

Recent Advances in Mitochondrial Fission/Fusion-Targeted Therapy in Doxorubicin-Induced Cardiotoxicity


Abstract
:1. Introduction
2. Molecular Insights into DOX-Induced Cardiotoxicity: Roles of Mitochondrial Dynamics Proteins from Recent In Vitro and In Vivo Reports
3. Roles of Pharmacological Interventions Targeting Mitochondrial Fission/Fusion Therapy in DOX-Induced Cardiotoxicity: A Report from Recent In Vitro and In Vivo Studies


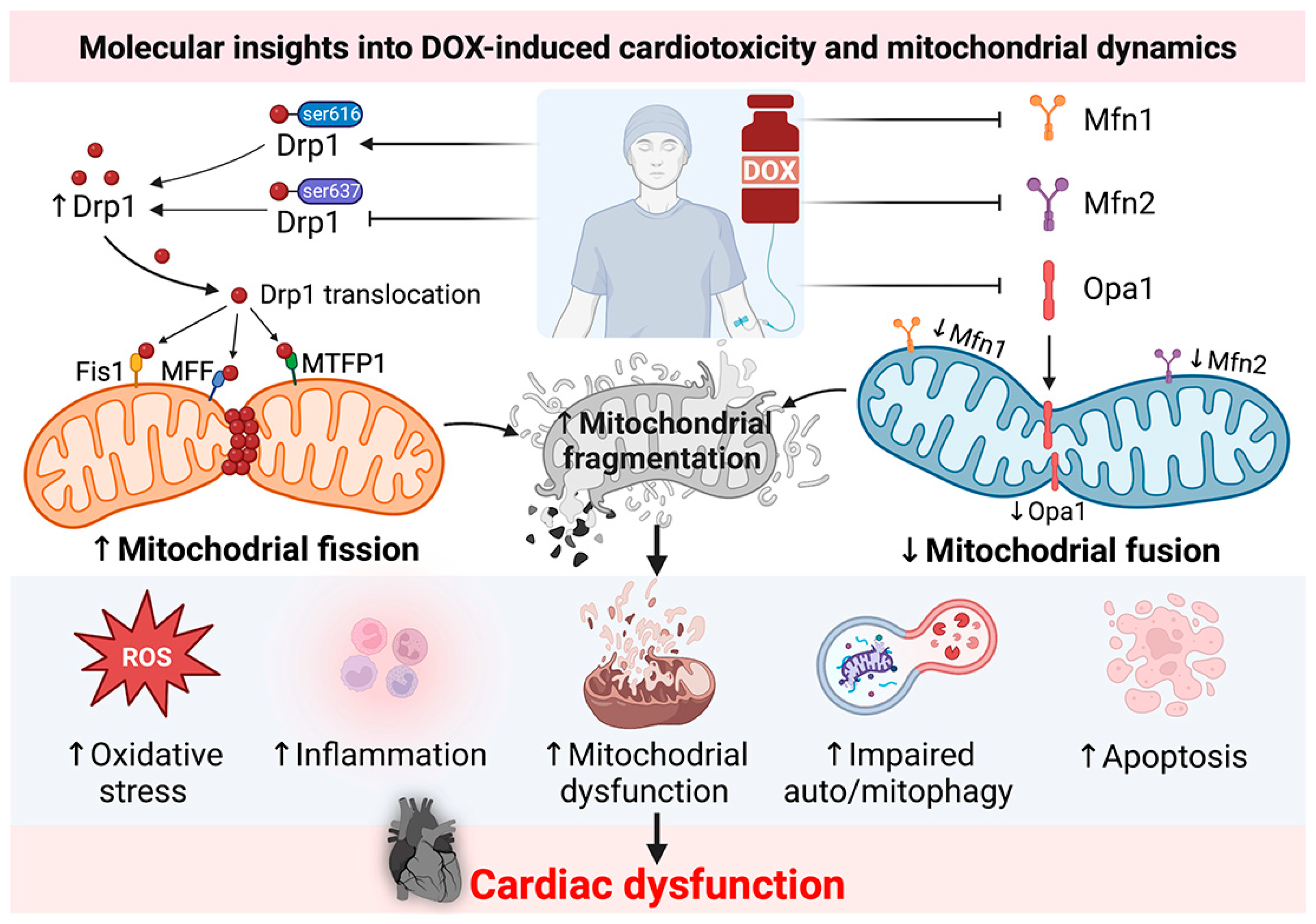{kind=link}
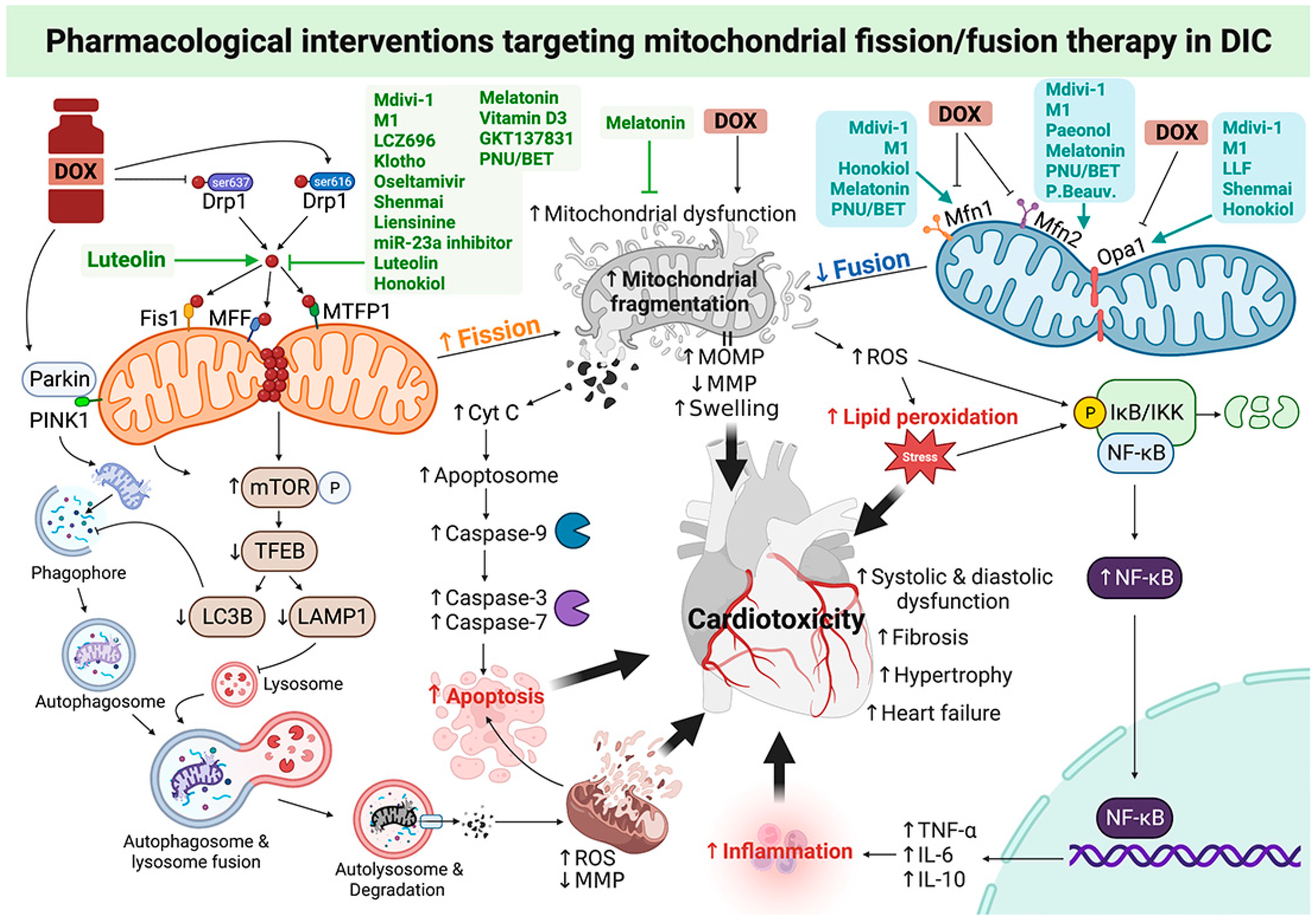{kind=link}
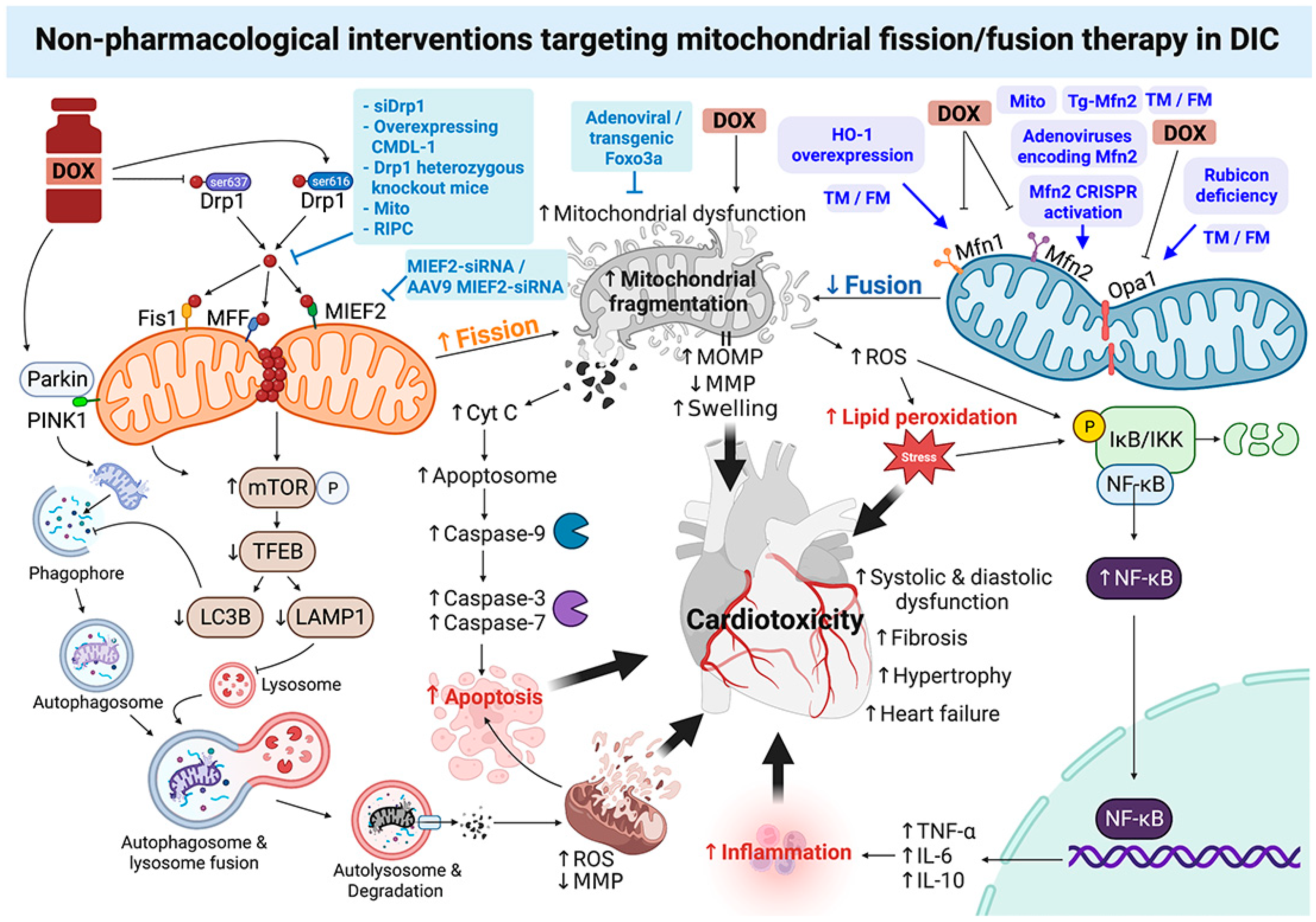{kind=link}
| Study Model | DOX Dosage | Intervention | Major Findings | Interpretation | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| Mitochondrial Dynamics/Morphology | Mitochondrial Function/Mitophagy/Autophagy | Cell Death/Inflammation/Oxidative Stress | Cell Viability/Cytotoxicity | Cardiomyocyte Function | |||||
| H9c2 cells | 0.3125 μM (48 h) | Mdivi-1 (5 μM, 48 h, co-treatment) | - | - | - | ↑Cell viability ↓LDH | - | Mdivi-1 increased cell viability with decreased cytotoxicity in H9c2 cells. | [16] |
| H9c2 cells | 5 μM (24 h) | Mdivi-1 (1 μM, 30 min, pre-treatment) | ↓p-Drp1ser616 | - | ↓Anexin5-positive cells ↓c-Caspase3 | - | - | Pre-treatment with Mdivi-1 and LCZ696 significantly inhibited mitochondrial fission, thus decreasing H9c2 cardiomyocyte apoptosis. | [28] |
| LCZ696 (20 μM, 30 min, pre-treatment) | |||||||||
| AC16 cells | 250 nM (24 h) | Mdivi-1 (24 h, co-treatment) | - | ↓ROS ↓MMP depolarization ↑CS ↑VDAC ↓LC3II-I ↓Beclin-1 ↑p62 ↓PINK1 ↓Parkin ↑PGC1-α ↑NRF1 ↑TFAM | - | ↑Cell viability | - | Mdivi-1 prevented DOX-induced mitochondrial superoxide overproduction and mitochondrial dysfunction by inhibiting mitophagy and preserving mitochondrial biogenesis. | [18] |
| H9c2 cells | 5 μM (24 h) | Klotho (0.1 μg/mL, 24 h, pre-treatment) | ↓p-Drp1ser616 | ↓MMP depolarization | ↓c-Caspase3 ↓TUNEL-positive cells ↓Annexin V- positive cells | - | ↑Number of cardiomyocytes ↑Peak shortening ↑+dp/dt ↑-dp/dt | Klotho pre-treatment alleviated DOX-induced cardiomyocyte dysfunction via inhibition of apoptosis, mitochondrial dysfunction, and fission in H9c2 cells. | [20] |
| H9c2 cells | 0.75 μmol/mL (24 h) | Loulu flowers (200 μg/mL, 2 h, pre-treatment) | ↑Opa1 ↑Mfn1 ↓MFF ↓Fis1 | ↓ROS ↓MMP depolarization | ↑SOD ↓p-p65 ↓p-IKK ↓p-IkBa ↓TUNEL-positive cells ↓Bax ↑Bcl-2 ↓c-Caspase3 ↓c-Caspase9 ↓c-PARP | - | - | Pre-treatment with LLF attenuated the DOX-induced decrease in viability, ROS production, apoptosis, inflammation, and aberrant expression of mitochondrial dynamics-related proteins in H9c2 cells. | [35] |
| H9c2 cells | 1 μM (12 h) | NEU1 inhibitor oseltamivir (10 μM, 2 h, pre-treatment) | ↓Drp1 | ↓ATG5 ↓Beclin-1 ↓LC3I/II ↓PINK1 ↓Parkin ↓p62 | ↓NEU1 ↓c-Caspase3 ↓c-Caspase9 ↓TUNEL-positive cells | - | - | Pre-treatment with a NEU1 inhibitor could suppress Drp1-dependent mitochondrial fission and PINK1/Parkin pathway-mediated mitophagy and alleviate cellular apoptosis in H9c2 cells. | [34] |
| H9c2 cells | 1 μM (16 h) | Shenmai injection (0.5%, 8 h, pre-treatment) | ↑Aspect ratio ↑Form factor ↑L-Opa1/S-Opa1 ↑p-Drp1ser637 ↑p-AMPKThr172 | ↓ROS ↓MMP depolarization ↓OCR | ↓Bax/Bcl-2 ↓c-Caspase3 ↓Annexin V-positive cells | ↑Cell viability | - | Shenmai rescued DOX-injured H9c2 cardiomyocytes from apoptosis and mitochondrial dysfunction by preventing DOX-induced excessive mitochondrial fission and insufficient mitochondrial fusion. | [33] |
| NMVMs | 5 μM (24 h) | Liensinine (20 μM, 24 h, co-treatment) | ↓Fragmented mitochondria ↓p-Drp1ser616 | ↓MMP depolarization ↓ROS ↑TOM20 ↑TIM23 ↓Rab7 ↓LRRK2 | ↓TUNEL-positive cells ↓p-ERK | - | - | Liensinine suppressed Drp1-mediated mitochondrial fragmentation, mitochondrial dysfunction, and apoptosis in NMVM cardiomyocytes. | [38] |
| H9c2 cells | 3 μM (24 h) | Melatonin (10 μM, 24 h, pre-treatment) | ↓Fragmented mitochondria | ↑ATP | - | ↑Cell viability | - | Pre-treatment with melatonin followed by DOX exposure decreased mitochondrial fragmentation and increased ATP production, resulting in preserved H9c2 rat cardiomyoblast viability. | [30] |
| NRVMs | 3 μM (24 h) | miR-23a inhibitor (100 nM, 48 h, pre-treatment) | ↔Mfn2 ↓p-Drp1 | ↓MMP depolarization ↓ROS ↑PGC1-α | ↓miR-23a ↓Dead cells ↓Cyt c ↓c-Caspase3 | ↑Cell viability | - | Inhibition of miR-23a attenuated NRVMs cardiomyocyte damage by directly targeting PGC-1α/p-Drp1, thereby inhibiting mitochondria-dependent apoptosis. | [40] |
| H9c2 cells | 1 μM (24 h) | Luteolin (20 μM, 24 h, co-treatment) | ↓Fragmented mitochondria ↓p-Drp1ser616 | ↓ROS | ↑SOD activity ↓Apoptotic cells ↓Bax ↑Bcl-2 ↑Bcl-xL ↓c-Caspase3 | - | - | Luteolin ameliorated DOX-induced toxicity in H9c2 cells via inhibiting mitochondrial fission, mitochondrial dysfunction, and apoptosis. | [32] |
| H9c2 cells | 0.3125 μM (48 h) | M1 (5 μM, 48 h) | - | - | - | ↑Cell viability ↓LDH | - | M1 increased cell viability with decreased cytotoxicity in H9c2 cells. | [16] |
| Primary cardiomyocytes | 3 μM (24 h) | Paeonol (50 μM, 24 h, co-treatment) | ↓Mitochondrial number ↑Mitochondrial size ↑Mfn2 | ↓MMP depolarization ↓ROS ↑OCR | ↓Apoptotic cells | ↑Cell viability ↓LDH | Pae enhanced Mfn2-mediated mitochondrial fusion, restored mitochondrial function and cardiomyocyte viability under the DOX conditions. | [43] | |
| NRVMs | 5 μM (24 h) | Honokiol (10 μM, 24 h, co-treatment) | ↓Fragmented mitochondria ↑Opa1 ↑Mfn1 ↓Drp1 | ↓MnSOD | ↓Apoptotic cells | - | - | Honokiol increased the activation of SIRT3 and Mfn1/Opa1-mediated fusion to prevent DOX-induced ROS production, mitochondrial damage, and cell death in rat neonatal cardiomyocytes. | [37] |
| AMCM cells | 1 μM (24 h) | Luteolin (10 μM, 24 h, co-treatment) | ↑p-Drp1ser616 ↓Mitochondrial elongation | ↓ROS ↓MMP depolarization ↑LAMP1 ↑TFEB ↓p-mTORser2448 | ↓TUNEL-positive cells ↓Bax ↓c-Caspase9 ↑Bcl2 ↑LC3BII ↑PINK1 ↑Parkin ↑Bnip3 | ↓LDH ↓CK | ↑Cell length ↑-dL/dt ↑+dL/dt | Luteolin reduced DOX-induced ROS accumulation, MMP collapse, and apoptosis by facilitating autophagosome formation and improving lysosomal function, possibly through a Drp1/mTOR/TFEB-dependent mechanism. | [19] |
| Study Model | DOX Dosage | Intervention | Major Findings | Interpretation | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| Mitochondrial Dynamics/Morphology | Mitochondrial Function/Mitophagy/Autophagy/ | Oxidative Stress/Inflammation/Cell Death | Heart Parameters | |||||
