Abstract
Meropenem is currently the most common carbapenem in clinical applications. Industrially, the final synthetic step is characterized by a heterogeneous catalytic hydrogenation in batch mode with hydrogen and Pd/C. The required high-quality standard is very difficult to meet and specific conditions are required to remove both protecting groups [i.e., p-nitrobenzyl (pNB) and p-nitrobenzyloxycarbonyl (pNZ)] simultaneously. The three-phase gas–liquid–solid system makes this step difficult and unsafe. The introduction of new technologies for small-molecule synthesis in recent years has opened up new landscapes in process chemistry. In this context, we have investigated meropenem hydrogenolysis using microwave (MW)-assisted flow chemistry for use as a new technology with industrial prospects. The reaction parameters (catalyst amount, T, P, residence time, flow rate) in the move from the batch process to semi-continuous flow were investigated under mild conditions to determine their influence on the reaction rate. The optimization of the residence time (840 s) and the number of cycles (4) allowed us to develop a novel protocol that halves the reaction time compared to batch production (14 min vs. 30 min) while maintaining the same product quality. The increase in productivity using this semi-continuous flow technique compensates for the slightly lower yield (70% vs. 74%) obtained in batch mode.
1. Introduction
Carbapenems are currently considered to be lifesaving drugs [1], with their broad-spectrum activity meaning that they play a vital role for human health. They have the greatest potency of all β-lactam antibiotics against Gram-positive and Gram-negative bacteria. Their medical use is so crucial and essential that they have been defined “last-line antibiotics” or “antibiotics of last resort”, as they are the most effective weapon against known and suspected multidrug-resistant (MDR) bacterial infections [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26].
Part of the carbapenem antibiotics subgroup, meropenem (1, Figure 1) is currently the most widely used in clinical treatment [27,28,29]. Its worldwide market was valued at USD 1.7 billion in 2021 and is expected to grow to USD 2.1 billion by 2027, making this drug a blockbuster [30]. Alone, it made up 43% of the USD 3.9 billion in global sales of the carbapenem family in 2021 [31]. It was discovered by Sumitomo Dainippon Pharma, now Sumitomo Pharma, in 1983 [32,33,34], and was approved by the Food and Drug Administration (FDA) in 1996 [35]. The drug is sold under the brand name Merrem™, as a generic drug substance, or in combination with vaborbactam, part of a new generation of β-lactamase inhibitors, under the brand name Vabomere™. It is administered intravenously in both forms, and is listed on the World Health Organization’s list of Essential Medicines thanks to its importance [36,37,38,39,40,41,42,43,44,45,46].
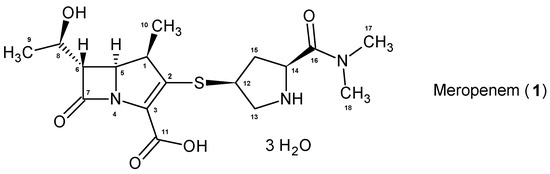
Figure 1.
Meropenem (1).
The literature reports different synthetic routes for its preparation [32,33,34,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68]. The most widely used industrial synthesis includes more than twenty steps characterized by protection with p-nitrobenzyl (pNB) and p-nitrobenzyloxycarbonyl (pNZ) on the carbapenem scaffold. This strategy, which allows the preparation of the drug with the correct stereochemical configuration (β-methyl in position 1), is still considered the most efficient and competitive in the world market. The high stability conferred to the β-lactam core during all chemical steps by these protecting groups with a high steric hindrance has been the main advantage of their use for more than 30 years. As depicted in Scheme 1, the condensation between 4-nitrobenzyl(4R,5S,6S)-3-[(diphenylphosphono)-oxy]-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3,2,0]hept-2-ene-2-carboxylate (2, MAP) and (2S,4S)-2-dimethylaminocarbonyl-4-mercapto-1-(4-nitrobenzyloxycarbonyl)pyrrolidine (3, cis-pNZ) gives bis-protected meropenem (4). Batch-mode hydrogenation with palladium over carbon (Pd/C) under pressure simultaneously removes the p-nitrobenzyl (pNB) and p-nitrobenzyloxycarbonyl (pNZ) protecting groups, according to mechanism reported in Figure 2. In the first step, the nitro group is reduced to the p-aminobenzyl(oxycarbonyl) intermediate, which spontaneously collapses to the quinonimine methide and the deprotected compound by 1,6-electron-pair shift [69]. The subtle equilibrium between the hydrogenation operating parameters and the stability of the carbapenem skeleton has always been the key to the success of this reaction, leading all manufacturers to balance these two factors. The catalytic system was characterized by the presence of N-methylmorpholine and acetic acid as buffers, which allowed it to operate at pH = 6.5–6.8 throughout the reaction time, ensuring the stability of both reagent and product. Water and isopropyl alcohol met the requirements of green chemistry and sustainability, and the waste was easy to handle. Catalyst filtration, workup, and subsequent first crystallization provides crude meropenem (1). The product is purified by a second crystallization to reach the quality standard before it is sterilized and becomes an injectable drug. Hydrogenation reactions pose significant risks for a production plant, and this can only be mitigated in part by heavy investment. The implementation of batch-mode manufacturing requires a bunker facility, and the engineered architecture must be designed to restrict uncontrolled events and any possible damage must be contained. The risk to people and the surrounding area has to be under control [70,71,72,73,74]. The considerable manufacturing capacity needed to support massive global demand, the operational accuracy needed to obtain a high-grade product and the high flammability of Pd/C are all real and routine issues that must be tackled in the commercial production of meropenem (1). Batch production is no longer adequate in the industrial manufacturing of pharmaceuticals, where a balance between streamlining and increased productivity is required to compete in the global active pharmaceutical ingredients (APIs) market. In addition, the high-quality standard required is very challenging to satisfy due to the instability of the carbapenem skeleton during the hydrogenation step.
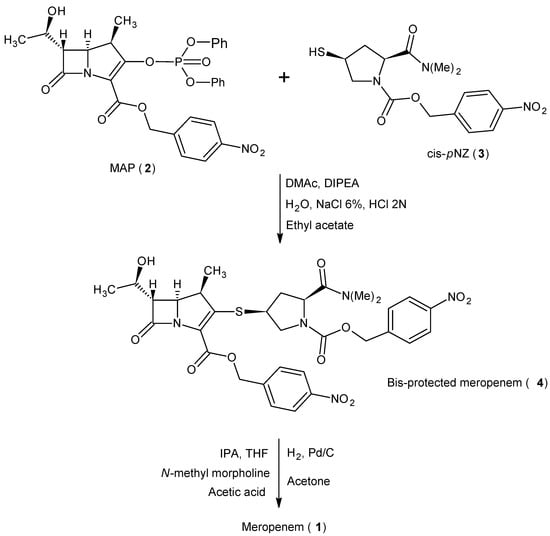
Scheme 1.
Meropenem (1) synthesis from MAP (2) and cis-pNZ (3).
