
A Physiologically Based Pharmacokinetic Model to Predict Determinants of Variability in Epirubicin Exposure and Tissue Distribution

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Chemical Information
2.2. Human Liver Microsomes
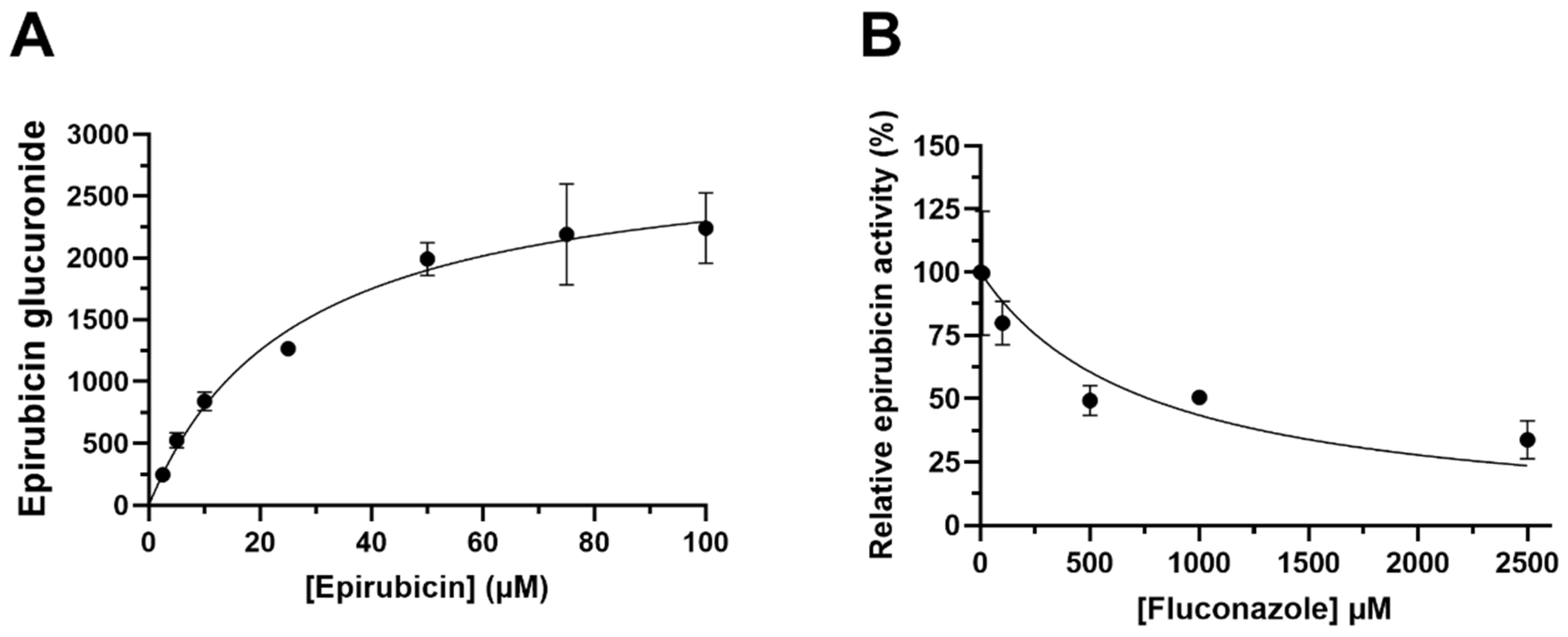
2.3. Epirubicin Glucuronidation Assay
2.4. Quantification of Epirubicin Glucuronide Formation
2.5. Data Analysis (In Vitro Kinetics)
2.6. Development and Verification of Epirubicin PBPK Model
2.6.1. Structural Model
2.6.2. Development of Epirubicin Compound Profile
2.6.3. Population Profile
2.6.4. Simulated Trial Design
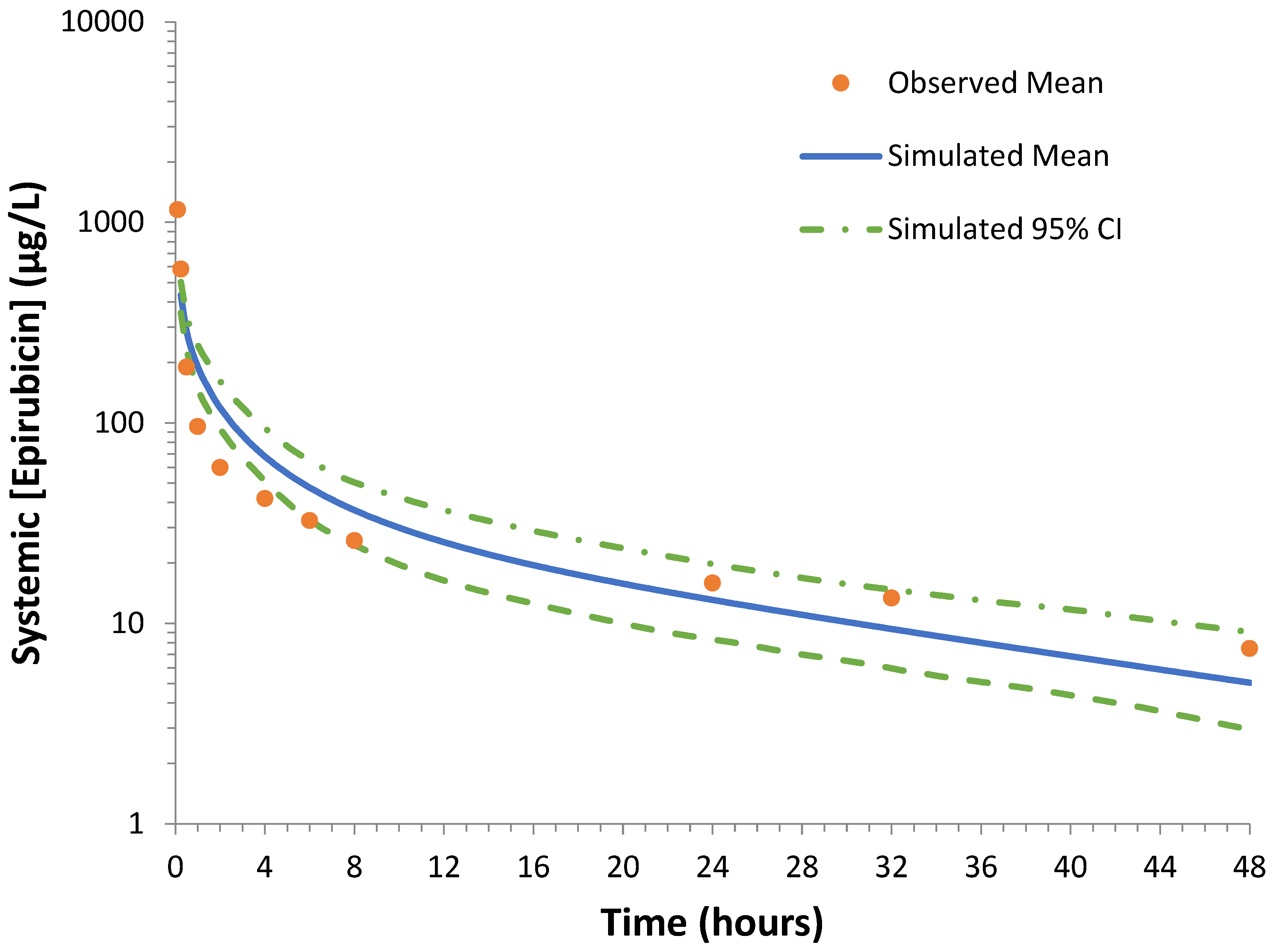
2.6.5. Observed Clinical Data and Compound File Verification
2.7. Population Characteristics Associated with Variability in Epirubicin Exposure
3. Results
3.1. Characterisation of In Vitro Epirubicin Glucuronidation
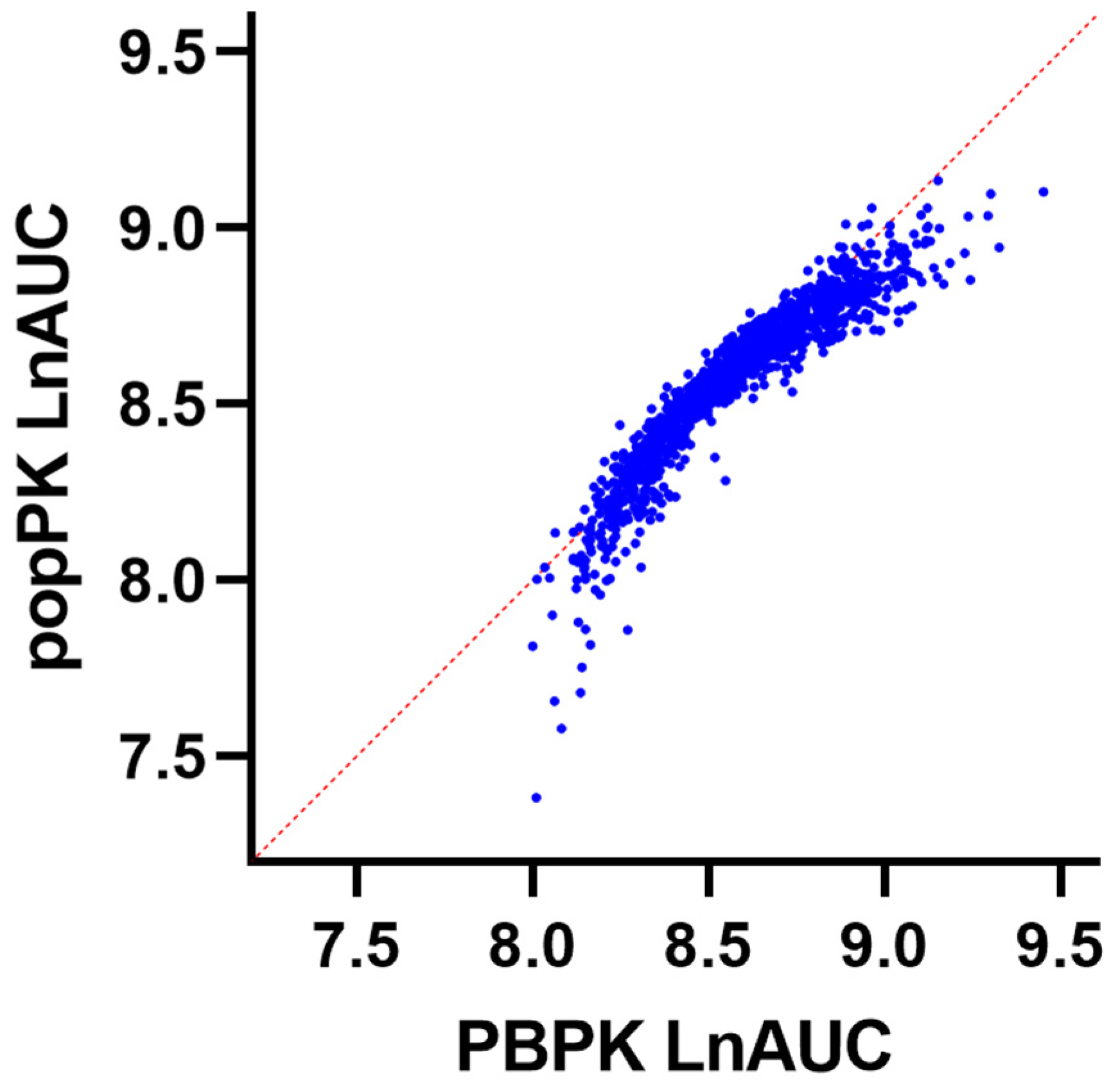
3.2. Verification of the Epirubicin PBPK Model
3.3. Epirubicin Exposure in Oncology Cohort
3.4. Epirubicin Clearance Pathways
3.5. Determination of Population Characteristics Affecting Epirubicin Clearance
3.6. Associations between Epirubicin Plasma and Tissue Concentration
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tariq, M.; Alam, M.A.; Singh, A.T.; Panda, A.K.; Talegaonkar, S. Improved oral efficacy of epirubicin through polymeric nanoparticles: Pharmacodynamic and toxicological investigations. Drug Deliv. 2016, 23, 2990–2997. [Google Scholar] [CrossRef] [Green Version]
- Forrest, R.A.; Swift, L.P.; Evison, B.J.; Rephaeli, A.; Nudelman, A.; Phillips, D.R.; Cutts, S.M. The hydroxyl epimer of doxorubicin controls the rate of formation of cytotoxic anthracycline-DNA adducts. Cancer Chemother. Pharmacol. 2013, 71, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Eksborg, S. Pharmacokinetics of anthracyclines. Acta Oncol. 1989, 28, 873–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wade, J.R.; Kelman, A.W.; Kerr, D.J.; Robert, J.; Whiting, B. Variability in the pharmacokinetics of epirubicin: A population analysis. Cancer Chemother. Pharmacol. 1992, 29, 391–395. [Google Scholar] [CrossRef]
- Gurney, H.P.; Ackland, S.; Gebski, V.; Farrell, G. Factors affecting epirubicin pharmacokinetics and toxicity: Evidence against using body-surface area for dose calculation. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1998, 16, 2299–2304. [Google Scholar] [CrossRef] [PubMed]