| Male Wistar rats | 18 mg/kg (3 mg/kg, 6 doses, days 0, 4, 8, 15, 22, and 29, IP) | Mdivi-1 (1.2 mg/kg, daily for 30 days, IP) | ↓Drp1 ↓p-Drp1ser616 ↑Mfn1 ↑Mfn2 ↑Opa1 ↓Mitochondrial volume density ↑Mitochondrial area | ↑RCR ↓ROS ↓MMP depolarization ↓Swelling ↓Parkin ↓Beclin-1 ↓p62 ↓LC3II/I | ↓MDA in serum and tissue ↓TNF-α ↓IL-6 ↓Bax ↓c-Caspase3 ↓Cyt c ↓TUNEL | ↑HR ↑LVESP ↑+dp/dt ↑SV ↑SBP ↑DBP ↑EF ↑FS ↑E/A ratio ↓LVEDP ↓-dp/dt ↓LF/HF ratio ↓NT-proBNP ↓Troponin-I | Mdivi-1 reduced mitochondrial dysfunction, oxidative stress, inflammation, and apoptosis, leading to improved cardiac function via modulation of mitochondrial fission/fusion proteins. | [16] |
| C57BL/6 mice | 12 mg/kg (4 mg/kg at days 0, 7, and 14, IP) | Klotho (0.01 mg/kg, every 48 h for 4 weeks, IP) | ↓p-Drp1ser616 ↑Mitochondrial length | ↑ATP content | ↓c-Caspase3 | ↑HW/BW ↓CK activity ↓Fibrosis ↓Disorganized myofibers ↑EF ↑FS ↓LVESD ↓LVEDD | Klotho alleviated DOX-induced cardiotoxicity by reducing apoptosis and mitochondrial fission through downregulation of Drp1 expression. | [20] |
| Male Sprague Dawley rats | 15 mg/kg (2.5 mg/kg, 6 times within 2 weeks, IP) | NEU1 inhibitor oseltamivir (20 mg/kg, 31 days, PO) | ↓Drp1 | ↓LC3II ↓Beclin-1 ↓ATG5 ↑p62 ↓PINK1 ↓Parkin | ↓NEU1 ↓LDH ↑T-AOC ↑GSH ↑SOD ↓H2O2 ↓c-Caspase3 ↓c-Caspase9 ↓Bax ↓Bad ↑Bcl-2 ↓TUNEL-positive cells | ↑EF ↑FS ↓LVEDs ↑Cardiomyocyte area ↓Fibrosis ↑Cross sectional area ↓c-TnT ↓CKMB | A NEU1 inhibitor effectively suppressed Drp1-dependent mitochondrial fission, autophagy, mitophagy, and apoptosis, leading to attenuation of cardiac dysfunction and remodeling in a rat model. | [34] |
| Female Sprague Dawley rats | 12 mg/kg (4 mg/kg, on days 4, 8 and 12, IP) | Melatonin (6 mg/rat/day, 14 days, drinking water) | ↑Mfn1 ↑Mfn2 ↓Drp1 ↓hFis1 | ↓PINK1 ↑PGC1-α ↑SIRT1 | ↓c-Caspase3 ↓c-PARP | ↑Cardiac output ↑Total work performance ↑HW | Melatonin effectively decreased Drp1-Fis1-mediated fission and apoptosis, increased Mfn1/2-mediated fusion, cellular ATP levels, and mitochondrial biogenesis, contributing to improved cardiac function in a rat model. | [30] |
| Female Balb/c mice | 30 mg/kg (10 mg/kg, on days 2, 8 and 15, IP) | Vitamin D3 (11,500 IU/kg, 14 days, pre-treatment) | ↓p-Drp1ser616 | - | ↓4-HNE ↓NQO1 ↓c-Caspase3 | ↑EF ↑SV ↑FS | Vitamin D3 pretreatment reduced mitochondrial fission-associated oxidative stress and apoptosis, resulting in improved cardiac function in a mouse model. | [45] |
| C57BL/6J mice | 12 mg/kg (4 mg/kg, 3 weekly injections at 0, 7, and 14 days, IP) | GKT137831 (dual inhibitor of NOX1 and NOX4) (60 mg/kg, once a day after the first DOX injection, PO) | ↓p-Drp1ser616 ↑p-Drp1ser637 ↑Mitochondrial length/width | - | ↓NLRP3 ↓c-Caspase1 ↓IL1-b ↓IL1-18 ↓p-NF-kB ↓NOX1 ↓NOX4 ↓GSDMD-NT | ↑EF ↑FS ↓LVESD ↓LVEDD ↑HW/BW | GKT137831 effectively suppressed DOX-induced Drp1-mediated mitochondrial fission and the consequent NLRP3 inflammasome activation and pyroptosis in a mouse model. | [29] |
| Male Wistar rats | 18 mg/kg (3 mg/kg, 6 doses, days 0, 4, 8, 15, 22, and 29, IP) | a7nAChR agonist (PNU: PNU-282987, 3 mg/kg, daily for 30 days, IP) | ↓Drp1 in mitochondria ↓p-Drp1ser616 ↑Mfn1 ↑Mfn2 | ↑RCR ↓ROS ↓MMP depolarization ↓Swelling ↓PINK1 ↓Parkin ↓Beclin-1 ↓p62 ↓LC3II/I | ↓MDA in serum and tissue ↓TNF-α ↓IL-6 ↓Bax ↓c-Caspase3 ↓TUNEL-positive cells | ↑HR ↑SV ↑SBP ↑DBP ↑EF ↑FS ↑E/A ratio ↓LF/HF ratio ↓NT-proBNP ↓Troponin-I | a7nAChR agonists and mAChR agonists effectively protected the heart against DOX cardiotoxicity via promoting Mfn1/2-induced mitochondrial fusion and inhibiting Drp1-induced mitochondrial fission, as well as reducing mitochondrial dysfunction, autophagy, mitophagy, inflammation, and apoptosis in rats. | [15] |
| mAChR agonist (BET: bethanechol, 12 mg/kg, daily for 30 days, IP) | ||||||||
| Male Wistar rats | 18 mg/kg (3 mg/kg, 6 doses, days 0, 4, 8, 15, 22, and 29, IP) | M1 (2 mg/kg, daily for 30 days, IP) | ↓Drp1 ↓p-Drp1ser616 ↑Mfn1 ↑Mfn2 ↑Opa1 ↓Mitochondrial volume density ↑Mitochondrial area | ↑RCR ↓ROS ↓MMP depolarization ↓Swelling ↓Parkin ↓Parkin ↓Beclin-1 ↓p62 ↓LC3II/I | ↓MDA in serum and tissue ↓TNF-α ↓IL-6 ↓Bax ↓c-Caspase3 ↓Cyt c ↓TUNEL | ↑HR ↑LVESP ↑+dp/dt ↑SV ↑SBP ↑DBP ↑EF ↑FS ↑E/A ratio ↓LVEDP ↓−dp/dt ↓LF/HF ratio ↓NT-proBNP ↓Troponin-I | M1 reduced mitochondrial dysfunction, oxidative stress, inflammation, and apoptosis, leading to improved cardiac function via modulation of mitochondrial fission/fusion proteins. | [16] |
| Male Sprague Dawley rats | 15 mg/kg (5 mg/kg, on days 1, 6 and 11, 3 times/2 weeks, IP) | Paeonol (150 mg/kg, daily for 3 days, PO, post-treatment) | ↑Mitochondrial size ↑Mfn2 | ↑Complex-III | ↓LDH ↓CK-MB | ↑EF ↑FS ↓LVESD ↑LVSP ↓LVEDP ↑+dp/dt ↑−dp/dt | Pae enhanced Mfn2-mediated mitochondrial fusion, restored mitochondrial function and cardiac performance in DIC rats. | [43] |
| Male BALB/c mice | 15 mg/kg (5 mg/kg, every 15 days for a total of three doses, IP) | Honokiol (0.2 mg/kg, daily, 45 days, IP) | ↓Fragmented mitochondria ↓Mitochondrial damage ↑Opa1 ↑Mfn1 | ↑Sirt3 ↑OGG1 ↓MnSOD ↓8-Oxo-dG ↓Mitochondrial citrate synthase ↑ATP | ↓TUNEL-positive cells ↑Bcl-2 | ↓HW/TL ↑FS ↓Fibrosis | Honokiol successfully blocked DIC in mice via promotion of Mfn1/Opa1-mediated fusion, reducing mitochondrial DNA damage, and improving mitochondrial function, leading to amelioration of cardiac dysfunction. | [37] |
| Male C57BL/6 mice | 20 mg/kg (5 mg/kg, at first, third, fifth and seventh day, IP) | Total flavonoids of Selaginella tamariscina (P.Beauv.) Spring (70–140 mg/kg, 8 days, PO) | ↑Mfn2 | ↑PPAR-α ↑PGC1-α ↑Sirt3 ↓PERK ↓ATF4 ↓CHOP | ↓LDH ↑SOD ↓MDA ↑GSH ↑CAT ↓Bcl-2 ↓Bax ↓Caspase9 ↓Cyt c | ↑EF ↑FS ↑E/A ratio ↓CKMB ↓BNP ↓cTnT ↓Fibrosis | Total flavonoids of Selaginella tamariscina effectively prevented DIC by enhancing Mfn2-mediated fusion, leading to alleviation of mitochondrial dysfunction and endoplasmic reticulum stress by activating Mfn2/PERK in a mouse model. | [46] |
4. Roles of Non-Pharmacological Interventions Targeting Mitochondrial Fission/Fusion Therapy in DOX-Induced Cardiotoxicity: A Report from Recent In Vitro and In Vivo Studies

| Study Model | DOX Dosage | Intervention | Major Findings | Interpretation | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| Mitochondrial Dynamics/Morphology | Mitochondrial Function/Mitophagy/Autophagy | Cell Death/Inflammation/Oxidative Stress | Cell Viability/Cytotoxicity | |||||
| H9c2 cells | 750 nM (24 h) | siDrp1 (knockdown of Drp1) | ↑Mitochondrial size ↑Form factor ↑Aspect ratio ↑Mitochondrial number | ↓Mitophagy foci | ↓PI-positive cells ↓c-PARP ↓c-Caspase3 | - | Drp1 knockdown decreased mitochondrial fragmentation, mitophagy flux, and apoptosis in H9c2 cardiomyocytes. | [36] |