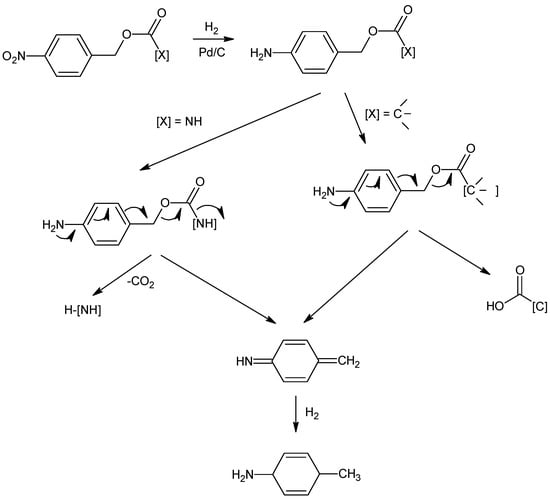
Figure 2.
Reduction mechanism of pNB/pNZ protecting groups.
In the context of the current drive towards the adoption of emerging technologies for the synthesis of APIs, functional intermediates and natural compounds, we have investigated the hydrogenation of bis-protected meropenem (4) under microwave (MW)-assisted flow chemistry. We have chosen this technology because of its well-known benefits to mass and heat transfer, miniaturization, safety, speed and sustainability. Semi and continuous flow chemistry have traditionally been used and developed as a safer and more efficient method when batch operations are deemed to be too dangerous. The small operational units minimize exposure to hazardous reagents and permit safe handling. The integration and modularization of operations allow for much easier study and production scale-up. Automated monitoring techniques and the reduction in the probability of human error are significant industrial advantages, making this technology flexible and agile. Although flow chemistry does not entirely eliminate safety concerns in industry, it does decrease risk factors to levels at which they are easier to manage. Furthermore, the use of MW irradiation makes this a highly efficient and green technology that can fulfill energy-saving and sustainability requirements. Faster heating rates and uniform heat distribution mean that MW chemistry is one of the most attractive of the emerging technologies [75,76,77,78,79,80,81,82,83,84,85,86].
MW-assisted flow chemistry has been widely applied over the last two decades to synthesize myriad organic compounds, demonstrating its versatility and potential. From unsaturated organic substrates to heterocycle synthesis and metal-catalyzed chemistry, this technology has played a central role in the development of sustainable multigram preparations that meet the dual requirement of process intensification and reduced production costs. It has proven to be a promising hybrid technique that can link lab-scale investigations and pilot processes, and thus meet industrial challenges [87,88,89,90,91,92,93,94]. In addition, the FDA and the European Medicines Agency (EMA) have strongly encouraged the study and implementation of this innovative, sustainable and environmentally friendly technology in API synthesis [95,96]. β-lactam chemistry has remained anchored to developments from many years ago and has only recently achieved renewal in a few cases [97,98]. With this revolution in place, MW-assisted flow chemistry could really upend acquired certainties in the near future.
The present work demonstrates the effective implementation of a new emerging technology in the synthesis of a blockbuster drug. It describes a new attractive protocol that is suitable for industrial application as an alternative to batch mode. MW-assisted flow chemistry fulfills the required high-quality standard, as hydrogenation is the final step in meropenem (1) manufacture. The quality product is comparable to commercial drug. The parameters have been investigated with an eye to process intensification and further scale-up. The carbapenem skeleton stability has been preserved by working under mild conditions and by exploiting its intrinsic characteristics. We believe that this work may be pioneering in API synthesis and indispensable to the development of this technology in the pharmaceutical panorama.
2. Materials and Methods
2.1. Chemistry
MAP (2), cis-pNZ (3), 10% Pd/C and the solvents were kindly provided by ACS Dobfar. All other reagents were purchased from Merck KGaA (Darmstadt, Germany).
The Agilent 1200 series HPLC system, NMR (Bruker 400, Munich, Germany), and Karl Fischer titration were the analytical instruments used to identify and analyze the products.
2.1.1. General Procedure for the Synthesis of (4R,5S,6S)-(p-Nitrobenzyl)-3-[[(3S,5S)-1-(p-nitrobenzyloxycarbonyl)-5-(dimethylaminocarbonyl)-3-pyrrolidinyl]thio]-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylate (4)
MAP (2, 94.0 g, 158.0 mmol) and cis-pNZ (3, 59.2 g, 167.6 mmol) were dissolved in 400 mL of dimethylacetamide and cooled to −10 °C. Over the course of 15 min, diisopropylethylamine (DIPEA) (88 mL, 505.3 mmol) was added dropwise at a temperature not exceeding −7 °C. The mixture was stirred for 90 min at −10 °C. An amount of 1200 mL of pre-cooled (0–5 °C) ethyl acetate was then added over 30 min with the temperature being maintained at −10 °C, and finally 800 mL of ice water was added, which raised the temperature to 0–5 °C. The aqueous phase was separated and extracted with 600 mL ethyl acetate at 0–5 °C. The combined organic phase was extracted twice, each time with a cold mixture of 320 mL 6% aqueous NaCl solution and 80 mL 2N hydrochloric acid. Finally, the organic phase was extracted with 400 mL phosphate buffer solution pH 7.0. The organic phase was separated, filtered to clarify, and the filter then washed with 40 mL of ethyl acetate. The filtrate was concentrated to 360 g at 40 °C and made up to 440 g with fresh ethyl acetate. The solution was stirred for 18 h at 20 °C in order to crystallize the crude bis-protected meropenem (4). In order to complete the crystallization, 140 mL of heptane were added dropwise, and the slurry was stirred for a further 30 min. The product was isolated by filtration, washed twice, each time with 200 mL of heptane, and dried for 16 h at 40 °C under vacuum to give the crude title compound (4) (101.2 g, 91.8% yield) as an off-white solid. The HPLC assay gave a result of 98.3%. Purification: crude bis-protected meropenem (4, 100 g, 143.3 mmol) was dissolved in 1680 mL ethyl acetate. The slurry was stirred at 20 °C for 90 min. An amount of 140 mL cyclohexane was added in order to complete the crystallization and the slurry was stirred for 2.5 h. The pure bis-protected meropenem (4) was isolated via filtration, washed with 200 mL cyclohexane, and dried for 16 h at 40 °C under vacuum to give the pure title compound (4) as a white solid (91.1 g, 91.1% yield). The HPLC assay gave a result of 99.6%.
Signals in the 1H- and 13C-NMR spectra are split due to the presence of pNZ rotamers in a ~56:44 ratio at 300 K.
1H NMR: (400 MHz, DMSO-d6, 300 K): δH 8.26, 8.25 (4H, 2 × d, 3J 8 Hz); 7.76 (2H, d, 3J 8 Hz); 7.68, 7.56 (2H, 2 × d, 3J 8 Hz); 5.50 & 5.34 (2H, AB syst, 2J 14 Hz); 5.30–5.05 (3H, m); 4.88, 4.80 (1H, 2 × t, 3J 8 Hz); 4.35–4.30 (1H, m); 4.28, 4.18 (1H, 2 × dd, 3J 10 Hz, 3 J 7 Hz); 4.10–4.00 (1H, m); 3.96–3.80 (1H, m); 3.70–3.60 (1H, m); 3.37–3.32 (1H, m); 3.28, 3.21 (1H, 2 × t, 3J 10 Hz); 3.07, 3.01 (3H, 2 × s); 2.98–2.80 (4H, m); 1.75–1.60 (1H, m); 1.21 (3H, d, 3J 7.5 Hz); 1.19 (3H, d, 3J 7.5 Hz), Figure S1. Underlined peaks are ascribed to the major rotamer.