- Robert, J.E. Clinical pharmacology and dose-effect relationship. Drugs 1993, 45 (Suppl. S2), 20–30. [Google Scholar] [CrossRef] [PubMed]
- Drooger, J.C.; van Pelt-Sprangers, J.M.; Leunis, C.; Jager, A.; de Jongh, F.E. Neutrophil-guided dosing of anthracycline-cyclophosphamide-containing chemotherapy in patients with breast cancer: A feasibility study. Med. Oncol. 2015, 32, 113. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Wu, J.; Lin, C.; Ding, S.; Lu, S.; Fang, Y.; Huang, J.; Hong, J.; Gao, W.; Zhu, S.; et al. The Comparative Safety of Epirubicin and Cyclophosphamide versus Docetaxel and Cyclophosphamide in Lymph Node-Negative, HR-Positive, HER2-Negative Breast Cancer (ELEGANT): A Randomized Trial. Cancers 2022, 14, 3221. [Google Scholar] [CrossRef]
- Kang, J.S.; Lee, M.H. Overview of therapeutic drug monitoring. Korean J. Intern. Med. 2009, 24, 1–10. [Google Scholar] [CrossRef]
- Ormrod, D.; Holm, K.; Goa, K.; Spencer, C. Epirubicin: A review of its efficacy as adjuvant therapy and in the treatment of metastatic disease in breast cancer. Drugs Aging 1999, 15, 389–416. [Google Scholar] [CrossRef]
- Mueller-Schoell, A.; Groenland, S.L.; Scherf-Clavel, O.; van Dyk, M.; Huisinga, W.; Michelet, R.; Jaehde, U.; Steeghs, N.; Huitema, A.D.; Kloft, C. Therapeutic drug monitoring of oral targeted antineoplastic drugs. Eur. J. Clin. Pharmacol. 2021, 77, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, A.; Hopkins, A.M.; Sorich, M.J.; Rowland, A. Evaluating the utility of therapeutic drug monitoring in the clinical use of small molecule kinase inhibitors: A review of the literature. Expert Opin. Drug Metab. Toxicol. 2021, 17, 803–821. [Google Scholar] [CrossRef] [PubMed]
- Wills, K.H.; Behan, S.J.; Nance, M.J.; Dawson, J.L.; Polasek, T.M.; Hopkins, A.M.; van Dyk, M.; Rowland, A. Combining therapeutic drug monitoring and pharmacokinetic modelling deconvolutes physiological and environmental sources of variability in clozapine exposure. Pharmaceutics 2022, 14, 47. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.; Van Dyk, M.; Hopkins, A.M.; Mounzer, R.; Polasek, T.M.; Rostami-Hodjegan, A.; Sorich, M.J. Physiologically Based Pharmacokinetic Modeling to Identify Physiological and Molecular Characteristics Driving Variability in Drug Exposure. Clin. Pharmacol. Ther. 2018, 104, 1219–1228. [Google Scholar] [CrossRef]