| Mice cardiomyocytes | 3 μM (24h) | Adenoviral Foxo3a (24h, pre-treatment) | ↓Fragmented mitochondria | - | ↑Foxo3a ↓TUNEL-positive cells ↓c-Caspase3 | - | Promoting Foxo3a and inhibiting MIEF2 attenuates DOX-induced mitochondrial fission and apoptosis in cardiomyocytes. | [42] |
| Adenoviral MIEF2-siRNA (24h, pre-treatment) | ↓Fragmented mitochondria ↓MIEF2 | - | ↓TUNEL-positive cells ↓c-Caspase3 | - | ||||
| H9c2 cells | 2 μM (24h) | Lentiviral overexpressing CMDL-1 (24h, pre-treatment) | ↑CMDL-1 ↓Fragmented mitochondria ↓Drp1 ↑p-Drp1ser637 | - | ↓Apoptotic cells | - | CMDL-1 played an anti-apoptotic role in DOX cardiotoxicity by promoting p-Drp1ser637-mediated mitochondrial fission and fragmentation. | [31] |
| NRVMs | 1.72 μM (4, 8, 24, and 48 h) | Mfn2 CRISPR activation plasmid (48 h, pre-treatment) | ↑Mfn2 (48 h) ↓Fragmented mitochondria (4 h) | ↓ROS (8 h) | ↓Caspase3 activity (8 h) ↓TUNEL-positive cells (24 h) | - | An increase in cardiomyocyte levels of Mfn2 attenuated the DOX-induced increase in mitochondrial fission and prevented mitochondrial ROS production, thus leading to the prevention of the DOX-induced cardiomyocyte apoptosis. | [41] |
| NRVMs | 3 μM (24 h) | Adenoviruses encoding the Mfn2 gene (Ad-Mfn2) (48 h, pre-treatment) | ↑Mitochondrial size ↓Mitochondrial number ↑Mfn2 | ↓MMP depolarization ↓ROS ↑OCR ↑ECAR ↑CI ↑CIII | ↓Caspase3 activity ↓TUNEL-positive cells ↓Cyt c ↓FoxO1 | ↑Cell viability ↓LDH | Restoration of Mfn2-mediated mitochondrial enhanced mitochondrial oxidative metabolism reduced cellular injury and apoptosis, and inhibited mitochondria-derived oxidative stress in the DOX-treated cardiomyocytes. | [39] |
| Study Model | DOX Dosage | Intervention | Major Findings | Interpretation | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| Mitochondrial Dynamics/Morphology | Mitochondrial Function/Mitophagy/Autophagy | Oxidative Stress/Inflammation/Cell Death | Heart Parameters | |||||
| Mice | 15 mg/kg (single dose, euthanized after 3 days, IP) | The Drp1 heterozygous knockout mice (Drp1+/−) | ↑Mitochondrial length ↑Mitochondrial width ↑Mitochondrial size | ↓LC3-II | ↓LDH ↓c-Caspase3 ↓Oxyblot ↓4HNE ↓p-PDH | ↓cTnI | Drp1-deficient mice were protected from DOX-induced cardiac damage via inhibiting Drp1-dependent mitochondrial fragmentation, autophagy, and apoptosis. | [36] |
| Male C57BL/6 mice | 20 mg/kg (5 mg/kg, weekly for 4 consecutive weeks, IP) | Foxo3a transgenic model (pre-treatment) | ↓Fragmented mitochondria | - | ↓TUNEL-positive cells | ↓LVIDd ↑FS | Foxo3a overexpression and knockdown of MIEF2 attenuated DOX-induced mitochondrial fission and apoptosis, leading to improved cardiac function in a mouse model. | [42] |
| AAV9 MIEF2-siRNA (2 × 1011 vector genomes (vg)/mouse, pre-treatment) | ||||||||
| FVB/NJ mice (Rubicon-deficient mice) | 20 mg/kg (single dose, euthanized on day 3, IP) | Rubicon-deficiency-generated by piggyBac transposition | ↓Fragmented mitochondria ↑Opa1 | ↓Rubicon ↑ATP ↓LC3II ↓P62 ↑Parkin | ↓LDH ↓ROS | ↓Fibrosis ↓CK-MB | Loss of Rubicon ameliorated DOX-induced cardiotoxicity through enhancement of Opa1-mediated mitochondrial fusion and improvement of autophagic flux and mitophagy in Rubicon-deficient mice. | [52] |
| Male and female mixed FVB/C57BL/6 mice | 18 mg/kg (6 mg/kg, every third day for a week, 14 days, IV) | Humanized HO-1 overexpressing (HBAC) | - | - | ↑HO-1 ↓CD11b+ | ↑EF ↓EDD ↓ESD ↓LVEDD ↓LVESD ↑LVPWtd ↑LVPWts | Mice globally overexpressing human HO-1 were protected from DOX-induced dilated cardiomyopathy, cardiac cytoarchitectural derangement, and infiltration of mononuclear phagocytes. Cardiac-specific overexpression of HO-1 ameliorated DOX-mediated dilation of the sarcoplasmic reticulum as well as mitochondrial fragmentation and increased numbers of damaged mitochondria in autophagic vacuoles. | [50] |
| Cardiac-specific HO-1 overexpression (cs-HO-1) | ↓Mitochondrial number ↑Mitochondrial area ↑Mfn1 ↑Mfn2 ↓Fis1 | ↑mtDNA ↑Pol-grammar ↑COX3 ↑Nrf1 ↑PGC1-α ↑TFAM ↑PINK1 | - | - | ||||
| Adult male Sprague Dawley rats | 12.5 mg/kg (3.125 mg/kg, 4 separated time points, once every 5 days, 20 days, IP) | Implantation of mitochondria (Mito) (500 μg/rat, day-21 after DOX induction, euthanized by day 60, intramyocardial injection) | ↓Drp1 ↑Mfn2 | ↑PGC1-α ↑TFAM ↑Nrf1 ↑Nrf2 ↑EER-a ↑mitoDNA expression ↑Beclin-1 ↑ATG-5 ↑LC3-II/I ↓CypD1 ↑OXPHOS | ↓γ-H2AX-positive cells ↓NOX1 ↓NOX2 ↓p22phox ↓Oxidative index ↓c-Caspase3 ↓Bax ↓c-PARP ↓Cyt c | ↑EF ↓HW/Tibial length ↓LW/Tibial length ↓Fibrosis ↓Hypertrophy ↑Cx43 ↓BNP+ | Implantation of mitochondria effectively inhibited Drp1-mediated fission and promoted Mfn2-mediated fusion, reducing oxidative stress, autophagy, apoptosis, and mitochondrial damage, leading to preservation of LVEF and myocardial remodeling in DIC rats. | [48] |
| Male C57BL/6 mice | 15 mg/kg (5 mg/kg, consecutive 3 weeks, IP) | Cardiac-specific Mfn2 Transgenic mice (Tg-Mfn2) | ↑Mfn2 ↓Mitochondrial number ↑Mitochondrial size | ↓ROS | ↓TUNEL-positive cells ↓Caspase3 activity ↓MDA ↑SOD ↓FoxO1 | ↑EF ↓LVESV ↓cTnT ↓Fibrosis | Restoration of Mfn2-mediated mitochondrial fusion reduced cardiac dysfunction, cellular injury/apoptosis, and mitochondria-derived oxidative stress in DOX-treated mice. | [39] |
| White pigs | 2.25 mg/kg (0.45 mg/kg, weeks 0, 2, 4, 6, and 8, IC) | RIPC (3 cycles of 5 min leg ischemia followed by 5 min reperfusion) | ↓Mitochondrial fragmentation ↓Drp1 | ↓Beclin-1 ↓p62 ↓mitDNA content | - | ↑LVEF ↑Wall thickening ↓Fibrosis | RIPC applied immediately before each DOX injection resulted in preservation of cardiac contractility with significantly higher long-term LVEF and less cardiac fibrosis through prevention of mitochondrial fragmentation and dysregulated autophagy in a pig model. | [44] |
| Male Sprague Dawley rats | 14 mg/kg (2 mg/kg, 7 weekly injections, IP) | Treadmill exercise (TM) (velocity from 18 to 27 m/min, 60 min/day, 5 days/week, 12 weeks, co-treatment) | ↑Mfn1 ↑Mfn2 ↑Opa1 ↔Drp1 | ↓Swelling ↓Beclin-1 ↓LC3-II ↔p62 ↓PINK1 | ↓Caspase3 ↓Caspase8 ↓Caspase9 ↓Bax/Bcl-2 | - | Endurance treadmill training and voluntary freewheel activity prevented DOX-increased mitochondrial dysfunction and apoptotic signaling, alterations in mitochondrial dynamics, and increases in autophagy and mitophagy signaling in a rat model. | [49] |
| Running wheel exercise (FW) (24 h/day, 12 weeks, co-treatment) | ||||||||