13C NMR: (100 MHz, DMSO-d6, 300 K): δC 175.02 (C), 174.90 (C), 171.51 (C), 171.15 (C), 160.73 (C), 153.75 (C), 153.48 (C), 151.15 (C), 151.08 (C), 147.86 (C), 147.72 (C), 145.46 (C), 145.50 (C), 145.46 (C), 145.50 (C), 129.08 (CH), 128.82 (CH), 128.56 (CH), 124.58 (C), 124.45 (C), 124.27 (CH), 124.18 (CH), 65.73 (CH2), 65.63 (CH2), 64.94 (CH), 64.89 (CH), 57.03 (CH), 56.67 (CH), 56.15 (CH), 43.83 (CH), 40.04 (CH), 37.15 (CH), 37.07 (CH), 36.54 (CH2), 35.82 (CH2), 22.51 (CH3), 22.46 (CH3), 17.84 (CH3), Figure S2.
2.1.2. General Procedure Applied in All Batch-Mode and MW-Assisted Flow-Mode Experiments for the Synthesis of Meropenem (1)
Bis-protected meropenem (4, 28.5 g, 40.8 mmol), acetic acid (1.4 g, 23.3 mmol), N-methyl morpholine (4.2 g, 41.5 mmol), 257 g tetrahydrofuran (THF), 282 g isopropyl alcohol and 428 g of water made up the starting solution, which was kept constant across all of the experiments. The initial concentration was about 40 mmol/L. N-methyl morpholine and acetic acid were used as buffers during the reaction, allowing it to work at pH = 6.5–6.8. The catalyst 10% Pd/C was set as indicated in Table 1. The total volume of slurry during hydrogenation was approximately 1000 mL, and had a density of 0.94 g/mL. After the hydrogenation experiments, the catalyst was filtered and the aqueous phase was extracted with 285 mL of dichloromethane at room temperature (solution pH = 5.3–5.5). An amount of 2850 mL of acetone was added to the aqueous phase for 3 h at 0–5 °C to crystallize the meropenem (1). The slurry was stirred for 60 min at 0–5 °C, filtered and washed with 30 mL of acetone. The crude product was dried for 30 min under vacuum with nitrogen flow. The yield values are listed in Table 1 and Table 4. The HPLC assay gave a result of 98%. Purification: crude meropenem (1, 5.0 g, 11.4 mmol) was dissolved in 150 mL water with NaHCO3 (0.1 g, 1.2 mmol) at 35 °C. After 5 min, the solution was cooled to 0–5 °C and treated with norite charcoal (0.5 g) for 1 h. The charcoal was removed by filtration and the filter cake was washed with 5 mL water. An amount of 0.5 g of seed was added, and the solution was stirred for 1 h at 0–5 °C. An amount of 450 mL acetone was added dropwise within 4 h and stirred for a further 30 min. The solid was collected by filtration, washed with 10 mL acetone and dried under vacuum for 1 h at room temperature to give pure meropenem (1) as a white solid (4.2 g, 84% yield). The HPLC assay gave a result of 99.5%.

Table 1.
Screening of hydrogenation conditions in the Parr reactor.
1H NMR (400 MHz, D2O): δH 1.15 (3H, d, 3J 7.6 Hz, 10-Me), 1.23 (3H, d, 3J 6.4 Hz, 9-Me), 1.91 (1H, appt quint, J 6.8 Hz, H-15α), 2.94§ (3H, s, 17-Me), 3.02§ (3H, s, 18-Me), 3.06 (1H, m, H-15β), 3.31 (1H, dq,2 J 14.8 Hz, 3J 7.2 Hz, H-1), 3.4–3.5 (2H, m, H-6/H-13α), 3.71 (1H, dd, 2J 12.4 Hz, 3J 6.4 Hz, H-13β), 4.00 (1H, appt quint, J 6.4 Hz, H-12), 4.15–4.25 (2H, m, H-5/H-8), 4.75 (1H, t, 3J 8.8 Hz, H-14), Figure S3.
13 C NMR (100 MHz, D2O): δC 176.75 (C-11), 168.0 (C-16), 167.75 (C-7), 137.6 (C-2), 134.0 (C-3), 65.3 (C-8), 59.0 (C-6), 58.4 (C-14), 56.1 (C-5), 52.35 (C-13), 42.65 (C-1), 40.6 (C-12), 36.8 (C-18)#, 36.0 (C-17)#, 20.9 (C-9), 16.0 (C-10), Figure S4.
§# may be interchanged, appt: apparent (first-order approximation).
2.2. Hydrogenation Reactions
2.2.1. Reaction in Batch Mode
A Parr reactor series 4578 High Pressure/High Temperature (Parr Instrument Company, Moline, IL, USA) was used for the batch-mode experiments. A floor stand with a pneumatic lift mounted the fixed-head reactor, which has a volume of 1800 mL and is capable of working at up to 345 bar pressure and 500 °C temperature. A mass flowmeter placed in the hydrogen line allowed the reactor pressure, which was measured with a pressure measurement on the top of the reactor, to be controlled. The temperature was kept constant using an electrically driven serpentine. An impeller performed slurry stirring correctly. Scheme 2 depicts the reactor configuration used for batch-mode hydrogenation.
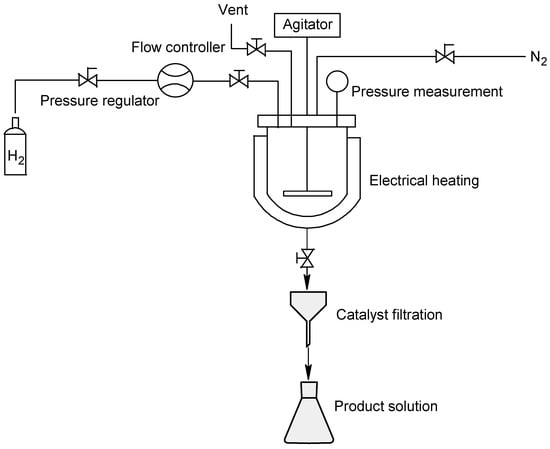
Scheme 2.
Batch-reactor configuration.
The bis-protected meropenem (4) solution was introduced into the reactor with the catalyst. Inertization was performed with three nitrogen purges at 5 bar, and the reactor was subsequently purged three times with hydrogen at 5 bar. The stirrer was set and the hydrogen valve opened. A pressure ramp from 0 bar to final pressure (3 bar/min) was applied, as was a temperature ramp (2.5 °C/min). The temperature was kept constant at the final reaction temperature, and the pressure was maintained using the hydrogen flowmeter (Table 1). The reaction was run until hydrogen uptake was completed, at which point the hydrogen flow was interrupted and the solution cooled to room temperature. The reactor was slowly vented to release the hydrogen and underwent inertization with three nitrogen purges at 5 bar. The slurry was filtered using nitrogen pressure on a Buchner funnel to remove the catalyst, allowing solution analysis to then be performed.
