

ADME, Pharmacokinetic Scaling, Pharmacodynamic and Prediction of Human Dose and Regimen of Novel Antiviral Drugs


Abstract
:1. Introduction
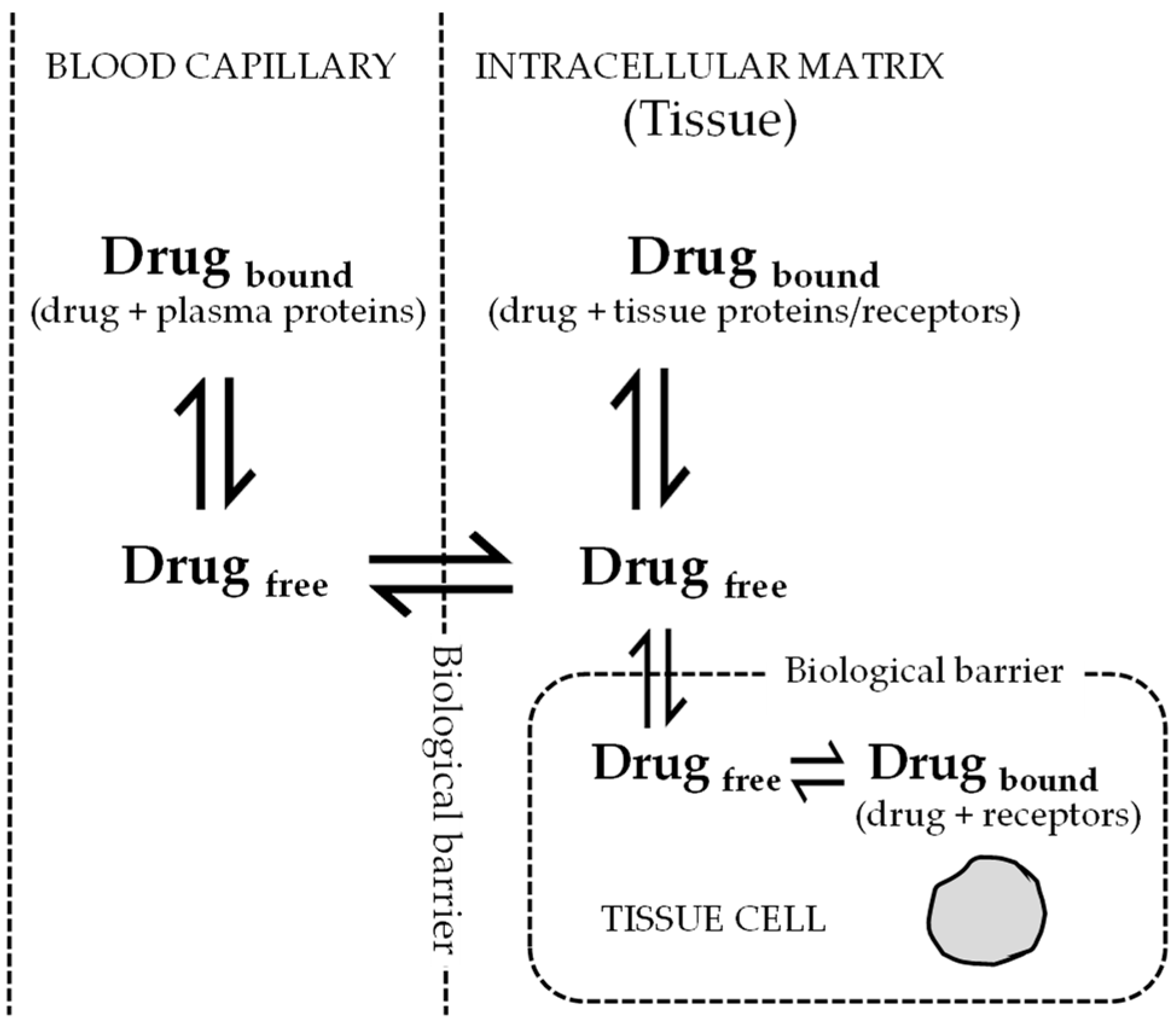
2. Role of Volume of Distribution for Antiviral Drug Development
3. Significance Clearance for Antiviral Drug Discovery
4. Variation in CYP Mediated Metabolism of Antiviral Drugs
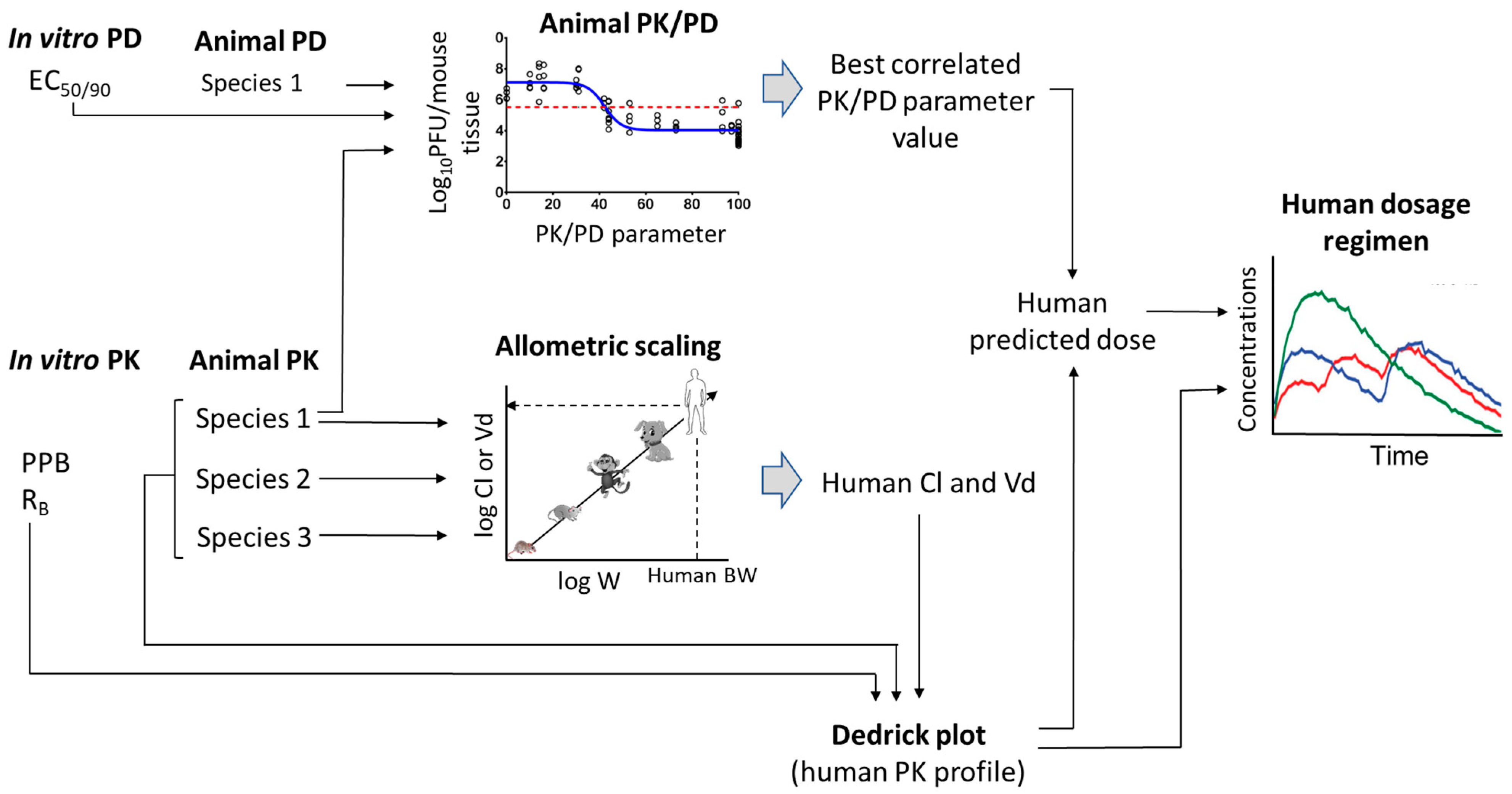
5. Scaling of Novel Antivirals from Preclinical to Clinical
5.1. Prediction of Human PK Parameters
5.2. Prediction of Human Plasma-Concentration Time Profiles: Species-Invariant Time Method (Complex Dedrick Plots)
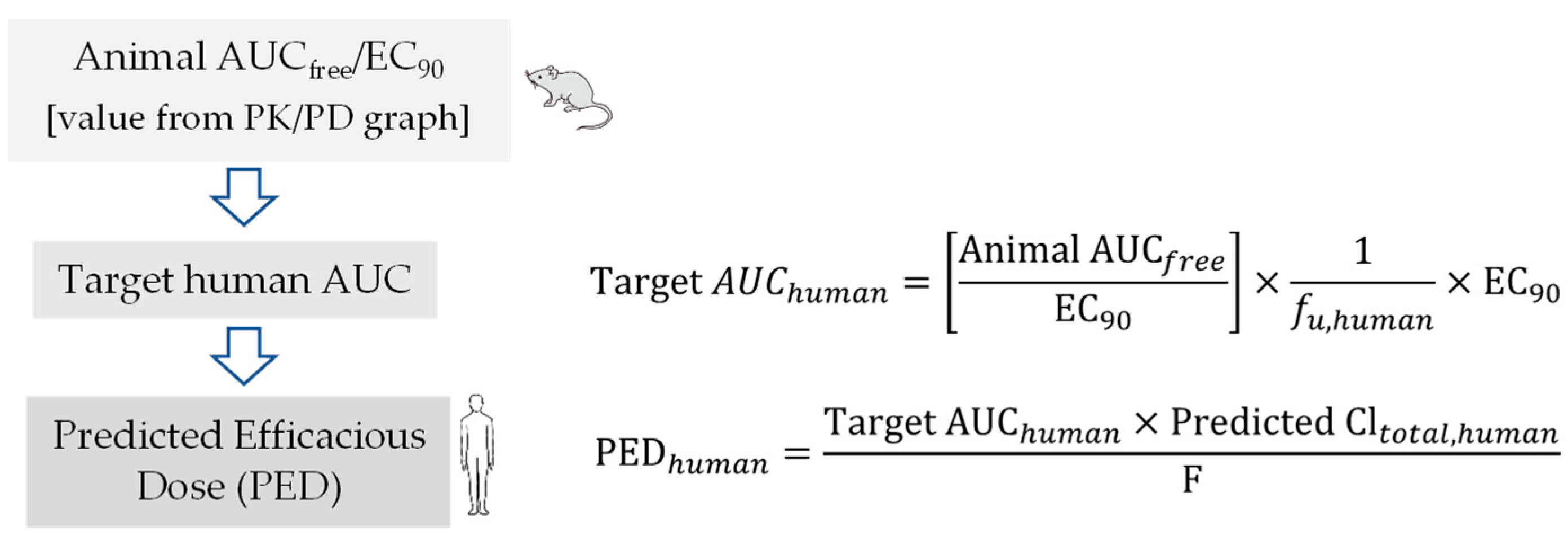
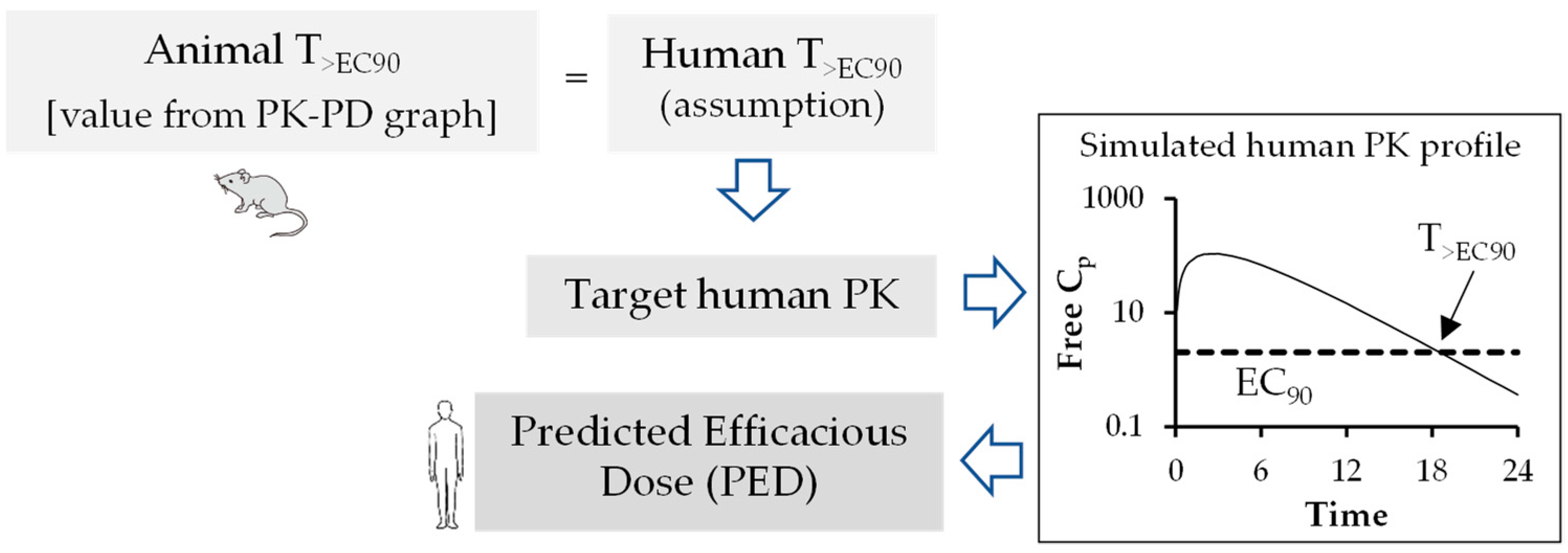
6. Pharmacokinetic-Pharmacodynamic (PK-PD) of Antiviral Drugs
Prediction of Human Efficacy Dose Using Different PK-PD Parameters
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kennedy, T. Managing the drug discovery/development interface. Drug Discov. Today 1997, 2, 436–444. [Google Scholar] [CrossRef]
- Prueksaritanont, T.; Tang, C. ADME of Biologics—What Have We Learned from Small Molecules? AAPS J. 2012, 14, 410–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez de Requena, D.; Bonora, S.; Garazzino, S.; Sciandra, M.; D’Avolio, A.; Raiteri, R.; Marrone, R.; Boffito, M.; De Rosa, F.G.; Sinicco, A.; et al. Nevirapine plasma exposure affects both durability of viral suppression and selection of nevirapine primary resistance mutations in a clinical setting. Antimicrob. Agents Chemother. 2005, 49, 3966–3969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boffito, M.; Back, D.; Stainsby-Tron, M.; Hill, A.; Di Perri, G.; Moyle, G.; Nelson, M.; Tomkins, J.; Gazzard, B.; Pozniak, A. Pharmacokinetics of saquinavir hard gel/ritonavir (1000/100 mg twice daily) when administered with tenofovir diproxil fumarate in HIV-1-infected subjects. Br. J. Clin. Pharm. 2005, 59, 38–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avery, L.B.; Zarr, M.A.; Bakshi, R.P.; Siliciano, R.F.; Hendrix, C.W. Increasing Extracellular Protein Concentration Reduces Intracellular Antiretroviral Drug Concentration and Antiviral Effect. Aids Res. Hum. Retrovir. 2013, 29, 1434–1442. [Google Scholar] [CrossRef] [Green Version]