| Male C57BL/6J mice | 20 mg/kg (5 mg/kg, weekly for 4 weeks, IP) | A motorized treadmill (speed of 13–15 m/min for 60 min/d for 4 wk, post-treatment) | ↑Drp1 ↓Mfn2 ↓Opa1 | ↑LC3II ↓p62 ↓ULK1ser757 ↓p-mTOR ↑PGC1-α ↓NRF1 ↓COXII ↓COXIII | ↓c-Caspase3 ↓TUNEL-positive cells ↓Protein carbonyl ↓4HNE ↓IL-1b ↓p22phox ↓p67phox ↓p47phox | ↓Abnormal morphology | A motorized treadmill exercise prevented DOX-induced apoptosis and mitigated tissue damage via increased mitophagy flux, increased Drp1-mitochondrial fission, and decreased fusion markers (Opa1 and Mfn2) in a mouse model. | [51] |
5. Existing Controversial Reports on Mitochondrial Fission/Fusion-Targeted Therapy in DOX-Induced Cardiotoxicity
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Nogueira, L.; Devasia, T.; Mariotto, A.B.; Yabroff, K.R.; Jemal, A.; Kramer, J.; Siegel, R.L. Cancer treatment and survivorship statistics, 2022. CA Cancer J. Clin. 2022, 72, 409–436. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, N.A.; Noronha, M.A.; Câmara, M.C.C.; Kurnik, I.S.; Feng, C.; Araujo, V.H.S.; Santos, J.; Feitosa, V.; Molino, J.V.D.; Rangel-Yagui, C.O.; et al. Doxorubicin nanoformulations on therapy against cancer: An overview from the last 10 years. Biomater. Adv. 2022, 133, 112623. [Google Scholar] [CrossRef] [PubMed]
- Jones, I.C.; Dass, C.R. Doxorubicin-induced cardiotoxicity: Causative factors and possible interventions. J. Pharm. Pharmacol. 2022, 74, 1677–1688. [Google Scholar] [CrossRef]
- Anjos, M.; Fontes-Oliveira, M.; Costa, V.M.; Santos, M.; Ferreira, R. An update of the molecular mechanisms underlying doxorubicin plus trastuzumab induced cardiotoxicity. Life Sci. 2021, 280, 119760. [Google Scholar] [CrossRef]
- Algieri, C.; Bernardini, C.; Oppedisano, F.; La Mantia, D.; Trombetti, F.; Palma, E.; Forni, M.; Mollace, V.; Romeo, G.; Troisio, I.; et al. The Impairment of Cell Metabolism by Cardiovascular Toxicity of Doxorubicin Is Reversed by Bergamot Polyphenolic Fraction Treatment in Endothelial Cells. Int. J. Mol. Sci. 2022, 23, 168977. [Google Scholar] [CrossRef]
- Li, Y.; Lin, R.; Peng, X.; Wang, X.; Liu, X.; Li, L.; Bai, R.; Wen, S.; Ruan, Y.; Chang, X.; et al. The Role of Mitochondrial Quality Control in Anthracycline-Induced Cardiotoxicity: From Bench to Bedside. Oxid. Med. Cell. Longev. 2022, 2022, 3659278. [Google Scholar] [CrossRef]
- Tahrir, F.G.; Langford, D.; Amini, S.; Mohseni Ahooyi, T.; Khalili, K. Mitochondrial quality control in cardiac cells: Mechanisms and role in cardiac cell injury and disease. J. Cell. Physiol. 2019, 234, 8122–8133. [Google Scholar] [CrossRef]
- Maneechote, C.; Palee, S.; Chattipakorn, S.C.; Chattipakorn, N. Roles of mitochondrial dynamics modulators in cardiac ischaemia/reperfusion injury. J. Cell. Mol. Med. 2017, 21, 2643–2653. [Google Scholar] [CrossRef]
- Cao, K.; Riley, J.S.; Heilig, R.; Montes-Gómez, A.E.; Vringer, E.; Berthenet, K.; Cloix, C.; Elmasry, Y.; Spiller, D.G.; Ichim, G.; et al. Mitochondrial dynamics regulate genome stability via control of caspase-dependent DNA damage. Dev. Cell. 2022, 57, 1211–1225.e1216. [Google Scholar] [CrossRef]
- Archer, S.L. Mitochondrial dynamics—Mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [Green Version]
- Yu, R.; Liu, T.; Ning, C.; Tan, F.; Jin, S.B.; Lendahl, U.; Zhao, J.; Nistér, M. The phosphorylation status of Ser-637 in dynamin-related protein 1 (Drp1) does not determine Drp1 recruitment to mitochondria. J. Biol. Chem. 2019, 294, 17262–17277. [Google Scholar] [CrossRef] [PubMed]
- Arinno, A.; Maneechote, C.; Khuanjing, T.; Ongnok, B.; Prathumsap, N.; Chunchai, T.; Arunsak, B.; Kerdphoo, S.; Shinlapawittayatorn, K.; Chattipakorn, S.C.; et al. Cardioprotective effects of melatonin and metformin against doxorubicin-induced cardiotoxicity in rats are through preserving mitochondrial function and dynamics. Biochem. Pharmacol. 2021, 192, 114743. [Google Scholar] [CrossRef]
- Khuanjing, T.; Ongnok, B.; Maneechote, C.; Siri-Angkul, N.; Prathumsap, N.; Arinno, A.; Chunchai, T.; Arunsak, B.; Chattipakorn, S.C.; Chattipakorn, N. Acetylcholinesterase inhibitor ameliorates doxorubicin-induced cardiotoxicity through reducing RIP1-mediated necroptosis. Pharmacol. Res. 2021, 173, 105882. [Google Scholar] [CrossRef] [PubMed]
- Prathumsap, N.; Ongnok, B.; Khuanjing, T.; Arinno, A.; Maneechote, C.; Apaijai, N.; Chunchai, T.; Arunsak, B.; Shinlapawittayatorn, K.; Chattipakorn, S.C.; et al. Acetylcholine receptor agonists provide cardioprotection in doxorubicin-induced cardiotoxicity via modulating muscarinic M(2) and α7 nicotinic receptor expression. Transl. Res. 2022, 243, 33–51. [Google Scholar] [CrossRef]
- Maneechote, C.; Khuanjing, T.; Ongnok, B.; Arinno, A.; Prathumsap, N.; Chunchai, T.; Arunsak, B.; Nawara, W.; Chattipakorn, S.C.; Chattipakorn, N. Promoting mitochondrial fusion in doxorubicin-induced cardiotoxicity: A novel therapeutic target for cardioprotection. Clin. Sci. 2022, 136, 841–860. [Google Scholar] [CrossRef]
- Gharanei, M.; Hussain, A.; Janneh, O.; Maddock, H. Attenuation of doxorubicin-induced cardiotoxicity by mdivi-1: A mitochondrial division/mitophagy inhibitor. PLoS ONE 2013, 8, e77713. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Guo, J.; Zhang, Q.; Cui, L.; Zhang, L.; Zhang, T.; Zhao, J.; Li, J.; Middleton, A.; Carmichael, P.L.; et al. Doxorubicin-induced mitophagy and mitochondrial damage is associated with dysregulation of the PINK1/parkin pathway. Toxicol. In Vitro 2018, 51, 1–10. [Google Scholar] [CrossRef]
- Xu, H.; Yu, W.; Sun, S.; Li, C.; Zhang, Y.; Ren, J. Luteolin Attenuates Doxorubicin-Induced Cardiotoxicity Through Promoting Mitochondrial Autophagy. Front. Physiol. 2020, 11, 113. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Sun, X.; Zhou, H.; Zhang, S.; Zhong, X.; Xu, X.; Guo, Y.; Xiong, Z.; Liu, M.; Lin, Y.; et al. Klotho attenuated Doxorubicin-induced cardiomyopathy by alleviating Dynamin-related protein 1—Mediated mitochondrial dysfunction. Mech. Ageing Dev. 2021, 195, 111442. [Google Scholar] [CrossRef]
- Wu, B.B.; Leung, K.T.; Poon, E.N. Mitochondrial-Targeted Therapy for Doxorubicin-Induced Cardiotoxicity. Int. J. Mol. Sci. 2022, 23, 31912. [Google Scholar] [CrossRef] [PubMed]
- Neganova, M.; Semakov, A.; Aleksandrova, Y.; Yandulova, E.; Pukhov, S.; Anikina, L.; Klochkov, S. N-Alkylation of Anthracycline Antibiotics by Natural Sesquiterpene Lactones as a Way to Obtain Antitumor Agents with Reduced Side Effects. Biomedicines 2021, 9, 50547. [Google Scholar] [CrossRef] [PubMed]