- Sorich, M.; Mulflib, F.; van Dyk, M.; Hopkins, A.; Polasek, T.; Marshall, J. Use of predictive analytics to identify physiological and molecular characteristics driving variability in axitinib exposure. J. Clin. Pharmacol. 2019, 59, 872–879. [Google Scholar] [CrossRef]
- Kluwe, F.; Michelet, R.; Mueller-Schoell, A.; Maier, C.; Klopp-Schulze, L.; Van Dyk, M.; Mikus, G.; Huisinga, W.; Kloft, C. Perspectives on Model-Informed Precision Dosing in the Digital Health Era: Challenges, Opportunities, and Recommendations. Clin. Pharmacol. Ther. 2020, 109, 29–36. [Google Scholar] [CrossRef]
- Tsamandouras, N.; Rostami-Hodjegan, A.; Aarons, L. Combining the ‘bottom up’ and ‘top down’ approaches in pharmacokinetic modelling: Fitting PBPK models to observed clinical data. Br. J. Clin. Pharmacol. 2015, 79, 48–55. [Google Scholar] [CrossRef]
- Shebley, M.; Sandhu, P.; Riedmaier, A.E.; Jamei, M.; Narayanan, R.; Patel, A.; Peters, S.A.; Reddy, V.P.; Zheng, M.; de Zwart, L.; et al. Physiologically Based Pharmacokinetic Model Qualification and Reporting Procedures for Regulatory Submissions: A Consortium Perspective. Clin. Pharmacol. Ther. 2018, 104, 88–110. [Google Scholar] [CrossRef] [Green Version]
- Sandström, M.; Lindman, H.; Nygren, P.; Lidbrink, E.; Bergh, J.; Karlsson, M.O. Model Describing the Relationship Between Pharmacokinetics and Hematologic Toxicity of the Epirubicin-Docetaxel Regimen in Breast Cancer Patients. J. Clin. Oncol. 2005, 23, 413–421. [Google Scholar] [CrossRef]
- Ralph, L.D.; Thomson, A.H.; Dobbs, N.A.; Twelves, C. A population model of epirubicin pharmacokinetics and application to dosage guidelines. Cancer Chemother. Pharmacol. 2003, 52, 34–40. [Google Scholar] [CrossRef]
- Innocenti, F.; Iyer, L.; Ramírez, J.; Green, M.D.; Ratain, M.J. Epirubicin glucuronidation is catalyzed by human UDP-glucuronosyltransferase 2B7. Drug Metab. Dispos. 2001, 29, 686–692. [Google Scholar] [PubMed]
- Sawyer, M.B.; Pituskin, E.; Damaraju, S.; Bies, R.R.; Vos, L.J.; Prado, C.M.; Kuzma, M.; Scarfe, A.G.; Clemons, M.; Tonkin, K.; et al. A Uridine Glucuronosyltransferase 2B7 Polymorphism Predicts Epirubicin Clearance and Outcomes in Early-Stage Breast Cancer. Clin. Breast Cancer 2016, 16, 139–144.e3. [Google Scholar] [CrossRef] [PubMed]