- Fischl, M.A.; Richman, D.D.; Flexner, C.; Fischl, M.A.; Richman, D.D.; Flexner, C.; Para, M.F.; Haubrich, R.; Karim, A.; Yeramian, P.; et al. Phase I/II study of the toxicity, pharmacokinetics, and activity of the HIV protease inhibitor SC-52151. J. Acquir. Immune Defic. Syndr. 1997, 15, 28–34. [Google Scholar] [CrossRef]
- Keogh, J.H.; Rynn, B.C.; Bruno Stieger, B.; Nicholls, G. Membrane Transporters: Fundamentals, Function and Their Role in ADME and Drug Development; Royal Society of Chemistry: London, UK, 2016; Volume 1. [Google Scholar]
- Suzuki, H.; Furukawa, H.; Miyazaki, H.; Hashimoto, M. Absorption, distribution, excretion and metabolism of SC-11800EE, a combined steroid preparation of SC-11800 (ethynodiol diacetate) and ethinyl estradiol in rats and mice. Radioisotopes 1977, 26, 152–157. [Google Scholar] [CrossRef] [Green Version]
- Bohnert, T.; Gan, L.S. Plasma protein binding: From discovery to development. J. Pharm. Sci. 2013, 102, 2953–2994. [Google Scholar] [CrossRef]
- Li, P.; Fan, Y.; Wang, Y.; Lu, Y.; Yin, Z. Characterization of plasma protein binding dissociation with online SPE-HPLC. Sci. Rep. 2015, 5, 14866. [Google Scholar] [CrossRef] [Green Version]
- Anusha, G.B.; Ahad, H.A.; Haranath, C.; Suryaprakash, R.C.; Kalpana, K. A technical view on transporters-the drug pharmacokinetics dictators. MOJ Bioequiv. Availab. 2019, 9, 47–52. [Google Scholar] [CrossRef]
- Liu, H.; Sahi, J. Role of Hepatic Drug Transporters in Drug Development. J. Clin. Pharm. 2016, 56 (Suppl. S7), S11–S22. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M. Pharmacogenetics of cytochrome P450 and its applications in drug therapy: The past, present and future. Trends Pharm. Sci. 2004, 25, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharm. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Fujikura, K.; Ingelman-Sundberg, M.; Lauschke, V.M. Genetic variation in the human cytochrome P450 supergene family. Pharm. Genom. 