- Reis-Mendes, A.; Padrão, A.I.; Duarte, J.A.; Gonçalves-Monteiro, S.; Duarte-Araújo, M.; Remião, F.; Carvalho, F.; Sousa, E.; Bastos, M.L.; Costa, V.M. Role of Inflammation and Redox Status on Doxorubicin-Induced Cardiotoxicity in Infant and Adult CD-1 Male Mice. Biomolecules 2021, 11, 111725. [Google Scholar] [CrossRef]
- Kabel, A.M.; Salama, S.A.; Adwas, A.A.; Estfanous, R.S. Targeting Oxidative Stress, NLRP3 Inflammasome, and Autophagy by Fraxetin to Combat Doxorubicin-Induced Cardiotoxicity. Pharmaceuticals 2021, 14, 111188. [Google Scholar] [CrossRef]
- Takemura, G.; Fujiwara, H. Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog. Cardiovasc. Dis. 2007, 49, 330–352. [Google Scholar] [CrossRef]
- Christidi, E.; Brunham, L.R. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell. Death Dis. 2021, 12, 339. [Google Scholar] [CrossRef] [PubMed]
- Maneechote, C.; Palee, S.; Apaijai, N.; Kerdphoo, S.; Jaiwongkam, T.; Chattipakorn, S.C.; Chattipakorn, N. Mitochondrial dynamic modulation exerts cardiometabolic protection in obese insulin-resistant rats. Clin. Sci. 2019, 133, 2431–2447. [Google Scholar] [CrossRef]
- Xia, Y.; Chen, Z.; Chen, A.; Fu, M.; Dong, Z.; Hu, K.; Yang, X.; Zou, Y.; Sun, A.; Qian, J.; et al. LCZ696 improves cardiac function via alleviating Drp1-mediated mitochondrial dysfunction in mice with doxorubicin-induced dilated cardiomyopathy. J. Mol. Cell. Cardiol. 2017, 108, 138–148. [Google Scholar] [CrossRef]
- Zeng, C.; Duan, F.; Hu, J.; Luo, B.; Huang, B.; Lou, X.; Sun, X.; Li, H.; Zhang, X.; Yin, S.; et al. NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol. 2020, 34, 101523. [Google Scholar] [CrossRef]
- Govender, J.; Loos, B.; Marais, E.; Engelbrecht, A.M. Melatonin improves cardiac and mitochondrial function during doxorubicin-induced cardiotoxicity: A possible role for peroxisome proliferator-activated receptor gamma coactivator 1-alpha and sirtuin activity? Toxicol. Appl. Pharmacol. 2018, 358, 86–101. [Google Scholar] [CrossRef]
- Aung, L.H.H.; Chen, X.; Cueva Jumbo, J.C.; Li, Z.; Wang, S.Y.; Zhao, C.; Liu, Z.; Wang, Y.; Li, P. Cardiomyocyte mitochondrial dynamic-related lncRNA 1 (CMDL-1) may serve as a potential therapeutic target in doxorubicin cardiotoxicity. Mol. Ther. Nucleic Acids 2021, 25, 638–651. [Google Scholar] [CrossRef]
- Shi, Y.; Li, F.; Shen, M.; Sun, C.; Hao, W.; Wu, C.; Xie, Y.; Zhang, S.; Gao, H.; Yang, J.; et al. Luteolin Prevents Cardiac Dysfunction and Improves the Chemotherapeutic Efficacy of Doxorubicin in Breast Cancer. Front. Cardiovasc. Med. 2021, 8, 750186. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, J.; Wang, Q.; Zhao, X.; Yang, D.; Niu, L.; Yang, Y.; Zheng, X.; Hu, L.; Li, Y. Shenmai Injection Protects Against Doxorubicin-Induced Cardiotoxicity via Maintaining Mitochondrial Homeostasis. Front. Pharmacol. 2020, 11, 815. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Lv, C.; Zhang, X.; Ruan, W.; Xu, X.; Chen, C.; Ji, X.; Lu, L.; Guo, X. Neuraminidase1 Inhibitor Protects Against Doxorubicin-Induced Cardiotoxicity via Suppressing Drp1-Dependent Mitophagy. Front. Cell. Dev. Biol. 2021, 9, 802502. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zhen, D.; Bai, M.; Xuan, T.; Wang, Y.; Liu, M.; Yu, L.; Bai, D.; Fu, D.; Wei, C. Ethanol extracts of Rhaponticum uniflorum (L.) DC flowers attenuate doxorubicin-induced cardiotoxicity via alleviating apoptosis and regulating mitochondrial dynamics in H9c2 cells. J. Ethnopharmacol. 2022, 288, 114936. [Google Scholar] [CrossRef] [PubMed]
- Catanzaro, M.P.; Weiner, A.; Kaminaris, A.; Li, C.; Cai, F.; Zhao, F.; Kobayashi, S.; Kobayashi, T.; Huang, Y.; Sesaki, H.; et al. Doxorubicin-induced cardiomyocyte death is mediated by unchecked mitochondrial fission and mitophagy. FASEB J. 2019, 33, 11096–11108. [Google Scholar] [CrossRef] [Green Version]
- Pillai, V.B.; Kanwal, A.; Fang, Y.H.; Sharp, W.W.; Samant, S.; Arbiser, J.; Gupta, M.P. Honokiol, an activator of Sirtuin-3 (SIRT3) preserves mitochondria and protects the heart from doxorubicin-induced cardiomyopathy in mice. Oncotarget 2017, 8, 34082–34098. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Wang, S.; Wang, L.; Ceylan, A.F.; Ren, J.; Zhang, Y. Mitophagy inhibitor liensinine suppresses doxorubicin-induced cardiotoxicity through inhibition of Drp1-mediated maladaptive mitochondrial fission. Pharmacol. Res. 2020, 157, 104846. [Google Scholar] [CrossRef]
- Ding, M.; Shi, R.; Cheng, S.; Li, M.; De, D.; Liu, C.; Gu, X.; Li, J.; Zhang, S.; Jia, M.; et al. Mfn2-mediated mitochondrial fusion alleviates doxorubicin-induced cardiotoxicity with enhancing its anticancer activity through metabolic switch. Redox Biol. 2022, 52, 102311. [Google Scholar] [CrossRef]
- Du, J.; Hang, P.; Pan, Y.; Feng, B.; Zheng, Y.; Chen, T.; Zhao, L.; Du, Z. Inhibition of miR-23a attenuates doxorubicin-induced mitochondria-dependent cardiomyocyte apoptosis by targeting the PGC-1α/Drp1 pathway. Toxicol. Appl. Pharmacol. 2019, 369, 73–81. [Google Scholar] [CrossRef]
- Tang, H.; Tao, A.; Song, J.; Liu, Q.; Wang, H.; Rui, T. Doxorubicin-induced cardiomyocyte apoptosis: Role of mitofusin 2. Int. J. Biochem. Cell. Biol. 2017, 88, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Li, R.; Liu, C.; Sun, T.; Htet Aung, L.H.; Chen, C.; Gao, J.; Zhao, Y.; Wang, K. Foxo3a inhibits mitochondrial fission and protects against doxorubicin-induced cardiotoxicity by suppressing MIEF2. Free. Radic. Biol. Med. 2017, 104, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Shi, R.; Fu, F.; Li, M.; De, D.; Du, Y.; Li, Z. Paeonol protects against doxorubicin-induced cardiotoxicity by promoting Mfn2-mediated mitochondrial fusion through activating the PKCε-Stat3 pathway. J. Adv. Res. 2022, 7, 2. [Google Scholar] [CrossRef]
- Galán-Arriola, C.; Villena-Gutiérrez, R.; Higuero-Verdejo, M.I.; Díaz-Rengifo, I.A.; Pizarro, G.; López, G.J.; Molina-Iracheta, A.; Pérez-Martínez, C.; García, R.D.; González-Calle, D.; et al. Remote ischaemic preconditioning ameliorates anthracycline-induced cardiotoxicity and preserves mitochondrial integrity. Cardiovasc. Res. 2021, 117, 1132–1143. [Google Scholar] [CrossRef]
- Lee, K.J.; Wright, G.; Bryant, H.; Wiggins, L.A.; Dal Zotto, V.L.; Schuler, M.; Malozzi, C.; Cohen, M.V.; Gassman, N.R. Cytoprotective Effect of Vitamin D on Doxorubicin-Induced Cardiac Toxicity in Triple Negative Breast Cancer. Int. J. Mol. Sci. 2021, 22, 147439. [Google Scholar] [CrossRef]