- Joy, A.A.; Vos, L.J.; Pituskin, E.; Cook, S.F.; Bies, R.R.; Vlahadamis, A.; King, K.; Basi, S.K.; Meza-Junco, J.; Mackey, J.R.; et al. Uridine Glucuronosyltransferase 2B7 Polymorphism-Based Pharmacogenetic Dosing of Epirubicin in FEC Chemotherapy for Early-Stage Breast Cancer. Clin. Breast Cancer 2021, 21, e584–e593. [Google Scholar] [CrossRef]
- Polasek, T.M.; Rayner, C.R.; Peck, R.W.; Rowland, A.; Kimko, H.; Rostami-Hodjegan, A. Toward Dynamic Prescribing Information: Codevelopment of Companion Model-Informed Precision Dosing Tools in Drug Development. Clin. Pharmacol. Drug Dev. 2018, 8, 418–425. [Google Scholar] [CrossRef]
- Rodrigues, A.; Rowland, A. From Endogenous Compounds as Biomarkers to Plasma-Derived Nanovesicles as Liquid Biopsy. Clin. Pharmacol. Ther. 2019, 105, 1407–1420. [Google Scholar] [CrossRef]
- Bowalgaha, K.; Elliot, D.J.; Mackenzie, P.I.; Knights, K.M.; Swedmark, S.; Miners, J.O. S-Naproxen and desmethylnaproxen glucuronidation by human liver microsomes and recombinant human UDP-glucuronosyltransferases (UGT): Role of UGT2B7 in the elimination of naproxen. Br. J. Clin. Pharmacol. 2005, 60, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Boase, S.; Miners, J.O. In vitro-in vivo correlations for drugs eliminated by glucuronidation: Investigations with the model substrate zidovudine. Br. J. Clin. Pharmacol. 2002, 54, 493–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowland, A.; Gaganis, P.; Elliot, D.J.; Mackenzie, P.I.; Knights, K.M.; Miners, J.O. Binding of inhibitory fatty acids is responsible for the enhancement of UDP-glucuronosyltransferase 2B7 activity by albumin: Implications for in vitro-in vivo extrapolation. J. Pharmacol. Exp. Ther. 2007, 321, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Miners, J.O.; Lillywhite, K.J.; Matthews, A.P.; Jones, M.E.; Birkett, D.J. Kinetic and inhibitor studies of 4-methylumbelliferone and 1-naphthol glucuronidation in human liver microsomes. Biochem. Pharmacol. 1988, 37, 665–671. [Google Scholar] [CrossRef]
- Rowland Yeo, K.; Jamei, M.; Yang, J.; Tucker, G.T.; Rostami-Hodjegan, A. Physiologically based mechanistic modelling to predict complex drug-drug interactions involving simultaneous competitive and time-dependent enzyme inhibition by parent compound and its metabolite in both liver and gut—The effect of diltiazem on the time-course of exposure to triazolam. Eur. J. Pharm. Sci. 2010, 39, 298–309. [Google Scholar]