2015, 25, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lauschke, V.M. The genetic landscape of major drug metabolizing cytochrome P450 genes—An updated analysis of population-scale sequencing data. Pharm. J. 2022, 22, 284–293. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M. Implications of polymorphic cytochrome p450-dependent drug metabolism for drug development. Drug Metab. Dispos. 2001, 29, 570–573. [Google Scholar]
- Zhou, S.F.; Liu, J.P.; Chowbay, B. Polymorphism of human cytochrome P450 enzymes and its clinical impact. Drug Metab. Rev. 2009, 41, 89–295. [Google Scholar] [CrossRef]
- Smolders, E.J.; de Kanter, C.T.M.M.; de Knegt, R.J.; van der Valk, M.; Drenth, J.P.H.; Burger, D.M. Drug–drug interactions between direct-acting antivirals and psychoactive medications. Clin. Pharm. 2016, 55, 1471–1494. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Haque, S.; Chowdhury, P.; Cory, T.J.; Kodidela, S.; Yallapu, M.M.; Norwood, J.M.; Kumar, S. Pharmacokinetics and pharmacodynamics of cytochrome P450 inhibitors for HIV treatment. Expert. Opin. Drug Metab. Toxicol. 2019, 15, 417–427. [Google Scholar] [CrossRef]
- Bahap, M.; Kara, E.; Sain Guven, G. Fighting on two fronts: Drug–drug interactions in people living with HIV infected with SARS-CoV-2. Eur. J. Hosp. Pharm. 2020, 28, e3. [Google Scholar] [CrossRef]
- Yagura, H.; Watanabe, D.; Kushida, H.; Tomishima, K.; Togami, H.; Hirano, A.; Takahashi, M.; Hirota, K.; Ikuma, M.; Kasai, D.; et al. Impact of UGT1A1 gene polymorphisms on plasma dolutegravir trough concentrations and neuropsychiatric adverse events in Japanese individuals infected with HIV-1. BMC Infect. Dis. 2017, 17, 622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belkhir, L.; Seguin-Devaux, C.; Elens, L.; Pauly, C.; Gengler, N.; Schneider, S.; Ruelle, J.; Haufroid, V.; Vandercam, B. Impact of UGT1A1 polymorphisms on Raltegravir and its glucuronide plasma concentrations in a cohort of HIV-1 infected patients. Sci. Rep. 2018, 8, 7359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalilieh, S.; Feng, H.; Hulskotte, E.G.J.; Wenning, L.A.; Butterton, J.R. Clinical pharmacology profile of boceprevir, a hepatitis C virus NS3 protease inhibitor: Focus on drug-drug interactions. Clin. Pharm. 