- Gao, L.; Yuan, P.; Wei, Y.; Fu, Y.; Hou, Y.; Li, P.; Chen, Y.; Ruan, Y.; Zhou, N.; Zheng, X.; et al. Total flavonoids of Selaginella tamariscina (P.Beauv.) Spring ameliorates doxorubicin-induced cardiotoxicity by modulating mitochondrial dysfunction and endoplasmic reticulum stress via activating MFN2/PERK. Phytomedicine 2022, 100, 154065. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.N.; Sung, P.H.; Chiang, J.Y.; Sheu, J.J.; Huang, C.R.; Chu, Y.C.; Chua, S.; Yip, H.K. Early treatment with combination of SS31 and entresto effectively preserved the heart function in doxorubicin-induced dilated cardiomyopathic rat. Biomed. Pharmacother. 2021, 141, 111886. [Google Scholar] [CrossRef]
- Yip, H.K.; Shao, P.L.; Wallace, C.G.; Sheu, J.J.; Sung, P.H.; Lee, M.S. Early intramyocardial implantation of exogenous mitochondria effectively preserved left ventricular function in doxorubicin-induced dilated cardiomyopathy rat. Am. J. Transl. Res. 2020, 12, 4612–4627. [Google Scholar] [PubMed]
- Marques-Aleixo, I.; Santos-Alves, E.; Torrella, J.R.; Oliveira, P.J.; Magalhães, J.; Ascensão, A. Exercise and Doxorubicin Treatment Modulate Cardiac Mitochondrial Quality Control Signaling. Cardiovasc. Toxicol. 2018, 18, 43–55. [Google Scholar] [CrossRef]
- Hull, T.D.; Boddu, R.; Guo, L.; Tisher, C.C.; Traylor, A.M.; Patel, B.; Joseph, R.; Prabhu, S.D.; Suliman, H.B.; Piantadosi, C.A.; et al. Heme oxygenase-1 regulates mitochondrial quality control in the heart. JCI Insight 2016, 1, e85817. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Kwon, I.; Jang, Y.; Cosio-Lima, L.; Barrington, P. Endurance Exercise Attenuates Doxorubicin-induced Cardiotoxicity. Med. Sci. Sport. Exerc. 2020, 52, 25–36. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, S.; An, L.; Wu, J.; Hu, X.; Lai, S.; Mazhar, H.; Zou, Y.; He, L.; Zhu, H. Loss of Rubicon ameliorates doxorubicin-induced cardiotoxicity through enhancement of mitochondrial quality. Int. J. Cardiol. 2019, 296, 129–135. [Google Scholar] [CrossRef]
- Zhao, Q.; Sun, Q.; Zhou, L.; Liu, K.; Jiao, K. Complex Regulation of Mitochondrial Function During Cardiac Development. J. Am. Heart Assoc. 2019, 8, e012731. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.B.; Sardão, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Shen, H. Advances in Cardiotoxicity Induced by Altered Mitochondrial Dynamics and Mitophagy. Front. Cardiovasc. Med. 2021, 8, 739095. [Google Scholar] [CrossRef] [PubMed]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell. 2017, 40, 583–594.e586. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Wang, J.; Bonamy, G.M.C.; Meeusen, S.; Brusch, R.G.; Turk, C.; Yang, P.; Schultz, P.G. A Small Molecule Promotes Mitochondrial Fusion in Mammalian Cells. Angew. Chem. Int. Ed. 2012, 51, 9302–9305. [Google Scholar] [CrossRef]

| Study Model | DOX Dosage | Major Findings | Interpretation | Ref. | |||
|---|---|---|---|---|---|---|---|
| Mitochondrial Dynamics/Morphology | Mitochondrial Function/Mitophagy/Autophagy | Oxidative Stress/Inflammation/ Cell Death | Cell Viability/Cytotoxicity/Function | ||||
| AC16 cells | 250 nM (24 h) | ↑Drp1 | ↑ROS ↑MMP depolarization ↓mtDNA ↓PGC1-α ↓NRF1 ↓TFAM ↑PINK1 ↑Parkin ↑LC3II-I ↑Beclin-1 ↓p62 | - | ↓Cell viability | DOX induced Drp1-mediated mitochondrial fission, leading to toxic effects in mitochondria and cytotoxicity in cardiomyocyte cell lines. | [18] |
| H9c2 cells | 750 nM (24 h) | ↓Opa1 ↓Mfn2 ↓Mitochondrial size ↓Form factor ↓Aspect ratio ↓Mitochondrial number | ↑Mitophagy foci | ↑PI-positive cells ↑c-PARP ↑c-Caspase3 | - | DOX impaired mitochondrial fusion and fragmentation, resulting in impaired mitophagy and apoptosis in cardiomyocytes. | [36] |
| H9c2 cells | 0.75 μM (24 h) | ↑MFF ↑Fis1 ↓Mfn1 ↓Opa1 | ↑ROS ↑MMP depolarization | ↓SOD ↑p-p65 ↑p-IKK ↓p-IkBα ↑TUNEL-positive cells ↑Bax ↓Bcl-2 ↑c-Caspase3 ↑c-Caspase9 ↑c-PARP | ↓Cell viability | DOX suppressed mitochondrial fusion and promoted fission, causing mitochondrial dysfunction, inflammation, and apoptosis, in addition to reduced cardiomyocyte viability. | [35] |
| H9C2 cells | 1 μM (12 h) | ↑Drp1 | ↑PINK1 ↑Parkin ↑ATG5 ↑Beclin-1 ↑LC3I/II ↓p62 | ↑NEU1 ↑c-Caspase3 ↑c-Caspase9 ↑TUNEL-positive cells | - | DOX increased mitochondrial fission, mitophagy, and autophagy, contributing to cardiomyocyte apoptosis. | [34] |
| H9c2 cells | 1 μM (16 h) | ↑p-Drp1ser616 ↓p-Drp1ser637 ↓L-Opa1/S-Opa1 ↓Aspect ratio ↓Form factor | ↑ROS ↑MMP depolarization ↓OCR ↓p-AMPKThr172 | ↑Bax/Bcl-2 ↑c-Caspase3 ↑Annexin V-positive cells | ↓Cell viability | DOX injured cardiomyocytes via increasing mitochondrial fragmentation, mitochondrial dysfunction, and apoptosis. | [33] |
| AMCM cells | 1 μM (24 h) | ↓p-Drp1ser616 ↑Mitochondrial elongation | ↑ROS ↑MMP depolarization ↓LAMP1 ↓TFEB ↑p-mTORser2448 ↓LC3B-II ↓PINK1 ↓Parkin ↓Bnip3 | ↑TUNEL-positive cells ↑Bax ↑c-Caspase9 ↓Bcl-2 | ↑LDH ↑CK ↓Cell length ↓−dL/dt ↓+dL/dt | DOX induced cardiomyocyte contractile dysfunction and apoptosis via impairing mitochondrial dynamics and dysfunction, mitophagy, and autophagy. | [19] |
| H9c2 cells/AC16 cells | 1 μM (24 h) | ↑p-Drp1ser616 ↑Fragmented mitochondria | ↑ROS | ↓SOD activity ↑Apoptotic cells ↑Bax ↓Bcl-2 ↓Bcl-xL ↑c-Caspase3 | - | DOX-mediated mitochondrial fission and fragmentation contribute to mitochondrial dysfunction and cardiomyocyte apoptosis. | [32] |
| NRVMs | 1.72 μM (4, 8, 24, and 48 h) | ↓Mfn2 (8/24 h) ↑Fragmented mitochondria (4 h) | ↑ROS (8 h) | ↑Caspase3 activity (8 h) ↑TUNEL-positive cells (24 h) | - | DOX reduced mitochondrial fusion, leading to mitochondrial fragmentation, dysfunction, and cardiomyocyte apoptosis. | [41] |
| H9c2 cells | 2 μM (24 h) | ↑Drp1 ↓p-Drp1ser637 ↓CMDL-1 ↑Fragmented mitochondria | - | ↑Apoptotic cells | - | DOX promoted mitochondrial fission and fragmentation, resulting in cardiomyocyte apoptosis. | [31] |
| Mouse cardiomyocytes | 3 μM (24 h) | ↑Fragmented mitochondria ↑MIEF2 | - | ↓Foxo3a ↑TUNEL-positive cells ↑c-Caspase3 | - | Foxo3a is downregulated in the cardiomyocytes in response to DOX treatment, leading to increased mitochondrial fission and apoptosis. | [42] |
| NRVMs | 3 μM (24 h) | ↑p-Drp1 ↓Mfn2 | ↑MMP depolarization ↑ROS ↓PGC1-α | ↑miR-23a ↑Cyt c ↑c-Caspase3 | ↓Cell viability | DOX increased mitochondrial fission while decreasing fusion, resulting in mitochondrial dysfunction, miR-23a upregulation, and cardiomyocyte apoptosis. | [40] |
| H9c2 cells | 3 μM (24 h) | ↑Fragmented mitochondria | ↓ATP | - | ↓Cell viability | DOX reduced cardiomyocyte viability via mitochondrial fragmentation and dysfunction. | [30] |
| NRVMs | 3 μM (24 h) | ↓Mfn2 ↓Mitochondrial size ↑Mitochondrial number | ↑MMP depolarization ↑ROS ↓OCR ↓ECAR ↓COM-I ↓COM-III | ↑FoxO1 ↑Caspase3 activity ↑TUNEL-positive cells ↑Cyt c | ↓Cell viability ↑LDH | DOX increased cardiomyocyte toxicity by inhibiting mitochondrial fusion-induced fragmentation, activating mitochondrial dysfunction, and inducing FoxO1-associated apoptosis. | [39] |