2.2.2. Reaction in MW-Assisted Flow Mode
MW-assisted flow hydrogenation was carried out in the FlowSYNTH reactor (Milestone Srl, Bergamo, Italy), which is a multimode system that operates at 2.45 GHz and is equipped with a vertical PTFE-TFM flow-through reactor that can work up to a maximum temperature of 200 °C and 20 bar of pressure, in open or closed loop modes. A three-way connection fitted with a non-return valve allows the starting solution and hydrogen feeds to be pumped simultaneously. A back-pressure valve placed on the top of the reactor, after a water-cooled heat exchanger, ensures pressure control and flow-stream decompression, allowing the solution to be collected. For a single cycle run, the slurry was run in the reactor (VR = 200 mL) using a hydraulic pump and was collected in an Erlenmeyer flask after catalyst filtration, and this was performed in loop for multiple-cycle runs. The starting solution and hydrogen flowed from the bottom to the top of the reactor, with the applied MW power being modified in real time to maintain the predefined temperature. The pressure was kept constant using a hydrogen mass flowmeter at the indicated pressure. Integrated reactor sensors continuously monitored the internal pressure, temperature and applied power inside the reactor for each run. Leak tests were successfully performed first with water and then with isopropyl alcohol and nitrogen to test the pressure control. A general scheme of the FlowSYNTH reactor is depicted in Scheme 3.
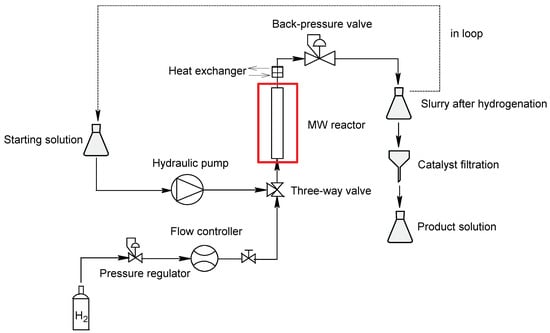
Scheme 3.
FlowSYNTH reactor configuration.
3. Results and Discussion
As a first step, we focused on investigating the preferred reaction parameters for batch-mode hydrogenation in order to subsequently upload them for flow-mode use. The key factors in determining successful pNB/pNZ removal are pressure, temperature and Pd/C amount. As reported in Table 1, catalyst load has a decisive impact on the meropenem (1) yield from hydrogenation, and influences how the process is run. Upon performing the deblock in the Parr reactor at 20 bar and 37 °C, we observed a drop in yield and an increase in the reaction time when the catalyst amount per test was halved. The same trend was observed when working at lower pressure (6.8 bar) and temperature (30 °C), but starting with a greater catalyst load. An inversely proportional relationship exists between these three parameters, and this had a significant impact on the choice of reaction conditions for the subsequent flow-mode experiments. In industry, reducing the amounts of the highly expensive catalysts would undoubtedly be impactful, but incompatible with the instability of the carbapenem nucleus under harsh conditions. We observed a reduction in yield even when the dry catalyst percentage and pressure value were kept constant, but the temperature increased (35 °C and 45 °C), demonstrating once again the need to work under moderate conditions. In all tests, the reagent conversion was complete, meaning that the reaction generated different undetectable impurities. Using a catalyst amount of 21% dry w/w for bis-protected meropenem (4) at 30 °C and 6.8 bar granted a meropenem (1) yield in solution, after catalyst filtration, of 89%. We decided to only crystallize the hydrogenated solution formed under moderate conditions to have a comparison with the products investigated in flow mode.
The work-up of the aqueous solution together with the purifying effect of acetone, used in crystallization, led to the isolation of crude meropenem (1) in Table 1 with an assay result of 98% and a yield of 74%. Compared to the data reported in literature [48], our system has proven to perform better. An LC-MS analysis (Supplementary Figure S5) of the product derived from Entry 4 identified two impurities, p-amino (5, 0.7%) and p-hydroxylamino (6, 0.3%) derivatives, which arose after the removal of the pNZ side chain from the stepwise reduction in the nitro group in the transient mono-protected pNB compound (Figure 3). These structures would seem to indicate the reaction mechanism proposed in Figure 2. The purification of the crude product purged these impurities and gave a pure product with 99.5% assay result, comparable to commercial product.
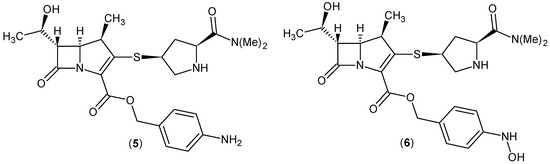
Figure 3.
Impurities in crude meropenem (1), from Entry 4, investigated using LC-MS.
Meropenem (1) hydrogenation under MW-assisted flow chemistry has never been investigated. Moreover, this technology appears to us to be the most suitable for a study into the potential industrial production of Meropenem (1) as the drug is a blockbuster and manufactured on a large scale.
Having identified the best conditions for carrying out the hydrogenation in batch mode, the reaction was investigated using the FlowSYNTH reactor. The best test (Entry 4) was taken as the reference point, and the catalyst amount for all experiments run in the MW-assisted flow reactor was not changed. As a first step, we focused on the influence of hydrogen pressure and reaction temperature, while keeping slurry-solution flow, residence time (tr) and, consequently, run time constant. The concept of developing a quick process supported by leak tests with the slurry solution directed us towards using a short residence time. The slurry-solution flow was set at 150 mL/min, the hydrogen flow rate between 150–200 NmL/min, with 80 s of residence time (considering only the liquid phase) and each run taking 7.5 min. The system was flushed with THF and nitrogen to clean the reactor and lines after each loop.
A pressure range of 4.0 bar to 8.0 bar was investigated, with MW irradiation being applied between 30–35 °C and the starting solution being flowed for one cycle. Under these conditions, we did not observe experimental evidence for the bis-protected meropenem (4) deblock in meropenem (1). Each analysis was performed on the aqueous solution after catalyst filtration.
Since pressure and temperature were observed to have no measurable impact, we decided to increase the residence time for the next experiments and use the same pressure conditions as Entry 4 (6.8 bar). This decision was taken to give more time to the hydrogen, with the catalyst, to react with the substrate. To ensure that the reactor, and consequently the slurry, are sufficiently pulsed with MW irradiation, the temperature was set at 35 °C; in order to achieve these new conditions, the slurry-solution flow was decreased and, accordingly, the run time was increased.
With the solution flow set at 105 mL/min and the hydrogen flow at 100 NmL/min to give 115 s of residence time and 10.5 min of run time, we obtained a decrease in bis-protected meropenem (4) concentration in a single cycle, as indicated in Table 2. Compared to the batch experiments, an 88% value of raw materials was still high; thus, the slurry was run until the complete disappearance of the substrate. After ten cycles and a total residence time of 1150 s (19 min), we obtained a meropenem (1) yield in solution of 69% with a bis-protected meropenem (4) residual of 3%. A progressive and constant decrease in the substrate has been observed without altering the catalyst activity.
Table 2.
Semi-continuous hydrogenation test with tr = 115 s for one loop at 6.8 bar.
The same experiment was repeated with a solution flow of 57 mL/min and a run time of 19 min, maintaining the hydrogen flow rate at 100 NmL/min, in order to have a residence time of 210 s to carry out the synthesis; substrate hydrogenation was completed in four cycles, as indicated in Table 3. A meropenem (1) yield in solution of 67% was obtained with a bis-protected meropenem (4) residual of 3% in a total residence time of 840 s (14 min). These semi-continuous tests clearly show the considerable impact of residence time on hydrogenation, making it a key parameter and necessary for reaction success. The synthesis was now performed in loop, and the total residence time was increased to allow the reaction between hydrogen and the starting materials to achieve protecting-group deblock. However, the slurry solution flow could not be reduced further to avoid the risks of clogging problems and pressure instability.