2015, 54, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Abel, S.; Russell, D.; Whitlock, L.A.; Ridgway, C.E.; Nedderman, A.N.R.; Walker, D.K. Assessment of the absorption, metabolism and absolute bioavailability of maraviroc in healthy male subjects. Br. J. Clin. Pharm. 2008, 65, 60–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Riviere, J.E. The application of allometric scaling principles to predict pharmacokinetic parameters across species. Expert. Opin. Drug Metab. Toxicol. 2014, 10, 1241–1253. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Jairam, R.K.; Mallurwar, S.R.; Sulochana, S.P.; Chandrasekhar, D.V.; Todmal, U.; Bhamidipati, R.K.; Richter, W.; Srinivas, N.R.; Mullangi, R. Prediction of Human Pharmacokinetics of Fomepizole from Preclinical Species Pharmacokinetics Based on Normalizing Time Course Profiles. AAPS PharmSciTech 2019, 20, 221. [Google Scholar] [CrossRef]
- Mahmood, I.; Balian, J.D. Interspecies scaling: Predicting clearance of drugs in humans. Three different approaches. Xenobiotica 1996, 26, 887–895. [Google Scholar] [CrossRef]
- Tang, H.; Mayersohn, M. A novel model for prediction of human drug clearance by allometric scaling. Drug Metab. Dispos. 2005, 33, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, I.; Martinez, M.; Hunter, R.P. Interspecies allometric scaling. Part I: Prediction of clearance in large animals. J. Vet. Pharm. Ther. 2006, 29, 415–423. [Google Scholar] [CrossRef]
- Sawada, Y.; Hanano, M.; Sugiyama, Y.; Iga, T. Prediction of the disposition of beta-lactam antibiotics in humans from pharmacokinetic parameters in animals. J. Pharm. Biopharm. 1984, 12, 241–261. [Google Scholar] [CrossRef]
- Mordenti, J. Man versus beast: Pharmacokinetic scaling in mammals. J. Pharm. Sci. 1986, 75, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Boxenbaum, H. Evolutionary biology, animal behavior, fourth-dimensional space, and the raison d’etre of drug metabolism and pharmacokinetics. Drug Metab. Rev. 1983, 14, 1057–1097. [Google Scholar] [CrossRef] [PubMed]