| Primary cardiomyocytes | 3 μM (24 h) | ↓Mfn2 ↑Mitochondrial number ↓Mitochondrial size | ↑MMP depolarization ↑ROS ↓OCR | ↑Apoptotic cells | ↓Cell viability ↑LDH | DOX reduced cardiomyocyte viability with increased toxicity via mitochondrial fusion-mediated fragmentation, leading to mitochondrial dysfunction and apoptosis. | [43] |
| H9c2 cells | 5 μM (24 h) | ↑p-Drp1ser616 | ↑MMP depolarization | ↑c-Caspase3 ↑TUNEL-positive cells ↑Annexin V-positive cells | ↓Cardiomyocyte number ↓Peak shortening ↓+dp/dt ↓−dp/dt | DOX induced cardiomyocyte dysfunction via promoting apoptosis, mitochondrial dysfunction, and fission. | [20] |
| NMVMs | 5 μM (24 h) | ↑p-Drp1ser616 ↑Fragmented mitochondria | ↑MMP depolarization ↑ROS ↓TOM20 ↓TIM23 ↑Rab7 ↑LRRK2 | ↑TUNEL-positive cells ↑p-ERK | - | DOX increased cardiomyocyte apoptosis by impairing excessive mitochondrial fission, fragmentation, and dysfunction. | [38] |
| H9c2 cells | 5 μM (24 h) | ↑p-Drp1ser616 ↓p-Drp1ser637 | ↑ROS | ↑c-Caspase1 | - | DOX induced mitochondrial fission and dysfunction and cardiomyocyte apoptosis. | [29] |
| H9c2 cells | 5 μM (24 h) | ↑p-Drp1ser616 | - | ↑Anexin5-positive cells ↑c-Caspase3 | - | DOX induced mitochondrial fission and cardiomyocyte apoptosis. | [28] |
| NRVMs | 5 μM (24 h) | ↑Drp1 ↓Opa1 ↓Mfn1 ↑Fragmented mitochondria | ↑MnSOD | ↑Apoptotic cells | - | DOX induced mitochondrial fission with impaired fusion, leading to mitochondrial dysfunction and cardiomyocyte apoptosis. | [37] |
| Study Model | DOX Dosage | Major Findings | Interpretation | Ref. | |||
|---|---|---|---|---|---|---|---|
| Mitochondrial Dynamics/Morphology | Mitochondrial Function/Mitophagy/Autophagy | Oxidative stress/Inflammation/Cell Death | Heart Parameters | ||||
| Male Sprague Dawley rats (Langendorff protocol) | 1 μM (120 min) | ↑p-Drp1 | - | ↑p-Akt ↑p-Erk1/2 ↑p-p53 | ↓LVDP ↓HR ↓Coronary flow ↓Time taken to depolarization ↓Time taken to hypercontracture | DOX caused a reduction in cardiac function by promoting mitochondrial fission and cell death. | [17] |
| White pigs | 2.25 mg/kg (0.45 mg/kg, weeks 0, 2, 4, 6, and 8, IC) | ↑Drp1 ↑Mitochondrial fragmentation | ↑Beclin-1 ↑p62 ↑mitDNA content | - | ↓LVEF ↓Wall thickening ↑Fibrosis | DOX induced cardiac dysfunction via upregulation of fragmented mitochondria with severe morphological abnormalities, together with upregulation of fission and autophagy proteins. | [44] |
| C57BL/6 mice | 12 mg/kg (4 mg/kg at days 0, 7, and 14, IP) | ↑p-Drp1ser616 ↓Mitochondrial length | ↓ATP content | ↑c-Caspase3 | ↓HW/BW ↑CK activity ↑Fibrosis ↑Disorganized myofibers ↓EF ↓FS ↑LVESD ↑LVEDD | DOX induced cardiotoxicity by promoting apoptosis and mitochondrial fission. | [20] |
| C57BL/6J mice | 12 mg/kg (4 mg/kg, 3 weekly injections at 0, 7, and 14 days, IP) | ↑p-Drp1ser616 ↓p-Drp1ser637 ↓Mitochondrial length/width | - | ↑NLRP3 ↑c-Caspase1 ↑IL1-b ↑IL1-18 ↑p-NF-kB ↑NOX1 ↑NOX4 ↑GSDMD-NT | ↓EF ↓FS ↑LVESD ↑LVEDD ↓HW/BW | DOX promoted mitochondrial fission and NLRP3 inflammasome hyperactivation to trigger pyroptotic cell death, which is causally linked to progressive myocardial dysfunction. | [29] |
| Female Sprague Dawley rats | 12 mg/kg (4 mg/kg, on days 4, 8 and 12, IP) | ↑Drp1 ↑hFis1 ↔Mfn1 ↓Mfn2 | ↑PINK1 ↑Parkin ↓PGC1-α ↓SIRT1 | ↑c-Caspase3 ↑c-PARP | ↓CO ↓Total work performance ↓HW | DOX increased mitochondrial fission while impairing fusion and mitophagy, resulting in apoptosis as well as mitochondrial and cardiac dysfunction. | [30] |
| Male Sprague Dawley rats | 12.5 mg/kg (3.125 mg/kg, once every 5 days, 4 separated time points within 20 days, sacrifice at day 60, IP) | ↑Drp1 ↑Mfn2 | ↑LC3BII/I ↑OXPHOS | ↑TLR-4 ↑MyD88 ↑p-NF-kb ↑IL-1B ↑TNF-α ↑NOX1 ↑NOX2 ↑Cyt c ↑c-Caspase3 ↑c-Caspase9 ↑Bax ↑Apoptotic cells | ↓EF ↑Fibrosis ↑Hypertrophy | DOX caused inflammation, oxidative stress, mitochondrial dysfunction, autophagic/apoptotic signaling pathways, and LV dysfunction. | [47] |
| Adult male Sprague Dawley | 12.5 mg/kg (3.125 mg/kg, 4 separated time points, once every 5 days, 20 days, IP) | ↑Drp1 ↑Mfn2 | ↓mitoDNA ↑PGC1-α ↓OXPHOS ↑CypD1 ↑Nrf1 ↑Beclin-1 ↑ATG-5 ↑LC3-II/I | ↑NOX1 ↑NOX2 ↑c-Caspase3 ↑Bax ↑c-PARP ↑Cyt c | ↓EF ↑HW/Tibial length ↑LW/Tibial length ↑Fibrosis ↑Hypertrophy ↓Cx43 ↑BNP+ | DOX induced inflammation, oxidative stress, mitochondrial damage, and impaired autophagic/apoptotic signaling pathways, all of which contributed to LV dysfunction. | [48] |
| Male Sprague Dawley rats | 14 mg/kg (2 mg/kg, 7 weekly injections, IP) | ↓Mfn1 ↓Mfn2 ↓Opa1 ↑Drp1 | ↑Swelling ↑Beclin-1 ↑LC3-II ↑p62 ↑PINK1 | ↑Caspase3 ↑Caspase8 ↑Caspase9 ↑Bax/Bcl-2 | - | DOX treatment resulted in augmentation of mPTP susceptibility and apoptotic signaling, decreased expression of fusion-related proteins, increased Drp1, and the activation of autophagy and mitophagy signaling. | [49] |
| Male Sprague Dawley rats | 15 mg/kg (2.5 mg/kg, 6 times within 2 weeks, IP) | ↑Drp1 | ↑LC3II ↑Beclin-1 ↑ATG5 ↓p62 ↑PINK1 ↑Parkin | ↑NEU1 ↑LDH ↓GSH ↓SOD ↑H2O2 ↑c-Caspase3 ↑c-Caspase9 ↑Bax ↑Bad ↓Bcl-2 ↑TUNEL-positive cells | ↓EF ↓FS ↑LVEDs ↓Cardiomyocyte area ↑Fibrosis ↓Cross sectional area ↑c-TnT ↑CKMB | Elevated NEU1 triggered by DOX increased mitochondrial fission, autophagy, and mitophagy, leading to a maladaptive feedback loop towards myocardial apoptosis, cell death, and cardiac dysfunction. | [34] |
| Mice | 15 mg/kg (single dose, euthanized after 3 days, IP) | ↓Mitochondrial length ↓Mitochondrial width ↓Mitochondrial size | ↑LC3-II | ↑LDH ↑4HNE ↑p-PDH ↑c-Caspase3 | ↑c-TnI | DOX induced myocardial injury via mitochondrial fragmentation, accelerated mitophagy flux, oxidative stress, and apoptosis. | [36] |
| Adult male Balb/c mice | 15 mg/kg (3 times/week for 2 weeks, IP) | ↑Drp1 ↑Mitochondrial length/width | ↓ATP | ↑TUNEL-positive cells ↑c-Caspase3 | ↑LVEDD ↑LVESD ↓EF ↑NT-proBNP ↑Fibrosis ↑Hypertrophy | Reduced cardiac function, mitochondrial morphology disturbance, reduced activity of mitochondrial respiration complex I and lowered ATP content were detected post-DOX stimulation in mice. | [28] |
| Male C57BL/6 mice | 15 mg/kg (5 mg/kg, 3 consecutive weeks, IP) | ↓Mfn2 ↑Mitochondrial number ↓Mitochondrial size | ↑ROS | ↑MDA ↓SOD ↑FoxO1 ↑TUNEL-positive cells ↑Caspase3 activity | ↓EF ↑LVESV ↑c-TnT ↑Fibrosis | DOX-induced upregulation of FoxO1 inhibited Mfn2-mediated mitochondrial fusion and promoted mitochondrial dysfunction and oxidative stress, resulting in cardiac dysfunction. | [39] |