- Mouridsen, H.T.; Alfthan, C.; Bastholt, L.; Bergh, J.; Dalmark, M.; Eksborg, S.; Hellsten, S.; Kjaer, M.; Peterson, C.; Skovsgård, T.; et al. Current Status of Epirubicin (Farmorubicin) in the Treatment of Solid Tumours. Acta Oncol. 1990, 29, 257–285. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, T.; Rowland, M. Physiologically based pharmacokinetic modelling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 2006, 95, 1238–1257. [Google Scholar] [CrossRef]
- Robert, J.; Vrignaud, P.; Nguyen-Ngoc, T.; Iliadis, A.; Mauriac, L.; Hurteloup, P. Comparative pharmacokinetics and metabolism of doxorubicin and epirubicin in patients with metastatic breast cancer. Cancer Treat. Rep. 1985, 69, 633–640. [Google Scholar]
- Robert, J. Clinical pharmacokinetics of epirubicin. Clin. Pharmacokinet. 1994, 26, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Gori, S.; Rulli, A.; Mosconi, A.M.; Sidoni, A.; Colozza, M.; Crinò, L. Safety of epirubicin adjuvant chemotherapy in a breast cancer patient with chronic renal failure undergoing hemodialytic treatment. Tumori J. 2006, 92, 364–365. [Google Scholar] [CrossRef] [PubMed]
- Nicolella, D.; Grimaldi, G.; Colantuoni, G.; Belli, M.; Frasci, G.; Perchard, J.; Comella, P. Weekly low dose epirubicin in elderly cancer patients. Tumori J. 1996, 82, 369–371. [Google Scholar] [CrossRef] [Green Version]
- Dobbs, N.A.; Twelves, C.J. What is the effect of adjusting epirubicin doses for body surface area? Br. J. Cancer 1998, 78, 662–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, M.; Wagner, A.D.; Kouvelakis, K.; Nanji, H.; Starling, N.; Chau, I.; Watkins, D.; Rao, S.; Peckitt, C.; Cunningham, D. Influence of sex on chemotherapy efficacy and toxicity in oesophagogastric cancer: A pooled analysis of four randomised trials. Eur. J. Cancer 2019, 121, 40–47. [Google Scholar] [CrossRef]
- Rodrigues, A.D.; Dyk, M.; Sorich, M.J.; Fahmy, A.; Useckaite, Z.; Newman, L.A.; Kapetas, A.J.; Mounzer, R.; Wood, L.S.; Johnson, J.G.; et al. Exploring the Use of Serum-Derived Small Extracellular Vesicles as Liquid Biopsy to Study the Induction of Hepatic Cytochromes P450 and Organic Anion Transporting Polypeptides. Clin. Pharmacol. Ther. 2021, 110, 248–258. [Google Scholar] [CrossRef]