- Boxenbaum, H. Interspecies pharmacokinetic scaling and the evolutionary-comparative paradigm. Drug Metab. Rev. 1984, 15, 1071–1121. [Google Scholar] [CrossRef]
- Chaira, T.; Barman, T.; Samuel Raj, V. In Vitro ADME, Preclinical Pharmacokinetics and Prediction of Human Pharmacokinetics of RBx14255, a Novel Ketolide with Pharmacodynamics Against Multidrug-Resistant Bacterial Pathogens. J. Pharm. Pharm. Sci. 2020, 23, 206–219. [Google Scholar] [CrossRef]
- Mahmood, I.; Yuan, R. A comparative study of allometric scaling with plasma concentrations predicted by species-invariant time methods. Biopharm. Drug Dispos. 1999, 20, 137–144. [Google Scholar] [CrossRef]
- Eagle, H.; Fleischman, R.; Musselman, A.D. The effective concentrations of penicillin in vitro and in vivo for streptococci, pneumococci, and Treponema pallidum. J. Bacteriol. 1950, 59, 625–643. [Google Scholar] [CrossRef] [Green Version]
- Eagle, H.; Fleischman, R.; Musselman, A.D. Effect of schedule of administration on the therapeutic efficacy of penicillin; importance of the aggregate time penicillin remains at effectively bactericidal levels. Am. J. Med. 1950, 9, 280–299. [Google Scholar] [CrossRef]
- Ambrose, P.G.; Bhavnani, S.M.; Rubino, C.M.; Louie, A.; Gumbo, T.; Forrest, A.; Drusano, G.L. Pharmacokinetics-pharmacodynamics of antimicrobial therapy: It’s not just for mice anymore. Clin. Infect. Dis. 2007, 44, 79–86. [Google Scholar] [CrossRef]
- Craig, W.A. Pharmacokinetic/pharmacodynamic parameters: Rationale for antibacterial dosing of mice and men. Clin. Infect. Dis. 1998, 26, 1–10. [Google Scholar] [CrossRef]
- Dudley, M.N.; Ambrose, P.G. Pharmacodynamics in the study of drug resistance and establishing in vitro susceptibility breakpoints: Ready for prime time. Curr. Opin. Microbiol. 2000, 3, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gascon, A.; Solinis, M.A.; Isla, A. The Role of PK/PD Analysis in the Development and Evaluation of Antimicrobials. Pharmaceutics 2021, 13, 833. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, P.G.; Grasela, D.M.; Grasela, T.H.; Passarell, J.; Mayer, H.B.; Pierce, P.F. Pharmacodynamics of fluoroquinolones against Streptococcus pneumoniae in patients with community-acquired respiratory tract infections. Antimicrob. Agents Chemother. 2001, 45, 2793–2797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asin-Prieto, E.; Rodriguez-Gascon, A.; Isla, A. Applications of the pharmacokinetic/pharmacodynamic (PK/PD) analysis of antimicrobial agents. J. Infect. Chemother. 2015, 21, 319–329. [Google Scholar] [CrossRef]
- Heffernan, A.J.; Sime, F.B.; Lipman, J.; Roberts, J.A. Individualising Therapy to Minimize Bacterial Multidrug Resistance. Drugs 2018, 78, 621–641. [Google Scholar] [CrossRef]
- Jorda, A.; Zeitlinger, M. Preclinical Pharmacokinetic/Pharmacodynamic Studies and Clinical Trials in the Drug Development Process of EMA-Approved Antibacterial Agents: A Review. Clin. Pharm. 2020, 59, 1071–1084. [Google Scholar] [CrossRef]
- Nicolau, D.P. Optimizing outcomes with antimicrobial therapy through pharmacodynamic profiling. J. Infect. Chemother. 2003, 9, 292–296. [Google Scholar] [CrossRef]
- Drusano, G.L. Antimicrobial pharmacodynamics: Critical interactions of ‘bug and drug’. Nat. Rev. Microbiol. 2004, 2, 289–300. [Google Scholar] [CrossRef]
- McSharry, J.J.; Drusano, G.L. Antiviral pharmacodynamics in hollow fibre bioreactors. Antivir. Chem. Chemother. 2011, 21, 183–192. [Google Scholar] [CrossRef]
- Widmer, N.; Meylan, P.; Ivanyuk, A.; Aouri, M.; Decosterd, L.A.; Buclin, T. Oseltamivir in seasonal, avian H5N1 and pandemic 2009 A/H1N1 influenza: Pharmacokinetic and pharmacodynamic characteristics. Clin. Pharm. 2010, 49, 741–765. [Google Scholar] [CrossRef]
- He, G.; Massarella, J.; Ward, P. Clinical pharmacokinetics of the prodrug oseltamivir and its active metabolite Ro 64-0802. Clin. Pharm. 1999, 37, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Mendel, D.B.; Tai, C.Y.; Escarpe, P.A.; Li, W.; Sidwell, R.W.; Huffman, J.H.; Sweet, C.; Jakeman, K.J.; Merson, J.; Lacy, S.A.; et al. Oral administration of a prodrug of the influenza virus neuraminidase inhibitor GS 4071 protects mice and ferrets against influenza infection. Antimicrob. Agents Chemother. 1998, 42, 640–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidwell, R.W.; Smee, D.F. In vitro and in vivo assay systems for study of influenza virus inhibitors. Antivir. Res. 2000, 48, 1–16. [Google Scholar] [CrossRef] [PubMed]