Table 3.
In-loop hydrogenation test with tr = 210 s for one loop and 6.8 bar.
Taking the process parameters used in the Table 3 experiments as the best conditions, the hydrogenation was carried out at a higher temperature induced by MW irradiation. A clear difference between MW-assisted flow hydrogenation and the batch-mode process emerges in the data reported in Table 4. The temperature increase from 35 °C to 45 °C improved the yield from 67% to 80%, while a 5% decrease in yield was observed at 55 °C. This trend provides useful information for future industrial application, as it clearly highlights the subtle balance between the operating parameters and the instability of the carbapenem skeleton. The combination of better mixing in the flow reactor with the uniform distribution of MW irradiation improved the yield of meropenem (1) in solution after catalyst filtration, while the opposite result was observed in experiments in the Parr reactor when the temperature was increased. A solution yield of 80% is slightly lower than the batch process data, as was the yield of the isolated product (70% vs. 74%). However, the benefits of running a semi-continuous process would enhance productivity and eliminate the small yield deviation.
Table 4.
In-loop hydrogenation test at 45 °C and 55 °C and 6.8 bar.
Table 4.
In-loop hydrogenation test at 45 °C and 55 °C and 6.8 bar.
| Entry | Cycles | Temperature (°C) | Total Residence Time (s) | Meropenem (1) Yield in Solution (%) after Catalyst Filtration | Crude Meropenem (1) Yields (%) |
|---|---|---|---|---|---|
| 23 | 4 | 45 | 840 | 80 | 70 |
| 24 | 4 | 55 | 840 | 75 | 65 |
Reaction conditions: The reaction conditions are reported in Materials and Methods in Section 2.1.2. The FlowSYNTH reactor is described in Materials and Methods in Section 2.2.2. The catalyst amount was 21% dry w/w with reference to bis-protected meropenem (4), as reported in Table 1, entry 4.
It is worth noting that the product quality obtained using MW-assisted flow hydrogenation is equivalent to that obtained using the batch process (98%). An LC-MS investigation of the crude meropenem (1), from Entry 23, showed the same impurities as reported in Figure 2 at the same values, confirming that they are characteristic of this specific reaction applied to this carbapenem.
4. Conclusions
An experimental study into the MW-assisted catalytic hydrogenation of bis-protected meropenem (4) has been carried out. The FlowSYNTH reactor has been used to this purpose, giving a meropenem (1) yield in solution after catalyst filtration of 80% with an isolated-product yield of 70%. The optimization of residence time (840 s) and cycle number (4) has led to the development of a novel protocol that has halved the reaction time (14 min vs. 30 min). Using MW irradiation, the deblock reaction was completed in only 14 min, whereas the batch process required 30 min. To achieve this result, the solution underwent four cycles with a residence time of 210 s per loop, with a solution flow of 57 mL/min at 45 °C and 6.8 bar. This technology has proven to be softer than the Parr reactor as it can achieve a similar yield result at a higher temperature. The carbapenem nucleus has been preserved and product quality is comparable to that of the batch product. Two characteristic impurities in the hydrogenation process have been found, and they are present both in the products of the MW-assisted flow process and the batch process, confirming that they are specific to this reaction. The results obtained using this technology show, once again, its potential and adaptability, even in the synthesis of an important carbapenem, such as meropenem. It is our hope that this work can pave the way for new opportunities for this emerging technology in the pharmaceutical world.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/pharmaceutics15051322/s1, Figure S1: 1H-NMR of bis-protected meropenem (4). Figure S2: 13C-NMR of bis-protected meropenem (4). Figure S3: 1H-NMR of meropenem (1). Figure S4: 13C-NMR of meropenem (1). Figure S5: LC-MS analysis of crude meropenem (1).
Author Contributions
Conceptualization, G.P., R.M. and G.C.; methodology, M.C., A.M. and S.T.; validation, M.C., R.M. and S.T.; formal analysis, G.P., A.M. and S.T.; investigation, M.C. and A.M.; data curation, S.T. and A.M.; writing—original draft preparation, G.P. and M.C.; writing—review and editing, S.T. and G.C.; supervision, G.P. and G.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within this article or Supplementary Materials.
Acknowledgments
ACS Dobfar SpA, the University of Turin, and the University of Insubria are warmly acknowledged for their financial and technical support.
Conflicts of Interest
All authors declare no conflict of interest.
References
- Queenan, A.M.; Bush, K. Carbapenemases: The versatile beta-lactamases. Clin. Microbiol. Rev. 2007, 20, 440–458. [Google Scholar] [CrossRef]
- Armstrong, T.; Fenn, S.J.; Hardie, K.R. JMM profile: Carbapenems: A broad-spectrum antibiotic. J. Med. Microbiol. 2021, 70, 001462. [Google Scholar] [CrossRef]
- Shah, P.M. Parenteral carbapenems. Clin. Microbiol. Infect. 2008, 14, 175–180. [Google Scholar] [CrossRef]
- Papp-Wallace, K.M.; Endimiani, A.; Taracila, M.A.; Bonomo, R.A. Carbapenems: Past, present, and future. Antimicrob. Agents Chemother. 2011, 55, 4943–4960. [Google Scholar] [CrossRef] [PubMed]
- Orsi, G.B.; Falcone, M.; Venditti, M. Surveillance and management of multidrug-resistant microorganisms. Expert Rev. Anti Infect. Ther. 2011, 9, 653–679. [Google Scholar] [CrossRef]
- Bradley, J.S.; Garau, J.; Lode, H.; Rolston, K.V.; Wilson, S.E.; Quinn, J.P. Carbapenems in clinical practice: A guide to their use in serious infection. Int. J. Antimicrob. Agents 1999, 11, 93–100. [Google Scholar] [CrossRef]
- El-Gamal, M.; Brahim, I.; Hisham, N.; Aladdin, R.; Mohammed, H.; Bahaaeldin, A. Recent updates of carbapenem antibiotics. Eur. J. Med. Chem. 2017, 5, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Paterson, D.L. Recommendation for treatment of severe infections caused by Enterobacteriaceae producing extended-spectrum beta-lactamases (ESBLs). Clin. Microbiol. Infect. 2000, 6, 460–463. [Google Scholar] [CrossRef]
- Paterson, D.L.; Bonomo, R.A. Extended-spectrum beta-lactamases: A clinical update. Clin. Microbiol. Rev. 2005, 18, 657–686. [Google Scholar] [CrossRef]
- Torres, J.A.; Villegas, M.V.; Quinn, J.P. Current concepts in antibiotic-resistant gram-negative bacteria. Expert Rev. Anti Infect. Ther. 2007, 5, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Bonfiglio, G.; Russo, G.; Nicoletti, G. Recent developments in carbapenems. Expert Opin. Investig. Drugs 2002, 11, 529–544. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Bradford, P.A. β-Lactams and β-lactamase inhibitors: An overview. Cold Spring Harb. Perspect. Med. 2016, 6, a025247. [Google Scholar] [CrossRef] [PubMed]