- Rowland, A.; Ruanglertboon, W.; Van Dyk, M.; Wijayakumara, D.; Wood, L.S.; Meech, R.; Mackenzie, P.I.; Rodrigues, A.D.; Marshall, J.; Sorich, M. Plasma extracellular nanovesicle (exosome)-derived biomarkers for drug metabolism pathways: A novel approach to characterize variability in drug exposure. Br. J. Clin. Pharmacol. 2019, 85, 216–226. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, A.D.; Wood, L.S.; Vourvahis, M.; Rowland, A. Leveraging Human Plasma-Derived Small Extracellular Vesicles as Liquid Biopsy to Study the Induction of Cytochrome P450 3A4 by Modafinil. Clin. Pharmacol. Ther. 2022, 111, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Useckaite, Z.; Rodrigues, A.D.; Hopkins, A.M.; Newman, L.A.; Johnson, J.G.; Sorich, M.J.; Rowland, A. Role of Extracellular Vesicle-Derived Biomarkers in Drug Metabolism and Disposition. Drug Metab. Dispos. Biol. Fate Chem. 2021, 49, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Achour, B.; Al-Majdoub, Z.M.; Grybos-Gajniak, A.; Lea, K.; Kilford, P.; Zhang, M.; Knight, D.; Barber, J.; Schageman, J.; Rostami-Hodjegan, A. Liquid Biopsy Enables Quantification of the Abundance and Interindividual Variability of Hepatic Enzymes and Transporters. Clin. Pharmacol. Ther. 2021, 109, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Achour, B.; Gosselin, P.; Terrier, J.; Gloor, Y.; Al-Majdoub, Z.M.; Polasek, T.M.; Daali, Y.; Rostami-Hodjegan, A.; Reny, J. Liquid Biopsy for Patient Characterization in Cardiovascular Disease: Verification against Markers of Cytochrome P450 and P-Glycoprotein Activities. Clin. Pharmacol. Ther. 2022, 111, 1268–1277. [Google Scholar] [CrossRef] [PubMed]
- Ruanglertboon, W.; Sorich, M.; Hopkins, A.; Rowland, A. Mechanistic Modelling Identifies and Addresses the Risks of Empiric Concentration-Guided Sorafenib Dosing. Pharmaceuticals 2021, 14, 389. [Google Scholar] [CrossRef]
- Polasek, T.M.; Tucker, G.T.; Sorich, M.J.; Wiese, M.D.; Mohan, T.; Rostami-Hodjegan, A.; Korprasertthaworn, P.; Perera, V.; Rowland, A. Prediction of olanzapine exposure in individual patients using PBPK modelling and simulation. Br. J. Clin. Pharmacol. 2018, 84, 462–476. [Google Scholar] [CrossRef] [Green Version]
- Polasek, T.M.; Rostami-Hodjegan, A. Virtual Twins: Understanding the Data Required for Model-Informed Precision Dosing. Clin. Pharmacol. Ther. 2020, 107, 742–745. [Google Scholar] [CrossRef]