- McSharry, J.J.; Weng, Q.; Brown, A.; Kulawy, R.; Drusano, G.L. Prediction of the pharmacodynamically linked variable of oseltamivir carboxylate for influenza A virus using an in vitro hollow-fiber infection model system. Antimicrob. Agents Chemother. 2009, 53, 2375–2381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drusano, G.L.; Preston, S.L.; Smee, D.; Bush, K.; Bailey, K.; Sidwell, R.W. Pharmacodynamic evaluation of RWJ-270201, a novel neuraminidase inhibitor, in a lethal murine model of influenza predicts efficacy for once-daily dosing. Antimicrob. Agents Chemother. 2001, 45, 2115–2118. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.N.; McSharry, J.J.; Weng, Q.; Adams, J.R.; Kulawy, R.; Drusano, G.L. Zanamivir, at 600 milligrams twice daily, inhibits oseltamivir-resistant 2009 pandemic H1N1 influenza virus in an in vitro hollow-fiber infection model system. Antimicrob. Agents Chemother. 2011, 55, 1740–1746. [Google Scholar] [CrossRef] [Green Version]
- Obach, R.S.; Baxter, J.G.; Liston, T.E.; Silber, B.M.; Jones, B.C.; MacIntyre, F.; Rance, D.J.; Wastall, P. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharm. Exp. Ther. 1997, 283, 46–58. [Google Scholar]
- Sharma, A.; Benbrook, D.M.; Woo, S. Pharmacokinetics and interspecies scaling of a novel, orally-bioavailable anti-cancer drug, SHetA2. PLoS ONE 2018, 13, e0194046. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antiviral Class [Reference] | Drug | Substrate of | Inhibitor of | BCS Class |
|---|---|---|---|---|
| Hepatitis C antiviral [19] | Simeprevir | CYP3A4 | CYP3A4/1A2 | IV |
| Ombitasvir | CYP2C8 | UGT1A1 | IV | |
| Paritaprevir | CYP3A4/3A5 | UGT1A1 | IV | |
| Boceprevir | CYP3A4/3A5 | CYP3A4/3A5 | IV | |
| Dasabuvir | CYP2C8/3A4 | UGT1A1 | IV | |
| Protease inhibitor [20] | Indinavir | CYP3A4, UGT1A1 | CYP3A4 | II/IV |