- Stewart, N.K.; Smith, C.A.; Frase, H.; Black, D.J.; Vakulenko, S.B. Kinetic and structural requirements of carbapenems activity in GES-type β-lactamases. Biochemistry 2015, 54, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-lactamases and β-lactamase inhibitors in the 21st century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef]
- Zhanel, G.G.; Wiebe, R.; Dilay, L.; Thomson, K.; Rubinstein, E.; Hoban, D.J.; Noreddin, A.M.; Karlowsky, J.A. Comparative review of the carbapenems. Drugs 2007, 67, 1027–1052. [Google Scholar] [CrossRef] [PubMed]
- Nicolau, D.P. Carbapenems: A potent class of antibiotics. Expert Opin. Pharmacother. 2008, 9, 23–37. [Google Scholar] [CrossRef]
- Kattan, J.N.; Villegas, M.V.; Quinn, J.P. New developments in carbapenems. Clin. Microbiol. Infect. 2008, 14, 1102–1111. [Google Scholar] [CrossRef]
- Baughman, R.P. The use of carbapenems in the treatment of serious infections. J. Intensive Care Med. 2009, 24, 230–241. [Google Scholar] [CrossRef]
- Breilh, D.; Texier-Maugein, J.; Allaouchiche, B.; Saux, M.C.; Boselli, E. Carbapenems. J. Chemother. 2013, 25, 1–17. [Google Scholar] [CrossRef]
- Moellering, R.C., Jr.; Eliopoulus, G.M.; Sentochnik, D.E. The carbapenems: New broad spectrum beta-lactam antibiotics. J. Antimicrob. Chemother. 1989, 24, 1–7. [Google Scholar] [CrossRef]
- Kahan, J.S.; Kahan, F.M.; Goegelman, R.; Currie, S.A.; Jackson, M.; Stapley, E.O.; Miller, T.W.; Miller, A.K.; Hendlin, D.; Mochales, S.; et al. Thienamycin, a new beta-lactam antibiotic. I. Discovery, taxonomy, isolation and physical properties. J. Antibiot. 1979, 32, 1–12. [Google Scholar] [CrossRef]
- Nordmann, P.; Dortet, L.; Poirel, L. Carbapenem resistance in Enterobacteriaceae: Here is the storm! Trends Mol. Med. 2012, 18, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Basker, M.J.; Boon, R.J.; Hunter, P.A. Comparative antibacterial properties in vitro of seven olivanic acid derivates: MM4550, MM13902, MM17880, MM22380, MM22381, MM22382 and MM22383. J. Antibiot. 1980, 33, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.R.; Turner, P.J.; Wannop, C.; Withnell, E.S.; Grindey, A.J.; Nairn, K. In vitro antibacterial activity of SM-7338, a carbapenem antibiotic with stability to dehydropeptidase I. Antimicrob. Agents Chemother. 1989, 33, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Liu, X.; Wang, S.; Fleming, J.; Wang, D.C.; Liu, W. The mechanism of NDM-1-catalyzed carbapenem hydrolysis is distinct from that of penicillin or cephalosporin hydrolysis. Nat. Commun. 2017, 8, 2242. [Google Scholar] [CrossRef]
- Woodward, R.B. Penems and related substances. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1980, 289, 239–250. [Google Scholar] [CrossRef]
- Raza, A.; Ngieng, S.C.; Sime, F.B.; Cabot, P.J.; Roberts, J.A.; Popat, A.; Kumeria, T.; Falconer, J.R. Oral meropenem for superbugs: Challenges and opportunities. Drug Discov. Today 2021, 26, 551–560. [Google Scholar] [CrossRef]
- Slama, T.G. Clinical review: Balancing the therapeutic, safety, and economic issues underlying effective antipseudomonal carbapenem use. Crit. Care 2008, 12, 233. [Google Scholar] [CrossRef]
- Cuzzolin, L.; Agostino, R. Antibiotic use in a cohort of extremely low birth weight neonates: Focus on off-label uses and prescription behaviour. Pharmacol. Pharm. 2018, 9, 382–394. [Google Scholar] [CrossRef]
- Meropenem Market Size with Emerging Trends 2022|Top Key Players Updates, Business Growing Strategies, Competitive Dynamics, Industry Segmentation and Forecast to 2027. Available online: https://www.marketwatch.com/press-release/meropenem-market-size-with-emerging-trends-2022-top-key-players-updates-business-growing-strategies-competitive-dynamics-industry-segmentation-and-forecast-to-2027-2022-11-07 (accessed on 4 February 2023).
- Carbapenem Market Share, Size, Trends, Industry Analysis Report. Available online: https://polarismarketresearch.com/industry-analysis/carbapenem-market#:~:text=The%20global%20carbapenem%20market%20was,serious%20or%20dangerous%20bacterial%20infections (accessed on 4 February 2023).
- Sunagawa, M.; Matsumura, H.; Inoue, T.; Fukasawa, M.; Kato, M. A novel carbapenem antibiotic, SM-7338 structure-activity relationships. J. Antibiot. 1990, 43, 519–532. [Google Scholar] [CrossRef]
- Sunagawa, M.; Matsumura, H.; Inoue, T.; Fukasawa, M.; Kato, M. Carboxylic Thio-Pyrrolidinyl Beta-Lactam Compounds and Production Thereof. EP 0,126,587, 9 May 1984. [Google Scholar]
- Sunagawa, M. β-Lactam Compounds. U.S. Patent 4,943,569, 24 July 1990. [Google Scholar]
- Highlights of Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/050706s037lbl.pdf (accessed on 5 February 2023).
- Thomas, C.; Priano, J.; Smith, T.L. Meropenem as an antidote for intentional valproic acid overdose. Am. J. Emerg. Med. 2020, 38, 690.e1–690.e2. [Google Scholar] [CrossRef] [PubMed]
- Steffens, N.A.; Zimmermann, E.S.; Nichelle, S.M.; Brucker, N. Meropenem use and therapeutic drug monitoring in clinical practice: A literature review. J. Clin. Pharm. Ther. 2021, 46, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.S. Meropenem. Pediatr. Infect. Dis. J. 1998, 17, 249–250. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, L.R.; Wagstaff, A.J.; Brogden, R.N.; Bryson, H.M. Meropenem. A review of its antibacterial activity, pharmacokinetic properties and clinical efficacy. Drugs 1995, 50, 73–101. [Google Scholar] [CrossRef]
- Pascale, R.; Giannella, M.; Bartoletti, M.; Viale, P.; Pea, F. Use of meropenem in treating carbapenem-resistant Enterobacteriaceae infections. Expert Rev. Anti Infect. Ther. 2019, 17, 819–827. [Google Scholar] [CrossRef]
- Dhillon, S. Meropenem/vaborbactam: A review in complicated urinary tract infections. Drugs 2018, 78, 1259–1270. [Google Scholar] [CrossRef]
- Novelli, A.; Del Giacomo, P.; Rossolini, G.M.; Tumbarello, M. Meropenem/vaborbactam: A next generation β-lactam β-lactamase inhibitor combination. Expert Rev. Anti Infect. Ther. 2020, 18, 643–655. [Google Scholar] [CrossRef]
- Jorgensen, S.C.J.; Rybak, M.J. Meropenem and vaborbactam: Stepping up the battle against carbapenem-resistant Enterobacteriaceae. Pharmacotherapy 2018, 38, 444–461. [Google Scholar] [CrossRef]
- Cho, J.C.; Zmarlicka, M.T.; Shaeer, K.M.; Pardo, J. Meropenem/vaborbactam, the first carbapenem/β-lactamase inhibitor combination. Ann. Pharmacother. 2018, 52, 769–779. [Google Scholar] [CrossRef]
- WHO Model List of Essential Medicines. Available online: https://en.wikipedia.org/wiki/WHO_Model_List_of_Essential_Medicines (accessed on 5 February 2023).