- Smith, L.A.; Cornelius, V.R.; Plummer, C.J.; Levitt, G.; Verrill, M.; Canney, P.; Jones, A. Cardiotoxicity of anthracycline agents for the treatment of cancer: Systematic review and meta-analysis of randomised controlled trials. BMC Cancer 2010, 10, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Precursor Ion (m/z) | Product Ion (m/z) | Dwell (s) | Fragmentor (V) | Collision Energy (V) | Cell Acceleration (V) | Polarity |
|---|---|---|---|---|---|---|
| 720.22 | 702.2 | 200 | 380 | 15 | 4 | Positive |
| 720.22 | 361.2 | 200 | 380 | 36 | 4 | Positive |
| 720.22 | 324.2 | 200 | 380 | 20 | 4 | Positive |
| 720.22 | 306 | 200 | 380 | 16 | 4 | Positive |
| Phys Chem | |
|---|---|
| Molecular Weight (g/mol) | 543.52 |
| log Po:w | 1.41 |
| Species | Diprotic Base |
| pKa (Strongest Acidic) | 8.010 |
| pKa 2 (Strongest Basic) | 10.030 |
| Blood Binding | |
| B/P | 0.729 |
| fu | 0.23 |
| Distribution (full PB-PK model) | |
| Vss (L/Kg) | 25.265 |
| Prediction Method | Rogers and Rowland [32] |
| Kp Scalar | 25 |
| Elimination | |
| HLM—UGT2B7 (Km; µM) | 26.2 |
| HLM—UGT2B7 (Vmax; pmol/min/mg protein) | 2897 |
| HLM—UGT2B7 (fu) | 1 |
| Additional clearance—CLR (L/h) | 9.0 |
| Interaction | |
| UGT2B7 (IndC50; µM) | 0.368 |
| UGT2B7 (Indmax) | 13.95 |
| Statistic | AUC (ng/mL·h) | CMax (ng/mL) | Dose (mg) | CL (Dose/AUC) (L/h) |
|---|---|---|---|---|
| Mean | 5374 | 12,683 | 213.2 | 41.6 |
| Median | 5197 | 12,653 | 212.1 | 40.5 |
| Geometric Mean | 5252 | 12,654 | 211.8 | 40.3 |
| 90% confidence interval (lower limit) | 5211 | 12,622 | 211.0 | 40.0 |
| 90% confidence interval (upper limit) | 5293 | 12,686 | 212.7 | 40.7 |
| 5th centile | 3776 | 11,296 | 176.4 | 26.7 |
| 95th centile | 7537 | 14,142 | 253.7 | 59.8 |
| Skewness | 0.99 | 0.12 | 0.29 | 0.50 |
| cv | 0.22 | 0.07 | 0.11 | 0.24 |
| Min Val | 2980 | 9678 | 149.8 | 14.8 |
| Max Val | 12,710 | 15,469 | 302.5 | 80.9 |
| Fold | 4.27 | 1.60 | 2.02 | 5.45 |
| Std Dev | 1191 | 864 | 24.1 | 10.2 |
| Variable | Estimated Ln AUC (ng/mL·h) | Standard Error | Range (95% CI) | R2 with Other Variables | p Value |
|---|---|---|---|---|---|
| Intercept (constant) | 8.211 | 0.02994 | 8.152 to 8.269 | <0.0001 | |
| Sex | −0.03709 | 0.003901 | −0.04474 to −0.02944 | 0.2216 | <0.0001 |