| Male Sprague Dawley rats | 15 mg/kg (5 mg/kg, on days 1, 6 and 11, 3 times/2 weeks, IP) | ↓Mitochondrial size ↓Mfn2 | ↓Complex-III | ↑LDH | ↓EF ↓FS ↑LVESD ↓LVSP ↑LVEDP ↓+dp/dt ↓-dp/dt ↑CKMB | DOX induced damage by inhibiting mitochondrial fusion via the PKCe-Stat3-Mfn2 pathway, leading to cardiac dysfunction. | [43] |
| Male BALB/c mice | 15 mg/kg (5 mg/kg, every 15 days for a total of three doses, IP) | ↓Opa1 ↓Mfn1 ↑Fragmented mitochondria ↑Mitochondrial damage | ↓Sirt3 ↓OGG1 ↑MnSOD ↑8-Oxo-dG ↓CS ↓ATP | ↑TUNEL-positive cells ↓Bcl-2 | ↑HW/TL ↓FS ↑Fibrosis | DOX-induced cardiotoxicity is associated with increased ROS production and consequent fragmentation of mitochondria and cell death. | [37] |
| Male Wistar rats | 18 mg/kg (3 mg/kg, 6 doses, days 0, 4, 8, 15, 22, and 29, IP) | ↑Drp1 in mitochondria ↑p-Drp1ser616 ↓Mfn1 ↓Mfn2 ↓Opa1 ↑Mitochondrial volume density ↓Mitochondrial area | ↓RCR ↑ROS ↑MMP depolarization ↑Swelling ↑Parkin ↑Beclin-1 ↑p62 ↑LC3II/I | ↑MDA in serum and tissue ↑TNF-a ↑IL-6 ↑Bax ↑c-Caspase3 ↑Cyt c ↑TUNEL-positive cells | ↓HR ↓LVESP ↓+dp/dt ↓SV ↓SBP ↓DBP ↓EF ↓FS ↓E/A ratio ↑LVEDP ↑-dp/dt ↑LF/HF ratio ↑NT-proBNP ↑cTn-I | DOX altered mitochondrial dynamics, mitochondrial dysfunction, autophagy and mitophagy activation, increased oxidative stress, inflammation, and apoptosis, leading to cardiac dysfunction. | [16] |
| Male Wistar rats | 18 mg/kg (3 mg/kg, 6 doses, days 0, 4, 8, 15, 22, and 29, IP) | ↑Drp1 in mitochondria ↑p-Drp1ser616 ↓Mfn1 ↓Mfn2 | ↓RCR ↑ROS ↑MMP depolarization ↑Swelling ↑PINK1 ↑Parkin ↑Beclin-1 ↑p62 ↑LC3II/I | ↑MDA in serum and tissue ↑TNF-a ↑IL-6 ↑Bax ↑c-Caspase3 ↑TUNEL-positive cells | ↓HR ↓SV ↓SBP ↓DBP ↓EF ↓FS ↓E/A ratio ↑LF/HF ratio ↑NT-proBNP ↑c-TnI ↓a7nAChR ↓M2AChR | DOX impaired both a7nAChR and M2AChR, leading to altered mitochondrial dynamics, mitochondrial dysfunction, autophagy and mitophagy activation, increased oxidative stress, inflammation, apoptosis, and cardiac dysfunction. | [15] |
| Male and female mixed FVB/C57BL/6 mice | 18 mg/kg (6 mg/kg, every third day for a week, 14 days, IV) | ↑Mfn1 ↑Fis1 ↑Mitochondrial number ↓Mitochondrial area | ↓mtDNA ↑Nrf1 ↑PINK1 ↑Parkin | ↓HO-1 ↑CD11b+ | ↓EF ↑EDD ↑ESD ↑LVEDD ↑LVESD ↓LVPWtd ↓LVPWts | DOX-induced dilated cardiomyopathy, cardiac cytoarchitectural derangement, infiltration of mononuclear phagocytes, dilation of the sarcoplasmic reticulum, mitochondrial fragmentation, and increased numbers of damaged mitochondria in autophagic vacuoles. | [50] |
| Male Wistar rats | 18 mg/kg (3 mg/kg, 6 doses, days 0, 4, 8, 15, 22, and 29, IP) | ↑Drp1 in mitochondria ↑p-Drp1ser616 ↓Mfn1 ↓Mfn2 ↓Opa1 ↑Mitochondrial volume density ↓Mitochondrial area | ↓RCR ↑ROS ↑MMP depolarization ↑Swelling ↑Parkin ↑Beclin-1 ↑p62 ↑LC3II/I | ↑MDA in serum and tissue ↑TNF-α ↑IL-6 ↑Bax ↑c-Caspase3 ↑Cyt c ↑TUNEL-positive cells | ↓HR ↓LVESP ↓+dp/dt ↓SV ↓SBP ↓DBP ↓EF ↓FS ↓E/A ratio ↑LVEDP ↑-dp/dt ↑LF/HF ratio ↑NT-proBNP ↑cTn-I | DOX caused cardiac dysfunction by altering mitochondrial dynamics, dysfunction, autophagy, mitophagy, oxidative stress, inflammation, and apoptosis. | [13] |
| Male Wistar rats | 18 mg/kg (3 mg/kg, 6 doses, days 0, 4, 8, 15, 22, and 29, IP) | ↑Drp1 in mitochondria ↑p-Drp1ser616 ↓Mfn1 ↓Mfn2 ↓Opa1 ↑Mitochondrial volume density ↓Mitochondrial area | ↓RCR ↑ROS ↑MMP depolarization ↑Swelling ↑Parkin ↑Beclin-1 ↑p62 ↑LC3II/I | ↑MDA in serum and tissue ↑TNF-α ↑IL-6 ↑Bax ↑c-Caspase3 ↑Cyt c ↑TUNEL-positive cells | ↓HR ↓LVESP ↓+dp/dt ↓SV ↓SBP ↓DBP ↓EF ↓FS ↓E/A ratio ↑LVEDP ↑-dp/dt ↑LF/HF ratio ↑NT-proBNP ↑cTn-I | DOX altered mitochondrial dynamics, mitochondrial dysfunction, autophagy and mitophagy activation, increased oxidative stress, inflammation, and apoptosis, leading to cardiac dysfunction. | [14] |
| Male C57BL/6 mice | 20 mg/kg (5 mg/kg, at first, third, fifth and seventh day, IP) | ↓Mfn2 | ↓PPAR-a ↓PGC1-a ↓Sirt3 ↑PERK ↑ATF4 ↑CHOP | ↑LDH ↓SOD ↑MDA ↓GSH ↓CAT ↓Nrf2 ↑Keap1 ↓Bcl-2 ↑Bax ↑Caspase9 ↑Cyt c | ↓EF ↓FS ↓E/A ratio ↑CKMB ↑BNP ↑c-TnT ↑Fibrosis | DOX induced cardiotoxicity by promoting mitochondrial dysfunction and ER stress by activating MFN2/PERK. | [46] |
| Male C57BL/6 mice | 20 mg/kg (5 mg/kg, weekly for 4 consecutive weeks, IP) | ↑Fragmented mitochondria ↑MIEF2 | - | ↓Foxo3a ↑TUNEL-positive cells | ↑LVIDd ↓FS | Foxo3a is downregulated in the mouse heart in response to DOX, leading to increased mitochondrial fission and apoptosis. | [42] |
| Male C57BL/6J mice | 20 mg/kg (5 mg/kg, weekly for 4 weeks, IP) | ↓Drp1 ↔Mfn2 ↔Opa1 | ↔LC3II ↔p62 ↔Beclin1 ↔ATG7 ↔LAMP2 ↔p-AMPK ↑ULK1ser757 ↑p-mTOR ↑Parkin ↓PGC1-a ↑NRF1 ↑COX1 ↑COX2 | ↑4HNE ↑IL-1b ↑c-Caspase3 ↑TUNEL-positive cells | ↑Abnormal morphology | DOX induced oxidative stress, apoptosis, and tissue damage without affecting autophagy or mitochondrial dynamics. | [51] |
| FVB/NJ mice | 20 mg/kg (single dose, euthanized at day 3, IP) | ↓Opa1 ↓Fis1 ↑Fragmented mitochondria | ↓ATP ↑LC3II ↑P62 ↓Parkin | ↑LDH ↑ROS | ↑Fibrosis ↑CKMB | DOX induced the accumulation of fragmented mitochondria, cytoplasmic vacuolization, an increase in oxidative stress, and an increase in the thickness of the dysfunctional LV wall. | [52] |
| 16 mg/kg (4 mg/kg, weekly for 4 weeks, IP) | - | - | - | ↓Systolic LVPW ↓Diastolic LVPW | |||
| Female Balb/c mice | 30 mg/kg (10 mg/kg, on days 2, 8 and 15, IP) | ↑p-Drp1ser616 | - | ↑4-HNE ↑NQO1 ↑c-Caspase3 | ↓CO ↓EF ↓SV ↓FS | DOX induced cardiotoxicity by increasing the reactive oxygen species, mitochondrial damage, and apoptosis. | [45] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maneechote, C.; Chattipakorn, S.C.; Chattipakorn, N. Recent Advances in Mitochondrial Fission/Fusion-Targeted Therapy in Doxorubicin-Induced Cardiotoxicity. Pharmaceutics 2023, 15, 1182. https://doi.org/10.3390/pharmaceutics15041182
Maneechote C, Chattipakorn SC, Chattipakorn N. Recent Advances in Mitochondrial Fission/Fusion-Targeted Therapy in Doxorubicin-Induced Cardiotoxicity. Pharmaceutics. 2023; 15(4):1182. https://doi.org/10.3390/pharmaceutics15041182
Chicago/Turabian StyleManeechote, Chayodom, Siriporn C. Chattipakorn, and Nipon Chattipakorn. 2023. "Recent Advances in Mitochondrial Fission/Fusion-Targeted Therapy in Doxorubicin-Induced Cardiotoxicity" Pharmaceutics 15, no. 4: 1182. https://doi.org/10.3390/pharmaceutics15041182
APA StyleManeechote, C., Chattipakorn, S. C., & Chattipakorn, N. (2023). Recent Advances in Mitochondrial Fission/Fusion-Targeted Therapy in Doxorubicin-Induced Cardiotoxicity. Pharmaceutics, 15(4), 1182. https://doi.org/10.3390/pharmaceutics15041182