| Nelfinavir | CYP2C19/3A4 | CYP3A4 | IV | |
| Saquinavir | CYP3A4 | CYP3A4 | IV | |
| Tipranavir | CYP3A4 | CYP3A4/2D6 | II | |
| Darunavir | CYP3A4 | CYP3A4 | II | |
| Lopinavir | CYP3A4 | CYP3A4 (minor) | IV | |
| Fosamprenavir | CYP3A4 | CYP3A4 | II | |
| Ritonavir | CYP3A4/2D6 | CYP3A4/2D6 (minor) | IV | |
| Atazanavir | CYP3A4 | CYP2C8/3A4 (minor) | II | |
| NRTIs [21] | Tenofovir disoproxil fumarate | Not CYP substrate | Not CYP inhibitor | III |
| Abacavir | Not CYP substrate, HLA-B | Not CYP inhibitor | III | |
| Zidovudine | hepatic glucuronidation, UGT | Not CYP inhibitor | I | |
| Lamivudine | OCT1, OCT2 | Not CYP inhibitor | III | |
| NNRTIs [20] | Efavirenz | CYP2B6 (major)/2A6/3A4 | CYP3A4 | II/IV |
| Nevirapine | CYP3A4/2B6 | Not CYP inhibitor | II | |
| Etravirine | CYP3A4/2C9/2C19 | CYP2C9/2C19 | IV | |
| Rilpivirine | CYP3A4 | Not CYP inhibitor | II | |
| Delavirdine | CYP3A4 | Not CYP inhibitor | III | |
| HIV integrase inhibitors [20,21,22,23] | Dolutegravir | 3A4 (minor), UGT1A1 | Not CYP inhibitor | II |
| Elvitegravir | CYP 3A4 | Not CYP inhibitor | II | |
| Raltegravir | Not CYP substrate | Not CYP inhibitor | II | |
| Nucleotide polymerase inhibitors [24] | Sofosbuvir | CYP 3A4/3A5 | Not CYP inhibitor | III |
| CCR5 antagonist [25] | Maraviroc | CYP3A4/3A5 | Not CYP inhibitor | III |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaira, T.; Subramani, C.; Barman, T.K. ADME, Pharmacokinetic Scaling, Pharmacodynamic and Prediction of Human Dose and Regimen of Novel Antiviral Drugs. Pharmaceutics 2023, 15, 1212. https://doi.org/10.3390/pharmaceutics15041212
Chaira T, Subramani C, Barman TK. ADME, Pharmacokinetic Scaling, Pharmacodynamic and Prediction of Human Dose and Regimen of Novel Antiviral Drugs. Pharmaceutics. 2023; 15(4):1212. https://doi.org/10.3390/pharmaceutics15041212
Chicago/Turabian StyleChaira, Tridib, Chandru Subramani, and Tarani Kanta Barman. 2023. "ADME, Pharmacokinetic Scaling, Pharmacodynamic and Prediction of Human Dose and Regimen of Novel Antiviral Drugs" Pharmaceutics 15, no. 4: 1212. https://doi.org/10.3390/pharmaceutics15041212