- Takeuchi, Y.; Inoue, T.; Sunagawa, M. Studies on the structures of meropenem (SM-7338) and it’s primary metabolite. J. Antibiot. 1993, 46, 827–832. [Google Scholar] [CrossRef]
- Sunagawa, M.; Sasaki, A.; Matsumura, H.; Goda, K.; Tamoto, K. Synthetic studies of carbapenem and penem antibiotics. V. Efficient synthesis of 1β-methylcarbapenem skeleton. Chem. Pharm. Bull. 1994, 42, 1381–1387. [Google Scholar] [CrossRef]
- Tewari, N.; Nizar, H.; Rai, B.P.; Singh, S.K.; George, V.; Prasad, M. An improved procedure for the preparation of carbapenem antibiotic: Meropenem. Org. Process Res. Dev. 2007, 11, 773–775. [Google Scholar] [CrossRef]
- Prashad, A.S.; Vlahos, N.; Fabio, P.; Feigelson, G.B. A highly refined version of the α-keto ester based carbapenem synthesis: The total synthesis of meropenem. Tetrahedron Lett. 1998, 39, 7035–7038. [Google Scholar] [CrossRef]
- Shih, D.H.; Baker, F.; Cama, L.; Christensen, B.G. Synthetic carbapenem antibiotics. I. 1-β-methylcarbapenem. Heterocycles 1984, 21, 29–40. [Google Scholar] [CrossRef]
- Iannazzo, L.; Soroka, D.; Triboulet, S.; Fonvielle, M.; Compain, F.; Dubèe, V.; Mainardi, J.L.; Hugonnet, J.E.; Braud, E.; Arthur, M.; et al. Routes of synthesis of carbapenems for optimizing both the inactivation of L,D-transpeptidase LdtMt1 of Mycobacterium tuberculosis and the stability toward hydrolysis by β-lactamse BlaC. J. Med. Chem. 2016, 59, 3427–3438. [Google Scholar] [CrossRef] [PubMed]
- Surulichamy, S.; Sekar, S.; Deshpande, P.N.; Ganpathy, P.; Sarangdhar, R.J.; Henry, S.S.; Karale, S.N.; Jangale, A.A.; Kaldate, R.D. An Improved Process for the Preparation of Beta-Lactam Antibiotic. WO 2007/031858, 22 March 2007. [Google Scholar]
- Sunagawa, M.; Isobe, Y.; Takeuchi, Y.; Matsumura, H.; Ozaki, Y.; Noguchi, Y. Carbapenem Compound and in Crystalline Form, and Its Production and Use. U.S. Patent 4,888,344, 19 December 1989. [Google Scholar]
- Nadenik, P.; Storm, O.; Kremminger, P. Meropenem Intermediate in Crystalline Form. WO 2005/118586, 15 December 2005. [Google Scholar]
- Tewari, N.; Meeran, H.N.P.N.; Rai, B.P.; Kumar, Y. A Process for the Preparation of Meropenem. WO 2006/035300, 6 April 2006. [Google Scholar]
- Williams, J.M.; Jobson, R.B. Process for Synthesizing Carbapenem Antibiotics. U.S. Patent 5,872,250, 16 February 1999. [Google Scholar]
- Nishino, K.; Koga, T. Improved Process for Producing Carbapenem Compound. EP 2,006,290, 24 December 2008. [Google Scholar]
- Song, Y.S.; Park, S.W.; Yoon, Y.J.; Yoon, H.K.; Moon, S.C.; Lee, B.G.; Choi, S.J.; Jun, S.A. Improved Method for Preparing Meropenem Using Zinc Powder. EP 2,407,468, 18 January 2012. [Google Scholar]
- Manca, A.; Monguzzi, R.A. Process for Synthesizing Carbapenem Using Raney Nichel. EP 2,141,167, 6 January 2010. [Google Scholar]
- Sunagawa, M.; Matsumura, H.; Inoue, T.; Fukasawa, M.; Kato, M. β-Lactam Compounds. U.S. Patent 5,122,604, 16 June 1992. [Google Scholar]
- Sunagawa, M.; Matsumura, H. Process for Preparing Carbapenem Compounds. U.S. Patent 5,578,722, 26 November 1996. [Google Scholar]
- Tewari, N.; Mane, A.; Rai, B.P.; Prasad, M. Process for the Preparation of Carbapenems. U.S. Patent 2007/0197781, 23 August 2007. [Google Scholar]
- Khemka, A.A.; Shejul, P.B.; Vyavahare, A.M.; Pandey, D.K.; Shete, S.N.; Jadhav, H.K.; Kadam, N.H. Meropenem Intermediate in Novel Crystalline Form and a Method of Manufacture of Meropenem. U.S. Patent 2009/0299057, 3 December 2009. [Google Scholar]
- Zhang, H. A Process for the Preparation of Meropenem. WO 2007/104221, 20 September 2007. [Google Scholar]
- Karale, S.N.; Jangale, A.A.; Kaldate, R.D. An Improved Process for the Preparation of Meropenem. WO 2011/141847, 17 November 2011. [Google Scholar]
- Gnanaprakasam, A.; Veermani, G.; Shahul Hameed, S.I.; Murugesan, K.; Thangavel, S.; Sekar Jeyaraj, M.; Thangavel, A.; Gautam Kumar, D. A Process for Preparation of Meropenem. WO 2012/160576, 29 November 2012. [Google Scholar]
- Cookson, J.; McNair, R.; Satoskar, D.V. Process for Preparing a Carbapenem Antibiotic. WO 2015/145161, 1 October 2015. [Google Scholar]
- Grünewald, E.; Weidlich, S.; Jantke, R. Process for the Deprotection of a Carbapenem by Heterogeneous Catalytic Hydrogenation with Hydrogen in the Presence of an Organic Amine. WO 2018/010974, 18 January 2018. [Google Scholar]
- Isidro-Llobet, A.; Guasch-Camell, J.; Alvarez, M.; Albericio, F. p-Nitrobenzyloxycarbonyl (pNZ) as a temporary Nα-protecting group in orthogonal solid-phase peptide synthesis—Avoiding diketopiperazine and aspartimide formation. Eur. J. Org. Chem. 2005, 14, 3031–3039. [Google Scholar] [CrossRef]
- Cossar, P.J.; Hizartzidis, L.; Simone, M.I.; McCluskey, A.; Gordon, C.P. The expanding utility of continuous flow hydrogenation. Org. Biomol. Chem. 2015, 13, 7119–7130. [Google Scholar] [CrossRef]
- Irfan, M.; Glasnov, T.N.; Kappe, C.O. Heterogeneous catalytic hydrogenation reactions in continuous-flow reactors. ChemSusChem 2011, 4, 300–316. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Jiano, J.; Song, P.; Nie, W.; Yi, C.; Zhang, Q.; Li, P. Recent progress in continuous-flow hydrogenation. ChemSusChem 2020, 13, 2876–2893. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, M.; O’Brien, M.; Ley, S.V.; Polyzos, A. Flow chemistry: Intelligent processing of gas-liquid transformations using a tube-in-tube reactor. Acc. Chem. Res. 2015, 48, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, B.; Cantillo, D.; Kappe, C.O. Continuous-flow technology-a tool for the safe manufacturing of active pharmaceutical ingredients. Angew. Chem. Int. Ed. Engl. 2015, 54, 6688–6728. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Baxendale, I.R. The synthesis of active pharmaceuticals ingredients (APIs) using continuous flow chemistry. Beilstein J. Org. Chem. 2015, 11, 1194–1219. [Google Scholar] [CrossRef] [PubMed]