| Age | 0.00251 | 0.000174 | 0.002169 to 0.002850 | 0.5024 | <0.0001 |
| BSA | 0.2669 | 0.01132 | 0.2447 to 0.2891 | 0.4266 | <0.0001 |
| Haematocrit | −0.00538 | 0.000373 | −0.006110 to −0.004649 | 0.005414 | <0.0001 |
| Albumin | 0.0111 | 0.000251 | 0.01060 to 0.01159 | 0.01572 | <0.0001 |
| GFR | −0.00178 | 0.000106 | −0.001983 to −0.001568 | 0.5261 | <0.0001 |
| Liver UGT2B7 | −9.77 × 10−8 | 1.34 × 10−9 | −1.00× 10−7 to −9.51 × 10−8 | 0.2798 | <0.0001 |
| Kidney UGT2B7 | −3.24 × 10−7 | 1.07 × 10−8 | −3.45 × 10−7 to −3.03 × 10−7 | 0.03006 | <0.0001 |
| Model | R | R2 | Adjusted R2 | Std. Error of the Estimate | R2 Change |
|---|---|---|---|---|---|
| a | 0.749 a | 0.561 | 0.561 | 0.141 | 0.561 |
| b | 0.821 b | 0.674 | 0.674 | 0.121 | 0.113 |
| c | 0.861 c | 0.741 | 0.740 | 0.108 | 0.067 |
| d | 0.886 d | 0.785 | 0.784 | 0.099 | 0.044 |
| e | 0.911 e | 0.830 | 0.830 | 0.087 | 0.046 |
| f | 0.922 f | 0.849 | 0.849 | 0.082 | 0.019 |
| g | 0.929 g | 0.863 | 0.863 | 0.079 | 0.014 |
| h | 0.932 h | 0.869 | 0.868 | 0.077 | 0.006 |
| Tissue | Mean Cmax (ng/mL) | Mean AUC (ng/mL·h) |
|---|---|---|
| Plasma | 979 | 4530 |
| Muscle | 2868 | 169,241 |
| Heart | 31,952 | 144,482 |
| Brain | 22,410 | 147,660 |
| Adipose | 1049 | 18,680 |
| Liver | 13,973 | 92,446 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ansaar, R.; Meech, R.; Rowland, A. A Physiologically Based Pharmacokinetic Model to Predict Determinants of Variability in Epirubicin Exposure and Tissue Distribution. Pharmaceutics 2023, 15, 1222. https://doi.org/10.3390/pharmaceutics15041222
Ansaar R, Meech R, Rowland A. A Physiologically Based Pharmacokinetic Model to Predict Determinants of Variability in Epirubicin Exposure and Tissue Distribution. Pharmaceutics. 2023; 15(4):1222. https://doi.org/10.3390/pharmaceutics15041222
Chicago/Turabian StyleAnsaar, Radwan, Robyn Meech, and Andrew Rowland. 2023. "A Physiologically Based Pharmacokinetic Model to Predict Determinants of Variability in Epirubicin Exposure and Tissue Distribution" Pharmaceutics 15, no. 4: 1222. https://doi.org/10.3390/pharmaceutics15041222
APA StyleAnsaar, R., Meech, R., & Rowland, A. (2023). A Physiologically Based Pharmacokinetic Model to Predict Determinants of Variability in Epirubicin Exposure and Tissue Distribution. Pharmaceutics, 15(4), 1222. https://doi.org/10.3390/pharmaceutics15041222