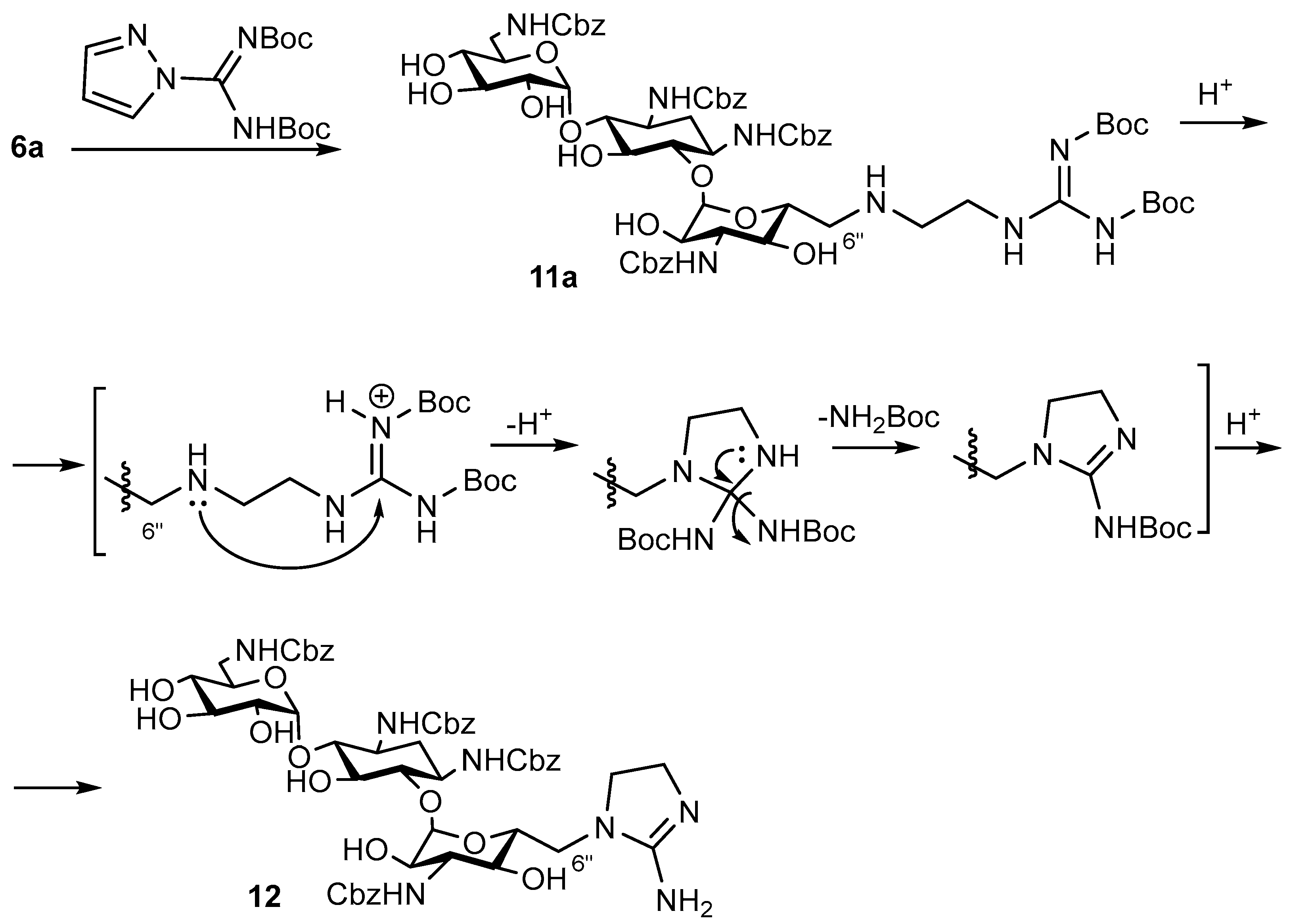
6″-(2-Amino-4,5-dihydro-1H-imidazol-1-yl)-1,3,6′,3″-tetra-N-Cbz-6″-deoxykanamycin A trifluoroacetate (12)
EDIA (120 µL, 0.66 mmol) was added to a mixture of 6″-(2-aminoethylamino)-1,3,6′,3″-tetra-N-Cbz-6″-deoxykanamycin A (6a) (234 mg, 0.221 mmol) and tert-butyl-(((tert-butyloxycarbonyl)amino)(1H-pyrazol-1-yl)methylen) carbamate (137 mg, 0.441 mmol) in DMF (4.5 mL). The reaction mixture was stirred at 50 °C for 24 h, then Et2O (150 mL) was added. The forming precipitate was filtered off, washed with Et2O (5 × 10 mL) and dried in a vacuum over P2O5. The resulting intermediate was further dissolved in CH2Cl2 (2 mL) and TFA (0.2 mL, 2.75 mmol) was added dropwise. The reaction mixture was stirred at rt for 24 h and then evaporated to dryness in a vacuum. The target compounds were purified by column chromatography on silica gel. The elution was carried out by the mixture CHCl3:MeOH:HCOOH (10:1.5:0.3). The fractions which contained the individual compounds were combined and evaporated to dryness, resulting in trifluoroacetate of 1,3,6′,3″-tetra-N-Cbz-6″-(2-guanidinoethylamino)-6″-deoxykanamycin A (white solid, 25 mg, 25%) and compound 12 in the form of trifluoroacetate (white solid, 20 mg, 23%).
Derivative 12. HRMS (ESI): calculated for [C53H66N7O18]+ = 1088.4386, found [M+H]+ = 1088.4459, Rf (CHCl3:MeOH:HCOOH 5:1:0.5) = 0.41, Rt (System A4)= 19.1 min.
1H NMR (δ, ppm, J/Hz): 1.50–3.59 (m, 7H, cHex), 3.06–4.98 (m, 7H, CH1″-6″), 3.09–4.86 (m, 7H, CH1′-6′), 3.47 (s, 2H, -NH-CH2-CH2-), 3.65 (s, 1H, -NH-CH2-CH2-), 3.72 (s, 1H), (s, 1H, -NH-CH2-CH2-), 4.45–5.1 (m, 8H, -OCH2), 6.80 (t, 1H, 3J = 4.8 Hz, -NH-CH2 CH6′-), 7.07 (d, 1H, 3J = 9.2 Hz, -NH-CH2 CH3″-), 7.17 (d, 1H, 3J = 8.2 Hz, -NH-CHcHex1-), 7.25–7.40 (m, 20H, 4×Ph), 7.36 (s, 1H, -NH-CHcHex3-), 7.55 (s, 1H, -NH-CH2-CH2-), 7.71 (s, 1H, -C-NH2).
13C NMR (δ, ppm, J/Hz): 34.3, 40.6, 41.5, 45.8, 48.5, 49.6, 50.4, 56.2, 65.1, 65.3, 69.3, 69.7, 70.0, 70.5, 70.7, 72.6, 73.0, 74.4, 79.8, 85.1, 97.9, 101.5, 127.6, 128.2, 136.9, 137.1, 137.2, 156.5, 156.6, 157.6, 157.9, 158.8.



 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