- Baraldi, P.T.; Hessel, V. Micro reactor and flow chemistry for industrial applications in drug discovery and development. Green Process. Synth. 2012, 1, 149–167. [Google Scholar] [CrossRef]
- Horàkovà, P.; Koči, K. Continuous-flow chemistry and photochemistry for manufacturing of active pharmaceutical ingredients. Molecules. 2022, 27, 8536. [Google Scholar] [CrossRef]
- Baumann, M.; Moody, T.S.; Smyth, M.; Wharry, S. A perspective on continuous flow chemistry in the pharmaceutical industry. Org. Process Res. Dev. 2020, 24, 1802–1813. [Google Scholar] [CrossRef]
- Burange, A.S.; Osman, S.M.; Luque, R. Understanding flow chemistry for the production of active pharmaceutical ingredients. iScience 2022, 25, 103892. [Google Scholar] [CrossRef]
- De Souza, J.M.; Galaverna, R.; De Souza, A.A.N.; Brocksom, T.J.; Pastre, J.C.; De Souza, R.O.M.A.; De Oliveira, K.T. Impact of continuous flow chemistry in the synthesis of natural products and active pharmaceutical ingredients. An. Acad. Bras. Ciệnc. 2018, 90, 1131–1174. [Google Scholar] [CrossRef]
- Lee, S.L.; O’Connor, T.F.; Yang, X.; Cruz, C.N.; Chatterjee, S.; Madurawe, R.D.; Moore, C.M.V.; Yu, L.X.; Woodcock, J. Modernizing pharmaceutical manufacturing: From batch to continuous production. J. Pharm. Innov. 2015, 10, 191–199. [Google Scholar] [CrossRef]
- Koenig, S.G.; Sneddon, H.F. Recent advances in flow chemistry in the pharmaceutical industry. Green Chem. 2017, 19, 1418–1419. [Google Scholar] [CrossRef]
- Plutschack, M.B.; Pieber, B.; Gilmore, K.; Seeberger, P.H. The Hitchhiker’s guide to flow chemistry. Chem. Rev. 2017, 117, 11796–11893. [Google Scholar] [CrossRef]
- Comito, M.; Monguzzi, R.; Tagliapietra, S.; Palmisano, G.; Cravotto, G. Towards antibiotic synthesis in continuous-flow processes. Molecules 2023, 28, 1421. [Google Scholar] [CrossRef] [PubMed]
- Bath, A.R.; Shalla, A.H.; Dongre, R.S. Microwave assisted one-pot catalyst free green synthesis of new methyl-7-amino-4-oxo-5-phenyl-2-thioxo-2,3,4,5-tetrahydro-1H-pyrano[2,3-d]pyrimidine-6-carboxylates as potent in vitro antibacterial and antifungal activity. J. Adv. Res. 2015, 6, 941–948. [Google Scholar] [CrossRef]
- Kappe, C.O.; Dallinger, D. The impact of microwave synthesis on drug discovery. Nat. Rev. Drug Discov. 2006, 5, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Calcio Gaudino, E.; Manzoli, M.; Carnaroglio, D.; Wu, Z.; Grillo, G.; Rotolo, L.; Medlock, J.; Bonrath, W.; Cravotto, G. Sonochemical preparation of alumina spheres loaded with Pd nanoparticles for 2-butyne-1,4-diol semi-hydrogenation in a continuous flow microwave reactor. RSC Adv. 2018, 8, 7029–7039. [Google Scholar] [CrossRef]
- Grillo, G.; Manzoli, M.; Bucciol, F.; Tabasso, S.; Tabanelli, T.; Cavani, F.; Cravotto, G. Hydrogenation of levulinic acid to γ-valerolactone via green microwave-assisted reactions either in continuous flow or solvent-free batch processes. Ind. Eng. Chem. Res. 2021, 60, 16756–16768. [Google Scholar] [CrossRef]
- Bucciol, F.; Tabasso, S.; Grillo, G.; Menegazzo, F.; Signoretto, M.; Manzoli, M.; Cravotto, G. Boosting levulinic acid hydrogenation to value-added 1,4-pentanediol using microwave-assisted gold catalysis. J. Catal. 2019, 380, 267–277. [Google Scholar] [CrossRef]
- Grillo, G.; Cintas, P.; Colia, M.; Calcio Gaudino, E.; Cravotto, G. Process intensification in continuous flow organic synthesis with enabling and hybrid technologies. Front. Chem. Eng. 2022, 4, 966541. [Google Scholar] [CrossRef]
- Martina, K.; Cravotto, G.; Varma, R.S. Impact of microwaves on organic synthesis and strategies toward flow processes and scaling up. J. Org. Chem. 2021, 86, 13857–13872. [Google Scholar] [CrossRef]
- Rinaldi, L.; Carnaroglio, D.; Rotolo, L.; Cravotto, G. A microwave-based chemical factory in the lab: From milligram to multigram preparations. J. Chem. 2015, 2015, 879531. [Google Scholar] [CrossRef]
- Tagliapietra, S.; Calcio Gaudino, E.; Martina, K.; Barge, A.; Cravotto, G. Microwave irradiation in micro-meso-fluidic systems; hybrid technology has issued the challenge. Chem. Rec. 2018, 18, 98–117. [Google Scholar] [CrossRef] [PubMed]
- Choedkiatsakul, I.; Ngaosuwan, K.; Assabumrungrat, S.; Mantegna, S.; Cravotto, G. Biodiesel production in a novel continuous flow microwave reactor. Renew. Energy 2015, 83, 25–29. [Google Scholar] [CrossRef]
- Quality Considerations for Continuous Manufacturing. Guidance for Industry. Available online: https://www.fda.gov/media/121314/download (accessed on 7 February 2023).
- ICH Guideline Q13 on Continuous Manufacturing of Drug Substances and Drug Products. Available online: https://wwwema.europe.eu/en/ich-guideline-q13-continuous-manufacturing-drug-substances-drug-products (accessed on 7 February 2023).
- Comito, M.; Monguzzi, R.; Tagliapietra, S.; Palmisano, G.; Cravotto, G. Cefonicid benzathine salt: A convenient, lean, and high-performance protocol to make an old cephalosporin shine. Antibiotics 2022, 11, 1095. [Google Scholar] [CrossRef] [PubMed]
- Comito, M.; Monguzzi, R.; Tagliapietra, S.; Palmisano, G.; Cravotto, G. Efficient pilot-scale synthesis of the key cefonicid intermediate at room temperature. Green Process. Synth. 2022, 11, 96–105. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).